

Abstract
Aberrant forms of the anaplastic lymphoma kinase (ALK) have been implicated in the pathogenesis of multiple human cancers, where ALK represents a rational therapeutic target in these settings. In this study, we report the identification and biological characterization of X-376 and X-396, two potent and highly specific ALK small molecule tyrosine kinase inhibitors (TKIs). In Ambit kinome screens, cell growth inhibition studies, and surrogate kinase assays, X-376 and X-396 were more potent inhibitors of ALK but less potent inhibitors of MET compared to PF-02341066 (PF-1066), an ALK/MET dual TKI currently in clinical trials. Both X-376 and X-396 displayed potent anti-tumor activity in vivo with favorable pharmacokinetic and toxicity profiles. Similar levels of drug sensitivity were displayed by the three most common ALK fusion proteins in lung cancer (EML4-ALK variants E13;A20, E20;A20, and E6b;A20) as well as a KIF5B-ALK fusion protein. Moreover, X-396 could potently inhibit ALK kinases engineered with two point mutations associated with acquired resistance to PF-1066, L1196M and C1156Y, when engineered into an E13;A20 fusion variant. Lastly, X-396 displayed synergistic growth inhibitory activity when combined with the mTOR inhibitor rapamycin. Our findings offer preclinical proof-of-concept for use of these novel agents to improve therapeutic outcomes of patients with mutant ALK-driven malignancies.
Keywords: ALK, ALK fusions, tyrosine kinase inhibitor, cancer, non-small cell lung cancer, rapamycin, targeted therapeutics, acquired resistance, gatekeeper mutation, PF-1066, X-376, X-396
Introduction
The anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase that is aberrant in a variety of malignancies. ALK was originally discovered in anaplastic large cell lymphoma (ALCL) as part of a chromosomal translocation t(2,5), which fuses the C-terminal kinase domain of ALK encoded on chromosome 2p23 to the N-terminus of nucleophosmin (NPM) on chromosome 5q35 (1). Subsequently, a variety of ALK fusion proteins have been found in multiple malignancies, including inflammatory myofibroblastic tumor (IMT) (2) and non-small cell lung cancer (NSCLC) (3-10). All ALK fusions tested biologically to date have demonstrated gain of function properties (1, 3, 4, 7). Activating mutations in wild-type ALK have also been identified in both familial and sporadic neuroblastoma. Most of these activating mutations occur within the tyrosine kinase domain and are transforming in vitro and in vivo (11-14).
Importantly, the activity of cancer-specific ALK variants is required for tumor maintenance. Thus, ALK mutants can serve as ‘Achilles heels’ to be exploited therapeutically. Multiple preclinical studies have shown that specific small molecule ALK tyrosine kinase inhibitors (TKIs) can delay tumor growth and/or induce tumor regression in xenograft and transgenic models (3-5, 15-19).
Based on such promising pre-clinical studies, ALK inhibitors have recently entered into clinical trials. The first agent in humans is PF-02341066 (crizotinib, Pfizer; hereafter referred to as ‘PF-1066′), an orally available small molecule ATP-mimetic compound. PF-1066 was originally designed as a MET inhibitor but was recognized to have ‘off-target’ anti-ALK activity (17). Strikingly, in a phase I study, patients with ALK fusion positive NSCLC demonstrated a 57% radiographic response rate (20). By contrast, chemotherapy response rates are <10% in previously treated patients with unselected NSCLC (21). A Phase III trial randomizing patients to crizotinib (PF-1066) vs. standard chemotherapy after disease progression on first-line treatment is now ongoing for patients with ALK fusion positive NSCLC.
Here, we report identification of X-376 and X-396, novel, more potent and specific ALK TKIs with potential therapeutic relevance. We compare the effectiveness of these ‘second-generation’ TKIs versus PF-1066 both in vitro and in vivo. We demonstrate the anti-tumor activity of these compounds against multiple ALK variants found in NSCLC, including two point mutations in the ALK tyrosine kinase domain which have been associated with acquired resistance to PF-1066. Finally, we show that these ALK TKIs display synergistic anti-tumor activity when combined with the mTOR inhibitor, rapamycin.
Materials and Methods
Compounds
X-376 and X-396 were synthesized according to procedures published in WO 2009/154769 and dissolved in DMSO. PF-1066 (ChemieTek, Indianapolis, IN) and TAE-684 (Selleck Chemicals, Houston, TX) were dissolved in DMSO. Rapamycin (cat # 9904, Cell Signaling, Danvers, MA) was dissolved in methanol. Ambit KINOMEscans (Ambit Biosciences, San Diego, CA) were performed for each ALK TKI against 96 distinct kinase targets using methods previously described (22, 23). Compounds (PF-1066, X-376, X-396) were formulated in 0.5% HPMC, 0.4% Tween80, 99.1% DI water for all in vivo studies (PK, efficacy, toxicity).
Computational Modeling
A model of X-376 in ALK was generated using the DS ViewerPro 5.0 program from Accelrys Inc. (San Diego, CA). The X-ray crystal structure of PF-1066 in the MET kinase domain (PDB code: 2WGJ) was used as a starting point.
Cell culture
All cell lines were maintained in a humidified incubator 5% CO2 at 37°C. The human lung adenocarcinoma cell lines H3122, H2228, PC-9, H2030, and HCC78 have been described previously (5, 24-26) and were verified to harbor their reported genetic alterations by direct sequencing of DNA or cDNA. The human anaplastic lymphoma cell line was a generous gift from Dr. S. Morris of St. Jude Children's Research Hospital (1). The human gastric carcinoma cell line, MKN-45, and the human hepatocellular carcinoma cell line, HepG2, have been described previously (27, 28). These cell lines were maintained in RPMI 1640 medium (Mediatech, Inc., Manassas, VA) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO) and penicillin-streptomycin (Mediatech, Inc., Manassas, VA) to final concentrations 100 U/ml and 100 μg/ml, respectively. The human neuroblastoma cell line SH-SY5Y (ATCC, Manassas, VA) was grown in a 1:1 mixture of EMEM and F12 (both from Mediatech, Inc., Manassas, VA) supplemented with 10% FBS and pen-strep. The human hepatocellular carcinoma cell line HepG2 (ATCC, Manassas, VA) was grown in EMEM supplemented with 10% FBS and pen-strep. 293 cells (ATCC, Manassas, VA) were cultivated in DMEM (Mediatech, Manassas, VA) supplemented with 10% FBS. 293s were transfected with various ALK cDNAs using Superfect reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions.
Ba/F3 cell line generation
EML4-ALK E13;A20 was cloned from pMA-3FLAG-EML4-ALK E13;A20 into the retroviral pBabe-puro backbone. The L1196M and C1156Y mutations were introduced into the cDNA encoding the E13;A20 variant using site-directed mutagenesis (Stratagene, La Jolla, CA) with mutant specific primers according to the manufacturer's instructions. Ba/F3 cell lines were established as described previously using purified IL-3 at 10ng/ml and puromycin at 1.5μg/mL (29). Transgene expression was assessed by Sanger sequencing and immunoblotting.
Antibodies and immunoblotting
The following antibodies were obtained from Cell Signaling Technology (Danvers, MA): ALK (cat # 3333), phospho-ALK tyrosine 1604 (cat # 3341), phospho-ALK tyrosine 1278/1282/1283 (cat # 3983), ribosomal protein S6 (cat # 2317), phospho-S6 (cat # 5364), phospho-ERK tyrosine 202/204 (cat # 9101), ERK (cat # 9102), phospho-AKT serine 473 (cat # 9271), AKT (cat # 9272), HRP-conjugated anti-mouse (cat # 7076), and HRP-conjugated anti-rabbit (cat # 7074). The Flag M2 antibody (cat # F1804) and the actin antibody (cat # A2066) were purchased from Sigma-Aldrich (St. Louis, MO).
For immunoblotting, cells were harvested, washed in PBS, and lysed in 50 mM Tris·HCl, pH 8.0/150 mM sodium chloride/5 mM magnesium chloride/1% Triton X-100/0.5% sodium deoxycholate/0.1% SDS/40 mM sodium fluoride/1 mM sodium orthovanadate and complete protease inhibitors (Roche Diagnostics, Indianapolis, IN). Lysates were subjected to SDS-PAGE followed by blotting with the indicated antibodies and detection by Western Lightening ECL reagent (PerkinElmer, Waltham, MA). Images were quantified using a Bio-Rad Gel Doc XR and Image Lab software (Bio-Rad, Hercules, CA).
Cell Viability and apoptosis assay
For viability experiments, cells were seeded in 96-well plates at 25%-33% confluency and exposed to drugs alone or in combination the following day. At 72 hours post drug addition, Cell Titer Blue Reagent (Promega, Madison, WI) was added and fluorescence was measured on a Spectramax spectrophotometer (Molecular Devices, Sunnyvale, CA) according to the manufacturer's instructions. All experimental points were set up in hextuplicate replicates and were performed at least two independent times. IC50's were calculated using GraphPad Prism version 5 for Windows. The curves were fit using a nonlinear regression model with a log (inhibitor) vs. response formula.
For apoptosis experiments, cells were seeded in 12 well plates at 25% confluency and treated in triplicate with ALK TKI. At 72h post drug addition, cells were collected, washed in PBS, and stained with annexin V and propidium iodide according to the manufacturer's instructions (Vybrant Apoptosis Assay Kit, Invitrogen, Carlsbad, CA). Data were collected on a FACSCanto II (BD Biosciences, San Jose, CA) and processed using WinList flow cytometry analysis software (Verity Software, Topsham, ME).
Expression constructs
cDNAs for EML4-ALK E13;A20 (variant 1), EML4-ALK E20;A20 (variant 2), EML4-ALK E6b;A20 (variant 3b), and KIF5B-ALK were synthesized by Geneart (Regensburg, Germany). The cDNAs were subcloned into a 3Flag-CMV vector (Sigma-Aldrich, St. Louis, MO). The L1196M and C1156Y mutations were introduced into the cDNA encoding the E13;A20 variant using site-directed mutagenesis (Stratagene, La Jolla, CA) with mutant specific primers according to the manufacturer's instructions. The cDNAs were fully re-sequenced in each case to ensure that no additional mutations were introduced. A cDNA encoding MET was obtained from Origene (Rockville, MD).
Xenograft studies
Nude mice (nu/nu; Harlan Laboratories) were used for in vivo studies and were cared for in accordance with guidelines approved by the Memorial Sloan-Kettering Cancer Center Institutional Animal Care and Use Committee and Research Animal Resource Center. 8 week old female mice were injected subcutaneously with 5 million H3122 cells together with matrigel. Once tumors reached an average volume of 290mm3, mice were randomized and dosed via oral gavage with either PF-1066, X-376, or X-396 at the indicated doses. A uniform volume for administration (200μl) was used for each group. Mice were observed daily throughout the treatment period for signs of morbidity/mortality. Tumors were measured twice weekly using calipers, and volume was calculated using the formula: length × width2 × 0.52. Body weight was also assessed twice weekly. The experiment was terminated after 3 weeks of treatment. Tumor samples were collected at either 2 or 8 hours after the last treatment. Each sample was cut in halves; half was preserved in 10% neutral buffered formalin and half was flash frozen in liquid nitrogen and stored at -80°C.
Pharmacokinetic and toxicity studies
Nude mice (nu/nu; Harlan Laboratories) were injected with H3122 cells as above. Once tumors reached an average volume of 450 mm3, a total of 27 athymic mice harboring H3122 tumors were randomized and dosed via oral gavage with either 50mg/kg X-376, 25mg/kg X-396, or the control vehicle. Two, five, and fifteen hours after the single treatment (3 tumors/timepoint/group), mice were sacrificed and serum was collected for assessment of drug concentration using an LC-MS based bioanalytical method by the DMPK group of the Scripps Research Institute (Jupiter, Florida).
To determine if PF-1066, X-376 and X-396 can cross the blood-brain-barrier (BBB), each compound was dosed to three male C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) at their respective efficacious doses (PF-1066: 50mg/kg, X-376 50mg/kg, and X-396 25mg/kg). Two hours after dosing, mice were sacrificed. Blood and brain tissues were collected and analyzed for drug concentration. The bioanalysis was done at the Scripps Research Institute (Jupiter, Florida) as described above.
For toxicity studies, the test article or vehicle was administered to all groups orally once a day for 10 consecutive days at a dose volume of 10ml/kg. Observations for morbidity, mortality, injury, and the availability of food and water were conducted twice daily for all animals. Clinical observations were conducted twice daily. Body weights were measured and recorded every other day during the study. Food consumption was measured and recorded daily during the study. Blood samples for clinical pathology evaluations were collected from all animals on day 11. Blood samples from an additional two animals/sex/group (minus vehicle group) were collected for determination of test article plasma concentrations at designated time points on day 10. The toxico-kinetic parameters were determined for the test articles from concentration-time data in the test species. At study termination, necropsy examinations were performed, organ weights were recorded, and selected tissues were microscopically examined. Studies were done in accordance with the current International Conference on Harmonization (ICH) harmonized tripartite guidelines, and the protocol was approved by the Medicilion Institutional Animal Care and Use Committee (Shanghai, China).
Results
X-376 and X-396 are aminopyridazine-based kinase inhibitors with increased in vitro potency against ALK compared to PF-1066
Based upon protein structure modeling of ALK, we designed a series of small molecule inhibitors that we hypothesized would have greater potency and specificity against ALK. While PF-1066 is an aminopyridine-based small molecule inhibitor, the lead compounds we identified, X-376 and X-396, are aminopyridazine-based structures. These agents all share a common hydrophobic 2,6-dichloro-3-fluoro-phenylethoxy group as well as a similar kinase hinge binding group compared to PF-1066, but differ significantly in their side chains (Fig. 1A). We next performed KINOMEscan (Ambit Biosciences) analysis to test the specificity of the ALK TKIs. Briefly, KINOMEscan is a competition binding assay that quantitatively measures the ability of a test compound to compete with an immobilized, active-site directed ligand (22, 23). Initial KINOMEscan screens on racemic forms of PF-1066 and X-376 showed different but overlapping binding constant specificity profiles (Fig. 1B). Table S1 summarizes the differential ability of these agents to inhibit the activity of 96 kinases; the vast majority of kinases are minimally affected. Additional Ambit-based dose-response profiling suggested that X-376 and X-396 were approximately 10-fold more potent against ALK but less specific for MET compared to PF-1066 (Table 1) (17).
Figure 1. Structure and specificity of ALK inhibitors.
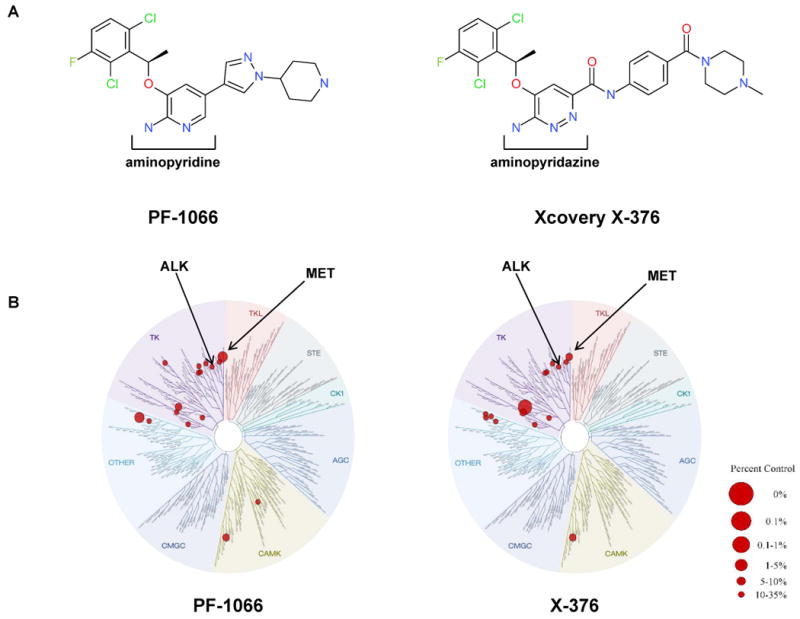
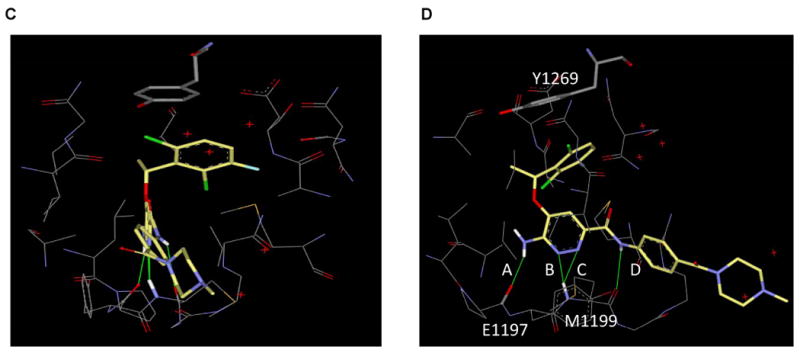
(A) Chemical structure of PF-1066 compared to X-376. (B) Kinase dendrograms for PF-1066 and X-376. Note that racemic mixtures were used in these specific assays. The size of the red circle is proportional to the degree of inhibition as depicted in the figure legend. Numerical data as well as IC50 values for X-376 against specific kinases are listed in Supplemental Table 1. (C) A model of X-376 in the ATP binding site of the ALK kinase domain (only residues within 5Å of X-376 are shown). (D) Rotated view highlighting hydrogen bonds between X-376 and amide peptides of hinge residues (E1197 and M1199). In addition to hydrogen bonds A and B, which are also present in the PF-1066/ALK complex, X-376 can form two more hydrogen bonds, C and D, with the receptor, thereby further increasing its binding affinity.
Table 1. In vitro potency and selectivity of ALK TKIs.
The degree of inhibition from Kinomescan is shown for each protein.
| Biochemical Potency and Selectivity (Ambit assays, IC50 in nM) | ||
|---|---|---|
| Compound | ALK | MET |
| PF-1066 | 4.5 | 0.51 |
| X-376 | 0.61 | 0.69 |
| X-396 | <0.4 | 0.74 |
To attempt to explain the increased potency for X-376 and X-396 against ALK on a structural level, we generated a model of X-376 within the ATP binding pocket of the ALK tyrosine kinase domain (Fig. 1C and 1D). This model was based upon the crystal structure of PF-1066 in the MET kinase domain (PDB code: 2WGJ). The hydrophobic interaction, especially the aromatic stacking, between Y1269 and the dichloroflorophenyl moiety, is conserved between PF-1066 and X-376 (Fig. 1D). However, for hydrogen bonds with hinge residues (E1197 and M1199), X-376 may be able to form two additional hydrogen bonds (C and D) compared to PF-1066. The hydrogen bond distances are 2.08Å, 1.95Å, 2.45Å, and 2.66Å for A, B, C, and D, respectively, in this model. Although the hydrogen bonds C and D are weaker than A and B, they may still contribute to the binding of X-376, thereby further increasing this compound's affinity for ALK.
X-376 and X-396 are more potent ALK inhibitors than PF-1066
To compare and contrast the in vitro effects of X-376 and X-396 versus PF-1066, we tested the ability of all three agents to inhibit the growth of four different cancer cell lines known to harbor ALK fusions or point mutations. In H3122 lung cancer cells harboring EML4-ALK E13;A20 (variant 1), X-376 and X-396 were 3-fold and 10-fold more potent, respectively, than PF-1066 (IC50: PF-1066 180nM, X-376 77nM, X-396 15nM) (Fig. 2A and Table 2). Similar results were obtained with H2228 lung cancer cells (Fig. 2B), SUDHL-1 lymphoma cells (Fig. 2C), and SY5Y neuroblastoma cells (Fig. 2D), which harbor an EML4-ALK E6a/b;A20 (variant 3a/b) fusion, an NPM-ALK fusion, and an activating point mutation within the ALK kinase domain (F1174L) (12), respectively. In H3122 cells, the relative decrease in cell growth seen with X-376 treatment correlated with increased apoptosis, as assessed by fluorescence-activated cell sorting for Annexin V and propidium iodide (Fig. 2E).
Figure 2. Potency of X-376 and X-396 in ALK mutant cell lines.

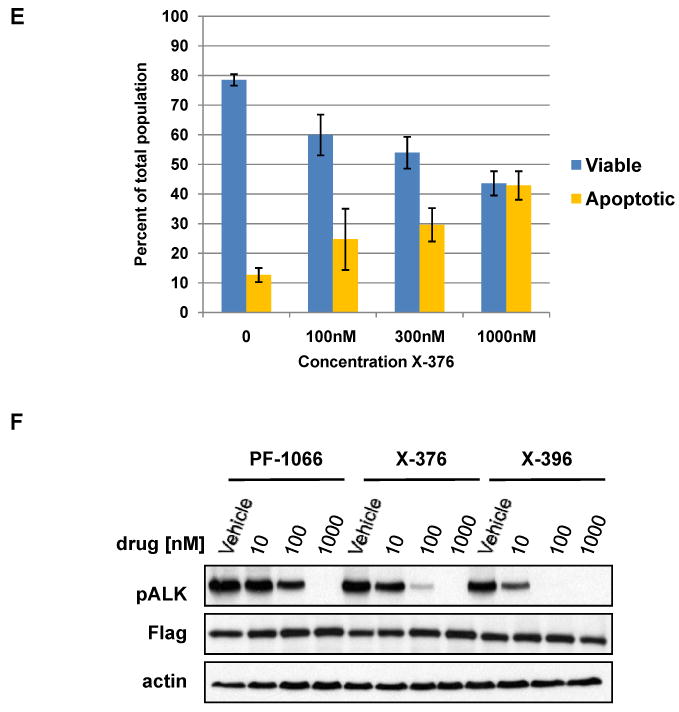
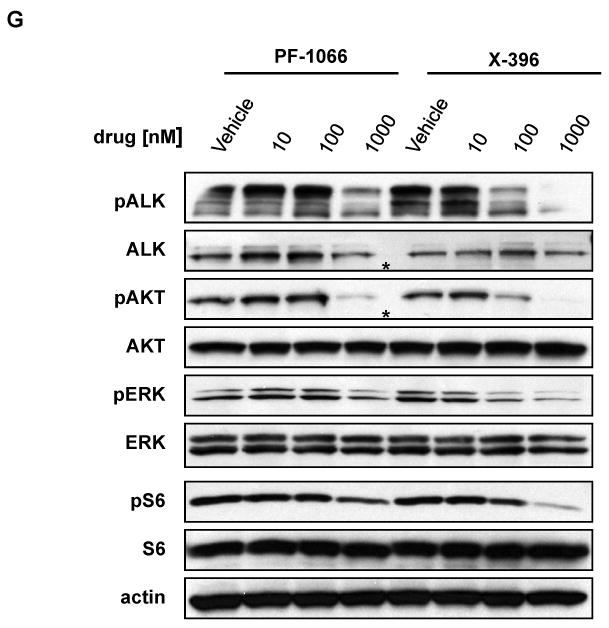
(A-D) H3122 lung cancer cells containing the EML4-ALK E13;A20 fusion, H2228 lung cancer cells harboring the EML4-ALK E6a/b;A20 fusion, SUDHL-1 lymphoma cells containing NPM-ALK fusion, and SY5Y neuroblastoma cells with an activating mutation within the ALK kinase domain (ALK F1174L) were treated with ALK TKIs or vehicle for 72h. Cell titer blue assays were performed to assess growth inhibition. Each point represents hextuplicate replicates. Data are presented as the percentage of viable cells compared to control (vehicle only treated) cells. See methods for details. (E) Apoptosis is induced by X-376 treatment. H3122 cells were treated with increasing concentrations of X-376 for 72h. Cells were stained with annexin V (AV) and propidium iodide (PI) and counted on a FACSCanto II machine. Viable cells are defined as the AV/PI double negative population. Apoptotic cells are defined as the sum of AV positive, PI negative plus AV/PI double positive cell populations. (F) 293 cells were transiently transfected with 3Flag-EML4-ALK E13;A20. At 48 hours post transfection, the cells were treated with increasing amounts of the ALK TKIs for 2 hours. Lysates were subjected to immunoblotting with antibodies specific for the indicated proteins. (G) H3122 lung cancer cells containing the EML4-ALK E13;A20 fusion were treated with increasing amounts of ALK TKIs for 1 hour as indicated. Lysates were subjected to immunoblotting with the specified antibodies. The asterisks (*) in the ALK and phospho-AKT blot indicate an empty lane (no lysate loaded) on the gel.
Table 2. IC50 values for PF-1066, X-376, and X-396 in a panel of cancer cell lines.
Cell lines were treated with ALK TKIs for 72h. Cell titer blue assays were performed to assess cell proliferation. IC50 values were calculated using GraphPad Prism software with a non-linear regression. Each experiment was performed at least 2 times. NT: not tested.
| Cell Line | Origin | IC50 (μM) | ||
|---|---|---|---|---|
| PF-1066 | X-376 | X-396 | ||
| H3122 | NSCLC, adenocarcinoma (EML4-ALK E13;A20) |
0.180 | 0.077 | 0.015 |
| H2228 | NSCLC, adenocarcinoma (EML4-ALK E6a/b; A20) |
0.150 | 0.057 | 0.045 |
| SUDHL-1 | ALCL (NPM-ALK) |
0.073 | 0.032 | 0.009 |
| SY5Y | Neuroblastoma (ALK F1174L) |
0.338 | 0.142 | 0.068 |
| MKN-45 | Gastric adenocarcinoma (MET dependent) |
0.051 | 0.150 | 0.156 |
| HepG2 | Hepatocellular carcinoma | 14.502 | 15.137 | 9.644 |
| H2030 | NSCLC, adenocarcinoma (KRAS G12C) |
2.474 | 1.427 | 0.757 |
| PC-9 | NSCLC, adenocarcinoma (EGFR exon 19 del) |
1.670 | 3.062 | 2.989 |
| HCC78 | NSCLC, adenocarcinoma (SLC34A2-ROS fusion) |
1.355 | >3 | >3 |
| Ba/F3 | + EML4-ALK E13;A20 WT | 0.250 | NT | 0.022 |
| + EML4-ALK E13;A20 L1196M | 1.815 | NT | 0.106 | |
| + EML4-ALK E13;A20 C1156Y | 0.458 | NT | 0.048 | |
By contrast, the ALK inhibitors did not significantly inhibit the growth of cells driven by other mutant kinases. For example, KRAS mutant (H2030) and EGFR mutant (PC-9) lung cancer cell lines were insensitive to treatment at low nanomolar concentrations of drug (Table 2; Figs. S1B and S1C). HCC78 cells, which contain a ROS1 fusion protein, have displayed moderate sensitivity to another ALK TKI, TAE684, which has ‘off-target’ anti-ROS activity (30). Our Ambit screen showed that the racemic forms of both PF-1066 and X-376 inhibited ROS1 in vitro (IC50: PF-1066 1.7nM; X-376: 19nM) (Table S1). However, we found HCC78 cells to be only modestly sensitive to PF-1066 (IC50 of 1.4 μM) and even less sensitive to treatment with X-376 or X-396 (IC50 of > 3μM for each drug) (Table 2; Fig. S1D. Finally, the ALK TKIs had minimal growth inhibitory effects against HepG2 (liver) cells (Table 2; Fig. S1A); this assay is often used as a measure for potential hepatotoxicity (31).
We also compared the efficacy of PF-1066 and X-396 to TAE-684 in H3122 cells. Unlike PF-1066 and X-396, TAE-684 has a diaminophenylpyrimidine scaffold and inhibits ALK, IGF-1R, and IR in biochemical assay (16). As previously reported in the literature, TAE-684 is a potent inhibitor of H3122 cell growth (5) with an IC50 of 5nM (Figs. S1E). However, this compound is not in clinical development.
To confirm the target specificity of X-376 and X-396, we assessed the ability of the compounds to inhibit ALK kinase activity using auto-phosphorylation of an ALK fusion protein as a surrogate measure. 293 cells were transiently transfected with an expression plasmid encoding flag-tagged EML4-ALK E13;A20 cDNAs and treated with PF-1066, X-376, or X-396. Immunoblotting of lysates showed that all three compounds inhibited the activity of ALK in a concentration-dependent manner. Consistent with data from the growth inhibition assays, X-376 and X-396 led to greater reductions in phospho-ALK at lower concentrations of drug (Fig. 2F).
We further evaluated the effects of PF-1066 and X-396 on endogenous ALK phosphorylation and potential downstream signaling pathways in H3122 lung cancer cells harboring the EML4-ALK E13;A20 fusion. Compared to PF-1066, X-396 inhibited ALK phosphorylation at lower concentrations of drug (Fig. 2G). This was accompanied by corresponding inhibition of the downstream targets, ERK and AKT. We also found that phosphorylation of the mTOR substrate, ribosomal protein S6, was inhibited with ALK TKI treatment (Fig. 2G). Collectively, these data show that X-376 and X-396 are more potent ALK inhibitors than PF-1066.
X-376 and X-396 display less activity against MET than PF-1066
PF-1066 has higher binding affinity for MET versus ALK in Ambit KINOMEscan assays (17). By contrast, X-376 and X-396 displayed similar binding constants for both kinases (Table 1). To assess if such differences in binding correlated with less activity against MET in cells, we determined the ability of X-376 and X-396 to inhibit the growth of MKN-45 gastric carcinoma cells, which are known to be highly sensitive to MET inhibitors (27). Compared to X-376 and X-396, PF-1066 was 3 fold more potent against MKN-45 cells (IC50s: PF-1066 51nM, X-376 150nM, X-396 156nM) (Fig. S2A and Table 2). Consistent with these data, PF-1066 more effectively reduced MET autophosphorylation in surrogate kinase assays (Fig. S2B). Thus, X-376 and X-396 are both more potent and more specific inhibitors of ALK compared to PF-1066.
Antitumor activity and pharmacokinetic properties of X-376 and X-396 in vivo
We next examined the effects of X-376 and X-396 in vivo against H3122 xenografts. A pharmacokinetic study revealed that both X-376 and X-396 showed substantial bioavailability and moderate half-lives in vivo (Table S2). Based on these data, we selected twice a day dosing for initial efficacy studies. We treated nude mice harboring H3122 xenografts with either X-376 at 50mg/kg bid or X-396 at 25mg/kg bid. Both agents significantly delayed the growth of tumors compared to vehicle alone (Fig. 3A). The efficacy was similar to that seen with PF-1066 dosed at 50mg/kg BID (Fig. S3). Additional doses and schedules of X-376 and X-396 have not yet been examined. However, based on our toxicity studies (below), we show that X-396 dosed at 25mg/kg is probably well below the maximum tolerated dose, suggesting that higher doses and greater efficacy can be achieved.
Figure 3. X-376 and X-396 effectively inhibit the growth of H3122 in vivo.

Athymic nu/nu female mice were injected subcutaneously with H3122 lung cancer cells harboring the EML4-ALK E13;A20 fusion. When tumors reached an average volume of 290mm3, mice were treated with X-376, X-396, or vehicle alone by oral gavage (n=6 per group). Tumor volumes (A) and mouse weights (B) were assessed every 2-3 days.
At the doses used in these xenograft experiments (PF-1066: 50mg/kg bid; X376: 50mg/kg bid; X-396 25mg/kg bid), plasma levels inversely correlated with cellular potency. For example, although PF-1066 and X-396 were the least and most potent ALK inhibitors, respectively, the plasma level at 2 hours after dosing was highest for PF-1066 (10.19 μM), lower for X-376 (6.27 μM), and lowest for X-396 (2.28 μM) (Table S2). The plasma levels followed the same trend at five and fifteen hours after dosing, respectively. These findings suggest that inhibitors like PF-1066 may need to be administered at higher doses in order to achieve the same efficacy of the more potent compounds.
In the xenograft experiments, both X-376 and X-396 appeared well-tolerated in vivo. Mouse weight was unaffected by either treatment (Fig. 3B). Drug-treated mice appeared healthy and did not display any signs of compound related toxicity. To further assess potential side effects of X-376 and X-396, we performed additional systemic toxicity and toxico-kinetic studies in Sprague Dawley (SD) rats. Following 10 days of repeated oral administration of X-376 at 25, 50, 100 mg/kg or X-396 at 20, 40, 80 mg/kg in SD rats, all animals survived to study termination. The no significant toxicity (NST) levels were determined to be 50 mg/kg for X-376 and 80mg/kg for X-396. The only statistically significant observation at NST levels was a <50% increase of the liver enzymes, AST and ALT. At NST levels, X-376 achieved an AUC of 41μM.hr and a Cmax of 5.04 μM, while X-396 had an AUC of 66 μM.hr and a Cmax of 7.19 μM (Table S2). These Cmax values are >50 and >400 times the drugs’ respective IC50's for H3122 cell growth inhibition. Notably, PF-1066 at the selected (tolerated) phase 3 dose results in a plasma level (∼500nM at 250mg BID) (32) that is only ∼3 times its IC50. The NST Cmax value of X-396 also compares favorably with the 2 hour plasma levels required for efficacy in mice (7.19μM vs. 2.28μM, Table S2). Taken together, these data suggest that X-396 is able to inhibit ALK effectively at doses far below those that are well tolerated.
Penetration of ALK TKIs in the brain
In EGFR mutant lung cancer, patients with acquired resistance to EGFR TKIs may relapse in the brain. One contributing factor is that the dose of drug in the brain is only one hundredth that in the blood (33). In anticipation of a similar phenomenon occurring in patients with ALK fusion positive lung cancers treated with ALK TKIs, we examined the penetration of PF-1066, X-376, and X-396 across the blood-brain-barrier (BBB) in mice. Each drug was dosed to three male C57BL/6J mice at their respective efficacious doses (PF-1066: 50mg/kg, X-376 50mg/kg, and X-396 25mg/kg). Two hours after dosing, mice were sacrificed. Blood and brain tissues were collected and analyzed for drug concentration. Brain penetration was defined as the ratio of compound concentration in brain tissues compared to that in plasma. We found that X-396 had comparable brain penetration to PF-1066, while X-376 had less (Table S3). Since PF-1066 at the selected Phase 3 dose achieves a plasma level of ∼500nM (32), projection of results from this animal study to humans would suggest that the concentration of PF-1066 in human brain would be only ∼80nM (16% of plasma level), which is below the IC50 (180nM) for inhibiting H3122 cell growth (Table S4). By contrast, if X-396 were also to achieve a plasma level of ∼500nM in humans, its brain concentration would be about 65nM (13% of plasma level), which is significantly higher than its IC50 of 15nM (Table S4). These data suggest that drugs like X-396 could lead to higher efficacy against ALK-fusion positive brain metastases. However, further studies with additional sampling times will be required to most accurately assess intracranial drug levels.
X-376 and X-396 are effective against multiple ALK variants found in NSCLC, including ALK mutations associated with acquired resistance to PF-1066
To date, at least 11 different ALK rearrangements have been reported in NSCLC (3-10). While all contain the ALK kinase domain starting at the region encoded by exon 20, most contain variable portions of EML4 at the N-terminus. Another fusion involves KIF5B (7). Whether these variants display differential sensitivity to ALK TKIs is unknown but could influence the outcome of clinical trials with ALK inhibitors. We therefore compared the efficacy of X-376 against 4 different ALK variants in surrogate kinase assays. 293 cells were transiently transfected with cDNAs encoding Flag-tagged versions of four ALK rearrangements: three of the most common EML4-ALK variants, EML4-ALK E13;A20 (variant 1), EML4-ALK E20;A20 (variant 2), EML4-ALK E6b;A20 (variant 3b) (6, 34), and KIF5B-ALK. After treatment with X-376, cell extracts were subjected to immunoblotting with antibodies against phospho-ALK and Flag. All fusions displayed dose-dependent inhibition of ALK kinase activity (Fig. 4A). Based upon quantitative examination of the immunoblotting results (data not shown), no major differences in sensitivity were observed among the four variants in these in vitro surrogate-kinase assays. However, additional studies in cell lines where these ALK variants are the oncogenic ‘drivers’ are needed to establish more subtle differences.
Figure 4. Effects of ALK TKIs against various ALK fusion variants and the EML4-ALK L1196M and C1156Y point mutations.
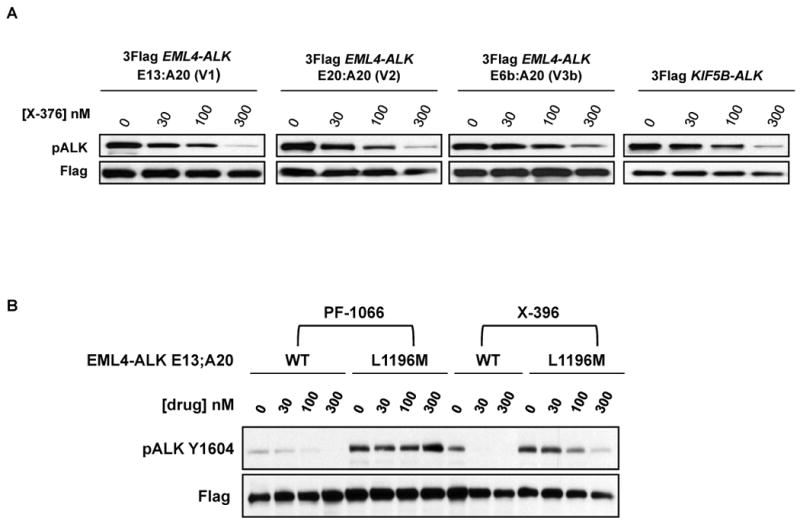
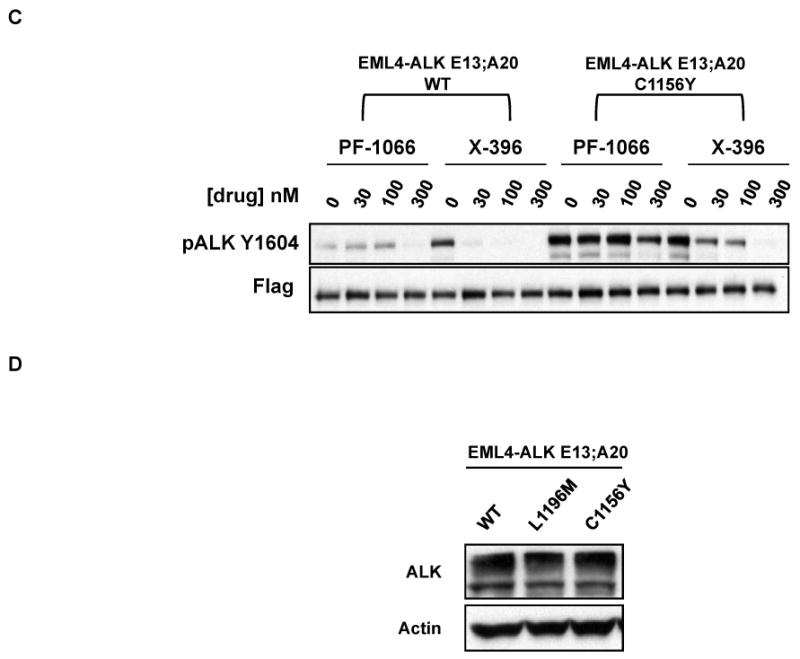

(A) 293 cells were transiently transfected with expression plasmids encoding various ALK fusions. At 48 hours post transfection, the cells were treated with increasing amounts of X-376 for 2 hours. Lysates were subjected to immunoblotting with antibodies specific for the indicated proteins. (B-C) 293 cells were transiently transfected with 3Flag-EML4-ALK E13;A20 WT alongside 3Flag-EML4-ALK E13;A20 L1196M (B) or 3Flag-EML4-ALK E13;A20 C1156Y (C). At 48 hours post transfection, the cells were treated with increasing amounts of the indicated ALK TKI for 2 hours. Lysates were subjected to immunoblotting with antibodies specific for the indicated proteins. The exposure of the pALK blot was selected to highlight the difference in baseline phosphorylation between wild-type and mutants. (D) Western blot demonstrating EML4-ALK E13;A20 WT, L1196M, and C1156Y expression in Ba/F3 cell lines. (E) Ba/F3 cells expressing EML4-ALK E13;A20 WT, L1196M, or C1156Y were treated with ALK TKIs or vehicle for 72h. Cell titer blue assays were performed to assess growth inhibition. Each point represents hextuplicate replicates. Data are presented as the percentage of viable cells compared to control (vehicle only treated) cells. See methods for details.
We next examined the efficacy of X-396 against two point mutations in EML4-ALK recently identified in patients with acquired resistance to PF-1066, L1196M and C1156Y (35). L1196 of ALK corresponds to the “gatekeeper” site, and C1156 is positioned adjacent to the N-terminal helix αC (35). To determine how the L1196M and C1156Y mutations affect ALK fusion proteins, we individually introduced the specific mutations into EML4-ALK E13;A20 cDNAs and performed surrogate kinase assays as described above. Introduction of L1196M and C1156Y into the EML4-ALK fusion protein led to a greater baseline level of phosphorylation (Figs. 4B and 4C), suggesting that these mutations lead to increased kinase activity. As expected, the L1196M and C1156Y mutants did reduce the sensitivity of the wildtype ALK fusion protein to both PF-1066 and X-396, however, the degree of reduction was less for X-396.
To expand on these results from the surrogate kinase assays, we evaluated the biological impact of each mutation on drug sensitivity in Ba/F3 cells. Expression of EML4-ALK E13;A20 WT, L1196M, and C1156Y (Fig. 4E) each led to IL3 independent growth of Ba/F3 cells. In accord with our data above from tumor cell lines with ALK genomic alterations, X-396 was ∼10 fold more potent than PF-1066 in inhibiting the growth of Ba/F3 cells expressing EML4-ALK E13;A20 wild-type (IC50 for WT: PF-1066 250nM, X-396 22nM, Fig. 4E, Table 2). As expected, the presence of the L1196M and C1156Y point mutants reduced the sensitivity of the EML4-ALK fusion to both PF-1066 and X-396 (Fig. 4E). However, the IC50 values for X-396 against the L1196M and C1156Y mutants were still in the ∼100nM or less range (IC50 for L1196M: PF-1066 1815nM, X-396 106nM; IC50 for C1156Y: PF-1066 458nM, X-396 48nM, Fig. 4E, Table 2). Since the therapeutic window is greater for X-396, these data suggest that X-396 has the potential to overcome these second-site drug resistance mutants.
Combined ALK/mTOR inhibition enhances growth inhibition of ALK fusion positive lung cancer cell lines
As shown above, treatment of H3122 cells with ALK TKIs led to a decrease in phosphorylation of the mTOR substrate, ribosomal protein S6 (Fig. 2G). Previous studies in NPM-ALK positive lymphoma cell lines have shown that the mTOR inhibitor, rapamycin, decreases proliferation and increases apoptosis in ALK fusion positive lymphoma cells (36). Given these two observations, we sought to determine if mTOR inhibition could synergize with ALK inhibition in EML4-ALK fusion positive NSCLC cell lines. Rapamycin alone caused a small but reproducible decrease in the proliferation of H3122 and H2228 cells (Fig. S4). Strikingly, when rapamycin was combined with X-396, we observed a synergistic decrease of cell growth in both H3122 (Fig. 5A) and in H2228 (Fig. 5B) cells. Drug synergism was assessed by both the Chou-Talalay and Mix-Low Methods (Table S5 and Fig. S5) (37, 38). Consistent with these data, phosphorylation of the mTOR substrate, S6, was decreased in the presence of rapamycin and was further diminished in the presence of both rapamycin and X-396 (Fig.5C). Overall, these data suggest that co-treatment with ALK and mTOR inhibitors may represent a strategy to increase the therapeutic efficacy for patients with ALK fusion positive lung cancer.
Figure 5. The mTOR inhibitor, rapamycin, is synergistic with X-396.
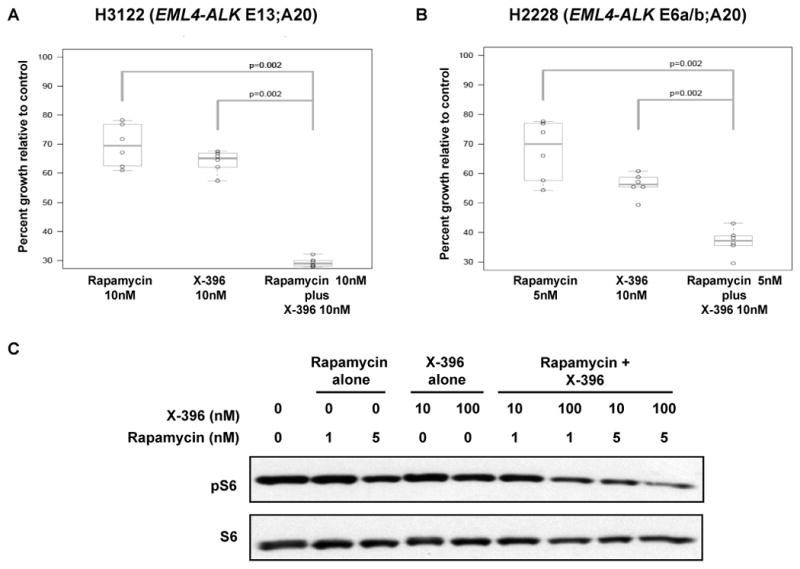
H3122 (A) and H2228 (B) were treated with rapamcyin alone, X-396 alone, rapamycin plus X-396 for 72 hours. Cell titer blue assays were performed to assess proliferation as per methods. Data are presented as the percentage of viable cells compared to control (vehicle only treated) cells. p values were calculated using the Wilcoxon rank-sum test. (C) H3122 cells were treated with rapamcyin alone, X-396 alone, or rapamycin plus X-396 at the indicated concentrations for 1 hour. Lysates were subjected to immunoblotting with antibodies specific for the indicated proteins.
Discussion
The advent of targeted therapies for specific cohorts of tumors defined by molecular criteria holds promise for improving therapeutic outcomes of cancer patients. Mutant ALK represents one clinically relevant molecular marker, found in a diverse spectrum of malignancies, including ALCL, NSCLC, IMT, and neuroblastoma. Constitutive activation of ALK, either through oncogenic fusions or point mutation, not only initiates tumorigenesis in these cancers but also renders them sensitive to treatment with ALK small molecule inhibitors. The first ALK inhibitor in clinical trials, PF-1066, has demonstrated promising results in NSCLCs harboring ALK fusions (20). However, despite displaying impressive radiographic regressions, patients have already demonstrated disease progression on drug. Thus, novel agents and drug combinations need to be developed to optimize the care of patients with tumors harboring ALK genomic alterations.
To this end, we report here identification and characterization of X-376 and X-396, two potent and highly specific ALK small molecule TKIs. In multiple different experiments, including Ambit kinome screens, in vitro growth inhibition studies using oncogene-dependent cancer cell lines, and surrogate kinase assays, X-376 and X-396 were more potent inhibitors of ALK and less potent inhibitors of MET compared to PF-1066. Both X-376 and X-396 also demonstrated potent anti-tumor activity in vivo with favorable pharmacokinetic and toxicity profiles and with broader therapeutic windows than PF-1066. The improved specificities of X-376 and X-396 could translate into improved anti-tumor responses as well as potentially decreased adverse drug reactions for patients treated with these agents.
Multiple ALK fusion variants have been described in NSCLC (3-10). The majority are comprised of EML4-ALK fusions. Previous work has shown that cells harboring the EML4-ALK E13;A20 fusion variant are highly sensitive to a small molecule ALK inhibitor (TAE684), whereas cells harboring the EML4-ALK E6a/b;A20 variant are less so (5). These observations raise the question as to whether different ALK fusion proteins demonstrate differential sensitivity to ALK TKIs, which could impact outcomes in clinical trials of patients with ALK fusion positive lung cancer. Here, using surrogate kinase assays on lysates from cells ectopically expressing three of the most common EML4-ALK variants (EML4-ALK E13;A20, EML4-ALK E20;A20, and EML4-ALK E6b;A20) (6, 34) as well as the KIF5B-ALK fusion, we did not find any major differences in sensitivity to X-376. These data suggest that current approaches such as fluorescent in situ hybridization for ALK fusions to identify patients eligible for treatment with ALK TKIs are reasonable even though such techniques do not distinguish among different variants. Further correlations among the different variants and clinical outcomes await detailed analysis of ongoing clinical trials.
Despite promising initial results with ALK TKIs, patients whose disease initially responded to therapy have already developed progressive disease. Mechanisms of “acquired resistance” to ALK TKIs are just beginning to be elucidated. Recently, Choi and colleagues reported the finding of L1196M and C1156Y point mutations in a patient with lung cancer who developed acquired resistance to PF-1066 (35). L1196, corresponds to the “gatekeeper” site in ALK. Mutations at this site alter drug binding (39) and are commonly found in tumor cells from cancer patients with acquired resistance to kinase inhibitors. C1156 is positioned adjacent to the N-terminal helix αC (35). The structural mechanism for how mutation at this site confers resistance to ALK TKIs remains to be elucidated. When the ALK L1196M and C1156Y mutations were introduced into the EML4-ALK E13;A20 fusion, kinase activity, as assessed by ALK autophosphorylation, was not inhibited by up to 300nM of PF-1066. By contrast, ALK autophosphorylation was significantly diminished by X-396, albeit at higher concentrations required to block autophosphorylation of the wild-type fusion. These results were recapitulated in Ba/F3 cells where introduction of the L1196M and C1156Y point mutants reduced the sensitivity of cells to both PF-1066 and X-396, however, the IC50 values for X-396 against these mutants were still ∼100nM or less.
Since both of these point mutations have been found in clinical samples from patients with acquired resistance to ALK TKIs, treatment with X-396 may represent one strategy to overcome them. In addition, our brain drug penetration studies suggest that X-396 can achieve high enough concentrations to treat intra-cerebral metastases effectively should they arise in patients.
Finally, an emerging paradigm in the design and implementation of targeted agents is the combination of such agents to improve therapeutic efficacy. The serine-threonine protein kinase, mTOR, plays an important role in protein translation and cell growth, and has been implicated as a therapeutic target in a variety of malignancies. Previous studies in NPM-ALK positive lymphoma cell lines have shown that the mTOR inhibitor, rapamycin, decreases proliferation and increases apoptosis in ALK fusion positive lymphoma cells (36). Interestingly, recent studies performed in an EML4-ALK mouse model showed only modest anti-tumor efficacy with combined PI3K/mTOR and MEK inhibition (40). However, the combination of an ALK inhibitor plus an mTOR inhibitor was not evaluated. Our results demonstrate that this combination can synergistically decrease cell proliferation in two different EML4-ALK lung cancer cell lines. These data indicate that mTOR signaling may play critical roles in ALK fusion signaling in general, which is currently not well characterized. Clinically, these data also suggest that trials of ALK TKIs in combination with rapamycin or analogous agents should be considered to enhance the antitumor efficacy of ALK inhibitors.
Supplementary Material
Acknowledgments
We thank Dr. Steve Morris at St. Jude Children's Research Hospital for the SUDHL-1 cells. We also thank Caroline Nebhan for assistance with plasmid preparation, Juliann Chmielecki for sharing sequence data from H3122, H2228, and HCC78 cell lines, and Nerina McDonald for assistance in culturing the Ba/F3 cells.
This work was supported by the VICC Cancer Center Core grant (P30-CA68485) and by a career development award from the Vanderbilt Specialized Program of Research Excellence in Lung Cancer grant (CA90949). CML was additionally supported by an NIH T32 training grant (5T32 CA119910-04) and a Uniting Against Lung Cancer grant. CL is a co-founder and the chief scientific officer of Xcovery.
References
- 1.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994 Mar 4;263(5151):1281–4. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 2.Lawrence B, Perez-Atayde A, Hibbard MK, Rubin BP, Dal Cin P, Pinkus JL, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol. 2000 Aug;157(2):377–84. doi: 10.1016/S0002-9440(10)64550-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007 Aug 2;448(7153):561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 4.Choi YL, Takeuchi K, Soda M, Inamura K, Togashi Y, Hatano S, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. 2008 Jul 1;68(13):4971–6. doi: 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- 5.Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008 Jul 1;14(13):4275–83. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeuchi K, Choi YL, Soda M, Inamura K, Togashi Y, Hatano S, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008 Oct 15;14(20):6618–24. doi: 10.1158/1078-0432.CCR-08-1018. [DOI] [PubMed] [Google Scholar]
- 7.Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S, Inamura K, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009 May 1;15(9):3143–9. doi: 10.1158/1078-0432.CCR-08-3248. [DOI] [PubMed] [Google Scholar]
- 8.Wong DW, Leung EL, So KK, Tam IY, Sihoe AD, Cheng LC, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009 Apr 15;115(8):1723–33. doi: 10.1002/cncr.24181. [DOI] [PubMed] [Google Scholar]
- 9.Horn L, Pao W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol. 2009 Sep 10;27(26):4232–5. doi: 10.1200/JCO.2009.23.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007 Dec 14;131(6):1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 11.Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008 Oct 16;455(7215):930–5. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.George RE, Sanda T, Hanna M, Frohling S, Luther W, 2nd, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008 Oct 16;455(7215):975–8. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janoueix-Lerosey I, Lequin D, Brugieres L, Ribeiro A, de Pontual L, Combaret V, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008 Oct 16;455(7215):967–70. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008 Oct 16;455(7215):971–4. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- 15.Sabbatini P, Korenchuk S, Rowand JL, Groy A, Liu Q, Leperi D, et al. GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol Cancer Ther. 2009 Oct;8(10):2811–20. doi: 10.1158/1535-7163.MCT-09-0423. [DOI] [PubMed] [Google Scholar]
- 16.Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci USA. 2007 Jan 2;104(1):270–5. doi: 10.1073/pnas.0609412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007 May 1;67(9):4408–17. doi: 10.1158/0008-5472.CAN-06-4443. [DOI] [PubMed] [Google Scholar]
- 18.Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007 Dec;6(12 Pt 1):3314–22. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- 19.Soda M, Takada S, Takeuchi K, Choi YL, Enomoto M, Ueno T, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci USA. 2008 Dec 16;105(50):19893–7. doi: 10.1073/pnas.0805381105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. Oct 28;363(18):1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanna N, Shepherd FA, Fossella FV, Pereira JR, De Marinis F, von Pawel J, et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol. 2004 May 1;22(9):1589–97. doi: 10.1200/JCO.2004.08.163. [DOI] [PubMed] [Google Scholar]
- 22.Fabian MA, Biggs WH, 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005 Mar;23(3):329–36. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 23.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008 Jan;26(1):127–32. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 24.Mukohara T, Engelman JA, Hanna NH, Yeap BY, Kobayashi S, Lindeman N, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005 Aug 17;97(16):1185–94. doi: 10.1093/jnci/dji238. [DOI] [PubMed] [Google Scholar]
- 25.Gandhi J, Zhang J, Xie Y, Soh J, Shigematsu H, Zhang W, et al. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS One. 2009;4(2):e4576. doi: 10.1371/journal.pone.0004576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009 May;41(5):521–3. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachleitner-Hofmann T, Sun MY, Chen CT, Tang L, Song L, Zeng Z, et al. HER kinase activation confers resistance to MET tyrosine kinase inhibition in MET oncogene-addicted gastric cancer cells. Mol Cancer Ther. 2008 Nov;7(11):3499–508. doi: 10.1158/1535-7163.MCT-08-0374. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz AL, Fridovich SE, Knowles BB, Lodish HF. Characterization of the asialoglycoprotein receptor in a continuous hepatoma line. J Biol Chem. 1981 Sep 10;256(17):8878–81. [PubMed] [Google Scholar]
- 29.Yuza Y, Glatt KA, Jiang J, Greulich H, Minami Y, Woo MS, et al. Allele-dependent variation in the relative cellular potency of distinct EGFR inhibitors. Cancer Biol Ther. 2007 May;6(5):661–7. doi: 10.4161/cbt.6.5.4003. [DOI] [PubMed] [Google Scholar]
- 30.McDermott U, Iafrate AJ, Gray NS, Shioda T, Classon M, Maheswaran S, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008 May 1;68(9):3389–95. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 31.Noor F, Niklas J, Muller-Vieira U, Heinzle E. An integrated approach to improved toxicity prediction for the safety assessment during preclinical drug development using Hep G2 cells. Toxicol Appl Pharmacol. 2009 Jun 1;237(2):221–31. doi: 10.1016/j.taap.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 32.Kwak EL, Camidge DR, Clark J, Shapiro GI, Maki RG, Ratain MJ, et al. Clinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066. J Clin Oncol. 2009;27(15S) Abstract 3509. [Google Scholar]
- 33.Clarke JL, Pao W, Wu N, Miller VA, Lassman AB. High dose weekly erlotinib achieves therapeutic concentrations in CSF and is effective in leptomeningeal metastases from epidermal growth factor receptor mutant lung cancer. J Neurooncol. Sep;99(2):283–6. doi: 10.1007/s11060-010-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sasaki T, Rodig SJ, Chirieac LR, Janne PA. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer. Jul;46(10):1773–80. doi: 10.1016/j.ejca.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. Oct 28;363(18):1734–9. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 36.Marzec M, Kasprzycka M, Liu X, El-Salem M, Halasa K, Raghunath PN, et al. Oncogenic tyrosine kinase NPM/ALK induces activation of the rapamycin-sensitive mTOR signaling pathway. Oncogene. 2007 Aug 16;26(38):5606–14. doi: 10.1038/sj.onc.1210346. [DOI] [PubMed] [Google Scholar]
- 37.Boik JC, Newman RA, Boik RJ. Quantifying synergism/antagonism using nonlinear mixed-effects modeling: a simulation study. Stat Med. 2008 Mar 30;27(7):1040–61. doi: 10.1002/sim.3005. [DOI] [PubMed] [Google Scholar]
- 38.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 39.Daub H, Specht K, Ullrich A. Strategies to overcome resistance to targeted protein kinase inhibitors. Nat Rev Drug Discov. 2004 Dec;3(12):1001–10. doi: 10.1038/nrd1579. [DOI] [PubMed] [Google Scholar]
- 40.Chen Z, Sasaki T, Tan X, Carretero J, Shimamura T, Li D, et al. Inhibition of ALK, PI3K/MEK and HSP90 in Murine Lung Adenocarcinoma Induced by EML4-ALK Fusion Oncogene. Cancer Res. Oct 15; doi: 10.1158/0008-5472.CAN-10-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.