

Summary
Macrophages promote tissue injury or repair depending upon on their activation status and the local cytokine milieu. It remains unclear whether the immunosuppressive effects of transforming growth factor beta (TGF-β) serve a non-redundant role in macrophage function in vivo. We generated macrophage-specific transgenic mice that express a truncated TGF-β receptor II under control of the CD68 promoter (CD68TGF-βDNRII)and subjected these animals to the dextran-sodium sulfate (DSS) model of colitis. CD68TGF-βDNRII mice have an impaired ability to resolve colitic inflammation as demonstrated by increased lethality, granulocytic inflammation, and delayed goblet cell regeneration compared to transgene negative littermates. CD68TGF-βDNRII mice produce significantly less interleukin 10, but have increased levels of IgE and IL-33+ macrophages than controls. These data are consistent with associations between ulcerative colitis and increased IL-33 production in humans and suggests that TGF-β may promote the suppression of intestinal inflammation, at least in part, through direct effects on macrophage function.
Keywords: inflammation, macrophages, intestine
Introduction
Damage within the gastrointestinal mucosa can be induced by a wide variety of physical, chemical and/or infectious stimuli [1]. Inflammatory bowel diseases (IBD) such as Crohn’s colitis and Ulcerative colitis generally result in epithelial cell death, loss of crypt architecture, submucosal edema, and mucosal ulceration [2]. The relapsing/remitting episodes of IBD[3] are associated with marked variations in pro-inflammatory cytokine production [4, 5], therefore mouse models of IBD have been used to investigate the regulatory mechanisms that reduce inflammation and restore intestinal homeostasis [6].
Dextran sodium sulfate (DSS)-induced colitis is a transient, myeloid-dependent gut injury model driven by epithelial cell damage [7]. The severity of DSS colitis may be controlled by anti-inflammatory cytokines such as IL-10 and TGF-β [8], but it is unclear whether these cytokines can directly modulate macrophage function(s) in ways that promote the resolution of inflammation following the termination of DSS-induced injury [9-14]. Furthermore, it is unknown whether IL-10 and TGF-β have redundant effects on macrophage function [15, 16].
TGF-β has multiple biological effects on hematopoietic and non-hematopoietic cells [17]. Binding of TGF-β to TGF-βRII phosphorylates SMAD transcription factors that are primarily immunosuppressive in function [17]. Genetic mutations in TGF-βRII are linked to ulcerative colitis (UC) and colitis-associated cancer in humans[18-20] and mice that express that lack TGF-β responsiveness in epithelial cells or T lymphocytes develop severe intestinal inflammation [21, 22]. Whether TGF-β suppresses colitic inflammation through direct effects on macrophages is unknown.
Herein, we employed the DSS colitis model to demonstrate that lack of TGF-β responsive macrophages impairs the normal resolution of colitic inflammation. CD68TGF-βDNRII mice produce high levels of IL-33, an IL-1 family cytokine that is over-expressed in the colonic mucosa of UC patients [23, 24] [25]. CD68TGF-βDNRII mice also produced significantly less IL-10 than littermate controls during colitis resolution. Taken together, these data show an important role for TGF-β in the specific regulation of intestinal macrophage function in vivo.
Results
Generation of Mϕ-specific TGF-β dominant negative receptor mice
A transgenic construct was generated to contain the human CD68 promoter (CD68-IVS1) [26, 27] followed by a human TGF-β receptor II lacking the cytoplasmic domain [28] (Fig. 1A). This truncated receptor binds its extra-cellular ligand (TGF-β1, TGF-β2, and TGF-β3) but does not signal; therefore it antagonizes TGF-β function in the cell by acting as a competitive inhibitor. This approach has been employed in a variety of tissue-specific promoter systems [21, 28-32]. Pronuclear injection of C57BL/6 oocytes allowed generation of a founder (designated CD68TGF-βDNRII) possessing a single integration of approximately 15-20 copies (Fig. 1B). Thioglycollate-elicited peritoneal exudates cells (PEC) were evaluated by flow cytometry to determine specificity of transgene expression. Compared to non-transgenic littermates, CD68TGF-βDNRII mice demonstrate TGF-βRII protein expression on CD11b+ myeloid cells (0.12% vs. 5.3 %), F4/80+ macrophages (0.27% vs. 7.9 %), but not on CD11c+ dendritic cells (0.15% vs. 0.32%), respectively (Fig. 1C). Transgene expression was not detected on granulocytes (GR-1+), B lymphocytes (B220+), or T lymphocytes (CD3+) from spleen (data not shown).
Figure 1.

Generation and characterization of CD68TGF-βDNRII transgenic mice (A) Model representation of transgenic construct. Primer binding sites for PCR genotyping are shown. (B) Southern blot hybridization of genomic DNA from a representative CD68TGF-βDNRII mouse (Tg mouse) or littermate control (Neg littermate) that was digested with EcoR1 and hybridized to a 250bp probe homologous to the junction between CD68-IVSI and 5’TGF-βRII. Approximation of transgene copy number was based on log-fold dilutions of control plasmid DNA. (C) Flow cytometry of thioglycollate-elicited peritoneal exudate cells (PEC) from WT and CD68TGF-βDNRII mice analyzed for human TGF-βRII expression on CD11b+, F4/80+ and CD11c+ populations. Representative dot plots are shown from pooled samples of 2 mice per group. Experiment repeated four times. (D) Comparison between IL-10 and TGF-β-mediated suppression of LPS-induced IL-12/23p40 production as determined by ELISA. (n= 5) Experiment repeated twice. (E) Comparison of IL-10 production from WT and CD68TGF-βDNRII PEC following treatment with LPS or TGF-β. (n=3) Mean ± SE shown. Experiment repeated four times. *=p<0.05, **=p<0.01, ***=p<0.001 as determined by Student t test.
To determine whether macrophages from CD68TGF-βDNRII mice had functionally impaired TGF-β responsiveness, the adherent fraction of thioglycollate-elicited peritoneal cells (PEC) (>90% macrophages) was tested for IL-10 vs. TGF-β-mediated suppression of endotoxin (LPS)-induced cytokine production. As expected, LPS induced a 1000-fold increase of IL-12/23p40 production within 24 h that was significantly suppressed by pre-treatment with IL-10 in both WT and CD68TGF-βDNRII groups (Fig. 1D). In contrast, LPS-induced 12/23p40 production was moderately suppressed in TGF-β-pre-treated WT PEC, which as not observed following treatment of CD68TGF-βDNRII PEC (Fig. 1D). IL-10 is induced in Mϕ following exposure to LPS [33] or TGF-β [34]. Fig. 1E demonstrates equivalent LPS-induced IL-10 production, but significantly impaired TGF-β-induced IL-10 production in CD68TGF-βDNRII PEC compared to WT. To determine whether over-expression of the mutant human TGF-βRII affected the endogenous murine TGF-β RII, lamina propria mononuclear cells from naïve WT and CD68TGF-βDNRII mice were evaluated by flow cytometry. Human TGF-βRII was detected on both CD11c+ F4/80+ and F4/80+ populations within the colon, but there were no differences between strains in the mean fluorescence intensity (MFI) of mouse TGF-βRII expression on any of the gated cell populations (Fig. 2). Transgene expression was specific, because CD3+CD4− and CD3+CD4+ lymphocytes showed no differences in staining for human or mouse TGF-βRII, although lymphocytes expressed comparatively higher levels of TGF-βRII than the myeloid cell populations (Supplemental Fig. 1). Thus, CD68TGF-βDNRII mice have a specific expression of a truncated human TGFβRII and impairment of TGF-β-dependent functions in macrophages.
Figure 2.
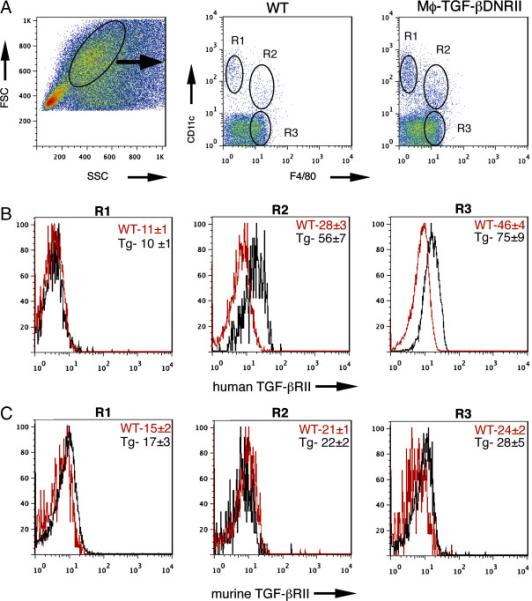
Expression of TGF-βRII in colonic Mϕ (A) FSCHi, SSC med colon lamina propria cells from naïve WT or CD68TGF-βDNRII mice were evaluated for expression of CD11c+, CD11c+F4/80+, and F4/80+ populations. (n=3) (B) Histograms show mean fluorescence intensity of staining for mAb specific for humanTGF-βRII on the gated populations indicated in “A” (n=3) (C) Histograms show mean fluorescence instensity for mAb specific for mouse TGF-βRII on the gated populations indicated in “A” (n=3) Experiments repeated 3 times.
Defective resolution of DSS-induced colitis in CD68TGF-βDNRII mice
Administration of 2.5% DSS ad libitum for 6 days to WT C57BL/6 mice causes a transient colitis that rapidly resolves following the return of mice to normal un-treated drinking water [3, 7]. CD68TGF-βDNRII mice administered 2% DSS lost weight at a slightly faster rate than WT littermates during the initial stages of colitis induction (Fig. 3A), but demonstrated impaired weight gain following the termination of DSS administration (Fig. 3A). Although there were no differences in mortality at this dose (Fig. 3B) there was increased severity of the clinical disease indicators (hunched posture, fecal blood, and diarrhea) in CD68TGF-βDNRII mice compared to controls (Fig. 3C). In contrast, CD68TGF-βDNRII mice administered 2.5% DSS rapidly lost >25% of their initial body weight (Fig. 3D) and 100% died 6 days following removal of DSS (Fig. 3E). Although littermate controls developed significant disease and 25% mortality within 10-12 days, most of the animals successfully return to their original weights by day 15 (Fig. 3D-F). No significant differences in mortality or disease activity were observed between strains administered 1.5% DSS (data not shown).
Figure 3.

Evaluation of DSS-induced colitis in WT and CD68TGF-βDNRII mice WT and CD68TGF-βDNRII mice were administered either 2% DSS (A, B, C) or 2.5% DSS (D, E, F) on day 0, returned to normal drinking water on d 6 and monitored until d 14. Weight changes (A, D) survival rates (B, E) and disease activity indices (DAI) were determined. (n=6-8). Closed circles (WT) and filled circles (transgenic) that represent mean ± SE are shown. Experiments repeated four times. *=p<0.05, **=p<0.01, ***=p<0.01 as determined by Student t test.
Representative images of distal colon demonstrate similar progression of DSS-induced epithelial cell necrosis and submucosal edema in both strains from day 0 to day 9 (Fig. 4). Although WT controls had resolved most of the granulocytic inflammation and edema by day 14, CD68TGF-βDNRII mice maintained granulocyte infiltrates and submucosal edema within the colon (Fig. 4A). This contributed to a significantly increased histopathological score (Fig. 4B) and decreased colon length (Fig. 4C) when compared to controls at day 9 and day 14. Recovery of goblet cell numbers within the colon was also markedly delayed in CD68TGF-βDNRII mice compared WT littermates (Fig. 4D).
Figure 4.

Histopathological analysis of naïve and colitic WT and CD68TGF-βDNRII mice. (A) Representative photomicrographs of H & E-stained colon tissue from naïve mice and mice treated with 2% DSS for six days. Images are representative of three independent experiments. (n=6) 100x magnification. Scale bar =150μm Distal colon segments at the time points indicated were evaluated for (B) colon length measurements (C) histopathological damage score (D) Quantitation of goblet cells from PAS stained sections. Closed circles (WT) and filled circles (transgenic) that represent mean ± SE are shown. *=p<0.05, **=p<0.01 as determined by Student t test.
CD68 TGF-βDNRII mice produce increased levels of pro-inflammatory cytokines and reduced levels of IL-10 during colitis resolution
TGF-β is a master regulator of both immunosuppressive and inflammatory cytokine production from a variety of cell types [35, 36]. To determine whether the delay in colitis resolution observed in CD68TGF-βDNRII mice was associated with broad defects in cytokine/chemokine production, we evaluated relative production within the colon of both strains at day 14 via protein array. Data expressed as the total pixel intensity (Supplemental Fig 2) or fold-difference in pixel intensity within the colonic tissue of CD68TGF-βDNRII mice compared to WT mice (Fig. 5A) revealed multiple abnormalities. Whereas granulocyte colony stimulating factor (G-CSF), I-309 (CCL1), IL-1-α, IP-10 (CXCL10) and MIP-2 (CXCL2) were highly elevated in CD68TGF-βDNRII mice, the production of IL-10 and MIG (CXCL9) were markedly reduced (Fig. 5A). This defect in IL-10 production from CD68TGF-βDNRII mice was observed in both the colon (Fig. 5B) and the sera (Fig. 5C) as compared to WT controls. CD68TGF-βDNRII mice also produced significantly less TGF-β in the serum and colon tissue during the resolution phase compared to WT (Supplemental Fig. 3).
Figure 5.

Measurement of cytokine production in WT and CD68TGF-βDNRII mice during colitis resolution. (A) Colon tissue lysates from WT and CD68TGF-βDNRII mice were obtained at day 14 following 2% DSS were evaluated by the mouse Proteome Prolifer Array™ (n=3) Data show the mean± SE fold-difference in CD68TGF-βDNRII expression compared to WT. (B) Levels of IL-10 produced in the colon tissue and (C) serum from WT and CD68TGF-βDNRII mice prior to and following 2% DSS administration as determined by ELISA. (n= 6-8) Closed circles (WT) and filled circles (transgenic) that represent mean ± SE are shown. Experiments repeated twice. *=p<0.05, **=p<0.01 as determined by Student t test.
CD68TGF-βDNRII mice only had a moderate increase of IFN-γ and no differences in IL-17A when compared to WT (Fig. 5A). Therefore, we asked whether the lack of IL-10 and TGF-β correlated with an increase of Type 2 responses. CD68TGF-βDNRII mice produced significantly greater levels of IgE than WT controls at day 14, although there were no differences between strains in IgE levels prior to colitis induction (Fig. 6A). Elevated IgE levels in CD68TGF-βDNRII mice were associated with the increased production of IL-33 within colon tissue (Fig. 6B). Furthermore, greater levels of IL-33 were detected within CD11b+ and CD11b+CD11c+ cells isolated from the lamina propria of CD68TGF-βDNRII mice compared to WT controls at day 14. Taken together, this suggests TGF-β responsiveness in macrophages serves an important role in limiting granulocyte recruitment and Type 2 inflammation during the resolution of DSS-induced colitis.
Figure 6.

Measurement of IgE levels and IL-33 within the colon following DSS induced colitis in production in WT and CD68TGF-βDNRII mice. (A) Serum IgE levels were measured in naïve and DSS-treated mice at day 14. (n=6) Experiments repeated twice. *=p<0.05, **=p<0.01 as determined by Student t test. (B) Measurement of IL-33 levels in colon tissue from WT and CD68TGF-βDNRII mice over the course of colitis induction. (n=6) Closed circles (WT) and filled circles (transgenic) that represent mean ± SE are shown. (C) Intracellular staining for IL-33 on CD11b+ and CD11b+CD11c+ gated populations from the colon lamina propria of naïve and DSS-treated mice at day 14. Histograms show mean fluorescence intensity of staining for IL-33 (n=3) Experiments repeated twice.
Discussion
Whether TGF-β serves a non-redundant role in macrophage immunoregulation within the mucosa has been unclear. Herein, we use the DSS induced model of colitis to demonstrate that CD68TGF-βDNRII mice, which specifically lack TGF-β responsiveness in macrophages, develop exacerbated gut immunopathology. Interestingly, the marked differences between WT and CD68TGF-βDNRII mice were primarily associated with the resolution of colitic inflammation. Impairment of TGF-β responsiveness in macrophages delayed the reduction of granulocytic inflammation, impaired IL-10 release, but increased the production of IL-33, a Type 2 cytokine that is produced at high levels in the mucosa of UC patients. Hence, TGF-β promotes the normal resolution of intestinal inflammation at least in part, through limiting the production of Type 2 cytokines from colonic macrophages.
CD68 (macrosialin) encodes a type 1 transmembrane protein in mononuclear phagocyte endosomes and its promoter drives Mϕ-specific transgene expression in mice [27, 37]. We demonstrate that the CD68 promoter drives transgene expression in colonic F4/80+ and F4/80+ CD11c+ populations, but is only marginally expressed in CD11c+ (specific for dendritic cells) or Gr-1+ cell populations (specific for neutrophils/granulocytes) (Fig. 2)(data not shown). This is distinct from all other myeloid-specific promoters such as: human CD11b, c-fms, and lysozyme that confer dendritic cell and neutrophil-specific expression [38-40]. Neutrophils promote oxidative tissue injury during DSS-induced colitis[41] and TGF-β is known to directly modulate neutrophil function in vivo [42], which makes the lack of transgene expression in granulocytes an important issue in this model system. Our data are consistent with prior evidence that the human CD68 promoter is primarily active in mature tissue-resident Mϕ populations [43, 44].
Prior to colitis induction, CD68 TGF-βDNRII mice do not have signs of overt inflammation or tissue injury. In contrast, mice that lack STAT-3 responsiveness in macrophages and neutrophils develop spontaneous colitis by 20 weeks of age [45]. Because STAT-3 is an important transcription factor for IL-10 responses [46], this may suggest distinct roles for IL-10 and TGF-β in the regulation of gastrointestinal inflammation. Exacerbated intestinal immunopathology following the cessation of DSS administration in CD68 TGF-βDNRII mice was associated with an extended period of granulocyte infiltration, G-CSF production, chemokine release, and myeloperoxidase (MPO) production (data not shown). This is consistent with prior evidence in this model that excess accumulation of activated Mϕ, neutrophils, eosinophils causes irreparable mucosal damage and lethality [47, 48].
Insufficient IL-10 production may partially explain the increased inflammation in CD68TGF-βDNRII mice, as IL-10-mediated suppression of colitis can be TGF-β dependent [49] and TGF-β induces Mϕ to produce IL-10 [34]. Furthermore, Mϕ from CD68TGF-βDNRII mice produced significantly less IL-10 following TGF-β stimulation in vitro (Fig. 1E) and in vivo (Fig. 5B-C). This link between TGF-β responsiveness in Mϕ and IL-10 production is consistent with evidence that TGF-β suppresses intestinal inflammation via regulatory Mϕ that produce IL-10 [50].
The intestinal injury caused by DSS in rodents shares some characteristics with UC in human patients, as both are characterized by diffuse mucosal inflammation, superficial ulceration, and goblet cell depletion. UC manifests as a TH2 cytokine (IL-4, 5, 13) driven erosion of the intestinal epithelium [23, 24, 51-53]. In contrast, Crohn’s colitis is driven by TH1 and TH17 cytokines (IFN-γ, IL-17A/F)[3, 54]. Although the etiology of UC remains unclear, recent studies have focused on the role of IL-33, an IL-1 family cytokine that instructs Type 2 inflammation [25].
In human UC patients, IL-33 expression is highly up-regulated within the intestinal mucosa and IL-33 deficient mice are protected from DSS-induced intestinal immunopathology [23, 24, 55]. Our data show that CD68TGFβDNRII mice produce high levels of IgE and IL-33 within the colon following DSS-induced gut injury. One source of IL-33 in CD68TGFβDNRII mice were intestinal Mϕ, which demonstrates that TGF-β serves an important role in limiting intestinal inflammation through suppression of IL-33. This may be an important mechanism that could partially explain how mutations in TGF-βRII in humans are associated with increased risk for UC and UC-associated cancer [19, 20]. Thus, it is tempting to speculate that blockade of IL-33 during UC may help to reduce the severity of colitis in these patients.
Overall, we demonstrate that mice engineered to have a specific impairment of TGF-β responsiveness in Mϕ develop increased severity of DSS-induced colitis during the resolution phase. This suggests that TGF-β mediated regulation of Mϕ function serves an important role in the suppression of intestinal inflammation following acute injury. In this regard, it will be important to determine whether CD68TGF-βDNRII mice develop altered susceptibility or resistance to infectious diseases or show defects in tissue repair mechanisms in other model systems.
Materials and Methods
Mice
The TGFβDNRII construct was obtained from Dr. Chung Lee at Northwestern University in a plasmid that encodes the extra-cellular and transmembrane domains, but lacks the cytoplasmic region for human TGF-β receptor II (−5 to 553), which blocks TGF-β responsiveness in vivo [56]. This region was sub-cloned into a modified pcDNA3.1™ (Invitrogen) using Not 1 and Xho 1. The 1kb promoter sequence from human CD68 (macrosialin) including the 89 bp intronic enhancer (provided by Peter Murray at St Jude Hospital) [26] was inserted 5′ to TGFβDNRII as a BamH1-EcoRV fragment and confirmed by restriction digest and DNA sequencing. CD68TGF-βDNRII mice were generated by pronuclear injection of fertilized C57BL/6 oocytes at the University of Cincinnati Transgenic core facility. Offspring were analyzed for genotype by PCR using primers specific for CD68IVS1 and human TGF-β type II. All mice used in the study were age matched male mice on a C57BL/6 background. All experiments were performed with age/sex-matched non-transgenic littermates used as controls. All procedures were approved by the institutional IACUC and performed in accordance with all governmental and institutional guidelines.
Southern Blot
Genomic DNA from tail biopsies was digested with EcoR1 overnight and 10μg of digested DNA was resolved in 1% agarose by electrophoresis. Serial dilutions of plasmid containing the CD68TGF-βDNRII were included as a positive control. Gels were denatured, neutralized, and cross-linked using standard protocols. 32P labeled probe was used for hybridization (49°C) and visualization via autoradiography.
DSS colitis model
Dextran sodium sulfate (41 kDa) (ICN Biomedical Inc) was used to supplement the drinking water of study animals for 6 days as 1.5%, 2%, or 2.5% (wt/vol) solution. Fresh solution was replaced at day 3. After day 6 mice were returned to normal water and monitored for an additional 8 days. Body weight, appearance, occult blood in feces Hem occult test (Beckman Coulter), stool consistency, and diarrhea were recorded daily from coded animals. At time of sacrifice, mice were evaluated for colon length. Disease activity index (DAI) was derived through evaluation of appearance/activity, diarrhea, and rectal bleeding. DAI = (appearance /activity) + (Diarrhea score) + (Rectal bleeding score). DAI has a maximum score of 5 determined as follows: Appearance/activity score (0-normal grooming and active vs. 1-lack of grooming and lacking normal activity), Diarrhea score (0-solid formed stool, 1-loose formed stool, and 2- watery fecal matter), rectal bleeding score (0-no blood, 1-positive hem occult test, 2-gross bleeding from rectum).
Colon histology and histopathology score
Approximately 1 length of distal colon was removed, fixed in 10% buffered formalin overnight and kept in 70% ETOH until processing. Tissue was embedded in paraffin and for each colon sample 5 μm sections were cut and stained with H&E or Periodic acid-Schiff (PAS) and examined by light microscopy. Colonic inflammation was evaluated in a blind manner by two observers that estimated the following: 1) percentage of involved area; 2) amount of follicles; 3) edema; 4) erosion/ulceration; 5) crypt loss; 6) infiltration of polymorphonuclear cells; and 7) infiltration of mononuclear cells. The percentage of area involved, erosion/ulceration, and the crypt loss were scored on a scale ranging from 0 to 4 as follows: 0, normal; 1, <10%; 2, 10 −25%; 3, 25–50%; and 4, >50%. Follicle aggregates were counted and scored as follows: 0, zero to one follicle; 1, two to three follicles; 2, four to five follicles; and 3, six follicles or more. The severity of the other parameters was scored on a scale from 0 to 3 as follows: 0, absent; 1, weak; 2, moderate; and 3, severe. All scores on the individual parameters together could result in a total score ranging from 0 to 24 [47].
Flow cytometry
Peritoneal Mϕ were harvested on d 4 following administration of 4% thioglycollate (Fisher scientific). Lamina propria mononuclear cells were isolated as follows: 1) the entire colon was surgically removed, opened longitudinally, denuded of the epithelial layer by incubation with 0.1% EDTA for 15 min with vigorous shaking at 37°C. 2) Tissues were washed several times with 1xPBS, minced, and digested with Liberase (Roche) in RPMI for 30min on an orbital shaker, 3) Tissue was passed repeatedly through a 16g syringe, pelleted via centrifugation, re-suspended in RPMI, and placed on 30%-70% Percoll gradient. 4) Cells were centrifuged at 2000rpm for 30min and mononuclear cells isolated from the interface. Cells were harvested, washed with 1xPBS and subjected to FACS staining protocols. FACS buffer (HBSS, 1%FBS and 0.2% sodium azide) supplemented with anti-FcγRII/RIII mAb (2.4G2) and goat gamma globulin (.5mg/ml) (Jackson Immunoresearch) was used to prevent non-specific binding. In some experiments, the isolated mononuclear cells were incubated with a polyclonal PE-labeled mouse anti-human TGF-βRII or anti-mouse TGF-βRII (R&D systems), anti-CD11c (clone N418), anti-CD11b (clone M1/70), or anti-F4/80 (clone BM8) (eBioscience). Anti-mouse IL-33 (clone 396118) from R &D systems was used for intracellular staining following the addition of Golgi-stop (BD pharmingen) for 2h to inhibit protein transport. In some experiments 7AAD was used to exclude dead cells from analyses. Acquisition was performed with a BD FACSCalibur and analysis was performed with Flojo 7.5.5 or Cellquest software.
Cytokine measurement
Colon tissue lysates were diluted in 1xPBS and subjected to the Proteome Profiler Array™ obtained from R& D systems according to manufacturer instructions. Densitometric evaluation of blots was performed with a Bio-Rad Molecular Imager® Gel Doc™ system. ELISA was used to quantify murine IL-10, TGF-β, and IL-33 (eBioscience).
Statistical analysis
Statistical significance was assessed by either one-tailed Students t test (two groups) or analysis of variance (ANOVA) for multiple groups with a post-hoc Tukey test to determine significance performed using Prism Graph Pad™.
Supplementary Material
Acknowledgements
We thank Amanda Roloson and Melissa Mingler for expert technical assistance and Marat Khodoun and Senad Divanovic for critical comments. Funding was provided by NIH grant R01GM083204 and the Department of Veterans Affairs. The British Heart Foundation supports D.R.G.
Footnotes
Conflict of interest: None
References
- 1.Ding LA, Li JS, Li YS, Zhu NT, Liu FN, Tan L. Intestinal barrier damage caused by trauma and lipopolysaccharide. World J Gastroenterol. 2004;10:2373–2378. doi: 10.3748/wjg.v10.i16.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 3.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2:541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 4.Monteleone G, Caruso R, Fina D, Peluso I, Gioia V, Stolfi C, Fantini MC, Caprioli F, Tersigni R, Alessandroni L, MacDonald TT, Pallone F. Control of matrix metalloproteinase production in human intestinal fibroblasts by interleukin 21. Gut. 2006;55:1774–1780. doi: 10.1136/gut.2006.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon JN, Pickard KM, Di Sabatino A, Prothero JD, Pender SL, Goggin PM, MacDonald TT. Matrix metalloproteinase-3 production by gut IgG plasma cells in chronic inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:195–203. doi: 10.1002/ibd.20302. [DOI] [PubMed] [Google Scholar]
- 6.Brown SJ, Mayer L. The immune response in inflammatory bowel disease. Am J Gastroenterol. 2007;102:2058–2069. doi: 10.1111/j.1572-0241.2007.01343.x. [DOI] [PubMed] [Google Scholar]
- 7.Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–1652. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 8.Huber S, Schramm C, Lehr HA, Mann A, Schmitt S, Becker C, Protschka M, Galle PR, Neurath MF, Blessing M. Cutting edge: TGF-beta signaling is required for the in vivo expansion and immunosuppressive capacity of regulatory CD4+CD25+ T cells. J Immunol. 2004;173:6526–6531. doi: 10.4049/jimmunol.173.11.6526. [DOI] [PubMed] [Google Scholar]
- 9.Hausmann M, Obermeier F, Schreiter K, Spottl T, Falk W, Scholmerich J, Herfarth H, Saftig P, Rogler G. Cathepsin D is up-regulated in inflammatory bowel disease macrophages. Clin Exp Immunol. 2004;136:157–167. doi: 10.1111/j.1365-2249.2004.02420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghia JE, Galeazzi F, Ford DC, Hogaboam CM, Vallance BA, Collins S. Role of M-CSF-dependent macrophages in colitis is driven by the nature of the inflammatory stimulus. Am J Physiol Gastrointest Liver Physiol. 2008;294:G770–777. doi: 10.1152/ajpgi.00453.2007. [DOI] [PubMed] [Google Scholar]
- 11.Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, Sakuraba A, Kitazume MT, Sugita A, Koganei K, Akagawa KS, Hibi T. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-γ axis. J Clin Invest. 2008;118:2269–2280. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Platt AM, Bain CC, Bordon Y, Sester DP, Mowat AM. An independent subset of TLR expressing CCR2-dependent macrophages promotes colonic inflammation. J Immunol. 2010;184:6843–6854. doi: 10.4049/jimmunol.0903987. [DOI] [PubMed] [Google Scholar]
- 13.Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci U S A. 2005;102:99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qualls JE, Kaplan AM, van Rooijen N, Cohen DA. Suppression of experimental colitis by intestinal mononuclear phagocytes. J Leukoc Biol. 2006;80:802–815. doi: 10.1189/jlb.1205734. [DOI] [PubMed] [Google Scholar]
- 15.Okamoto R, Watanabe M. Cellular and molecular mechanisms of the epithelial repair in IBD. Dig Dis Sci. 2005;50(Suppl 1):S34–38. doi: 10.1007/s10620-005-2804-5. [DOI] [PubMed] [Google Scholar]
- 16.Smith PD, Ochsenbauer-Jambor C, Smythies LE. Intestinal macrophages: unique effector cells of the innate immune system. Immunol Rev. 2005;206:149–159. doi: 10.1111/j.0105-2896.2005.00288.x. [DOI] [PubMed] [Google Scholar]
- 17.Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 18.Becker C, Fantini MC, Schramm C, Lehr HA, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Galle PR, Blessing M, Rose-John S, Neurath MF. TGF-β suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity. 2004;21:491–501. doi: 10.1016/j.immuni.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 19.Souza RF, Garrigue-Antar L, Lei J, Yin J, Appel R, Vellucci VF, Zou TT, Zhou X, Wang S, Rhyu MG, Cymes K, Chan O, Park WS, Krasna MJ, Greenwald BD, Cottrell J, Abraham JM, Simms L, Leggett B, Young J, Harpaz N, Reiss M, Meltzer SJ. Alterations of transforming growth factor-beta 1 receptor type II occur in ulcerative colitis-associated carcinomas, sporadic colorectal neoplasms, and esophageal carcinomas, but not in gastric neoplasms. Hum Cell. 1996;9:229–236. [PubMed] [Google Scholar]
- 20.Souza RF, Lei J, Yin J, Appel R, Zou TT, Zhou X, Wang S, Rhyu MG, Cymes K, Chan O, Park WS, Krasna MJ, Greenwald BD, Cottrell J, Abraham JM, Simms L, Leggett B, Young J, Harpaz N, Meltzer SJ. A transforming growth factor beta 1 receptor type II mutation in ulcerative colitis-associated neoplasms. Gastroenterology. 1997;112:40–45. doi: 10.1016/s0016-5085(97)70217-8. [DOI] [PubMed] [Google Scholar]
- 21.Beck PL, Rosenberg IM, Xavier RJ, Koh T, Wong JF, Podolsky DK. Transforming growth factor-beta mediates intestinal healing and susceptibility to injury in vitro and in vivo through epithelial cells. Am J Pathol. 2003;162:597–608. doi: 10.1016/s0002-9440(10)63853-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, Powrie F. T cells that cannot respond to TGF-β escape control by CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:737–746. doi: 10.1084/jem.20040685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobori A, Yagi Y, Imaeda H, Ban H, Bamba S, Tsujikawa T, Saito Y, Fujiyama Y, Andoh A. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol. 2010 doi: 10.1007/s00535-010-0245-1. [DOI] [PubMed] [Google Scholar]
- 24.Seidelin JB, Bjerrum JT, Coskun M, Widjaya B, Vainer B, Nielsen OH. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol Lett. 2010;128:80–85. doi: 10.1016/j.imlet.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, Gorman DM, Bazan JF, Kastelein RA. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 26.Lang R, Rutschman RL, Greaves DR, Murray PJ. Autocrine deactivation of macrophages in transgenic mice constitutively overexpressing IL-10 under control of the human CD68 promoter. J Immunol. 2002;168:3402–3411. doi: 10.4049/jimmunol.168.7.3402. [DOI] [PubMed] [Google Scholar]
- 27.Gough PJ, Gordon S, Greaves DR. The use of human CD68 transcriptional regulatory sequences to direct high-level expression of class A scavenger receptor in macrophages in vitro and in vivo. Immunology. 2001;103:351–361. doi: 10.1046/j.1365-2567.2001.01256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee GT, Hong JH, Kwak C, Woo J, Liu V, Lee C, Kim IY. Effect of dominant negative transforming growth factor-beta receptor type II on cytotoxic activity of RAW 264.7, a murine macrophage cell line. Cancer Res. 2007;67:6717–6724. doi: 10.1158/0008-5472.CAN-06-4263. [DOI] [PubMed] [Google Scholar]
- 29.Hahm KB, Im YH, Parks TW, Park SH, Markowitz S, Jung HY, Green J, Kim SJ. Loss of transforming growth factor beta signalling in the intestine contributes to tissue injury in inflammatory bowel disease. Gut. 2001;49:190–198. doi: 10.1136/gut.49.2.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ince MN, Elliott DE, Setiawan T, Metwali A, Blum A, Chen HL, Urban JF, Flavell RA, Weinstock JV. Role of T cell TGF-β signaling in intestinal cytokine responses and helminthic immune modulation. Eur J Immunol. 2009;39:1870–1878. doi: 10.1002/eji.200838956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frugier T, Koishi K, Matthaei KI, McLennan IS. Transgenic mice carrying a tetracycline-inducible, truncated transforming growth factor beta receptor (TGF-βRII) Genesis. 2005;42:1–5. doi: 10.1002/gene.20115. [DOI] [PubMed] [Google Scholar]
- 32.Kang SS, Bloom SM, Norian LA, Geske MJ, Flavell RA, Stappenbeck TS, Allen PM. An antibiotic-responsive mouse model of fulminant ulcerative colitis. PLoS Med. 2008;5:e41. doi: 10.1371/journal.pmed.0050041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chanteux H, Guisset AC, Pilette C, Sibille Y. LPS induces IL-10 production by human alveolar macrophages via MAPKinases- and Sp1-dependent mechanisms. Respir Res. 2007;8:71. doi: 10.1186/1465-9921-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maeda H, Kuwahara H, Ichimura Y, Ohtsuki M, Kurakata S, Shiraishi A. TGF-β enhances macrophage ability to produce IL-10 in normal and tumor-bearing mice. J Immunol. 1995;155:4926–4932. [PubMed] [Google Scholar]
- 35.Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kriegel MA, Li MO, Sanjabi S, Wan YY, Flavell RA. Transforming growth factor-beta: recent advances on its role in immune tolerance. Curr Rheumatol Rep. 2006;8:138–144. doi: 10.1007/s11926-006-0054-y. [DOI] [PubMed] [Google Scholar]
- 37.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- 38.Greaves DR, Gordon S. Macrophage-specific gene expression: current paradigms and future challenges. Int J Hematol. 2002;76:6–15. doi: 10.1007/BF02982713. [DOI] [PubMed] [Google Scholar]
- 39.Herbert DR, Holscher C, Mohrs M, Arendse B, Schwegmann A, Radwanska M, Leeto M, Kirsch R, Hall P, Mossmann H, Claussen B, Forster I, Brombacher F. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity. 2004;20:623–635. doi: 10.1016/s1074-7613(04)00107-4. [DOI] [PubMed] [Google Scholar]
- 40.Sasmono RT, Oceandy D, Pollard JW, Tong W, Pavli P, Wainwright BJ, Ostrowski MC, Himes SR, Hume DA. A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood. 2003;101:1155–1163. doi: 10.1182/blood-2002-02-0569. [DOI] [PubMed] [Google Scholar]
- 41.Buanne P, Di Carlo E, Caputi L, Brandolini L, Mosca M, Cattani F, Pellegrini L, Biordi L, Coletti G, Sorrentino C, Fedele G, Colotta F, Melillo G, Bertini R. Crucial pathophysiological role of CXCR2 in experimental ulcerative colitis in mice. J Leukoc Biol. 2007;82:1239–1246. doi: 10.1189/jlb.0207118. [DOI] [PubMed] [Google Scholar]
- 42.Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–194. doi: 10.1016/j.ccr.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin AA, Tripathi PK, Sholl A, Jordan MB, Hildeman DA. Gamma interferon signaling in macrophage lineage cells regulates central nervous system inflammation and chemokine production. J Virol. 2009;83:8604–8615. doi: 10.1128/JVI.02477-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lykens JE, Terrell CE, Zoller EE, Divanovic S, Trompette A, Karp CL, Aliberti J, Flick MJ, Jordan MB. Mice with a selective impairment of IFN-γ signaling in macrophage lineage cells demonstrate the critical role of IFN-γ-activated macrophages for the control of protozoan parasitic infections in vivo. J Immunol. 184:877–885. doi: 10.4049/jimmunol.0902346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S. Enhanced TH1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39–49. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 46.Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci U S A. 2005;102:8686–8691. doi: 10.1073/pnas.0500419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahrens R, Waddell A, Seidu L, Blanchard C, Carey R, Forbes E, Lampinen M, Wilson T, Cohen E, Stringer K, Ballard E, Munitz A, Xu H, Lee N, Lee JJ, Rothenberg ME, Denson L, Hogan SP. Intestinal macrophage/epithelial cell-derived CCL11/eotaxin-1 mediates eosinophil recruitment and function in pediatric ulcerative colitis. J Immunol. 2008;181:7390–7399. doi: 10.4049/jimmunol.181.10.7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Munitz A, Waddell A, Seidu L, Cole ET, Ahrens R, Hogan SP, Rothenberg ME. Resistin-like molecule alpha enhances myeloid cell activation and promotes colitis. J Allergy Clin Immunol. 2008;122:1200–1207. e1201. doi: 10.1016/j.jaci.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fuss IJ, Boirivant M, Lacy B, Strober W. The interrelated roles of TGF-β and IL-10 in the regulation of experimental colitis. J Immunol. 2002;168:900–908. doi: 10.4049/jimmunol.168.2.900. [DOI] [PubMed] [Google Scholar]
- 50.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat Immunol. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 51.Pastorelli L, Garg RR, Hoang SB, Spina L, Mattioli B, Scarpa M, Fiocchi C, Vecchi M, Pizarro TT. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proc Natl Acad Sci U S A. 2010;107:8017–8022. doi: 10.1073/pnas.0912678107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beltran CJ, Nunez LE, Diaz-Jimenez D, Farfan N, Candia E, Heine C, Lopez F, Gonzalez MJ, Quera R, Hermoso MA. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1097–1107. doi: 10.1002/ibd.21175. [DOI] [PubMed] [Google Scholar]
- 53.Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, Yang Z, Exley M, Kitani A, Blumberg RS, Mannon P, Strober W. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C, Pouly S, Murphy AJ, Valenzuela DM, Yancopoulos GD, Becher B, Littman DR, Neurath MF. RORγ-Expressing Th17 Cells Induce Murine Chronic Intestinal Inflammation via Redundant Effects of IL-17A and IL-17F. Gastroenterology. 2008 doi: 10.1053/j.gastro.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 55.Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, Nambu A, Abe T, Kiyonari H, Matsumoto K, Sudo K, Okumura K, Saito H, Nakae S. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci U S A. 2010;107:18581–18586. doi: 10.1073/pnas.1003059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shah AH, Tabayoyong WB, Kimm SY, Kim SJ, Van Parijs L, Lee C. Reconstitution of lethally irradiated adult mice with dominant negative TGF-β type II receptor-transduced bone marrow leads to myeloid expansion and inflammatory disease. J Immunol. 2002;169:3485–3491. doi: 10.4049/jimmunol.169.7.3485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.