

Abstract
Polycystic kidney disease is the defining condition of a group of common life-threatening genetic disorders characterized by the bilateral formation and progressive expansion of renal cysts that lead to end stage kidney disease. Although a large body of information has been acquired in the past years about the cellular functions that characterize the cystic cells, the mechanism(s) triggering the cystogenic conversion are just starting to emerge. Recent findings link defects in ciliary functions, planar cell polarity pathway, and centrosome integrity in early cystic development. Many of the signals dysregulated during cystogenesis may converge on the centrosome for its central function as a structural support for cilia formation and a coordinator of protein trafficking, polarity, and cell division. Here, we will discuss the contribution of proliferation, cilium and planar cell polarity to the cystic signal and will analyze in particular the possible role that the basal bodies/centrosome may play in the cystogenetic mechanisms.
1. Introduction
Hereditary cystic kidney diseases comprise a heterogeneous group of monogenic disorders [1]. In some instances the bilateral development of multiple fluid-filled cysts in kidneys is part of a more complex syndromic clinical manifestation, whereas in others it is a distinctive feature of the disease and an important cause of end stage kidney disease. We will focus on the latter disorders, hereafter referred to as polycystic kidney disease.
Polycystic kidney disease is characterized by the hyperproliferation of tubular epithelial cells, the alterations of their fluid secretion functions, and changes in the extracellular matrix deposition and fibrosis, all of which profoundly alter the organ architecture and impair renal function. Autosomal dominant and autosomal recessive forms of polycystic kidney disease have been recognized with an incidence of 1:800 and 1:20,000, respectively.
Autosomal dominant polycystic kidney disease (ADPKD) is caused by the dysregulation of the PKD1 or PKD2 genes, which code for polycystin-1 (PC1) and polycystin-2 (PC2), respectively. PC1 and PC2 may form a complex through the interaction of the respective carboxyl termini, thus establishing reciprocal regulatory functions. Consequently, regardless of the genotype, the clinical manifestations of ADPKD largely overlap, with few notable exceptions: on average, individuals with mutation in the PKD1 gene reach end stage kidney disease 20 years earlier than patients carrying mutations in the PKD2 gene, and PKD2 mutations result in more severe disease in males than in females.
Autosomal Recessive Polycystic Kidney Disease (ARPKD) results from mutations in the polycystic kidney and hepatic disease 1 gene (PKHD1), encoding fibrocystin/polyductin (FPC) [2, 3]. ARPKD generally manifests earlier in life with the most severe cases resulting in perinatal or neonatal death. In addition, collecting duct ectasia results in cysts that remain connected with the nephrons of origin. Unlike ADPKD, in which cysts are prevalent in the collecting ducts but may develop everywhere along the nephron, in ARPKD cystogenesis is restricted to the collecting ducts.
PC1 is a large integral membrane protein with receptor-like structural characteristics [4], which undergoes a complex Notch-like processing [5, 6]. Abundant evidence supports the role of the PC1 carboxyl terminus in signaling mechanisms. The C terminal tail of PC1 contains phosphorylation sites for different tyrosine and serine/threonine kinases [7] and a domain for the interaction with G proteins and the activation of the JNK/AP1 pathway [8, 9]. Importantly, in response to changes in mechanical stimulation, the carboxyl terminal tail undergoes regulated intramembrane proteolysis and translocates into the nucleus to activate the AP1 pathway through a process negatively regulated by PC2 [10].
PC2 is a Ca2+ regulated, non-selective cation channel that shares sequence and structure similarities with the superfamily of transient receptor potential channels [11-15]. PC2 is expressed predominantly in the ER, but it is also found in the Golgi, the plasma membrane, and on the cilium where with PC1, and likely FPC, it forms a mechanosensor complex that controls Ca2+ influx in response to flow [16, 17]. On the plasma membrane, PC2 only partially co-localizes with PC1 and adhesion complexes, suggesting that it may function independently as homodimer or participate in different complexes with other members of the TRP family, thus expanding the functional characteristics of these channels [16]. The loose interaction of PC2 with PC1 and adhesion complexes may be important to confer PC2 more dynamic mechanosensorial properties independent of or opposed to PC1. For example, situs inversus, the phenotype with reversed orientation of visceral organs, is associated with Pkd2 but not Pkd1 knockout mouse models, indicating the independent mechanosensing function of PC2 in the nodal cilia [18]. In the case of stretch-activated ion channels PC1 and PC2 exert opposing effects with PC2 inhibiting channel opening and PC1 reverting this suppression [19]. Though many aspects of the regulation PC2 function remain unclear, the growing evidence of its multiple interactions with cytoskeleton organizing proteins supports its Ca2+-dependent mechanosensorial role at different cellular compartments (for a comprehensive review see references 15, 20, 21). Interestingly, the subcellular localization of PC1 at the cell adherens, desmosomes, focal adhesions, and cilia provides the proximity with cytoskeletal components suggesting a possible role of PC1 in the control of cytoskeleton rearrangement (Figure1) [22-25].
Figure 1.
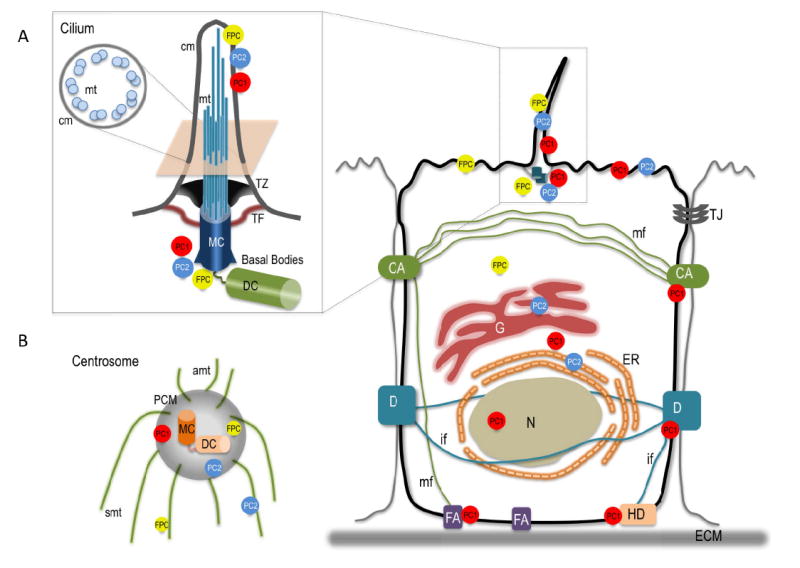
Subcellular localization of PC1, PC2, and FPC. A) Polycystic proteins localize to multiple compartment within the cell including the cilium in which they form a Ca2+ non-selective channel whose activity is essential during renal morphogenesis. In the kidney, the cilium protrudes from the apical side of renal epithelial cells into the lumenal space. The cilium is supported by nine doublets of microtubules that nucleate from the basal body, a specialization of the mother centriole (MC), at the base of the cilium (1A, inset). B) Following cilium resorption, pericentriolar material (PCM) organizes around the centrioles to form the centrosome. Microtubules emanating from the centrosome maintain cellular structure and are required for multiple cellular functions including spindle organization and cytokinesis. Polycystic proteins also localize to the centrosome and both PC2 and FPC are found to associate with the spindle microtubules during cell division. While the ciliary localization of polycystic proteins is important for fluid flow sensing, their function on the centrosome and mitotic spindle remains obscure. Similarly unclear is whether the localization at cell-cell and cell matrix contacts plays a role in tension sensing and cytoskeletal rearrangement. N, nucleus; G, Golgi apparatus; ER, endoplasmic reticulum; CA, cell adherens; D, desmosomes; HD, hemidesmosomes; FA, focal adherens; ECM, extracellular matrix; TJ, tight junction; mt, microtubules; mf, actin microfilaments; if, intermediate filaments; MD, mother centriole; DC, daughter centriole; TZ, transition zone; TF, transition fiber; PCM, pericentriolar matrix; cm, ciliary membrane; amt, astral microtubules; smt, spindle microtubules.
Similar to PC1, FPC undergoes a complex proteolytic process at the ciliary membrane [26, 27]. The large ectodomain is cleaved by a proprotein convertase and remains tethered to the carboxyl stalk via disulfide bonds. Shedding of the ectodomain occurs concomitantly to the regulated intramembrane proteolysis that releases the intracellular cytoplasmic tail that then traffics to the nucleus or nucleolus. This process appears to be dependent on intracellular Ca2+ release, but it remains unknown whether a ligand or a mechanical change triggers the shedding of the ectodomain [26]. Similarly, the possible paracrine signaling function of the shed domain and the implication of the nuclear translocation on gene regulation remain unclear, though the Notch-like regulation and the ciliary localization of the process suggest that it may be involved in the maintenance of nephron architecture.
Excellent reviews have presented and discussed the characteristics and functions of polycystic genes and their encoded proteins in detail [28-31]. Here we will review the early events of renal cystogenesis and the relationship of polycystic proteins with the centrosome, its association with the cilium and its function in cell cycle control.
2. Proliferation of cystic cells
Cell hyperproliferation underlying continuous expansion of the cysts and renal enlargement is a hallmark of ADPKD and ARPKD and a determinant of renal failure [32, 33]. Under normal conditions the mitotic index of the adult kidney is very low. However, in renal tissues from ADPKD or ARPKD patients, as well as from Pkd1 or Pkd2 mutant animal models, nuclei positive for proliferating cell nuclear antigen (PCNA) and Ki67 mitotic markers are readily detectable [34-36]. In fact, multiple mitogenic pathways may be constitutively activated in polycystic kidney diseaseas a consequence of altered Ca2+ homeostasis or abnormal protein trafficking.
Defects in PC2 Ca2+ channel activity that lead to low intracellular Ca2+ concentration, aberrant G-protein signaling by PC1 dysregulation [8, 37], and decreased cyclic nucleotide catabolism [38] may contribute to the accumulation of cAMP and the abnormal activation of the Ca2+ inhibitable adenyl cyclase 5/6. Cystic cells proliferate in response to increased cAMP levels [39, 40] and the activation of the PKA/B-Raf/MAPK pathway [41], in contrast to normal primary renal epithelial cells, whose growth is inhibited by cAMP [42]. Cyst expansion then accelerates partly through a mechanism promoting chloride-driven fluid secretion [43, 44].
Altered protein trafficking may also contribute to cystogenic signals as in the case of the mislocalization of EGF receptors in renal epitelia. The EGF receptor (EGFR/HER1) is normally expressed apically during the embryonic mammalian kidney development, but its localization shifts to the basal side in the adult organ. In ADPKD and ARPKD, however, the EGFR/HER1 expression is increased and mislocalized to the apical membrane where it results in a paracrine loop of persistent stimulation by its ligand released in the filtrate or in the cystic fluid [45-47]. Other dedifferentiating processes characteristic of cystic cells may further reinforce this autostimulatory mechanism. For instance, the expression of ErbB2/Neu/HER2, a member of the EGFR superfamily, is developmentally regulated and restricted to the embryonic kidneys. However, the re-expression ErbB2/Neu/HER2 in the adult ADPKD renal epithelia allows it to heterodimerize with EGFR/HER1 on the apical membrane [48]. The interference with the autocrine/paracrine EGF/EGFR stimulatory loop reduced cystic lesions in organ culture [49], slowed down cyst expansion and ameliorated polycystic kidney disease in different, though not all, animal models [50-52] [53].
Other proliferative pathways may also be activated. In particular, the evidence of the activation of the mammalian target of rapamycin (mTOR) signaling in the cyst lining cells of the kidneys from different mouse models of renal cystic disease (MAL, overexpressing myelin and lymphocyte protein; and the IFT88 hypomorph, orpk,) and in human ADPKD specimens suggests that this may be a common pathway underlying cystic proliferation [54] (for an extensive review see [55]).
The serine/threonine kinase mTOR is the key component of the multiprotein complexes mTORC1, which positively controls protein translation, cell metabolism and proliferation, and mTORC2, which is involved in actin cytoskeleton organization and cell survival [56, 57]. The activation of mTORC1 is suppressed by the heterodimer of hamartin and tuberin, encoded by the TSC1 and TSC2 genes, respectively. Stimulation of the PI3kinase/Akt or ERK pathways leads to the phosphorylation-mediated inhibition of TSC2/tuberin and the activation of mTORC1 [58, 59]. Tuberin and PC1 functionally cooperate to regulate the mTOR pathway. PC1 interacts with tuberin [54], sequesters it on the membrane and protects it from Akt phosphorylation, thus suppressing the activation of mTORC1 [60]. Conversely, tuberin is necessary for the proper localization PC1, as seen in Eker rats that carry a homozygous mutation of the Tsc2 gene. In the absence of functional tuberin, PC1 accumulates in the Golgi and fails to properly traffic to the lateral cell membrane, demonstrating that tuberin is necessary for proper PC1 localization [61].
In various animal models of polycystic kidney disease, a significant reduction of cystic growth has been obtained by pharmacologically preventing the cAMP increase, Ca2+ imbalance, EGF stimulation, mTOR activation [62-67] or by inhibiting cell cycle progression with the cyclin-dependent kinase inhibitor, roscovitine [68]. These in vivo results have provided the rationale for different experimental therapeutic approaches that are currently under investigation [69]. However, recently concluded clinical trials that tested the efficacy of mTOR inhibitors (rapamycin/sirolimus and the analog everolimus) on ADPKD patients at different stages of the disease yielded disappointing results. Treatment with these inhibitors showed no improvement in the renal function, despite a transient reduction in total kidney volume in patients with a more advanced stage of disease [70, 71]. Unlike the findings from a shorter study with sirolimus on fewer patients [72], these trials also indicated that both mTOR inhibitors presented considerable side effects that severely limit their therapeutic value for ADPKD, even when administered at doses far lower than those used in the animal models.
A further detailed analysis of these studies may help explain some of the differences within the human studies and the discrepancies with the experimental data on animal models as commented in [73, 74]. In light of the remarkably promising preclinical results, it would be premature to interpret the discouraging results of the human trials to confute the validity of targeting the mTOR pathway in cystic diseases. Rather, it may be necessary to explore alternative strategies in which mTOR inhibition is part of a combination therapy or in which mTOR inhibitors could be specifically targeted to the kidney.
A smaller clinical trial on ADPKD patients was also conducted to assess the efficacy of octreotide, a long-lasting somatostatin analog, that inhibits the intracellular accumulation of cAMP in renal epithelia [75]. In this 12-month study, results similar to those with mTOR inhibitors were obtained: octreotide arrested the increase of kidney volume but failed at improving kidney function. Differently from mTOR inhibitors, however, octreotide appeared to be well tolerated with no serious adverse effects.
Overall, these clinical trials underscore the complexity and variability of the disease progression, and question the use of kidney volume change as a surrogate marker of organ function [32, 33, 73]. They also suggest that at advanced stages of the disease, cell proliferation is dissociated from cellular and organ function. As such, proliferation may have different roles at different stages of cystogenesis. Findings of proliferating cells in normal tubular epithelia surrounding cysts suggest that cell growth is an early event in the cystogenic transformation. Nevertheless, no cysts derive from the active proliferation during normal organ morphogenesis, and active growth of renal carcinoma cells does not necessarily result in cyst formation. Therefore, it remains difficult to establish whether the activation of these pathways represents the cystogenic trigger or if it supports cyst expansion.
3. Cilium and cystogenesis
The observation that cystic proteins localize on the primary cilium and basal body [76-78] provided new insights into the mechanisms of renal cystic diseases. The intense focus on the cilium that followed unveiled the genetic determinants of numerous complex diseases that define a new class of disorders collectively referred to as ciliopathies (for comprehensive reviews, see references 79-82).
The primary cilium is a highly compartmentalized organelle present in most cell types that functions as a sensor of extracellular environmental cues. It is formed as a single protrusion of the plasma membrane supported by the axoneme, a cytoskeletal component that is assembled as a ring of 9 microtubule doublets arranged tangentially to the center in a configuration known as 9+0 (Figure 1, inset A). Defects in cilia formation result in complex phenotypes, which invariably include cystic kidneys [83]. In renal epithelia, cilia convert mechanical force of fluid flow into cellular functions [84]. PC1, PC2 and FPC are expressed in renal primary cilia where they are a part of a mechanosensor complex that translates the ciliary bending induced by flow into Ca2+ influx [85-87]. Their functional role was supported by observation that STAT6, whose ciliary localization depends on flow stimulation, is part of a complex that includes the cleaved carboxyl terminus of PC1 and the transcriptional coactivator P100. As the carboxyl tail of PC1 is proteolytically cleaved, the complex translocates into the nucleus and activates gene expression, thereby linking mechanical stimulation of the cilium by urine flow and cellular responses [88]. However, impaired mechanosensation of cilium as a primary defect in cystogenesis was challenged by the work on conditional knockout models of the intraflagellar transport Ift88 (polaris) and the Kif3a subunit of kinesin-2 genes, which are essential for ciliogenesis [89]. While the deletion of Ift88 or Kif3a during gestation prevented cilia formation and resulted in severe cystic disease within two weeks after birth, deletion of either gene in the adult animals did not immediately result in detectable cystic phenotype, despite the cilia ablation. Eventually, mild renal cyst formation was observed six months after the knockout, revealing different requirements of ciliary function during renal development and in the maintenance of adult kidney [89]. These results indicated that cilia are dispensable in adult mice and that other components may participate in the cystic process.
Interestingly, the conditional models of Pkd1 inactivation similarly displayed greater susceptibility of young mice to develop severe cystic kidney disease as compared to the adult mice. These mouse models offered the opportunity to investigate the early cystogenetic events following the depletion of PC1 [90-92]. A detailed analysis of perinatal Pkd1 inactivation demonstrated that the deletion of the gene within day P13 led to extensive cystogenesis and kidney enlargement, whereas inactivation of Pkd1 from day P14 onward resulted in late onset cystic kidney disease [91]. These observations uncovered a window of susceptibility, which corresponds with the completion of mouse nephrogenesis when proliferation is actively ongoing and a specifically timed brake point. However, the study also showed that although proliferation abruptly decreased after day P14, it remained significantly higher at P16 as compared to the adult kidney. Nevertheless the course of cystic disease was comparable in the P16 and older mice, suggesting that proliferation per se may not be sufficient to trigger the cystogenic change [91]. As the brake point was characterized molecularly by a change in gene expression pattern consistent with a developmental switch, it was proposed that components of an early developmental program could in fact be the cystogenic triggers. Such a program may be recalled during the re-epithelization process that follows renal injury. In support of this notion, in adult kidneys in which Pkd1 is conditionally inactivated or in which no cilia can be formed because of the conditional Kif3a excision, the cystic phenotype can be accelerated by the induction of ischemia/reperfusion injury or pharmacological nephrotoxicity [36, 93, 94]. In addition, although cell growth occurs rapidly following injury, it reverts to control levels before cystic expansion, again suggesting that proliferation cannot be the only cystogenic switch [94]. Nevertheless, even though proliferation may not be sufficient as the sole cystogenic trigger, it may yet provide the necessary context for such a trigger to arise as indicated in recent experiments on the conditional inactivation of the Hnf1b gene, which encodes a transcription factor involved in the expression of genes that include Pkd2, Pkhd1 and UMOD (encoding uromodulin). Similar to the IFT88, Kif3a, and Pkd1 models, the pre- or perinatal conditional inactivation of Hnf1b leads to rapid polycystic kidney disease, while the ablation of Hfn1b in the adult leads to slow onset cystic disease that can turn into rapidly progressing disease following renal injury. Careful analysis of proliferating cells using BrdU showed that tubular dilatation coincided with the regenerative proliferation burst and the loss of mitotic orientation only in the mutant dividing cells and not in those of wild type kidneys [95]. Therefore, cell proliferation may create the conditions for the cystogenic switch, which may include defects in oriented cell division, planar cell polarity (PCP), and changes in the centrosome positioning [93-95].
4. Role of planar cell polarity in cystogenesis
The organization and asymmetric distribution of protein content that cells maintain in parallel to the epithelial plane is called planar cell polarity (PCP). The mechanisms of PCP are fundamental for the developmental patterning of both invertebrates and vertebrates [96] and are regulated by the non-canonical Wnt pathway (for a comprehensive review of the Wnt signaling in cystic diseases, see reference 97).
During kidney development, the spindle of the dividing cell organizes with an orientation parallel to the axis of the elongating tubule, revealing an intrinsic cell polarity. The evidence of a link between cilia and PCP came from the observation that the ciliary protein inversin, the product of the NPHP2 gene whose mutations cause nephronophtisis, functioned as a switch from the canonical to the non-canonical Wnt pathway [98]. Whether PCP in turn played a role in cystic disease was first observed in kidneys in two rodent renal cystic models: the mouse with inactivation of the Tcf2/HNF1β transcription factor [99], and the pck rat, which carries a mutated PKHD1 gene ortholog [100]. In both cystic models a significant number of spindles in the dividing cells of the kidneys were misaligned, suggesting that the loss of proper spindle orientation and planar cell polarity are linked to cystogenesis [101].
Recently, a direct proof of the role of PCP in renal cystic development was provided by the knockout mouse model of Fat4 gene, which encodes a PCP protein of the proto-cadherins family [102]. Homozygous Fat4-/- mutants died at birth but displayed multiple characteristics of PCP protein defects including anomalies in the elongation of the cochlea and disruption of hair cell organization in the organ of Corti. Fat4-/- mutants also displayed smaller kidneys with dilated and shorter tubules and significant defects in oriented cell division. Crossing Fat4-/- mice with mutants for other PCP components, Vangl2 and Fjx1, exacerbated the cystic phenotype [102]. Together with the ciliary localization of FAT4, these findings further strengthened the link between PCP and cilium during cystogenesis. The interdependence of PCP and ciliary function is also supported by observations with other PCP core proteins, Dishevelled and Vangl2. Dishevelled is involved in the docking of the centrioles/basal bodies to the apical membrane that precedes ciliogenesis [103], and Vangl2 is required for the asymmetric positioning of motile cilia in cells of zebrafish neural tube [104]. Furthermore, the fluid flow influences centrioles’ movement and contributes to the orientation of motile cilia in conjunction with PCP in ependymal cells [105, 106].
It should be noted, however, that more recent reports question the role of oriented cell division as a primary cause of cystogenesis. In the hypomorphic mutant for Wnt9b, whose expression is required for renal morphogenesis, cystogenesis starts in utero, leading to the development of grossly cystic kidneys within a month of age [107]. The analysis of the embryonic renal development revealed that during the period from E13.5 to P1, tubules lengthen through the movement of the cells that assume an elongated shape parallel to the tubule axis in a process of convergent extension, which is dependent on PCP and the activation of the Rho/Jnk signaling pathway. The impairment of this process in the Wnt9b mutants alters tubule diameter and triggers cyst formation. Interestingly, until P1, cell division appeared similarly misoriented in both Wnt9b mutants and wild type mice, suggesting that defects in oriented cell division alone cannot account for in utero cystogenesis [107]. Moreover, a study in Pkd1, Pkd2 and Phkd1 mouse mutants showed that changes in oriented cell division did not precede cystogenesis, but rather followed the cystic transformation [108]. While challenging the defects of oriented cell division as a driver of cystogenesis, these results nevertheless emphasize the role of PCP in cystogenesis.
5. Centrosome and cell cycle
The basal bodies located at the base of the cilium are a morphological specialization of the centrioles/centrosome, specifically the mother centriole from which the axoneme emanates to support the formation of the primary cilium (Figure 1). Functionally, the basal bodies participate in the intraflagellar transport (IFT) through the organization of the transition zone and the control of vesicles trafficking to and from the cilium [109], thereby coupling the cilium and centrosome functions. The essential role played by the centrosome in coordinating the ciliary and PCP crosstalk is further emphasized by the alteration of the Wnt signaling following the disruption of basal bodies in zebrafish bbs4 morphants [110].
The centrioles/centrosome serve as the microtubule-organizing center (MTOC), and thus play a major role in the spatial organization of the microtubular network required for not only the formation of primary cilia, but also cell polarity, migration, trafficking of cytoplasmic organelles, and organization of the mitotic spindle [111]. Because of these essential functions that it underlies, the centrosome integrity and duplication are tightly controlled. In most cells, under normal conditions the centrosome divides only once per cell cycle through a mechanism coupled to the cell cycle progression, so that each daughter cell receives only one centrosome [112, 113]. Reciprocal interactions exist between IFT and centrosomal proteins to regulate their trafficking and localization. For example, IFT20 shuttles between the Golgi and the cilium, and is required for the localization of pericentrin to the centrosome [114, 115]. Conversely, reduced expression of pericentrin also lowers the levels of IFT20, IFT88, IFT57 and PC2 in centrioles and inhibits cilia formation [115].
In cells preparing to cycle, the cilium is reabsorbed, leaving the basal bodies/centrioles free to anchor to the cell cortex and to be ready for centrosome duplication and the subsequent organization of the microtubule rearrangement that is required for the assembly of the spindle, mitosis, and cytokinesis. Cilium resorption may allow redistribution of ciliary components to the centrosome that can affect the cell cycle progression. For instance, IFT88/polaris remains tightly associated with the centrosome and modulates the G1-S transition by titering out Che-1, an inhibitor of the growth suppressor function of Rb [116]. Consequently, interfering with various centrosome proteins leads to the p53-dependent block of cell cycle progression from G1 to S and failure to assemble cilia [117, 118]. p53 is also a centrosomal protein, and its depletion increases centrosome amplification [119]. The control of cell cycle progression and restriction of centrosome overduplication by p53 is exerted partly via the transactivation of p21 and the direct association of p53 with the centrosome [120-122].
Both PC1 and PC2 exert a direct effect on cell cycle and centrosome duplication. The heterologous expression of PC1 or PC2 arrests the cell cycle in G1 through different mechanisms that converge on the induction of the cyclin dependent kinase (Cdk) inhibitor p21 and the inhibition of Cdk2 activity [123, 124]. In the case of PC1, the expression of p21 results from the activation of the JAK2-dependent phosphorylation of STAT1, but not of p53 [123]. In contrast, PC2 functions by binding to Id2, a member of the helix–loop–helix (HLH) family of transcriptional regulators that antagonize basic HLH transcription factors that are involved in the control of cell cycle progression. The interaction with PC2 sequesters Id2 in the cytosol, thus preventing its translocation into the nucleus where it suppresses p21 transcription [124]. PC1 and PC2 exert a reciprocal control on the activation of these pathways. The physical interaction of PC1 with JAK2 is dependent on the presence of PC2 as a cofactor, whereas PC1 phosphorylation of PC2 is required for its interaction with Id2. Conversely, depletion of PC1 or PC2 results in faster G1 to S progression [124-126] and reduced expression of p53 in HEK293 cells [125]. Lowered p53 expression is also observed in embryonic kidneys of Pkd1-/- mice [127], albeit in this case it is difficult to determine whether such downregulation is an effector or a consequence of the cystogenic transformation.
Polycystic proteins localize on the centrosome and are important to maintain centrosome integrity (Figure 2). The inhibition of PC1 expression induces centrosome amplification in vitro, and supernumerary centrosomes were observed both in the kidneys of Pkd1 conditional knockout animal model and in human renal tissue from ADPKD patients in vivo [128]. These centrosomes appeared fully functional, as they were able to organize multipolar spindles. However, the cells dividing with aberrant mitotic spindles entered mitotic catastrophe or produced genetically unstable progeny, characterized by significant apoptosis and aneuploidy [129, 130]. Amplified centrosomes were noted on seemingly normal tubular cells, suggesting that centrosome aberrations may be an early event in the cystic conversion [128]. Similarly, centrosome amplification was also reported in fibroblast cell lines derived from Pkd2 transgenic mice and in mesenchymal cells of Pkd2 knockout embryos [131], indicating that PC2 dysregulation also affects centrosome integrity. Polycystins’ broad tissue distribution and the effects of interference of PC1 or PC2 in centrosome integrity in non-renal cells suggest that polycystins play a fundamental role in the mechanisms controlling centrosome duplication and that centrosomal aberrations may be important in cystic development.
Figure 2.
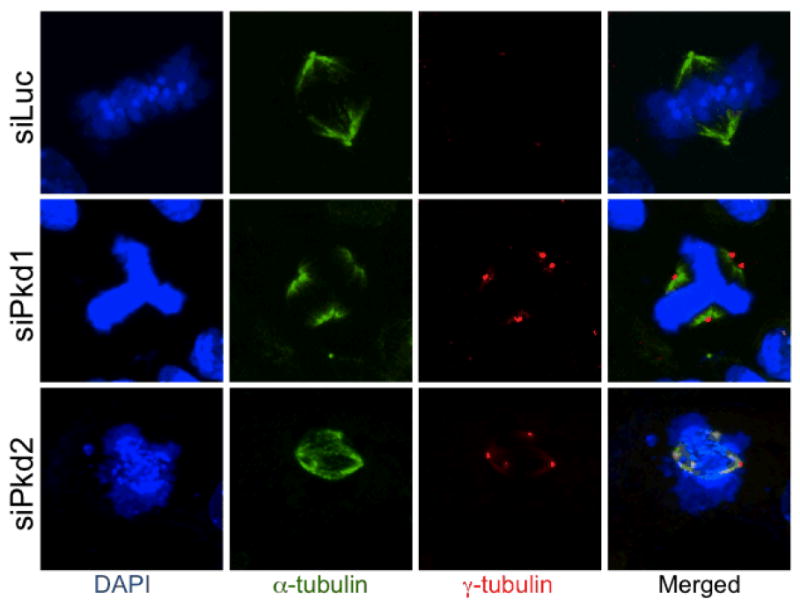
Centrosome amplification following the suppression PC1 or PC2 expression. Centrosome amplification and mitotic spindle abnormalities occur rapidly after the knockdown of Pkd1 or Pkd2. Shown are MDCK cells and IMCD3 cells three days following the transduction with a lentivirus constitutively expressing the shRNAs specific for Pkd1 and Pkd2, respectively. IMCD3 cells (or MDKC cells, not shown) transduced with control lentivector expressing shLuc against luciferase (siLuc) maintain normal mitosis. Cells were immunostained with anti-α-tubulin and anti-γ-tubulin to specifically detect microtubules (green) and centrosomes (red), respectively, and then counterstained with DAPI to visualize DNA (blue).
More recently, at least some FPC isoforms have also been shown to be required for the maintenance of centrosome integrity and proper spindle assembly [132]. Similarly to PC2, FPC is found on the spindle during cell division, but the mechanisms controlling its localization remain unknown. The spindle localization of PC2, however, was shown to require the interaction with Diaphanous (mDia)-related formin 1, mDia1 [133], a protein involved in actin polymerization and microtubule stabilization [134]. Depletion of mDia1 coincides with the loss of PC2ocalization from the spindle and a decreased Ca2+ release in mitotic cells. The function of PC2 on the spindle is unclear, but the interaction of PC2 with the actin bundling protein α-actin in and with the microtubule-dependent motor kinesin-2 subunit KIF3A, both of which activate PC2 channel activity in vitro, lends support to the intriguing possibility that PC2-mediated Ca2+ transport may function in the cytoskeletal remodeling required for cell division [21, 135, 136]. Although the spindle localization of PC1 is unclear, its presence on the centrosome along with FPC may be important in the reconstitution and regulation of the PC2 Ca2+ channel activity [87, 132, 137]. Overall, these observations underline the interdependence of cilium, centrosome, and cytoskeletal rearrangement.
The mechanisms contributing to centrosome amplification remain speculative, but it might involve the altered expression of p53 and/or cyclin-A, as observed in PC1-deficient cells [125, 138], as well as imbalanced Ca2+ homeostasis. Centrosome amplification can occur following cytokinesis failure or by reiterated centriole duplication within the same cycle. Evidence of multinucleation and enlarged nuclei in PC1-or PC2-deficient cells suggests that supernumerary centrosomes may result from endoreduplication. Cytokinesis depends on the accumulation of Ca2+ stores to the furrow and on the proper Ca2+ release before abscission [139]. It remains to be determined whether the reciprocal interaction of PC2 (or a polycystins complex) with cytoskeletal components has any function on this Ca2+ regulation. Furthermore, both centrosome duplication and cell growth processes depend on increased Ca2+ transients from internal Ca2+ stores [140], a requirement that seems to be at odds with the decreased intracellular Ca2+ content in polycystic cells [141, 142]. However, experiments on HEK293 cells indicate that PC1 negatively regulates non-capacitative Ca2+ entry (NCCE) channels and that cell proliferation upon PC1 knockdown is sustained by an NCCE-dependent increase in Ca2+ oscillations [143]. Hence, it is possible that changes in the frequency and amplitude of Ca2+ oscillations may support also centrosomal amplification or centriole reduplication [144].
The centrosomal defects extend to other diseases with renal cystic manifestations. Loss of hamartin, the product of the TSC1 gene whose mutations cause tuberosclerosis, also leads to centrosome amplification [145]. The depletion of the centrosomal Mks1 or Mks3/meckelin proteins, which are mutated in the autosomal recessive Meckel-Gruber syndrome, results in centrosome amplification and, in the case of Mks3, in multiciliation [146]. Renal cysts develop following the loss of IFT20, which results in cilia ablation, centrosome amplification with loss of centrosome positioning, and mitotic spindle misorientation [147]. It will be of interest to determine whether centrosome defects are common to other renal cystic diseases.
Centrosome aberrations occur early after the inhibition of polycystic proteins and, similar to ciliary defects, they may be a common denominator in renal cystic disease. A causative role of centrosome defects in cystogenesis is difficult to establish, but its expected consequences are consistent with all the findings characteristic of ADPKD cells. Errors in centrosome duplication may result in the formation of monopolar or multipolar spindles, aberrations associated with chromosome missegregation, genomic instability, and apoptosis. Cells that accumulate excessive genomic damage/imbalance become apoptotic, whereas others may survive carrying abnormal karyotypes [148, 149], and altered physiological functions. A kidney specific interference with effectors of the centrosome duplication process will be required to establish a causal link between centrosome anomalies and renal cystic development.
6. Conclusions
As intense research has focused on cystic cells, we have a better understanding of the mechanisms that support cystic expansion, including alterations of calcium homeostasis and changes in protein trafficking and interactions, which sustain the constitutive activation of mitogenic pathways. On the other hand, the signals (or lack thereof) that trigger the cystic conversion are unknown and the mechanisms underlying the early cystogenic events are just emerging in a picture of increasing complexity.
The view of the cilium as sensor of fluid flow has expanded to the regulation of planar cell polarity and defects in PCP-controlled mechanisms, convergent extension, and oriented cell division have been indicated as possible cystogenic triggers. However, it seems likely that ciliary functions and PCP in the cystogenic conversion cannot be clearly separated, as they exert a reciprocal regulation. Docking of centrioles/centrosome to the cortex is essential for the formation of the basal bodies and ciliogenesis as well as for the establishment of the spindle pole position [150]. Therefore, a cystogenic trigger driven by centrosome amplification is also conceivable as the presence of supernumerary centrioles, caused by the depletion or malfunction of different cystic proteins, can produce conflicting cues leading to improper attachment, misalignment of the spindle axis, or altered cilium positioning. These effects may be exposed by the dysregulation of cell cycle progression in cells with amplified centrosomes.
Very important has been the finding that a developmental switch limits the cystogenic susceptibility to ciliary defects to a period of time largely overlapping with the completion of murine renal morphogenesis [91]. Whether PCP mutants are similarly constrained remains to be demonstrated, and experiments with conditional inactivation of PCP genes may provide a clue on whether and how ciliary and PCP functions follow an order of succession in cystogenesis. In the adult, tubular epithelia injury reestablishes the susceptibility to cystogenesis, although it has not been determined whether this depends on the reactivation of the same renal morphogenetic developmental program. Both early development and repair processes are characterized by the need for cell proliferation. Since proliferation shows a biphasic curve, that is, it subsides before starting again in the cystic cells, it cannot be the main cystogenic trigger [94]. Nevertheless, proliferation appears necessary for the trigger to be set off [95].
While we are gaining a better understanding of multiple cellular processes and cell components that play a role in cystogenesis, a unifying pathogenetic mechanism is still missing, largely due to our incomplete knowledge of the workings of polycystic proteins. Further efforts will be necessary to integrate the functions of cilium/centrosome, PCP, and cell proliferation and to determine the sequence of early events that initiate the cystogenic signal.
Acknowledgments
We thank Carlo Iomini and Debbie Hyink for the helpful comments. Work in the authors’ laboratory is supported by the National Institutes of Health (5R01DK78231-2) and the Department of Defense (W81XWH-09-1-0269).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bisceglia M, Galliani CA, Senger C, Stallone C, Sessa A. Renal cystic diseases: a review. Adv Anat Pathol. 2006;13:26–56. doi: 10.1097/01.pap.0000201831.77472.d3. [DOI] [PubMed] [Google Scholar]
- 2.Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, Rudnik-Schöneborn S, Mrug M, Sweeney W, Avner ED, Zerres K, Guay-Woodford LM, Somlo S, Germino GG. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet. 2002;70:1305–1317. doi: 10.1086/340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet. 2002;30:259–269. doi: 10.1038/ng833. [DOI] [PubMed] [Google Scholar]
- 4.Malhas AN, Abuknesha RA, Price RG. Interaction of the leucine-rich repeats of polycystin-1 with extracellular matrix proteins: possible role in cell proliferation. J Am Soc Nephrol. 2002;13:19–26. doi: 10.1681/ASN.V13119. [DOI] [PubMed] [Google Scholar]
- 5.Qian F, Boletta A, Bhunia AK, Xu H, Liu L, Ahrabi AK, Watnick TJ, Zhou F, Germino GG. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc Natl Acad Sci USA. 2002;99:16981–16986. doi: 10.1073/pnas.252484899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei W, Hackmann K, Xu H, Germino G, Qian F. Characterization of cis-autoproteolysis of polycystin-1, the product of human polycystic kidney disease 1 gene. J Biol Chem. 2007;282:21729–21737. doi: 10.1074/jbc.M703218200. [DOI] [PubMed] [Google Scholar]
- 7.Wilson PD. Polycystin: new aspects of structure, function, and regulation. J Am Soc Nephrol. 2001;12:834–845. doi: 10.1681/ASN.V124834. [DOI] [PubMed] [Google Scholar]
- 8.Parnell S, Magenheimer B, Maser R, Rankin C, Smine A, Okamoto T, Calvet J. The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochem Biophys Res Commun. 1998;251:625–631. doi: 10.1006/bbrc.1998.9514. [DOI] [PubMed] [Google Scholar]
- 9.Parnell SC, Magenheimer BS, Maser RL, Zien CA, Frischauf A-M, Calvet JP. Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins. J Biol Chem. 2002;277:19566–19572. doi: 10.1074/jbc.M201875200. [DOI] [PubMed] [Google Scholar]
- 10.Chauvet V, Tian X, Husson H, Grimm DH, Wang T, Hiesberger T, Hieseberger T, Igarashi P, Bennett AM, Ibraghimov-Beskrovnaya O, Somlo S, Caplan MJ. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J Clin Invest. 2004;114:1433–1443. doi: 10.1172/JCI21753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai Y, Maeda Y, Cedzich A, Torres VE, Wu G, Hayashi T, Mochizuki T, Park JH, Witzgall R, Somlo S. Identification and characterization of polycystin-2, the PKD2 gene product. The Journal of biological chemistry. 1999;274:28557–28565. doi: 10.1074/jbc.274.40.28557. [DOI] [PubMed] [Google Scholar]
- 12.Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich BE, Somlo S. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol. 2002;4:191–197. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 13.Alenghat FJ, Nauli SM, Kolb R, Zhou J, Ingber DE. Global cytoskeletal control of mechanotransduction in kidney epithelial cells. Exp Cell Res. 2004;301:23–30. doi: 10.1016/j.yexcr.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Köttgen M. TRPP2 and autosomal dominant polycystic kidney disease. Biochim Biophys Acta. 2007;1772:836–850. doi: 10.1016/j.bbadis.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Tsiokas L, Kim S, Ong E-C. Cell biology of polycystin-2. Cell Signal. 2007;19:444–453. doi: 10.1016/j.cellsig.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheffers MS, Le H, van der Bent P, Leonhard W, Prins F, Spruit L, Breuning MH, de Heer E, Peters DJM. Distinct subcellular expression of endogenous polycystin-2 in the plasma membrane and Golgi apparatus of MDCK cells. Hum Mol Genet. 2002;11:59–67. doi: 10.1093/hmg/11.1.59. [DOI] [PubMed] [Google Scholar]
- 17.Luo Y, Vassilev PM, Li X, Kawanabe Y, Zhou J. Native polycystin 2 functions as a plasma membrane Ca2+-permeable cation channel in renal epithelia. Mol Cell Biol. 2003;23:2600–2607. doi: 10.1128/MCB.23.7.2600-2607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karcher C, Fischer A, Schweickert A, Bitzer E, Horie S, Witzgall R, Blum M. Lack of a laterality phenotype in Pkd1 knock-out embryos correlates with absence of polycystin-1 in nodal cilia. Differentiation. 2005;73:425–432. doi: 10.1111/j.1432-0436.2005.00048.x. [DOI] [PubMed] [Google Scholar]
- 19.Sharif-Naeini R, Folgering JHA, Bichet D, Duprat F, Lauritzen I, Arhatte M, Jodar M, Dedman A, Chatelain FC, Schulte U, Retailleau K, Loufrani L, Patel A, Sachs F, Delmas P, Peters DJM, Honoré E. Polycystin-1 and -2 dosage regulates pressure sensing. Cell. 2009;139:587–596. doi: 10.1016/j.cell.2009.08.045. [DOI] [PubMed] [Google Scholar]
- 20.Tsiokas L. Function and regulation of TRPP2 at the plasma membrane. Am J Physiol Renal Physiol. 2009;297:F1–9. doi: 10.1152/ajprenal.90277.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X-Z, Li Q, Wu Y, Liang G, Lara CJ, Cantiello HF. Submembraneous microtubule cytoskeleton: interaction of TRPP2 with the cell cytoskeleton. FEBS J. 2008;275:4675–4683. doi: 10.1111/j.1742-4658.2008.06616.x. [DOI] [PubMed] [Google Scholar]
- 22.Huan Y, van Adelsberg J. Polycystin-1, the PKD1 gene product, is in a complex containing E-cadherin and the catenins. J Clin Invest. 1999;104:1459–1468. doi: 10.1172/JCI5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geng L, Burrow C, Li H, Wilson P. Modification of the composition of polycystin-1 multiprotein complexes by calcium and tyrosine phosphorylation. Biochim Biophys Acta. 2000;1535:21–35. doi: 10.1016/s0925-4439(00)00079-x. [DOI] [PubMed] [Google Scholar]
- 24.Boca M, D’Amato L, Distefano G, Polishchuk R, Germino G, Boletta A. Polycystin-1 induces cell migration by regulating phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements and GSK3beta-dependent cell cell mechanical adhesion. Mol Biol Cell. 2007;18:4050–4061. doi: 10.1091/mbc.E07-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao H, Sellin LK, Pütz M, Nickel C, Imgrund M, Gerke P, Nitschke R, Walz G, Kramer-Zucker AG. A short carboxy-terminal domain of polycystin-1 reorganizes the microtubular network and the endoplasmic reticulum. Exp Cell Res. 2009;315:1157–1170. doi: 10.1016/j.yexcr.2009.01.027. [DOI] [PubMed] [Google Scholar]
- 26.Hiesberger T, Gourley E, Erickson A, Koulen P, Ward CJ, Masyuk TV, Larusso NF, Harris PC, Igarashi P. Proteolytic cleavage and nuclear translocation of fibrocystin is regulated by intracellular Ca2+ and activation of protein kinase C. The Journal of biological chemistry. 2006;281:34357–34364. doi: 10.1074/jbc.M606740200. [DOI] [PubMed] [Google Scholar]
- 27.Kaimori J-Y, Nagasawa Y, Menezes LF, Garcia-Gonzalez MA, Deng J, Imai E, Onuchic LF, Guay-Woodford LM, Germino GG. Polyductin undergoes notch-like processing and regulated releasefrom primary cilia. Human molecular genetics. 2007;16:942–956. doi: 10.1093/hmg/ddm039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris PC. 2008 Homer W. Smith Award: insights into the pathogenesis of polycystic kidney disease from gene discovery. J Am Soc Nephrol. 2009;20:1188–1198. doi: 10.1681/ASN.2009010014. [DOI] [PubMed] [Google Scholar]
- 30.Zhou J. Polycystins and primary cilia: primers for cell cycle progression. Annu Rev Physiol. 2009;71:83–113. doi: 10.1146/annurev.physiol.70.113006.100621. [DOI] [PubMed] [Google Scholar]
- 31.Chapin HC, Caplan MJ. The cell biology of polycystic kidney disease. J Cell Biol. 2010;191:701–710. doi: 10.1083/jcb.201006173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grantham J, Torres V, Chapman A, Guay-Woodford L, Bae K, King B, Wetzel L, Baumgarten D, Kenney P, Harris P, Klahr S, Bennett W, Hirschman G, Meyers C, Zhang X, Zhu F, Miller J. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354:2122–2130. doi: 10.1056/NEJMoa054341. [DOI] [PubMed] [Google Scholar]
- 33.Grantham JJ, Chapman AB, Torres VE. Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clinical journal of the American Society of Nephrology : CJASN. 2006;1:148–157. doi: 10.2215/CJN.00330705. [DOI] [PubMed] [Google Scholar]
- 34.Nadasdy T, Laszik Z, Lajoie G, Blick KE, Wheeler DE, Silva FG. Proliferative activity of cyst epithelium in human renal cystic diseases. J Am Soc Nephrol. 1995;5:1462–1468. doi: 10.1681/ASN.V571462. [DOI] [PubMed] [Google Scholar]
- 35.Chang MY, Parker E, Ibrahim S, Shortland JR, Nahas ME, Haylor JL, Ong ACM. Haploinsufficiency of Pkd2 is associated with increased tubular cell proliferation and interstitial fibrosis in two murine Pkd2 models. Nephrol Dial Transplant. 2006;21:2078–2084. doi: 10.1093/ndt/gfl150. [DOI] [PubMed] [Google Scholar]
- 36.Takakura A, Contrino L, Zhou X, Bonventre JV, Sun Y, Humphreys BD, Zhou J. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Human molecular genetics. 2009;18:2523–2531. doi: 10.1093/hmg/ddp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delmas P, Nomura H, Li X, Lakkis M, Luo Y, Segal Y, Fernández-Fernández JM, Harris P, Frischauf A-M, Brown DA, Zhou J. Constitutive activation of G-proteins by polycystin-1 is antagonized by polycystin-2. J Biol Chem. 2002;277:11276–11283. doi: 10.1074/jbc.M110483200. [DOI] [PubMed] [Google Scholar]
- 38.Wang X, Ward CJ, Harris PC, Torres VE. Cyclic nucleotide signaling in polycystic kidney disease. Kidney Int. 2010;77:129–140. doi: 10.1038/ki.2009.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mangoo-Karim R, Uchic M, Lechene C, Grantham JJ. Renal epithelial cyst formation and enlargement in vitro: dependence on cAMP. Proc Natl Acad Sci USA. 1989;86:6007–6011. doi: 10.1073/pnas.86.15.6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11:1179–1187. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- 41.Yamaguchi T, Nagao S, Wallace DP, Belibi FA, Cowley BD, Pelling JC, Grantham JJ. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003;63:1983–1994. doi: 10.1046/j.1523-1755.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 42.Sutters M, Yamaguchi T, Maser RL, Magenheimer BS, St John PL, Abrahamson DR, Grantham JJ, Calvet JP. Polycystin-1 transforms the cAMP growth-responsive phenotype of M-1 cells. Kidney Int. 2001;60:484–494. doi: 10.1046/j.1523-1755.2001.060002484.x. [DOI] [PubMed] [Google Scholar]
- 43.Belibi FA, Reif G, Wallace DP, Yamaguchi T, Olsen L, Li H, Helmkamp GM, Grantham JJ. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004;66:964–973. doi: 10.1111/j.1523-1755.2004.00843.x. [DOI] [PubMed] [Google Scholar]
- 44.Montesano R, Ghzili H, Carrozzino F, Rossier BC, Féraille E. cAMP-dependent chloride secretion mediates tubule enlargement and cyst formation by cultured mammalian collecting duct cells. Am J Physiol Renal Physiol. 2009;296:F446–457. doi: 10.1152/ajprenal.90415.2008. [DOI] [PubMed] [Google Scholar]
- 45.Cai Y, Anyatonwu G, Okuhara D, Lee K-B, Yu Z, Onoe T, Mei C-L, Qian Q, Geng L, Wiztgall R, Ehrlich BE, Somlo S. Calcium dependence of polycystin-2 channel activity is modulated by phosphorylation at Ser812. J Biol Chem. 2004;279:19987–19995. doi: 10.1074/jbc.M312031200. [DOI] [PubMed] [Google Scholar]
- 46.Du J, Wilson PD. Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am J Physiol. 1995;269:C487–495. doi: 10.1152/ajpcell.1995.269.2.C487. [DOI] [PubMed] [Google Scholar]
- 47.Orellana SA, Sweeney WE, Neff CD, Avner ED. Epidermal growth factor receptor expression is abnormal in murine polycystic kidney. Kidney Int. 1995;47:490–499. doi: 10.1038/ki.1995.62. [DOI] [PubMed] [Google Scholar]
- 48.Wilson S, Amsler K, Hyink D, Li X, Lu W, Zhou J, Burrow C, Wilson P. Inhibition of HER-2(neu/ErbB2) restores normal function and structure to polycystic kidney disease (PKD) epithelia. Biochim Biophys Acta. 2006;1762:647–655. doi: 10.1016/j.bbadis.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 49.Sweeney WE, Futey L, Frost P, Avner ED. In vitro modulation of cyst formation by a novel tyrosine kinase inhibitor. Kidney Int. 1999;56:406–413. doi: 10.1046/j.1523-1755.1999.00577.x. [DOI] [PubMed] [Google Scholar]
- 50.Sweeney W, Hamahira K, Sweeney J, Garcia-Gatrell M, Frost P, Avner E. Combination treatment of PKD utilizing dual inhibition of EGF-receptor activity and ligand bioavailability. Kidney Int. 2003;64:1310–1319. doi: 10.1046/j.1523-1755.2003.00232.x. [DOI] [PubMed] [Google Scholar]
- 51.Sweeney WE, Chen Y, Nakanishi K, Frost P, Avner ED. Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor. Kidney Int. 2000;57:33–40. doi: 10.1046/j.1523-1755.2000.00829.x. [DOI] [PubMed] [Google Scholar]
- 52.Torres V, Sweeney W, Wang X, Qian Q, Harris P, Frost P, Avner E. EGF receptor tyrosine kinase inhibition attenuates the development of PKD in Han:SPRD rats. Kidney Int. 2003;64:1573–1579. doi: 10.1046/j.1523-1755.2003.00256.x. [DOI] [PubMed] [Google Scholar]
- 53.Torres V, Sweeney W, Wang X, Qian Q, Harris P, Frost P, Avner E. Epidermal growth factor receptor tyrosine kinase inhibition is not protective in PCK rats. Kidney Int. 2004;66:1766–1773. doi: 10.1111/j.1523-1755.2004.00952.x. [DOI] [PubMed] [Google Scholar]
- 54.Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, Walz G, Piontek KB, Germino GG, Weimbs T. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA. 2006;103:5466–5471. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boletta A. Emerging evidence of a link between the polycystins and the mTOR pathways. PathoGenetics. 2009;2:6. doi: 10.1186/1755-8417-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. The Journal of biological chemistry. 2010;285:14071–14077. doi: 10.1074/jbc.R109.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao N, Flynn DC, Zhang Z, Zhong X-S, Walker V, Liu KJ, Shi X, Jiang B-H. G1 cell cycle progression and the expression of G1 cyclins are regulated by PI3K/AKT/mTOR/p70S6K1 signaling in human ovarian cancer cells. Am J Physiol, Cell Physiol. 2004;287:C281–291. doi: 10.1152/ajpcell.00422.2003. [DOI] [PubMed] [Google Scholar]
- 59.Distefano G, Boca M, Rowe I, Claas W, Ma L, Piontek K, Germino G, Pandolfi P, Boletta A. Polycystin-1 Regulates ERKs-dependent Phosphorylation of Tuberin to Control Cell Size through mTOR and its Downstream Effectors S6K and 4EBP1. Mol Cell Biol. 2009 doi: 10.1128/MCB.01259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dere R, Wilson PD, Sandford RN, Walker CL. Carboxy terminal tail of polycystin-1 regulates localization of TSC2 to repress mTOR. PLoS ONE. 2010;5:e9239. doi: 10.1371/journal.pone.0009239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kleymenova E, Ibraghimov-Beskrovnaya O, Kugoh H, Everitt J, Xu H, Kiguchi K, Landes G, Harris P, Walker C. Tuberin-dependent membrane localization of polycystin-1: a functional link between polycystic kidney disease and the TSC2 tumor suppressor gene. Mol Cell. 2001;7:823–832. doi: 10.1016/s1097-2765(01)00226-x. [DOI] [PubMed] [Google Scholar]
- 62.Meijer E, de Jong P, Peters D, Gansevoort R. Better understanding of ADPKD results in potential new treatment options: ready for the cure? J Nephrol. 2008;21:133–138. [PubMed] [Google Scholar]
- 63.Gattone VH, Chen NX, Sinders RM, Seifert MF, Duan D, Martin D, Henley C, Moe SM. Calcimimetic inhibits late-stage cyst growth in ADPKD. J Am Soc Nephrol. 2009;20:1527–1532. doi: 10.1681/ASN.2008090927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Torres VE, Harris PC. Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J Intern Med. 2007;261:17–31. doi: 10.1111/j.1365-2796.2006.01743.x. [DOI] [PubMed] [Google Scholar]
- 65.Chapman AB. Autosomal dominant polycystic kidney disease: time for a change? J Am Soc Nephrol. 2007;18:1399–1407. doi: 10.1681/ASN.2007020155. [DOI] [PubMed] [Google Scholar]
- 66.Shillingford JM, Piontek KB, Germino GG, Weimbs T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J Am Soc Nephrol. 2010;21:489–497. doi: 10.1681/ASN.2009040421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sweeney WE, von Vigier RO, Frost P, Avner ED. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 2008;19:1331–1341. doi: 10.1681/ASN.2007060665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949–952. doi: 10.1038/nature05348. [DOI] [PubMed] [Google Scholar]
- 69.Torres VE. Treatment strategies and clinical trial design in ADPKD. Adv Chronic Kidney Dis. 2010;17:190–204. doi: 10.1053/j.ackd.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, Scheffel H, Weishaupt D, Wüthrich RP. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:820–829. doi: 10.1056/NEJMoa0907419. [DOI] [PubMed] [Google Scholar]
- 71.Walz G, Budde K, Mannaa M, Nürnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Hörl WH, Obermüller N, Arns W, Pavenstädt H, Gaedeke J, Büchert M, May C, Gschaidmeier H, Kramer S, Eckardt K-U. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363:830–840. doi: 10.1056/NEJMoa1003491. [DOI] [PubMed] [Google Scholar]
- 72.Perico N, Antiga L, Caroli A, Ruggenenti P, Fasolini G, Cafaro M, Ondei P, Rubis N, Diadei O, Gherardi G, Prandini S, Panozo A, Bravo RF, Carminati S, De Leon FR, Gaspari F, Cortinovis M, Motterlini N, Ene-Iordache B, Remuzzi A, Remuzzi G. Sirolimus therapy to halt the progression of ADPKD. J Am Soc Nephrol. 2010;21:1031–1040. doi: 10.1681/ASN.2009121302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watnick T, Germino GG. mTOR inhibitors in polycystic kidney disease. N Engl J Med. 2010;363:879–881. doi: 10.1056/NEJMe1006925. [DOI] [PubMed] [Google Scholar]
- 74.Perico N, Remuzzi G. Polycystic kidney disease: Do mTOR inhibitors still have a future in ADPKD? Nat Rev Nephrol. 2010;6:696–698. doi: 10.1038/nrneph.2010.153. [DOI] [PubMed] [Google Scholar]
- 75.Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, Kim B, King BF, Glockner J, Holmes DR, Rossetti S, Harris PC, LaRusso NF, Torres VE. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010;21:1052–1061. doi: 10.1681/ASN.2009121291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151:709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hou X, Mrug M, Yoder BK, Lefkowitz EJ, Kremmidiotis G, D’Eustachio P, Beier DR, Guay-Woodford LM. Cystin, a novel cilia-associated protein, is disrupted in the cpk mouse model of polycystic kidney disease. J Clin Invest. 2002;109:533–540. doi: 10.1172/JCI14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13:2508–2516. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 79.Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 80.Lancaster MA, Gleeson JG. The primary cilium as a cellular signaling center: lessons from disease. Curr Opin Genet Dev. 2009;19:220–229. doi: 10.1016/j.gde.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies, American journal of medical genetics Part C. Seminars in medical genetics. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231. [DOI] [PubMed] [Google Scholar]
- 82.Deltas C, Papagregoriou G. Cystic diseases of the kidney: molecular biology and genetics. Arch Pathol Lab Med. 2010;134:569–582. doi: 10.5858/134.4.569. [DOI] [PubMed] [Google Scholar]
- 83.Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18:1381–1388. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]
- 84.Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science. 2006;313:629–633. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- 85.Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184:71–79. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- 86.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AEH, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 87.Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, Roberts KA, Zhou J. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Molecular and Cellular Biology. 2007;27:3241–3252. doi: 10.1128/MCB.00072-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Low SH, Vasanth S, Larson CH, Mukherjee S, Sharma N, Kinter MT, Kane ME, Obara T, Weimbs T. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev Cell. 2006;10:57–69. doi: 10.1016/j.devcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 89.Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, Yoder BK. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol. 2007;17:1586–1594. doi: 10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lantinga-van Leeuwen IS, Leonhard WN, van der Wal A, Breuning MH, de Heer E, Peters DJM. Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum Mol Genet. 2007;16:3188–3196. doi: 10.1093/hmg/ddm299. [DOI] [PubMed] [Google Scholar]
- 91.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med. 2007;13:1490–1495. doi: 10.1038/nm1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Takakura A, Contrino L, Beck AW, Zhou J. Pkd1inactivation induced in adulthood produces focal cystic disease. J Am Soc Nephrol. 2008;19:2351–2363. doi: 10.1681/ASN.2007101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet. 2008;17:1578–1590. doi: 10.1093/hmg/ddn045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Happé H, Leonhard WN, van der Wal A, van de Water B, Lantinga-van Leeuwen IS, Breuning MH, de Heer E, Peters DJM. Toxic tubular injury in kidneys from Pkd1-deletion mice accelerates cystogenesis accompanied by dysregulated planar cell polarity and canonical Wnt signaling pathways. Hum Mol Genet. 2009;18:2532–2542. doi: 10.1093/hmg/ddp190. [DOI] [PubMed] [Google Scholar]
- 95.Verdeguer F, Le Corre S, Fischer E, Callens C, Garbay S, Doyen A, Igarashi P, Terzi F, Pontoglio M. A mitotic transcriptional switch in polycystic kidney disease. Nature Medicine. 2010;16:106–110. doi: 10.1038/nm.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klein TJ, Mlodzik M. Planar cell polarization: an emerging model points in the right direction. Annu Rev Cell Dev Biol. 2005;21:155–176. doi: 10.1146/annurev.cellbio.21.012704.132806. [DOI] [PubMed] [Google Scholar]
- 97.Lancaster MA, Louie CM, Silhavy JL, Sintasath L, Decambre M, Nigam SK, Willert K, Gleeson JG. Impaired Wnt-beta-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nature Medicine. 2009;15:1046–1054. doi: 10.1038/nm.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Krönig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gresh L, Fischer E, Reimann A, Tanguy M, Garbay S, Shao X, Hiesberger T, Fiette L, Igarashi P, Yaniv M, Pontoglio M. A transcriptional network in polycystic kidney disease. EMBO J. 2004;23:1657–1668. doi: 10.1038/sj.emboj.7600160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Masyuk TV, Huang BQ, Masyuk AI, Ritman EL, Torres VE, Wang X, Harris PC, LaRusso NF. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol. 2004;165:1719–1730. doi: 10.1016/S0002-9440(10)63427-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fischer E, Legue E, Doyen A, Nato F, Nicolas J-F, Torres V, Yaniv M, Pontoglio M. Defective planar cell polarity in polycystic kidney disease. Nat Genet. 2006;38:21–23. doi: 10.1038/ng1701. [DOI] [PubMed] [Google Scholar]
- 102.Saburi S, Hester I, Fischer E, Pontoglio M, Eremina V, Gessler M, Quaggin SE, Harrison R, Mount R, McNeill H. Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nature Genetics. 2008;40:1010–1015. doi: 10.1038/ng.179. [DOI] [PubMed] [Google Scholar]
- 103.Park TJ, Haigo SL, Wallingford JB. Ciliogenesis defects in embryos lacking inturned or fuzzy function are associated with failure of planar cell polarity and Hedgehog signaling. Nature Genetics. 2006;38:303–311. doi: 10.1038/ng1753. [DOI] [PubMed] [Google Scholar]
- 104.Borovina A, Superina S, Voskas D, Ciruna B. Vangl2 directs the posterior tilting and asymmetric localization of motile primary cilia. Nat Cell Biol. 2010;12:407–412. doi: 10.1038/ncb2042. [DOI] [PubMed] [Google Scholar]
- 105.Kotsis F, Nitschke R, Doerken M, Walz G, Kuehn EW. Flow modulates centriole movements in tubular epithelial cells. Pflugers Arch. 2008;456:1025–1035. doi: 10.1007/s00424-008-0475-8. [DOI] [PubMed] [Google Scholar]
- 106.Guirao B, Meunier A, Mortaud S, Aguilar A, Corsi J-M, Strehl L, Hirota Y, Desoeuvre A, Boutin C, Han Y-G, Mirzadeh Z, Cremer H, Montcouquiol M, Sawamoto K, Spassky N. Coupling between hydrodynamic forces and planar cell polarity orients mammalian motile cilia. Nat Cell Biol. 2010;12:341–350. doi: 10.1038/ncb2040. [DOI] [PubMed] [Google Scholar]
- 107.Karner CM, Chirumamilla R, Aoki S, Igarashi P, Wallingford JB, Carroll TJ. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nature Genetics. 2009;41:793–799. doi: 10.1038/ng.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nishio S, Tian X, Gallagher AR, Yu Z, Patel V, Igarashi P, Somlo S. Loss of Oriented Cell Division Does not Initiate Cyst Formation. J Am Soc Nephrol. 2009 doi: 10.1681/ASN.2009060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- 110.Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S, Badano JL, Katsanis N. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nature Genetics. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- 111.Doxsey S, Zimmerman W, Mikule K. Centrosome control of the cell cycle. Trends Cell Biol. 2005;15:303–311. doi: 10.1016/j.tcb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 112.Hoffmann I. Linking the cell cycle to the centrosome cycle. 2005 [Google Scholar]
- 113.Meraldi P, Nigg E. The centrosome cycle. FEBS Lett. 2002;521:9–13. doi: 10.1016/s0014-5793(02)02865-x. [DOI] [PubMed] [Google Scholar]
- 114.Follit JA, Tuft RA, Fogarty KE, Pazour GJ. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol Biol Cell. 2006;17:3781–3792. doi: 10.1091/mbc.E06-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jurczyk A, Gromley A, Redick S, San Agustin J, Witman G, Pazour GJ, Peters DJM, Doxsey S. Pericentrin forms a complex with intraflagellar transport proteins and polycystin-2 and is required for primary cilia assembly. J Cell Biol. 2004;166:637–643. doi: 10.1083/jcb.200405023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Robert A, Margall-Ducos G, Guidotti J-E, Brégerie O, Celati C, Bréchot C, Desdouets C. The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. Journal of Cell Science. 2007;120:628–637. doi: 10.1242/jcs.03366. [DOI] [PubMed] [Google Scholar]
- 117.Srsen V, Gnadt N, Dammermann A, Merdes A. Inhibition of centrosome protein assembly leads to p53-dependent exit from the cell cycle. J Cell Biol. 2006;174:625–630. doi: 10.1083/jcb.200606051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mikule K, Delaval B, Kaldis P, Jurcyzk A, Hergert P, Doxsey S. Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat Cell Biol. 2007;9:160–170. doi: 10.1038/ncb1529. [DOI] [PubMed] [Google Scholar]
- 119.Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude G. Abnormal centrosome amplification in the absence of p53. Science. 1996;271:1744–1747. doi: 10.1126/science.271.5256.1744. [DOI] [PubMed] [Google Scholar]
- 120.Tarapore P, Fukasawa K. Loss of p53 and centrosome hyperamplification. Oncogene. 2002;21:6234–6240. doi: 10.1038/sj.onc.1205707. [DOI] [PubMed] [Google Scholar]
- 121.Tarapore P, Horn H, Tokuyama Y, Fukasawa K. Direct regulation of the centrosome duplication cycle by the p53-p21Waf1/Cip1 pathway. Oncogene. 2001;20:3173–3184. doi: 10.1038/sj.onc.1204424. [DOI] [PubMed] [Google Scholar]
- 122.Shinmura K, Bennett R, Tarapore P, Fukasawa K. Direct evidence for the role of centrosomally localized p53 in the regulation of centrosome duplication. Oncogene. 2007;26:2939–2944. doi: 10.1038/sj.onc.1210085. [DOI] [PubMed] [Google Scholar]
- 123.Bhunia A, Piontek K, Boletta A, Liu L, Qian F, Xu P, Germino F, Germino G. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell. 2002;109:157–168. doi: 10.1016/s0092-8674(02)00716-x. [DOI] [PubMed] [Google Scholar]
- 124.Li X, Luo Y, Starremans PG, McNamara CA, Pei Y, Zhou J. Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat Cell Biol. 2005;7:1202–1212. doi: 10.1038/ncb1326. [DOI] [PubMed] [Google Scholar]
- 125.Kim H, Bae Y, Jeong W, Ahn C, Kang S. Depletion of PKD1 by an antisense oligodeoxynucleotide induces premature G1/S-phase transition. Eur J Hum Genet. 2004;12:433–440. doi: 10.1038/sj.ejhg.5201136. [DOI] [PubMed] [Google Scholar]
- Battini L, Fedorova E, Macip S, Li X, Wilson PD, Gusella GL. Stable knockdown of polycystin-1 confers integrin-α2β1-mediated anoikis resistance. J Am Soc Nephrol. 2006;17:3049–3058. doi: 10.1681/ASN.2006030234. [DOI] [PubMed] [Google Scholar]
- 127.Nishio S, Hatano M, Nagata M, Horie S, Koike T, Tokuhisa T, Mochizuki T. Pkd1 regulates immortalized proliferation of renal tubular epithelial cells through p53 induction and JNK activation. J Clin Invest. 2005;115:910–918. doi: 10.1172/JCI22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Battini L, Macip S, Fedorova E, Dikman S, Somlo S, Montagna C, Gusella GL. Loss of polycystin-1 causes centrosome amplification and genomic instability. Human molecular genetics. 2008;17:2819–2833. doi: 10.1093/hmg/ddn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Castedo M, Perfettini J, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 130.Castedo M, Perfettini J, Roumier T, Valent A, Raslova H, Yakushijin K, Horne D, Feunteun J, Lenoir G, Medema R, Vainchenker W, Kroemer G. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene. 2004;23:4362–4370. doi: 10.1038/sj.onc.1207572. [DOI] [PubMed] [Google Scholar]
- 131.Burtey S, Riera M, Ribe E, Pennenkamp P, Rance R, Luciani J, Dworniczak B, Mattei MG, Fontés M. Centrosome overduplication and mitotic instability in PKD2 transgenic lines. Cell Biol Int. 2008;32:1193–1198. doi: 10.1016/j.cellbi.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 132.Zhang J, Wu M, Wang S, Shah J, Wilson PD, Zhou J. Polycystic kidney disease protein fibrocystin localizes to the mitotic spindle and regulates spindle bipolarity. Human molecular genetics. 2010 doi: 10.1093/hmg/ddq233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rundle DR, Gorbsky G, Tsiokas L. PKD2 interacts and co-localizes with mDia1 to mitotic spindles of dividing cells: role of mDia1 IN PKD2 localization to mitotic spindles. The Journal of biological chemistry. 2004;279:29728–29739. doi: 10.1074/jbc.M400544200. [DOI] [PubMed] [Google Scholar]
- 134.Palazzo AF, Cook TA, Alberts AS, Gundersen GG. mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat Cell Biol. 2001;3:723–729. doi: 10.1038/35087035. [DOI] [PubMed] [Google Scholar]
- 135.Li Q, Montalbetti N, Shen PY, Dai X-Q, Cheeseman CI, Karpinski E, Wu G, Cantiello HF, Chen X-Z. Alpha-actinin associates with polycystin-2 and regulates its channel activity. Human molecular genetics. 2005;14:1587–1603. doi: 10.1093/hmg/ddi167. [DOI] [PubMed] [Google Scholar]
- 136.Li Q, Montalbetti N, Wu Y, Ramos A, Raychowdhury MK, Chen X-Z, Cantiello HF. Polycystin-2 cation channel function is under the control of microtubular structures in primary cilia of renal epithelial cells. J Biol Chem. 2006;281:37566–37575. doi: 10.1074/jbc.M603643200. [DOI] [PubMed] [Google Scholar]
- 137.Hanaoka K, Qian F, Boletta A, Bhunia A, Piontek K, Tsiokas L, Sukhatme V, Guggino W, Germino G. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature. 2000;408:990–994. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- 138.De Boer L, Oakes V, Beamish H, Giles N, Stevens F, Somodevilla-Torres M, Desouza C, Gabrielli B. Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene. 2008;27:4261–4268. doi: 10.1038/onc.2008.74. [DOI] [PubMed] [Google Scholar]
- 139.Mitsuyama F, Sawai T. The redistribution of Ca2+ stores with inositol 1,4,5-trisphosphate receptor to the cleavage furrow in a microtubule-dependent manner. Int J Dev Biol. 2001;45:861–868. [PubMed] [Google Scholar]
- 140.Matsumoto Y, Maller JL. Calcium, calmodulin, and CaMKII requirement for initiation of centrosome duplication in Xenopus egg extracts. Science. 2002;295:499–502. doi: 10.1126/science.1065693. [DOI] [PubMed] [Google Scholar]
- 141.Qian Q, Li M, Cai Y, Ward C, Somlo S, Harris P, Torres V. Analysis of the polycystins in aortic vascular smooth muscle cells. J Am Soc Nephrol. 2003;14:2280–2287. doi: 10.1097/01.asn.0000080185.38113.a3. [DOI] [PubMed] [Google Scholar]
- 142.Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc Natl Acad Sci USA. 2007;104:6454–6459. doi: 10.1073/pnas.0610324104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Aguiari G, Trimi V, Bogo M, Mangolini A, Szabadkai G, Pinton P, Witzgall R, Harris PC, Borea PA, Rizzuto R, del Senno L. Novel role for polycystin-1 in modulating cell proliferation through calcium oscillations in kidney cells. Cell Prolif. 2008;41:554–573. doi: 10.1111/j.1365-2184.2008.00529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Uhlén P, Fritz N. Biochemistry of calcium oscillations. Biochemical and biophysical research communications. 2010;396:28–32. doi: 10.1016/j.bbrc.2010.02.117. [DOI] [PubMed] [Google Scholar]
- 145.Astrinidis A, Senapedis W, Henske EP. Hamartin, the tuberous sclerosis complex 1 gene product, interacts with polo-like kinase 1 in a phosphorylation-dependent manner. Hum Mol Genet. 2006;15:287–297. doi: 10.1093/hmg/ddi444. [DOI] [PubMed] [Google Scholar]
- 146.Tammachote R, Hommerding CJ, Sinders RM, Miller CA, Czarnecki PG, Leightner AC, Salisbury JL, Ward CJ, Torres VE, Gattone VH, Harris PC. Ciliary and centrosomal defects associated with mutation and depletion of the Meckel syndrome genes MKS1 and MKS3. Human molecular genetics. 2009;18:3311–3323. doi: 10.1093/hmg/ddp272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jonassen JA, San Agustin J, Follit JA, Pazour GJ. Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J Cell Biol. 2008;183:377–384. doi: 10.1083/jcb.200808137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Brasier JL, Henske EP. Loss of the polycystic kidney disease (PKD1) region of chromosome 16p13 in renal cyst cells supports a loss-of-function model for cyst pathogenesis. J Clin Invest. 1997;99:194–199. doi: 10.1172/JCI119147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gogusev J, Murakami I, Doussau M, Telvi L, Stojkoski A, Lesavre P, Droz D. Molecular cytogenetic aberrations in autosomal dominant polycystic kidney disease tissue. J Am Soc Nephrol. 2003;14:359–366. doi: 10.1097/01.asn.0000046963.60910.63. [DOI] [PubMed] [Google Scholar]
- 150.Marshall WF. Basal bodies platforms for building cilia. Curr Top Dev Biol. 2008;85:1–22. doi: 10.1016/S0070-2153(08)00801-6. [DOI] [PubMed] [Google Scholar]