

Abstract
This study was undertaken to determine if the ketogenic diet could be useful for glioblastoma patients. The hypothesis tested was whether glioblastoma cells can metabolize ketone bodies. Cerebellar astrocytes and C6 glioblastoma cells were incubated in glutamine and serum free medium containing [2,4-13C]β-hydroxybutyrate (BHB) with and without glucose. Furthermore, C6 cells were incubated with [1-13C]glucose in the presence and absence of BHB. Cell extracts were analyzed by mass spectrometry and media by 1H magnetic resonance spectroscopy and HPLC. Using [2,4-13C]BHB and [1-13C]glucose it could be shown that C6 cells, in analogy to astrocytes, had efficient mitochondrial activity, evidenced by 13C labeling of glutamate, glutamine and aspartate. However, in the presence of glucose, astrocytes were able to produce and release glutamine, whereas this was not accomplished by the C6 cells, suggesting lack of anaplerosis in the latter. We hypothesize that glioblastoma cells kill neurons by not supplying the necessary glutamine, and by releasing glutamate.
Keywords: C6 cells, Astrocytes, β-hydroxybutyrate, Glucose, Anaplerosis, Glutamate, Glutamine, glioblastoma
Introduction
Glioblastoma is the most malignant brain tumour forming 50% of all primary brain carcinomas. Surgery, radiotherapy and chemotherapy have been the available therapies. However, despite improvements in these treatment methods overall survival of patients with glioblastoma remains extremely poor [20]. Detailed understanding of the metabolic pathways of glial tumour cells and their differences to normal cells could provide potential targets for treatment. These can be directed to block or manipulate specific pathways in cancer cells to inhibit their metabolism and thus, growth.
Glucose is the primary energy substrate for the adult mammalian brain; however lactate and ketone bodies can be used as alternative energy substrates under stressful and pathological conditions [28]. Ketone bodies include β-hydroxybutyrate (BHB), acetoacetate and acetone, of which the latter is generally considered of little metabolic significance. Under normal physiological conditions there is a minimal accumulation of cerebral ketones with rapid utilization, implying blood–brain barrier control of ketone oxidation in the nonfasted adult human brain [24]. However, in a hypoglycaemic state transport across the blood brain barrier increases and the brain metabolizes a larger amount of ketone bodies which bypass cytoplasmic glycolysis and directly enter the TCA cycle as acetyl-CoA [35]. It has been postulated that glioma cells lack the ability to metabolize ketone bodies due to defects in the mitochondrial TCA cycle and electron transport chain [31, 32]. The ketogenic diet (KD) is a high-fat, low-protein, low-carbohydrate diet used to treat children with drug-resistant epilepsy [9, 32] and it has been reported as beneficial treatment for advanced glial tumors [17]. However, the latter study consists of two case reports only. Animal studies have been published showing positive effects of the KD and hypoglycemia on glial tumours [32].
Bouzier et al. [1] have reported that for C6 cells exogenous lactate was the major substrate for oxidative metabolism and that glucose was entering a different compartment. However, there is a lack of studies that can delineate the detailed metabolic pathways in glioma tumor cells especially the ability of tumor cells to shift between metabolic pathways under different conditions. Incubating C6 cells (glioblastoma cell line) with [2,4-13C]BHB followed by biochemical analysis can give information about C6 cells and normal animal cells metabolism. Using this method the present study was performed to explore the mitochondrial metabolic pathways in the C6 glioblastoma cell line and cerebellar astrocytes in the presence and absence of glucose. C6 cells were also incubated with [1-13C]glucose in the presence and absence of unlabeled BHB to explore if BHB can substitute for glucose labelling.
Materials and Methods
Materials
The C6-glioma cell line was obtained from the ATCC-LGC Promochem (Sweden) and stock samples were kept in liquid nitrogen. Plastic tissue culture dishes were purchased from Nunc A/S (Roskilde, Denmark) and fetal calf serum (FCS) from Seralab Ltd. (Sussex, UK). Dulbecco’s minimum essential medium, unlabeled BHB and D(+)-glucose were from Sigma Chemical Co. (St. Louis, MO, USA). Sodium [2,4-13C]BHB (99%) and [1-13C]glucose (99%) were from Cambridge Isotope Laboratories (Woburn, MA, USA), and the GC/MS derivatization reagent MTBSTFA (N-methyl-N-(tert-butyldimethylsilyl)trifluoroacetamide) + 1% t-BDMS-Cl (tert-butyldimethylchlorosilane) was purchased from Regis Technologies, Inc. (Morton Grove, IL, USA). F-12 Kraighn's Nutrient mixture and penicillin-streptomycin liquid were purchased from Invitrogen Ltd (Paisley, UK). All other chemicals were of the purest grade available from regular commercial sources.
Cell Cultures
NMRI mice were obtained from Taconic M&B (Copenhagen, Denmark). Cerebellar astrocytes were cultured as described earlier [10] in 75 cm2 flasks. Briefly, cerebellum was taken from 7-day-old mice and passed through Nitex nylon netting (80 μm pore size) into Dulbecco’s minimum essential medium (DMEM) containing 20% (v/v) fetal calf serum (FCS). Medium was changed 2 days after plating and subsequently twice a week for 3 weeks gradually decreasing the FCS concentration to 10%. Experiments were performed with three-week-old cultures.
C6 cells were used after 36 passages. On day one 5 × 106 cells were seeded in 75 cm2 flasks and cultured with F-12 media containing penicillin (10,000 units/ml), streptomycin (10,000 μg/ml) and 15% (v/v) fetal calf serum. Experiments were performed 2 days after seeding.
Experiments using [2,4-13C]BHB or [1-13C]glucose
Culture medium was replaced with 15 ml serum and glutamine free DMEM containing 1 mM [2,4-13C]BHB with or without 3 mM glucose or 3 mM [1-13C]glucose with and without BHB. After 4 h the experimental medium was removed and lyophilized. The cells were extracted with 70% ethanol and supernatants were lyophilized.
Protein Analysis
Cellular protein in the ethanol pellets was determined after dissolving in 1 M KOH at 37°C for 60 min, using the Pierce BCA (Pierce, Rockford, IL, USA) protein assay with bovine serum albumin as standard.
HPLC
Amino acids in medium and C6 cell extracts were quantified by HPLC on a Hewlett Packard 1100 system (Agilent Technologies, Palo Alto, CA, USA). The amino acids were pre-column derivatized with o-phthaldialdehyde [8] and subsequently separated on a ZORBAX SB-C18 (4.6 × 150 mm, 3.5 μm) column from Agilent using a phosphate buffer (50 mM, pH = 5.9) and a solution of methanol (98.75%) and tetrahydrofurane (1.25%) as eluents. The separated amino acids were detected with fluorescence and compared to a standard curve derived from standard solutions of amino acids. Total glutathione (i.e., glutathione plus glutathione disulfide) was determined by HPLC as in [8] after derivatization with OPA after reducing the sample with 5% (v/v) mercaptoethanol.
GC/MS
Lyophilized cell extracts were re-dissolved in HCl, adjusted to pH < 2 and dried under atmospheric air. The amino acids were in multiple steps extracted into an organic phase of ethanol and benzene and dried again under atmospheric air before derivatization with MTBSTFA in the presence of 1% t-BDMS-Cl [14]. The samples were analyzed on a Agilent 6890 gas chromatograph linked to a Agilent 5975B mass spectrometer. Atom percent excess (13C) was determined after allowing for naturally abundant 13C and silicon (from silyl groups).
1H Magnetic Resonance Spectroscopy (MRS)
Lyophilized media samples were dissolved in 0.4 ml 99% D2O containing 0.1% ethylene glycol. Proton MR spectra were accumulated on Bruker DRX500 spectrometer (BRUKER Analytik GmbH, Rheinstetten, Germany). The following acquisition parameters were applied: 90° pulse angle, an acquisition time of 1.36 s and a relaxation delay of 10 s, 32 scans were accumulated for each sample. Water suppression was achieved by applying a low-power presaturation pulse at the water frequency.
Data Analysis
The glucose- and BHB consumption was calculated by subtracting the amount of glucose or BHB (quantified from 1H spectra) present in the media from the known concentration of the added glucose and BHB. Lactate production was calculated from the 1H spectra.
Peaks from GC/MS spectra were integrated, and atom percent excess values were calculated by comparison with unlabeled standard solutions. Results are presented as means ± standard deviation (SD). It was not considered useful to compare metabolites between astrocytes and C6 cells. Thus, even though the experiment was carried out with four groups, only same culture groups (two) were analyzed statistically and thus Student’s t-test was the appropriate test, and P < 0.05 was considered statistically significant.
Results
The labeling pattern in glutamate, glutamine, aspartate and malate from [2,4-13C]BHB and [1-13C]glucose after the first turn of the TCA cycle is shown in Fig. 1a. [2,4-13C]BHB enters the TCA cycle as [2-13C]acetyl-CoA after conversion to [2,4-13C]acetoacetate and subsequently [2,4-13C]acetoacetyl-CoA. [2-13C]Acetyl-CoA may condense with unlabeled oxaloacetate. The subsequent steps yield [4-13C]α-ketoglutarate, which may be transaminated to [4-13C]glutamate and [4-13C]glutamine giving rise mono labeling (M + 1) in the mass spectrum. If [4-13C]α-ketoglutarate is converted to succinyl-CoA, due to the symmetry of succinate, labeling of malate and oxaloacetate will occur in either the C-2 or C-3 position. [2/3-13C]Oxaloacetate can be transaminated to [2/3-13C]aspartate which is detected by GC–MS as a singlet (M + 1). Alternatively (Fig. 1b), [2/3-13C]oxaloacetate may be condensed with either labeled acetyl-CoA to form citrate, giving rise to double labeling of the metabolites (M + 2) or with unlabled acetyl-CoA to give mono labeled metabolites. Figure 2 shows the metabolism of pyruvate which can be generated from glucose but not BHB. Pyruvate can be carboxylated and can enter the TCA cycle as oxaloactate.
Fig. 1.
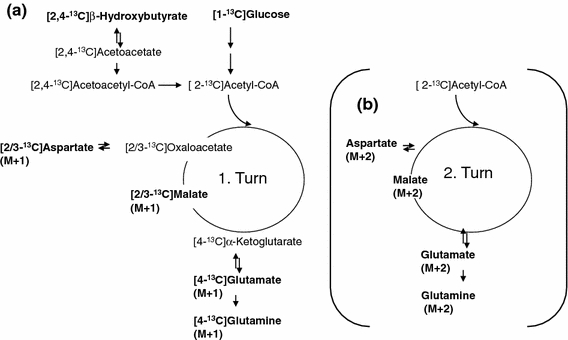
Schematic presentation of labeling of glutamate, glutamine, aspartate and malate from [2,4-13C]β-hydroxybutyrate and [1-13C]glucose from the first turn of the TCA cycle (a). Condensation of [2-13C]acetyl-CoA with unlabeled oxaloacetate generates mono label in the metabolites which is detected as M + 1. The fate of the 13C from the second turn of the TCA cycle is indicated in (b). If oxaloacetate is labeled in the C3 or C2 position and condenses with [2-13C]acetyl-CoA, the resulting metabolites are double labeled and thus are detected by mass spectroscopy as M + 2
Fig. 2.
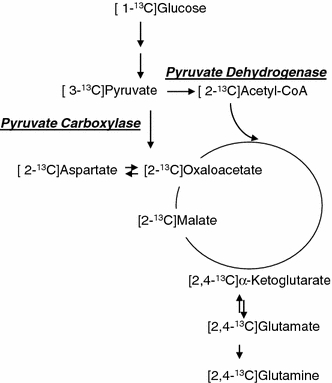
Metabolism of [1-13C]glucose which can be carboxylated to yield net production of glutamate, glutamine and aspartate. 13C labeling from the first turn of the TCA cycle when both acetyl-CoA and oxaloacetate are labeled is indicated only. Labeling from pyruvate carboxylase will result in labeling in the C2 positions and that from pyruvate dehydrogenase in the C4 positions of α-ketoglutarate, glutamate and glutamine
Both astrocytes and C6 cells metabolized glucose and BHB efficiently (Table 1). Whereas astrocytes decreased BHB consumption in the presence of glucose, the opposite was the case for C6 cells. As expected in the presence of glucose lactate production increased in both cell types (Table 1). The intracellular concentration of glutamate increased in C6 cells in the presence of glucose whereas glutamine and aspartate amounts remained unchanged (Table 1).
Table 1.
Lactate production, glucose and β-hydroxybutyrate (BHB) consumption in cerebellar astrocytes and C6 glioma cells and amounts of glutamate, glutamine and aspartate in C6 glioma cells in the presence and absence of glucose
| µmol/mg protein/4h | Astrocytes BHB | C6 BHB | Astrocytes BHB+glucose | C6 + BHB+glucose |
|---|---|---|---|---|
| Glucose consumption | 11.9 ± 2.2 | 15.3 ± 3.0 | ||
| BHB consumption | 1.9 ± 0.6 | 1.7 ± 0.7 | 1.4 ± 0.2* | 2.4 ± 1.1* |
| Lactate production | 2.9 ± 0.6 | 3.3 ± 0.5 | 10.5 ± 1.0* | 13.4 ± 2.5* |
| nmol/mg protein/4 h | ||||
| Glutamate | n.d. | 4.7 ± 0.6 | n.d. | 11.5 ± 4.2* |
| Glutamine | n.d. | 1.2 ± 0.6 | n.d. | 0.7 ± 0.3 |
| Aspartate | n.d. | 3.3 ± 1.0 | n.d. | 2.4 ± 1.2 |
Cells (n = 6) were incubated for 4 h with serum and gluamine free DMEM containing 1 mM [2,4-13C]hydroxybutyrate with or without 3 mM glucose. Results for glucose, BHB and lactate were derived from 1H MRS and amounts of glutamate, glutamine and aspartate from HPLC and are presented as means ± SD, and P < 0.05 was considered statistically significant. BHB = β-hydroxybutyrate, n.d. = not determined. * Significantly different from same group without glucose
Labelling from [2,4-13C]BHB was decreased in glutamate and malate in the presence of glucose in astrocytes and C6 cells and in aspartate in astrocytes (Fig. 3). This was evidenced by an increase in M (molecules without 13C) and a decrease in both M + 1 and M + 2. Glutamine labeling in both cell types and aspartate labeling in C6 cells was only slightly affected by the presence of glucose (Fig. 3). [1-13C]glucose labeling of C6 cells in the presence and absence of BHB is shown in Fig. 4. It is apparent that BHB has little influence on labeling from [1-13C]glucose, only M + 2 labeling in glutamate and malate are slightly decreased.
Fig. 3.
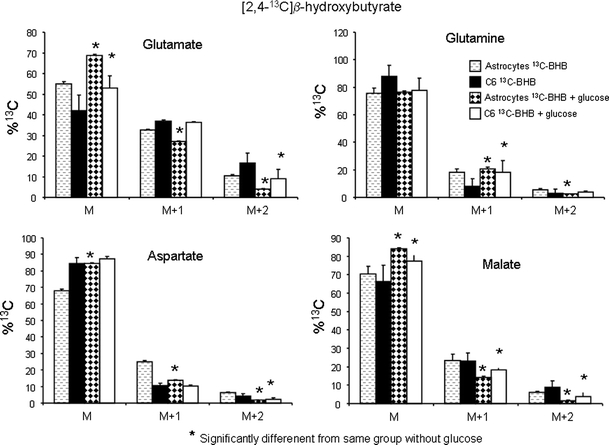
Distribution of 13C label (M, no label above natural abundance; M + 1, one 13C atom; M + 2, two 13C atoms) in glutamate, glutamine, aspartate and malate detected by mass spectrometry in cell extracts of cultured cerebellar astrocytes (n = 6) and C6 cells (n = 6) after incubation for 4 h with serum and glutamine free DMEM containing 1 mM [2,4-13C]β-hydroxybutyrate with or without 3 mM glucose. Results are presented as means ± SD, and P < 0.05 was considered statistically significant, for details see Methods
Fig. 4.
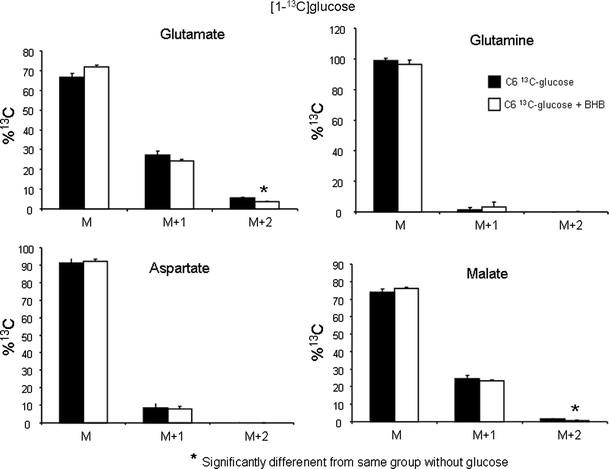
Distribution of 13C label (M, no label above natural abundance; M + 1, one 13C atom; M + 2, two 13C atoms) in glutamate, glutamine, aspartate and malate detected by mass spectrometry in cell extracts of cultured C6 cells (n = 3) after incubation for 4 h with serum and glutamine free DMEM containing 3 mM [1-13C]glucose with or without 1 mM BHB. Results are presented as means ± SD, and P < 0.05 was considered statistically significant, for details see Methods
Figure 5 shows the amount of glutamine, glutamate, aspartate and glutathione in the cell culture medium after the 4 h incubation. The amounts of glutamine released to the medium increased in astrocytes in the presence of glucose whereas that of aspartate and glutathione decreased (Fig. 5). The C6 cells released very little to the medium except for glutamate, which was not quantifiable from astrocytes in the absence of glucose.
Fig. 5.
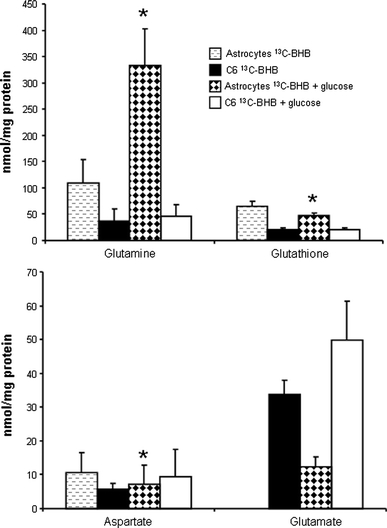
Content of glutamate, aspartate, glutamine and glutathione detected by HPLC in medium of cultured cerebellar astrocytes and C6 cells. Cells were incubated for 4 h with serum and glutamine free DMEM containing 1 mM [2,4-13C]β-hydroxybutyrate with or without 3 mM glucose. Results are presented as means ± SD, and P < 0.05 was considered statistically significant, for details see Methods. * Significantly different from same group without glucose
Discussion
This study was undertaken to determine if the ketogenic diet could be useful for glioblastoma patients. The hypothesis tested was that whereas astrocytes (and neurons) can function in the presence of ketone bodies [11, 15], this would not be the case for glioblastoma cells.
It is believed that cancer cells are dependent on glycolysis to meet their high energy demand and the results of a number of studies have shown that this is also the case for glioblastoma cells [21, 26]. The function of the TCA cycle and oxidative metabolism in glioma cells is the subject of controversy. It has been suggested that these cells have a defective tricarboxylic acid (TCA) cycle and electron transport chain and a reduced number of mitochondria as well as a lower mitochondrial density [21]. Many investigators have stressed the importance of glutamine as a source of reduced nitrogen for maintenance of nucleotide biosynthesis and that of nonessential amino acids and proteins. In fact it has been shown that the human glioblastoma cell line SF188 avidly metabolizes glutamine to lactate and this catabolism was also accompanied by secretion of nitrogen as ammonia and alanine [4].
In mammals glucose and glutamine are two of the most abundant nutrients in plasma. Neurons, protected but also isolated by the blood brain barrier, are particularly dependent on the availability of both substrates. The blood usually supplies the glucose, whereas astrocytes supply the glutamine to neurons. The latter was confirmed in the present study. The C6 glioblastoma cells did not release glutamine in agreement with an earlier study [6, 16, 23]. Moreover, in vivo in a glioma-bearing rat model it was demonstrated that C6 cells do not synthesize glutamine either and avidly take up glutamine directly from the blood circulation to provide nitrogen needed during the glioma growth [2].
In the present study the glioblastoma cells and astrocytes were cultured in glucose and glutamine free environment to test if they could survive under those conditions. The ketone body BHB was added since neural cells with intact mitochondria can survive under these conditions. Surprisingly, both cell types tolerated the absence of glucose well and mitochondrial metabolism was evidenced by avid catabolism of [2,4-13C]BHB. The lactate release observed in the glucose free medium is possibly due to residual glucose inside the cells and also intracellular alanine and pyruvate conversion to lactate. The similarity in the lactate production in the presence and absence of glucose between astrocytes and C6 cells is also worth mentioning. It might be related to the fact that the isoenzyme pattern of lactate dehydrogenase in astrocytes in culture is not identical to that of astrocytes in the adult brain [18]. Rodrigues et al. [27] reported lactate recycling in C6 cells incubated with glucose and lactate or pyruvate but not in the presence of glucose alone. In the present study it was not possible to determine such recycling.
The astrocytes released glutamine even in the absence of glucose whereas C6 cells secreted glutamate. It has been shown that elevated levels of extracelluler glutamate can stimulate tumor cell proliferation and motility via autocrine or paracrine activation of glutamate receptors and enhance invasion of the surrounding tissue by causing excitotoxic damage to normal brain and necrosis in glioblastoma [5, 13, 19]. Elevated extracellular glutamate levels have been shown to be caused by increased synthesis and release and impaired glutamate uptake in glioblastoma [36]. Glutamate, mainly from glutamine, is released via the cystine-glutamate exchanger found on glioma cell membranes [36].
Cystine, which was present in the incubation medium, is an essential precursor for the biosynthesis of glutathione that protects cells (including glioma) from endogenously produced oxidative damage [3, 12, 34]. In the present study, experimental medium from astrocytes contained glutathione, whereas almost none was found in that from C6 cells indicating that astrocytes can help protect neurons by release of glutathione [7] but this is not the case for the C6 cells.
Metabolism of [1-13C]glucose by astrocytes is well documented and glutamine production and pyruvate carboxylation is prevalent [22, 33, 37]. Incubating C6 cells with [1-13C]glucose, however, showed that they have very little label in glutamine, showing that glutamine synthesis is extremely small in C6 cells as also shown by Portais et al. [23] and the same holds for aspartate. Glutamate and malate labeling is only slightly smaller from [1-13C]glucose than [2,4-13C]BHB.
In the presence of glucose, astrocytes and C6 cells reacted differently. Whereas astrocytes decreased their BHB consumption slightly, the opposite was the case for the C6 cells. Astrocytes used glucose in their TCA cycle as evidenced by decreased labelling of glutamate and aspartate from [2,4-13C]BHB which was not the case in C6 cells. Furthermore, astrocytes were able to increase the amounts of glutamate, glutamine and aspartate inside the cells and their release of glutamine to the medium. This is an indication of their capacity for anaplerosis and thus pyruvate carboxylation most likely via pyruvate carboxylase [25]. Only approximately half of the glucose was released as lactate both from astrocytes and C6 cells. This is surprising since in the absence of BHB most of the glucose is used for lactate production at a 3 mM glucose concentration in the incubation medium [30]. It appears that BHB increases mitochondrial metabolism of glucose in both cell types. C6 cells did not show any indication for carboxylation, which can be accomplished via pyruvate carboxylase or malic enzyme. This is in agreement with previous studies [4, 23] which have reported entry of pyruvate into the TCA cycle via pyruvate dehydrogenase but not pyruvate carboxylase. Taken together these results show clearly that C6 cells have a functioning TCA cycle that can utilize ketone bodies but do not perform anaplerosis. C6 cells release glutamate whereas astrocytes release glutamine and glutathione.
Acknowledgment
The excellent technical assistance of Bente Urfjell and Lars Evje is gratefully acknowledged.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- 1.Bouzier AK, Voisin P, Goodwin R, Canioni P, Merle M. Glucose and lactate metabolism in C6 glioma cells: evidence for the preferential utilization of lactate for cell oxidative metabolism. Dev Neurosci. 1998;20(4–5):331–338. doi: 10.1159/000017328. [DOI] [PubMed] [Google Scholar]
- 2.Bouzier AK, Quesson B, Valeins H, Canioni P, Merle M. [1–13C]glucose metabolism in the tumoral and nontumoral cerebral tissue of a glioma-bearing rat. J Neurochem. 1999;72(6):2445–2455. doi: 10.1046/j.1471-4159.1999.0722445.x. [DOI] [PubMed] [Google Scholar]
- 3.Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25(31):7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci. 2007;104(49):19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Groot JF, Liu TJ, Fuller G, Yung WK. The excitatory amino acid transporter-2 induces apoptosis and decreases glioma growth in vitro and in vivo. Cancer Res. 2005;65(5):1934–1940. doi: 10.1158/0008-5472.CAN-04-3626. [DOI] [PubMed] [Google Scholar]
- 6.Dolińska M, Dybel A, Hilgier W, Zielińska M, Zabłocka B, Buzańska L, Albrecht J. Glutamine transport in C6 glioma cells: substrate specificity and modulation in a glutamine deprived culture medium. J Neurosci Res. 2001;66(5):959–966. doi: 10.1002/jnr.10047. [DOI] [PubMed] [Google Scholar]
- 7.Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384(4):505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- 8.Geddes JW, Wood JD. Changes in the amino acid content of nerve endings (synaptosomes) induced by drugs that alter the metabolism of glutamate and gamma-aminobutyric acid. J Neurochem. 1984;42:16–24. doi: 10.1111/j.1471-4159.1984.tb09691.x. [DOI] [PubMed] [Google Scholar]
- 9.Greene AE, Todorova MT, Seyfried TN. Perspectives on the metabolic management of epilepsy through dietary reduction of glucose and elevation of ketone bodies. J Neurochem. 2003;86(3):529–537. doi: 10.1046/j.1471-4159.2003.01862.x. [DOI] [PubMed] [Google Scholar]
- 10.Hertz L, Juurlink BHJ, Hertz E, Fosmark H, Schousboe A. Preparation of primary cultures of mouce (rat): astrocytes. In: Shahar A, De Vellis J, Vernadakis A, Haber B, editors. A dissection and tissue culture manual for the nervous system. New York: Alan R. Liss; 1989. pp. 105–108. [Google Scholar]
- 11.Kunnecke B, Cerdan S, Seelig J. Cerebral metabolism of [1, 2–13C2]glucose and [U-13C4]3-hydroxybutyrate in rat brain as detected by 13C NMR spectroscopy. NMR Biomed. 1993;6(4):264–277. doi: 10.1002/nbm.1940060406. [DOI] [PubMed] [Google Scholar]
- 12.Lewerenz J, Klein M, Methner A. Cooperative action of glutamate transporters and cystine/glutamate antiporter system Xc- protects from oxidative glutamate toxicity. J Neurochem. 2006;98(3):916–925. doi: 10.1111/j.1471-4159.2006.03921.x. [DOI] [PubMed] [Google Scholar]
- 13.Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67(19):9463–9471. doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mawhinney TP, Robinett RS, Atalay A, Madson MA. Analysis of amino acids as their tert.-butyldimethylsilyl derivatives by gas-liquid chromatography and mass spectrometry. J Chromatogr. 1986;358:231–242. doi: 10.1016/S0021-9673(01)90333-4. [DOI] [PubMed] [Google Scholar]
- 15.Melo TM, Nehlig A, Sonnewald U. Neuronal-glial interactions in rats fed a ketogenic diet. Neurochem Int. 2006;48(6–7):498–507. doi: 10.1016/j.neuint.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 16.Merle M, Pianet I, Canioni P, Labouesse J. Comparative 31P and 1H NMR studies on rat astrocytes and C6 glioma cells in culture. Biochimie. 1992;74(9–10):919–930. doi: 10.1016/0300-9084(92)90076-Q. [DOI] [PubMed] [Google Scholar]
- 17.Nebeling LC, Miraldi F, Shurin SB, Lerner E. Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: two case reports. J Am Coll Nutr. 1995;14(2):202–208. doi: 10.1080/07315724.1995.10718495. [DOI] [PubMed] [Google Scholar]
- 18.Nissen C, Schousboe A. Activity and isoenzyme pattern of lactate dehydrogenase in astroblasts cultured from brains of newborn mice. J Neurochem. 1979;32(6):1787–1792. doi: 10.1111/j.1471-4159.1979.tb02292.x. [DOI] [PubMed] [Google Scholar]
- 19.Noch E, Khalili K (2009) Molecular mechanisms of necrosis in glioblastoma: the role of gutamate excitotoxicity. Cancer Biol Ther 8(19):1791–1797 [DOI] [PMC free article] [PubMed]
- 20.Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, Burkhard C, Schüler D, Probst-Hensch NM, Maiorka PC, Baeza N, Pisani P, Yonekawa Y, Yasargil MG, Lütolf UM, Kleihues P. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64(19):6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 21.Oudard S, Boitier E, Miccoli L, Rousset S, Dutrillaux B, Poupon MF. Gliomas are driven by glycolysis: putative roles of hexokinase, oxidative phosphorylation and mitochondrial ultrastructure. Anticancer Res. 1997;17(3C):1903–1911. [PubMed] [Google Scholar]
- 22.Portais JC, Pianet I, Allard M, Merle M, Raffard G, Kien P, Biran M, Labouesse J, Caille JM, Canioni P. Magnetic resonance spectroscopy and metabolism. Applications of proton and 13C NMR to the study of glutamate metabolism in cultured glial cells and human brain in vivo. Biochimie. 1991;73(1):93–97. doi: 10.1016/0300-9084(91)90080-K. [DOI] [PubMed] [Google Scholar]
- 23.Portais JC, Schuster R, Merle M, Canioni P. Metabolic flux determination in C6 glioma cells using carbon-13 distribution upon [1–13C]glucose incubation. Eur J Biochem. 1993;217(1):457–468. doi: 10.1111/j.1432-1033.1993.tb18265.x. [DOI] [PubMed] [Google Scholar]
- 24.Pan JW, de Graaf RA, Petersen KF, Shulman GI, Hetherington HP, Rothman DL. [2, 4–13C2]-beta-Hydroxybutyrate metabolism in human brain. J Cereb Blood Flow Metab. 2002;22(7):890–898. doi: 10.1097/00004647-200207000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qu H, Eloqayli H, Unsgård G, Sonnewald U. Glutamate decreases pyruvate carboxylase activity and spares glucose as energy substrate in cultured cerebellar astrocytes. J Neurosci Res. 2001;66(6):1127–1132. doi: 10.1002/jnr.10032. [DOI] [PubMed] [Google Scholar]
- 26.Roslin M, Henriksson R, Bergstrom P, Ungerstedt U, Bergenheim AT. Baseline levels of glucose metabolites, glutamate and glycerol in malignant glioma assessed by stereotactic microdialysis. J Neurooncol. 2003;61(2):151–160. doi: 10.1023/A:1022106910017. [DOI] [PubMed] [Google Scholar]
- 27.Rodrigues TB, Gray HL, Benito M, Garrido S, Sierra A, Geraldes CF, Ballesteros P, Cerdán S. Futile cycling of lactate through the plasma membrane of C6 glioma cells as detected by (13C, 2H) NMR. J Neurosci Res. 2005;79(1–2):119–127. doi: 10.1002/jnr.20308. [DOI] [PubMed] [Google Scholar]
- 28.Schurr A, Payne RS, Miller JJ, Rigor BM. Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: an in vitro study. Brain Res. 1997;744(1):105–111. doi: 10.1016/S0006-8993(96)01106-7. [DOI] [PubMed] [Google Scholar]
- 30.Schousboe A, Westergaard N, Waagepetersen HS, Larsson OM, Bakken IJ, Sonnewald U. Trafficking between glia and neurons of TCA cycle intermediates and related metabolites. Glia. 1997;21(1):99–105. doi: 10.1002/(SICI)1098-1136(199709)21:1<99::AID-GLIA11>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 31.Seyfried TN, Kiebish M, Mukherjee P, Marsh J. Targeting energy metabolism in brain cancer with calorically restricted ketogenic diets. Epilepsia. 2008;49(Suppl 8):114–116. doi: 10.1111/j.1528-1167.2008.01853.x. [DOI] [PubMed] [Google Scholar]
- 32.Seyfried TN, Sanderson TM, El-Abbadi MM, McGowan R, Mukherjee P. Role of glucose and ketone bodies in the metabolic control of experimental brain cancer. Br J Cancer. 2003;89(7):1375–1382. doi: 10.1038/sj.bjc.6601269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sonnewald U, Westergaard N, Hassel B, Müller TB, Unsgård G, Fonnum F, Hertz L, Schousboe A, Petersen SB. NMR spectroscopic studies of 13C acetate and 13C glucose metabolism in neocortical astrocytes: evidence for mitochondrial heterogeneity. Dev Neurosci. 1993;15(3–5):351–358. doi: 10.1159/000111355. [DOI] [PubMed] [Google Scholar]
- 34.Sontheimer H. A role for glutamate in growth and invasion of primary brain tumors. J Neurochem. 2008;105(2):287–295. doi: 10.1111/j.1471-4159.2008.05301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF., Jr Ketone bodies, potential therapeutic uses. IUBMB Life. 2001;51(4):241–247. doi: 10.1080/152165401753311780. [DOI] [PubMed] [Google Scholar]
- 36.Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59:4383–4391. [PubMed] [Google Scholar]
- 37.Zwingmann C, Leibfritz D. Regulation of glial metabolism studied by 13C-NMR. NMR Biomed. 2003;16(6–7):370–399. doi: 10.1002/nbm.850. [DOI] [PubMed] [Google Scholar]