

Abstract
Mitochondrial apoptosis-inducing factor (AIF) is a FAD-containing protein that under certain conditions translocates to the nucleus and causes a programmed cell death, apoptosis. The apoptogenic action of AIF is redox controlled as the NADH-reduced AIF dimer has lower affinity for DNA than the oxidized monomer. To gain further insights into the mechanism of AIF, we investigated its interaction with a series of quinone oxidants, including a number of anticancer quinones. Our data indicate that the NADH:quinone oxidoreduction catalyzed by AIF follows a “ping-pong” scheme, with the reductive half-reaction being rate-limiting and the FADH--NAD+ charge-transfer complex serving as an electron donor. AIF is equally reactive toward benzo- and naphthoquinones, but may discriminate structures with a higher number of aromatic rings. The reactivity of quinones is mainly defined by their one-electron reduction potential, whereas the size and nature of the substituents play a minor role. AIF is unlikely to significantly contribute to bioreductive activation of low-potential quinoidal anticancer quinones. However, high-potential quinones, e.g. a toxic natural compound naphthazarin, maintain AIF in the oxidized state when a significant excess of NADH is present. Thus, these compounds may prevent the accumulation of the reduced form of AIF in vivo, and enhance AIF-mediated apoptosis.
Keywords: Apoptosis-inducing factor, quinone, apoptosis induction, oxidative stress, bioreductive activation
Apoptosis-inducing factor (AIF) is a FAD-containing protein tethered to the inner membrane of mitochondria in normal cells. There is compelling evidence that AIF plays a vital role in mitochondrial bioenergetics but its specific redox function is still unknown [1]. Another well established role of AIF is induction of caspase-independent programmed cell death, apoptosis [2,3]. Upon outer mitochondrial membrane permeabilization, mature AIF undergoes proteolysis [4-6] and the truncated fragment (AIF(Δ1-101/102)) translocates to the cytosol and subsequently to the nucleus to trigger chromatin condensation and DNA degradation [2,7,8].
AIF has a glutathione reductase (GR) fold and is most similar to bacterial oxygenase-coupled ferredoxin reductases [9,10]. This and the presence of the FAD cofactor suggested that AIF is a redox active protein. The earlier studies showed that when refolded and reconstituted with FAD, AIF possesses modest NAD(P)H:acceptor oxidoreductase activities, but the flavin cofactor is not required for the apoptotic action [2,3,9,11]. However, recent functional and structural studies performed on naturally folded AIF demonstrated that its oligomeric and conformational states change upon FAD oxidoreduction, which in turn could affect the apoptogenic action [12, 13]. In particular, it was found that reduction of AIF by NAD(P)H leads to formation of a dimer with a tight FADH--NAD(P)+ charge-transfer (CT) complex, resistant to oxidation by oxygen and interacting with DNA weaker than the oxidized monomer. Furthermore, the CT complex has a differently folded regulatory loop and an inaccessible nuclear leading sequence, through which AIF is predominantly transported to the nucleus. Altogether, these findings indicated that the apoptogenic function of AIF might be redox controlled [12,13]. It is important therefore to further investigate the redox properties and reactions catalyzed by AIF, especially with the redox active cytotoxic compounds, as they may affect the cell-death-inducing potency of the flavoprotein.
Since translocation of AIF from mitochondria is facilitated under the ‘oxidative stress’ conditions [14,15], it is logical to suggest that interaction of AIF with reactive oxidative species could shift the redox equilibrium toward the oxidized monomeric state, thereby enhancing proapoptotic properties of AIF. This possibility prompted us to reexamine reactions of AIF with quinoidal compounds, which have previously been poorly and randomly characterized [2,3,9,11,12], and include in the study a series of quinoidal antitumour agents. It is well known that quinones can become toxic upon reduction by flavoenzymes, and induce apoptotic and necrotic cell death [16-18]. The ‘oxidative stress’-type cytotoxicity arises from the redox cycling of their radical forms produced after one-electron reduction by flavin-containing electrontransferases, e.g. NADPH:cytochrome P450 reductase [16,19,20]. On the other hand, two-electron reduction of antitumour aziridinyl-substituted quinones by mammalian NAD(P)H: quinone oxidoreductase (NQO1), leads to formation of the corresponding hydroquinones that alkylate DNA more readily than the parent oxidized compounds [20,21]. Apart from the possible role of the aforementioned reactions in AIF-mediated apoptosis, investigation of the AIF-quinone interaction is interesting from a mechanistic viewpoint, because the reduced active center of AIF contains a tightly bound NAD+ (Kd = 5–80 nM [12], and Churbanova and Sevrioukova, unpublished data) which is sandwiched between the near-coplanar isoalloxazine ring of FADH- and aromatic ring of Phe309 [13]. This makes AIF an interesting experimental model for studying flavin-dependent transformations of redox active compounds.
In this study, we characterized the quinone reductase mechanism and substrate specificity of a model form (AIF(Δ1-77)) of mature mouse AIF, which possesses the same redox potential and NADH:acceptor reductase activities as a recombinant mature AIF [12], by measuring steady- and pre-steady-state kinetics of electron transfer to various quinones, including toxic natural compounds and quinoidal antitumour agents with different electron-accepting potency and steric parameters.
Materials and methods
Expression and purification of recombinant mouse AIF(Δ1-77) were carried out as described previously [12]. AIF concentration was determined spectrophotometrically using ε452 = 13.2 mM-1cm-1. NADH, NAD+, cytochrome c, superoxide dismutase, ubiquinone-6, and model quinones (compounds 1, 2, 4-6, 10-15, 18-21, 24; Fig. 1) were obtained from Sigma-Aldrich. Daunorubicin was from Minmedprom (Russia). Antitumour aziridinyl-substituted 1,4-benzoquinones AZQ, MeDZQ, RH1 and BZQ (compounds 7, 16, 17, 23; Fig. 1) were synthesized as described previously [21-23]. t-Butyl-substituted 1,4-benzoquinones (compounds 3, 8, 9; Fig. 1) were synthesized as reported elsewhere [24,25]. The purity of synthesized compounds was checked according to their melting point, elemental analysis, IR and NMR spectra.
Fig. 1.

Chemical structures of the studied compounds.
Steady-state kinetic measurements were carried out in 0.1 M K-phosphate (pH 7.0) containing 1 mM EDTA at 25 °C using a Hitachi-557 UV-VIS spectrophotometer. The reaction was monitored spectrophotometrically by following consumption of 200 μM NADH (Δε340 = 6.2 mM-1 cm-1) at various electron acceptor concentrations in 1.0 cm optical path cells. In some cases, higher concentrations of NADH and 0.2 cm optical path cells were used. Experiments with ubiquinone-6 (50 μM maximal concentration) were performed in the presence of 0.1% Triton X-100. Reduction of 50 μM cytochrome c was monitored at 550 nm (Δε550 = 20 mM-1 cm-1). Catalytic (kcat) and bimolecular rate constants (kcat/Km) for the quinone reduction at fixed NADH concentrations, or NADH oxidation at fixed quinone concentrations correspond to the reciprocal intercepts and slopes of the Lineweaver-Burk plots, [E]/v vs. 1/[oxidant] or [E]/v vs. [NADH], where [E] is the enzyme concentration, and v is the reaction rate. kcat is the number of NADH molecules oxidized by a single enzyme molecule per second. The calculated rates were corrected for the intrinsic NADH-oxidase activity of AIF, 0.0003 s-1. In case of 1,4-benzoquinone and 2-methyl-1,4-benzoquinone, the reaction rates were also corrected for non-enzymatic oxidation of NADH by these compounds. In control experiments, kinetic measurements were performed in a sealed anaerobic cuvette under argon atmosphere. Kinetic parameters were obtained by Quasi-Newton or Simplex-Quasi-Newton nonlinear estimation methods using Statistica (version 4.30, StatSoft, 1993).
Pre-steady-state kinetic studies on AIF were performed under aerobic conditions at 25 °C in 0.1 M K-phosphate (pH 7.0) containing 1 mM EDTA, using a SX.17 MV stopped-flow spectrophotometer (Applied Photophysics). Absorbance changes were monitored at 452 and 700 nm to follow FAD reduction and FADH--NAD+ charge-transfer complex formation, respectively [12]. In the AIF reduction experiments, 40-50 μM protein was rapidly mixed with an equal volume of an excess of NADH, and the absorbance changes at 452 and 700 nm were recorded. In the AIF reoxidation experiments, 30-50 μM AIF was first mixed with sub-equimolar NADH (95 %) and when the 452 and 700 nm absorbance stabilized (35-40 min), the reduced AIF-NAD+ complex was rapidly mixed with an equal volume of 2,6-dimethyl-1,4-benzoquinone solution, and absorbance changes at 452 and 700 nm were recorded. In multiple turnover experiments, AIF was rapidly mixed with an equal volume of solution containing both NADH and quinone. Control reoxidation experiments were performed under anaerobic conditions, where the solutions of quinones and NADH before the introduction of AIF were purged with ultrapure argon for 60 min and loaded into the stopped flow apparatus in gas-tight syringes. Kinetic data were analyzed using the Applied Photophysics software.
Results
Reduction of AIF by NADH is accompanied by a decrease in absorbance at 452 nm and appearance of a long-wavelength absorbance band (Fig. 2), indicative of the FADH--NAD+ CT complex formation. The latter species is produced as a result of π-π interactions between the reduced isoalloxazine ring of FAD and electron-defficient nicotinamide ring of the reaction product, NAD+. This type of spectra has been observed for the FADH- -NAD(P)+ complexes of reduced lipoamide dehydrogenase and glutathione reductase, formed in the presence of alkylating agents which modify the catalytic thiols of the aforementioned enzymes [26]. The semiquinone form of AIF is thermodynamically unstable and does not accumulate during the reoxidation of NADH-reduced AIF at pH 7.0 [12]. However, during the reoxidation of reduced AIF-NAD+ complex by oxygen at pH ≥8.0, a small amount (10-15%) of anionic semiquinone (FAD.-, λ max = 380 nm) was formed (Churbanova and Sevrioukova, unpublished data). Due to high affinty of reduced AIF for NAD+ (Kd = 5-80 nM [12]; Churbanova and Sevrioukova, unpublished data) and low reactivity of the FADH--NAD+ species toward oxygen (half-lifetime under air atmosphere >120 min [12]), all experiments described below were performed under aerobic conditions, unless specified otherwise.
Fig. 2.

Absorbance spectra of 14 μM AIF recorded before (1) and after (2) addition of 140 μM NADH in 100 mM K-phosphate, pH 7.0, 1 mM EDTA at 25 °C.
Steady-State Kinetics
Steady-state reduction of 2,6-dimethyl-1,4-benzoquinone by AIF follows a “ping-pong” scheme, as evidenced by parallel Lineweaver-Burk plots at fixed concentrations of NADH and different quinone concentrations (Fig. 3). The calculated bimolecular rate constant (kcat/Km) of AIF reduction by NADH was equal to 280±20 M-1s-1, whereas the kcat value at saturating concentrations of NADH and quinone oxidant was estimated to be 0.24±0.02 s-1, consistent with the previous findings [12]. The reaction rates measured under anaerobic conditions were the same, whithin experimental error.
Fig. 3.

Lineweaver-Burk plots of the initial rate constants for NADH oxidation catalyzed by AIF, which were determined at different concentrations of 2,6-dimethyl-1,4-benzoquinone and 216 μM (1), 320 μM (2), 466 μM (3), and 700 μM (4) NADH. Kinetic measurements were conducted in 100 mM K-phosphate, pH 7.0, 1 mM EDTA at 25 °C.
In order to evaluate the specificity of AIF for quinones, we examined a series of compounds with a broad range of quinone/semiquinone redox potential (E17, 0.09 ÷ -0.41 V), and different number of aromatic rings (benzo-, naphtho-, and anthraquinones) and size of substituents, as reflected by the Van der Waals volume (VdWvol) (Fig. 1, Table 1). The studied compounds included antitumour aziridinyl-substituted 1,4-benzoquinones AZQ, BZQ, MeDZQ, and RH1, and daunorubicin, as well as quinoidal products of oxidation of t-butyl-substituted-1,4-hydroquinone antioxidants (Fig. 1, Table 1). The presence of high concentrations of NAD+ (5.0 mM) had a weak inhibiting effect, decreasing kcat/Km for the quinone reduction by less than 15%. When plotted against the E17 value for benzo- and naphthoquinones (Fig. 4), log kcat/Km linearly increased with an increase in the electron-accepting potency of the compounds (r2 = 0.776, F(1.18) = 62.34). On the other hand, tricyclic anthraquinones and daunorubicin were less reactive toward AIF as might be expected based on their E17 values (Fig. 4). Comparison of reactivities of RH1, MeDZQ, and 1,4-dihydroxy-9,10-anthraquinone, which have similar Van der Waals volumes (Table 1), suggests that the low reactivity of tricyclic quinones is not attributed to their larger size. However, when VdWvol was used as a second variable in a two-parameter regression analysis, statistics for the kinetic data on benzo- and naphthoquinones (compounds 1-17, 19, 23, 24; Table 1) was slightly improved (r2 = 0.782, F(2.17) = 35.06) and a weak negative influence of VdWvol on their reactivity became evident:
Table 1. Molecular and redox properties of the quinoidal compounds.
One-electron reduction potentials (E17) and Van der Waals volumes (VdWvol) of the investigated quinones were determined previously [16-19]. The catalytic (kcat) and second order (kcat/Km) rate constants for the AIF-catalyzed quinone reduction reactions were measured in the presence of 200 μM NADH as described in Materials and Methods.
| No. | Compound | kcat/Km (M-1 s-1 | kcat (s-1) | E17 (V) | VdWvol (Ǻ3) |
|---|---|---|---|---|---|
| 1. | 1,4-Benzoquinone | (7.6 ± 0.8) × 103 | 0.030 ± 0.005 | 0.09 | 124 |
| 2. | 2-Methyl-1,4-benzoquinone | (7.3 ± 0.7) × 103 | 0.041 ± 0.003 | 0.01 | 143 |
| 3. | 2-t-Butyl-1,4-benzoquinone | (1.3 ± 0.1) × 104 | 0.040 ± 0.004 | 0.01 | 194 |
| 4. | 2,5-Dimethyl-1,4-benzoquinone | (2.9 ± 0.4) × 103 | 0.030 ± 0.003 | -0.07 | 161 |
| 5. | 2,3-Dimethyl-1,4-benzoquinone | (1.0 ± 0.1) × 104 | 0.042 ± 0.002 | -0.07 | 161 |
| 6. | 2,6-Dimethyl-1,4-benzoquinone | (1.2 ± 0.2) × 104 | 0.044 ± 0.001 | -0.07 | 161 |
| 7. | 2,5-Bis(carboethoxyamino)- 3,6-diaziridinyl-1,4-benzoquinone (AZQ) | (1.7 ± 0.2) × 103 | n.d. | -0.07 | 361 |
| 8. | 2,5-Di-t-butyl-1,4-benzoquinone | (1.2 ± 0.1) × 103 | 0.025 ± 0.003 | -0.08 | 264 |
| 9. | 2,6-Di-t-butyl-1,4-benzoquinone | (9.2 ± 1.0) × 102 | 0.050 ± 0.004 | -0.08 | 264 |
| 10. | 5-Hydroxy-1,4-naphthoquinone | (2.0 ± 0.4) × 103 | 0.047 ± 0.003 | -0.09 | 180 |
| 11. | 5,8-Dihydroxy-1,4-naphthoquinone | (3.9 ± 0.3) × 103 | 0.044 ± 0.001 | -0.11 | 192 |
| 12. | 1,4-Naphthoquinone | (1.5 ± 0.2) × 103 | 0.037 ± 0.002 | -0.15 | 168 |
| 13. | 2-Methyl-5-hydroxy-1,4-naphthoquinone | (1.1 ± 0.1) × 103 | 0.041 ± 0.004 | -0.16 | 200 |
| 14. | Trimethyl-1,4-benzoquinone | (1.7 ± 0.1) × 103 | 0.054 ± 0.003 | -0.17 | 178 |
| 15. | 2-Methyl-1,4-naphthoquinone | (4.7 ± 0.3) × 102 | n.d. | -0.20 | 187 |
| 16. | 2,5-Diaziridinyl -3-hydroxymethyl-6-methyl-1,4-benzoquinone (RH1) | (4.3 ± 0.1) × 102 | n.d. | -0.23 | 247 |
| 17. | 2,5-Dimethyl-3,6-diaziridinyl-1,4-benzoquinone (MeDZQ) | (2.3 ± 0.3) × 103 | n.d. | -0.23 | 238 |
| 18. | 1,4-Dihydroxy-9,10-anthraquinone | < 10 | n.d. | -0.25 | 245 |
| 19. | Tetramethyl-1,4-benzoquinone | (1.5 ± 0.1) × 102 | n.d. | -0.26 | 198 |
| 20. | 1,8-Dihydroxy-9,10-anthraquinone | < 10 | n.d. | -0.33 | 245 |
| 22. | Daunorubicin | < 10 | n.d. | -0.34 | 600 |
| 23. | 2,5-Bis(2-hydroxyethylamino)-3,6-diaziridinyl-1,4-benzoquinone (BZQ) | (3.9 ± 0.4) × 101 | n.d. | -0.38 | 327 |
| 24. | 2-Hydroxy-1,4-naphthoquinone | (6.2 ± 0.1) × 101 | n.d. | - |
n.d. - not determined.
Fig. 4.

Dependence of the reactivity of quinones (log kcat/Km) toward AIF on their one-electron reduction potential (E17). Quinone numbering is according to Figure 1. The regression line is a linear fit reflecting correlation between the reactivities and E17 values of benzo- and naphthoquinones.
| (1) |
Similarly, a large VdWvol might be a reason for the lack of reactivity of ubiquinone-6 (E17 = -0.24 V, VdWvol = 727 Ǻ3), which we tested as a possible physiological oxidant of AIF. It can be concluded, therefore, that (i) AIF is equally specific toward benzo- and naphthoquinones but may discriminate structures with a higher number of aromatic rings, and (ii) the reactivity of benzo- and naphthoquinones is mainly defined by their oxidative potency, while the size and nature of their substituents play a less important role. Similar log k vs. E17 dependences are characteristic for a number of flavoenzymes which reduce quinones either in one-electron way (NADPH:cytochrome P-450 reductase, ferredoxin:NADP+ reductase), or in two-electron way (Enterobacter cloacae nitroreductase, E. cloacae PB2 pentaerythritol reductase) [20,27].
Quantitatively, the percentage of a one-electron flux during NAD(P)H:quinone oxidoreduction can be estimated using the 1,4-benzoquinone-mediated enzymatic reduction of cytochrome c. At pH < 7.2, cytochrome c is reduced by 1,4-benzohydroquinone with a negligibly low rate, whereas 1,4-benzosemiquinone delivers electrons to the hemoprotein very rapidly (k = 106 M-1s-1 [28]). Hence, the percentage of a one-electron flux can be expressed as a ratio of the cytochrome c reduction rate and the doubled rate of enzymatic oxidation of NAD(P)H in the presence of 1,4-benzoquinone [28]. AIF was found to reduce quinones in a mixed one- and two-electron way, with 34±5.0 % of a one-electron flux.
Pre-steady-state Kinetics
We also examined the pre-steady-state kinetics of AIF reactions. A 452 nm absorbance decrease and an increase at 700 nm observed during reduction of AIF by NADH (Fig. 2) proceed with similar rates (Fig. 5A). The rate constant for AIF reduction reaches the limiting value at high concentrations of NADH (Fig. 5B), which was equal to 0.31±0.03 s-1 under studied conditions. The estimated apparent bimolecular reduction rate constant was 251±20 M-1s-1, close to the kcat/Km value derived from the steady-state kinetic data (Fig. 3). Besides, the rate constant for the pre-steady-state AIF reduction with 200 μM NADH (Fig. 5B) is in the same range as the kcat values for the quinone reduction reactions measured under steady-state conditions with a similar NADH concentration (Table 1). Again, the presence of 5.0 mM NAD+ had no feasible effect on the kinetics of AIF reduction (Fig. 5B).
Fig. 5.

Pre-steady-state kinetics of AIF reduction by NADH. A) Kinetic traces of the absorbance change at 452 (1) and 700 nm (2) observed during reduction of 22 μM AIF by 200 μM NADH. The obtained first-order reduction rate constants were 0.034±0.002 s-1 (1) and 0.0372±0.027 s-1 (2). B) Dependence of the first-order rate constants for AIF reduction on the concentration of NADH in the absence (open circles) and presence of 5.0 mM NAD+ (filled circles).
During pre-steady-state oxidation of reduced AIF by 2,6-dimethyl-1,4-benzoquinone, an increase in absorbance at 452 nm and a decrease at 700 nm proceed with similar rates (Fig. 6A). This means that NAD+ dissociation/CT complex decay occur simultaneously with the FAD oxidation. The rate constant for AIF oxidation depends linearly on the 2,6-dimethyl-1,4-benzoquinone concentration, reaching values that exceed the maximal rate for the NADH-dependent AIF reduction by one order of magnitude (Figs. 5B and 6B). The bimolecular oxidation rate constant for the 2,6-dimethyl-1,4-benzoquinone, in turn, is close to the corresponding value obtained in the steady-state kinetic experiments (8.0±0.5 × 103 M-1s-1 and 1.2 ±0.5 × 104 M-1s-1, respectively). Although formation of the FAD semiquinone may be anticipated during the single-electron oxidation of AIF, we observed neither a biphasic nature of the absorbance changes nor a transient increase at 380 nm, indicative of formation of FAD.- as a reaction intermediate. The reoxidation reactions carried out under anaerobic conditions gave identical within error results at 452, 700, and 380 nm, comparing to those obtained under aerobic conditions.
Fig. 6.

Pre-steady-state kinetics of AIF oxidation by 2,6-dimethyl-1,4-bezoquinone. A) Kinetic traces of the absorbance change at 452 (1,3) and 700 nm (2) during oxidation of 14 μM AIF by 100 μM 2,6-dimethyl-1,4-benzoquinone (1,2), or after mixing of reduced AIF with a pure buffer solution (3). The obtained first-order oxidation rate constants were 0.890±0.002 s-1 (1) and 1.013±0.012 s-1 (2). B) Dependence of the observed rate constant for AIF oxidation on the concentration of 2,6-dimethyl-1,4-benzoquinone.
Next we examined how the AIF redox state changes during turnover with NADH under conditions imitating the cytotoxic action of quinones in mammalian cells. It was found that even at low cytotoxic concentration of quinones, 20 μM [16], and significant excess of NADH over quinone, 5,8-dihydroxy-1,4-naphthoquinone and 2,6-dimethyl-1,4-benzoquinone significantly delay formation of the reduced form of the flavoprotein, as judged from the kinetics of the absorbance decay at 452 nm (Fig. 7). In the presence of RH1, on the other hand, the course of the reaction was only weakly perturbed, with AIF being predominantly in the reduced state (Fig. 7). Although a stoichiometric quinone:AIF ratio was used, AIF performed multiple turnovers (Fig. 7). This is due to regeneration of the oxidants under aerobic conditions. Free radicals and, presumably, two-electron reduced 5,8-dihydroxy-1,4-naphthoquinone formed during a mixed one- and two-electron reduction of quinones by AIF, are rapidly reoxidized by oxygen [16]. In the absence of external oxygen supply, reoxidation of AIF in the reaction mixture terminates after oxygen depletion.
Fig. 7.

Reduction of 20 μM AIF with 1.0 mM NADH in the absence (1) and presence of 20 μM 2,6-dimethyl-1,4-benzoquinone (2), 5,8-dihydroxy-1,4-naphthoquinone (3), and RH1 (4). Kinetics of FAD reduction was monitored in 100 mM K-phosphate, pH 7.0, 1 mM EDTA at 452 nm and 25°C.
Discussion
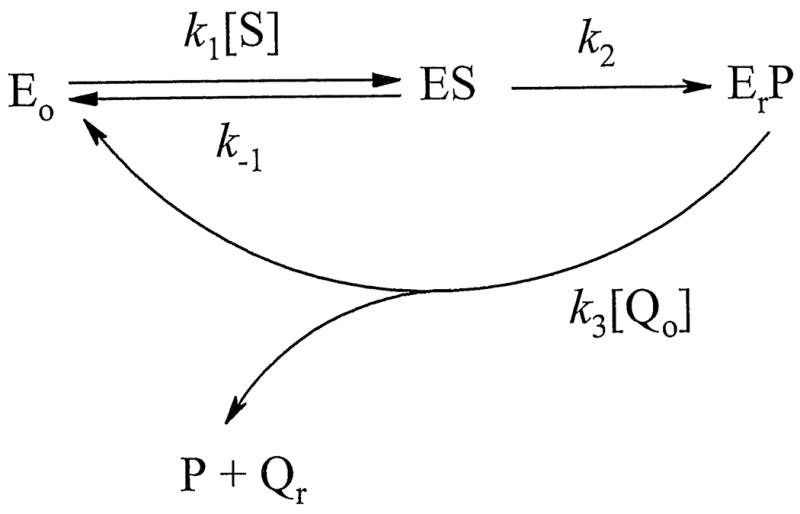
The goal of this study was to characterise the mechanism of reduction of quinoidal xenobiotics and drugs by mitochondrial AIF with emphasis on its possible contribution to apoptosis or other toxic events in the cell. First, we characterized the catalytic cycle of NADH:quinone oxidoreduction mediated by AIF. The steady-state reactions catalyzed by AIF follow a “ping-pong” mechanism (Fig. 3) and involve the reductive and oxidative half-reactions. One notable observation is that the maximal pre-steady-state rate of NADH-dependent FAD reduction (0.31±0.03 s-1, Fig. 5B) is close to the maximal turnover rate under steady-sate conditions (0.24±0.02 s-1) but considerably lower than the rate of AIF oxidation by 2,6-dimethyl-1,4-benzoquinone (Fig. 6B). Our results suggest that (i) the reductive half-reaction is rate-limiting, (ii) quinones are predominantly reduced by the FADH--NAD+ CT complex, and (iii) FAD oxidation coincides with NAD+ dissociation. That the oxidized pyridine nucleotide weakly binds to oxidized AIF and rapidly and irreversibly dissociates upon FAD oxidation is evidenced by the finding that high concentrations of NAD+ do not interfere with AIF reduction with NADH (Fig. 5A). The proposed reaction course is shown in Scheme 1 (Eo and Er correspond to the oxidized and reduced forms of AIF; S, P, Qo and Qr stand for NADH, NAD+, oxidized and reduced quinone, respectively), while the steady-state reaction rate consistent with the “ping-pong” mechanism can be described by Equation 2:
Scheme 1.
| (2) |
Correlation between the reactivity and redox potentials of the investigated quinones (Fig. 4) suggests that AIF has no pronounced specificity for any particular quinone structure, but may discriminate tricyclic quinones and bulky ubiquinone-6 possibly due to sterical hindrances with the active site. The mixed single- and two-electron reduction of quinones by AIF is another indication of instability of the FAD semiquinone [9,12], because two-electron or mixed reduction of quinones is usually performed by the enzymes that do not stabilize flavin radicals [20,29]. The partial single-electron character of quinone reduction does not contradict to the lack of a transient formation of FAD.- in the pre-steady-state oxidation of AIF. This shows that the oxidation of two-electron reduced flavin by the single-electron oxidant is slower than the oxidation of flavin semiquinone by a second molecule of the oxidant, the former reaction being the rate-limiting step in the reoxidation [30].
Secondly, our data demonstrate that the quinone reductase activity of AIF (expressed as a kcat/Km ratio in Table 1) is 102-104-fold lower than that of cytochrome P450 reductase and NAD(P)H:quinone oxidoreductase (NQO1) [19-21], the two major producers of one- and two-electron reduced forms of quinone xenobiotics, respectively, that promote coresponding ways of quinone cytotoxicity in mammalian cells [16,20,21]. The quinone reductase activity of AIF is also 10-100-fold lower than that of mammalian thioredoxin reductase [31], but is comparable with the activity of glutathione reductase from various sources [32,33]. This implies that the quinone reductase reactions catalyzed by AIF are unlikely to play a significant role in the bioreductive activation of quinone drugs and xenobiotics in vivo. Nonetheless, the interaction of AIF with quinones may be physiologically important, because some of these compounds at low concentrations could significantly delay reduction of FAD even when NADH is in significant excess (Fig. 7). Consequently, by maintaining AIF in the oxidized form, quinones may enhance its proapoptotic properties under reducing conditions. This outcome can be mediated only by the relatively high-potential quinones that oxidize AIF relatively fast (e.g., 5,8-dihydroxy-1,4-naphthoquinone and 2,6-dimethyl-1,4-benzoquinone), but not by the slowly reacting anticancer compounds with the lower E17 values, such as RH1 (Fig. 7, Table 1).
The low quinone reductase activity of AIF is unlikely to be due to the low redox potential of the FAD/FADH- couple (E07 = -0.341 V; determined in the absence of NAD(H) [12]), which in general should favor quinone reduction. One possible factor that could affect the AIF reactivity toward oxidants is the FAD redox potential change induced by binding of NAD+. In homologous to AIF putidaredoxin reductase, for example, association of NAD+ elevates E07 by 170 mV [34]. Taking into account the estimated Kd values for the complex between NAD+ and reduced (5–80 nM [12]; Churbanova and Sevrioukova, unpublished data) and oxidized AIF (>10 mM; Fig. 5B), the NAD+-dependent redox potential change calculated from the Nernst equation would be within the 0.165-0.183 V range, giving the E07 value for the reduced AIF-NAD+ complex of -0.158 ÷ -0.176 V. Such drastic E07 change could certainly lower the electron donating ability of the FADH--NAD+ CT complex. Interestingly, the His453Leu mutant of AIF, which possesses a 56 mV higher E07 (determined in the absence of NAD(H)) but does not form CT complex with NAD+, catalyzes electron transfer reactions more effectively than wild type AIF [13]. Another reason for the low reactivity of AIF could be significant conformational changes occuring during its oxidoreduction. Displacement of Phe309 and the accompanying rearrangement of hydrophobic and H-bonding interactions in the active site during AIF reduction [13] could be energy-consuming and affect the rate of reductive half-reaction. This possibility may explain, in part, why the bimolecular rate constant of AIF reduction with NADH is 103-105-fold lower compared to that reported for other NAD(P)H-dependent flavoenzymes [21,26,29,31]. In the the oxidative half-reaction, quinones oxidize reduced isoalloxazine ring of FADH- paralelly stacked with the nicotinamide ring of NAD+, which in turn is shielded by the Phe309 ring [13]. The stacking of three aromatic rings might impede an access of the oxidant to the re-face of the isoalloxazine ring and slow down the reaction. The conformational changes in AIF taking place after reduction result in the formation of an aromatic tunnel (Tyr559, Phe507, Tyr 346, Trp350, Trp 578, Tyr491, Phe309, and Phe481) stretching from the active site to the surface [13]. However, this tunnel is unlikely to serve as an access channel for the oxidants because it is too long (>20 Ǻ) and tightly packed. Alternatively, quinones may access the si-face of FAD flanked by Pro172, Pro173, Trp195, and Trp482 [13]. This side of the molecule becomes solvent accessible after AIF reduction, although the access to the active site is somewhat hindered by the adjacent 199-202 β-hairpin [13].
In summary, our investigation of the NADH:quinone oxidoreduction catalyzed by AIF revealed that the mitochondrial protein is most reactive toward the high-potential benzo- or naphthoquinones and least reactive toward the low-potential anticancer quinoidal drugs. The quinone reductase activity of AIF is too low to play a significant role in bioreductive activation of quinones in vivo. However, some high-potential quinone model compounds or toxic natural compounds like 5,8-dihydroxy-1,4-naphthoquinone (naphthazarin) may affect the redox status of AIF and, thus, modulate its apoptogenic potency by oxidizing the FADH--NAD+ CT complex. Such potentially harmful AIF-quinone interactions can be exacerbated during oxidative stress and lead to accelerated cell death.
Research Highlights.
In this work, we for the first time characterized the catalytic cycle of NADH:quinone reductase reaction of apoptosis-inducing factor (AIF) and characterized its quinone-substrate specificity, which may be important in the potentiation of AIF apoptosis-inducing potency by quinones. We found that low cytotoxic concentrations of quinones may prevent the reduction of AIF even at large excess NADH, thus preventing the formation of reduced AIF, whose dimeric form possesses decreased apoptosis-inducing potency.
Acknowledgments
This work was supported by the Science and Studies Foundation of Lithuania. I.F.S. was supported by the United States National Institutes of Health Grant GM67637. We thank Prof. Saulius Klimašauskas (Institute of Biotechnology, Vilnius) for the access to stopped-flow facility.
Abbreviations used
- AIF
apoptosis-inducing factor
- E17
redox potential of a quinone/semiquinone couple at pH 7.0
- E07
redox potential of a quinone/hydroquinone couple (standard potential) at pH 7.0
- GR
glutathione reductase
- kcat/Km
bimolecular rate constant
- kcat
catalytic constant
- AZQ
2,5-bis(carboethoxyamino)-3,6-diaziridinyl-1,4-benzoquinone
- BZQ
2,5-bis(2′-hydroxyethylamino)-3,6-diaziridinyl-1,4-benzoquinone
- MeDZQ
2,5-dimethyl-3,6-diaziridinyl-1,4-benzoquinone
- RH1
2,5-diaziridinyl-3-hydroxymethyl-6-methyl-1,4-benzoquinone
- VdWvol
Van der Waals volume
Footnotes
This paper is dedicated to Professor Liuda Rasteikienė on the occasion of her 85th birthday
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hangen E, Blomgren K, Benit P, Kroemer G, Modjtahedi N. Trends Biochem Sci. 2010;35:278–287. doi: 10.1016/j.tibs.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Magion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderowski DP, Penninger JM, Kroemer G. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 3.Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prevost MC, Brothers G, Mak TW, Peninger J, Earnshaw WC, Kroemer G. J Exp Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Otera H, Ohsakaya S, Nagaura ZI, Ishihara N, Mihara K. EMBO J. 2005;17:1–12. doi: 10.1038/sj.emboj.7600614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG. J Biol Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- 6.Yuste VJ, Moubarak RS, Delettre C, Bras M, Sancho P, Robert N, d'Alayer J, Susin SA. Cell Death Differ. 2005;12:1445–1448. doi: 10.1038/sj.cdd.4401687. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Yang C, Chai J, Shi Y, Xue D. Science. 2002;298:1587–1592. doi: 10.1126/science.1076194. [DOI] [PubMed] [Google Scholar]
- 8.Cande C, Vahsen N, Kouranti I, Schmidt E, Daugas E, Spahr C, Luban J, Kroemer RT, Giordanetto F, Garrido C, Penninger JM, Kroemer G. Oncogene. 2004;23:1514–1521. doi: 10.1038/sj.onc.1207279. [DOI] [PubMed] [Google Scholar]
- 9.Miramar MD, Costantini P, Ravagnan L, Saraiva LM, Haouzi D, Brothers G, Penninger JM, Peleato ML, Kroemer G, Susin SA. J Biol Chem. 2001;276:16391–16398. doi: 10.1074/jbc.M010498200. [DOI] [PubMed] [Google Scholar]
- 10.Mate MJ, Ortiz-Lombardia M, Boitel B, Haouz A, Tello D, Susin SA, Penninger J, Kroemer G, Alzari PM. Nat Struct Biol. 2002;9:442–449. doi: 10.1038/nsb793. [DOI] [PubMed] [Google Scholar]
- 11.Loeffler M, Dougas E, Susin SA, Zamzami N, Metivier D, Nieminen AL, Brothers G, Penninger JM, Kroemer G. FASEB J. 2001;15:758–767. doi: 10.1096/fj.00-0388com. [DOI] [PubMed] [Google Scholar]
- 12.Churbanova IY, Sevrioukova IF. J Biol Chem. 2008;283:5622–5631. doi: 10.1074/jbc.M709147200. [DOI] [PubMed] [Google Scholar]
- 13.Sevrioukova IF. J Mol Biol. 2009;390:924–938. doi: 10.1016/j.jmb.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murahashi H, Azuma H, Zamzami N, Furuya KJ, Ikebuchi K, Yamaguchi M, Yamada Y, Sato N, Fujihara M, Kroemer G, Ikeda H. J Leucocyte Biol. 2003;73:399–406. doi: 10.1189/jlb.0702335. [DOI] [PubMed] [Google Scholar]
- 15.Srinivas P, Gopinath G, Banerji A, Dinakar A, Srinavas G. Mol Carcinogenesis. 2004;40:201–211. doi: 10.1002/mc.20031. [DOI] [PubMed] [Google Scholar]
- 16.O'Brien PJ. Chem-Biol Interact. 1991;80:1–41. doi: 10.1016/0009-2797(91)90029-7. [DOI] [PubMed] [Google Scholar]
- 17.Öllinger K, Kågedal K. Subcell Biochem. 2002;36:151–170. [PubMed] [Google Scholar]
- 18.Verrax J, Taper H, Buc Calderon P. Curr Mol Pharmacol. 2008;1:80–92. [PubMed] [Google Scholar]
- 19.Čėnas N, Anusevičius Ž, Bironaitė D, Bachmanova GI, Archakov AI, Öllinger K. Arch Biochem Biophys. 1994;315:400–406. doi: 10.1006/abbi.1994.1517. [DOI] [PubMed] [Google Scholar]
- 20.Čėnas N, Anusevičius Ž, Nivinskas H, Misevičienė L, Šarlauskas J. Methods Enzymol. 2004;382B:258–277. doi: 10.1016/S0076-6879(04)82015-9. [DOI] [PubMed] [Google Scholar]
- 21.Anusevičius Ž, Šarlauskas J, Čėnas N. Arch Biochem Biophys. 2002;404:254–262. doi: 10.1016/s0003-9861(02)00273-4. [DOI] [PubMed] [Google Scholar]
- 22.Winski SL, Hargreaves RHJ, Butler J, Ross D. Clin Cancer Res. 1998;4:3083–3088. [PubMed] [Google Scholar]
- 23.Chou F, Khan AH, Driscoll JS. J Med Chem. 1976;19:1302–1308. doi: 10.1021/jm00233a010. [DOI] [PubMed] [Google Scholar]
- 24.Karpov VV, Khidekel ML. Zh Org Khim. 1967;3:1669–1674. [Google Scholar]
- 25.McClure JD. J Org Chem. 1963;28:69–71. [Google Scholar]
- 26.Williams CH., Jr . In: The Enzymes. 3rd. Boyer PD, editor. Vol. 13. Academic Press; New York: 1976. pp. 89–173. [Google Scholar]
- 27.Misevičienė L, Anusevičius Ž, Šarlauskas J, Harris RJ, Scrutton NS, Čėnas N. Acta Biochim Polon. 2007;54:379–385. [PubMed] [Google Scholar]
- 28.Iyanagi T, Yamazaki Y. Biochim Biophys Acta. 1970;216:284–294. doi: 10.1016/0005-2728(70)90220-3. [DOI] [PubMed] [Google Scholar]
- 29.Anusevičius Ž, Misevičienė L, Medina M, Martinez-Julvez M, Gomez-Moreno C, Čėnas N. Arch Biochem Biophys. 2005;437:1444–150. doi: 10.1016/j.abb.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 30.Čėnas N, Diep Le KH, Terrier M, Lederer F. Biochemistry. 2007;46:4661–4670. doi: 10.1021/bi602634y. [DOI] [PubMed] [Google Scholar]
- 31.Čėnas N, Nivinskas H, Anusevičius Ž, Šarlauskas J, Lederer F, Arner ESJ. J Biol Chem. 2004;279:2583–2592. doi: 10.1074/jbc.M310292200. [DOI] [PubMed] [Google Scholar]
- 32.Čėnas NK, Rakauskienė GA, Kulys JJ. Biochim Biophys Acta. 1989;973:399–404. doi: 10.1016/s0005-2728(89)80381-0. [DOI] [PubMed] [Google Scholar]
- 33.Grellier P, Marozienė A, Nivinskas H, Šarlauskas J, Aliverti A, Čėnas N. Arch Biochem Biophys. 2010;494:32–39. doi: 10.1016/j.abb.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 34.Reipa V, Holden MJ, Vilker VL. Biochemistry. 2007;46:13235–13244. doi: 10.1021/bi7012118. [DOI] [PubMed] [Google Scholar]