

Abstract
Besides fluorine, oxygen is the most electronegative element with the highest reduction potential in biological systems. Metabolic pathways in mammalian cells utilize oxygen as the ultimate oxidizing agent to harvest free energy. They are very efficient, but not without risk of generating various oxygen radicals. These cells have good antioxidative defense mechanisms to neutralize these radicals and prevent oxidative stress. However, increased oxidative stress results in oxidative modifications in lipid, protein, and nucleic acids, leading to mitochondrial dysfunction and cell death. Oxidative stress and mitochondrial dysfunction have been implicated in many neurodegenerative disorders including Alzheimer's disease, Parkinson's disease, and stroke-related brain damage. Research has indicated mitochondria play a central role in cell suicide. An increase in oxidative stress causes mitochondrial dysfunction, leading to more production of reactive oxygen species and eventually mitochondrial membrane permeabilization. Once the mitochondria are destabilized, cells are destined to commit suicide. Therefore, antioxidative agents alone are not sufficient to protect neuronal loss in many neurodegenerative diseases. Combinatorial treatment with antioxidative agents could stabilize mitochondria and may be the most suitable strategy to prevent neuronal loss. This review discusses recent work related to oxidative toxicity in the central nervous system and strategies to treat neurodegenerative diseases.
1. Sensitivity of Neurons to Oxidative Stress
Neuronal cells in the brain are highly sensitive to oxidative stress due to their large dependence on oxidative phosphorylation for energy as compared to other cells. The demand for oxygen consumption is extremely high with 1-2% of the oxygen being converted into superoxide anion radicals (O2 ∙−) and hydrogen peroxide, leading to oxidative stress [1]. Oxidative stress exists when there is an imbalance of reactive oxygen species (ROS) production and antioxidant activity. Since there are high levels of metals such as iron in the brain, metal toxicity is also a problem leading to oxidative stress. One way that the brain combats stress is by employing the aerobic isoenzymatic form of lactate dehydrogenase when glucose is metabolized [2]. Previous reports have indicated that these isoenzymatic forms of lactate dehydrogenase play a significant role in the metabolic functions of neurons [3]. Since neurons in the brain also strongly depend on mitochondrial driven aerobic respiration, when the mitochondria become dysfunctional, these neurons become much more susceptible to oxidative stress. Mitochondria already have a high level of oxidative stress and, therefore, any increase in internal or external reactive oxygen species (ROS) leads to dysfunctional mitochondria, which in turn produces more ROS leading to a vicious and detrimental cycle (Figure 1).
Figure 1.
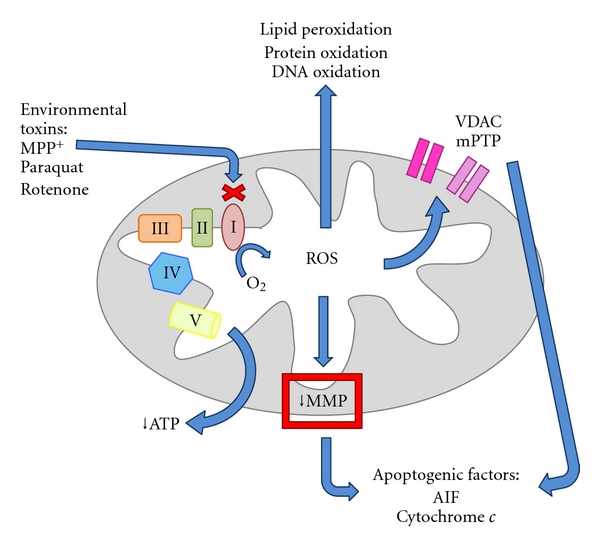
Environmental toxins cause the production of ROS by inhibiting complex I of the electron transport chain (ETC) and decrease the production of ATP. This ROS contributes to a loss in the mitochondrial membrane potential and well as disruption of mitochondrial permeability transition pores and voltage-dependant anion channels contributing to apoptosis. ROS also moves to the cytosol where it oxidizes proteins, DNA, and lipids.
Mitochondria have their own enzyme for combating ROS production by converting superoxide radicals to hydrogen peroxide by manganese superoxide dismutase (MnSOD), which are further broken down into water by peroxidases [4]. With heightened levels of oxidative stress, however, these combatants are not enough, and as we age, our defenses against oxidative stress decrease.
1.1. Factors Leading to Oxidative Stress
Generation of ROS at complex I, coined “complex I syndrome,” in the mitochondrial electron transport chain (ETC) has been linked to age-associated modifications in the central nervous system [4, 5]. When mitochondrial DNA is the target of oxidation, it can lead to mutations, rearrangements, and transcriptional errors that impair important mitochondrial components leading to more oxidative stress and eventual cell death. This has been shown to be more sensitive in cerebellar granule neuronal cells due to their deficiency in repairing mitochondrial DNA damaged by oxidative stress [4].
Lipid peroxidation causes a collapse of plasma and mitochondrial membranes, releasing cytochrome c, and inducing apoptosis. The brain is most affected by lipid peroxidation because of its high oxidizable lipid and metal content in comparison with other tissue [5].
Superoxide radicals and hydrogen peroxide can also create further oxidative stress by metal-catalyzed reactions [6]. Under oxidative stress, superoxide radicals can oxidize iron molecules. The released iron then takes part in the Fenton reaction and generates hydroxyl [6]. It has been shown that inactivation of mitochondrial aconitase (an enzyme involved in the citric acid cycle) by ROS contributes to the release of free iron and hydrogen peroxide leading to neuronal cell [7].
As a result of the reactions mentioned above, there are increased levels of oxidized glutathione (GSSG) with a concomitant decrease in reduced glutathione (GSH), oxidized protein, and increased lipid peroxidation, all of which are commonly used as markers of oxidative stress and the extent of damage caused by it. We have shown a decrease in GSH levels when rats are challenged with the herbicide paraquat, known to cause neurotoxicity and depletion of substantia nigra neurons due to oxidative stress (Figure 2).
Figure 2.
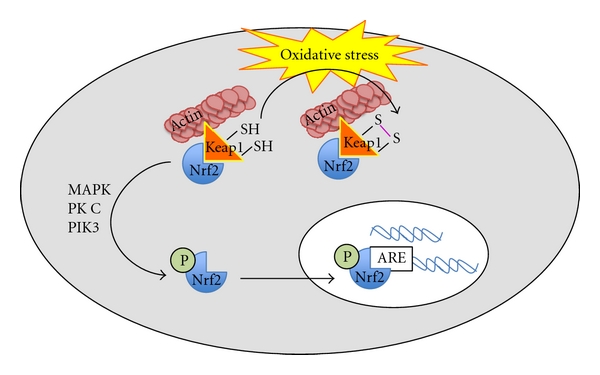
Oxidative stress induces Nrf2 dissociation from Keap1. Nrf2 is activated by phosphorylation and translocated into the nucleus where it may act as a transcription factor for antioxidant response genes.
Direct oxidative stress by hydrogen peroxide has been shown to induce inflammation by NF-κB activation and interleukins and is involved in the stress activated protein kinase (JNK) pathway [8]. Recent studies on chronic exposure of neuronal cells to hydrogen peroxide elicit dynamic responses, including changes in cytoskeletal structure, energy metabolic shifts (aerobic to glycolysis), and transmembrane receptor activity [9]. In other studies, chronic exposure to hydrogen peroxide has been shown to have a protective role by inducing the upregulation of antioxidant enzymes such as catalase and superoxide dismutase [10, 11].
1.2. Oxidative Stress in Neurodegenerative Diseases Induced by Environmental Toxins
Oxidative stress has been linked to aging and two of the most common neurodegenerative diseases, namely, Alzheimer's disease (AD) and Parkinson's disease (PD). AD is characterized by the loss of neurons, synapses, and neurotransmitters throughout the brain, but especially in the hippocampus and cerebral cortex. Mitochondrial dysfunction may be the underlying reason for the loss, marked by an increase in ROS, lipid peroxidation, and protein oxidation, which are found in AD brains, thus contributing to oxidative stress [28, 29]. Amyloid beta (Aβ), one of the hallmarks of AD, is also involved in oxidative stress and mitochondrial dysfunction by reducing the mitochondrial membrane potential. As an age-related disease, this reduction is intensified in the brain of aging animal models compared to younger animals [28].
In PD, the oxidative stress is most damaging and selective to the mitochondria, specifically in the substantia nigra region of the midbrain. Susceptibility to this disease can be due to genetics, environmental toxins (including most pesticides and herbicides), or a combination of the two, which can cause mitochondrial damage leading to oxidative stress [30].
Many cell culture models have been used to establish the role of oxidative stress in PD in hopes of translating the observed results to an in vivo model. For example, glutamate excitotoxicity on mixed neuronal-glial cell cultures along with hypoxia-induced neuronal cell death decreased ATP production and increased ROS [31]. Direct hydrogen peroxide insult has been shown to induce all the same negative factors mentioned above in conjunction with PD [32] and is also associated with the proapoptotic protein Bax [33].
One well-established in vivo model for studying PD is the use of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a byproduct found in synthetic heroine. MPTP crosses the blood-brain barrier and is metabolized into 1-methyl-4-phenylpyridinium (MPP+) where it has been shown to block complex I of the ETC and increase ROS, lipid peroxidation, and protein oxidation [13, 34]. As mentioned previously, genetics and environmental toxins can provide negative synergistic effects contributing to PD. Recently, it has been found that DJ-1 deficient mice challenged with MPTP have an increase in both oxidative stress and dopaminergic neuronal cell death [35]. DJ-1 is a gene associated with an early onset form of PD.
Exposure to paraquat, which is structurally similar to MPP+, has been correlated with the risk of PD in multiple studies [36–38]. Paraquat treatment on human neuronal cell culture causes mitochondrial permeabilization and oxidative stress [39]. It is believed to induce symptoms of PD by reacting in its reduced form with oxygen to produce a superoxide radical [40, 41]. Although banned in Europe in 2007, this herbicide is still the most used worldwide [42, 43]. In addition, rotenone, a pesticide and complex I inhibitor, induces the symptoms of PD similar to paraquat and MPP+.
Exposure to paraquat has also been established as an in vivo model of PD. Both paraquat-injected rats and mice show parkinsonian symptoms, oxidative stress, and dopaminergic neural loss in the substantia nigra region of the midbrain [13, 44, 45]. These symptoms could also be attenuated by water-soluble coenzyme Q10, an ETC component and antioxidant [13].
It was recently shown that the mechanism in which paraquat and rotenone induce dopaminergic cell death might be through the JNK pathway. This is believed to be due to increased phosphorylation of JNK as demonstrated in primary cultured dopaminergic neurons under paraquat and rotenone insult [46, 47] and caspase-3-dependent apoptosis [48]. Another mechanistic pathway of dopaminergic cell death by both paraquat and MPP+ is believed to be through the activation of NADPH oxidase-1 (a superoxide-generating enzyme complex), [49, 50].
It was also observed that rotenone induced dopaminergic neurodegeneration in an animal model by means of microglial activation, causing NADPH oxidase-derived superoxides to be formed [51]. However, recent studies show that human cell line microglia, although activated, only produce extracellular ROS and, therefore, do not contribute to neurodegeneration when exposed to chronic, low doses of rotenone [52]. Paraquat has been shown to induce oxidative stress in the cytosol, whereas MPP+ and rotenone induce oxidative stress in the mitochondria [48]. Although all these chemicals induce symptoms of PD, their mechanism of neuronal cell death varies, therefore, providing multiple approaches to not only study the mechanisms associated with PD but also to develop innovative therapeutic interventions for combating this disease.
1.3. Role of Stress-Responsive Transcription Factor Nrf2 (NF-E2-Related Factor 2) in Protection against Oxidative Toxicity
Role of Nrf2, an important stress-responsive transcription factor of the “cap-and-collar” β-leucine zipper family, is now considered instrumental to several neurodegenerative disorders [53–55]. Expression of a number of Phase II enzymes (e.g., NQO1, GSTs) and antioxidant proteins (e.g., GCL, HO-1, thioredoxin) are regulated by this gene. It is believed that this process is driven by the association of Nrf2 to the antioxidant responsive element (ARE) consensus sequence (5′-TGACnnnGCA-3′) on the promoter region of these genes [56–59]. Considerable efforts are being made to locate some of its downstream effector genes, including thioredoxin reductase and MafG [60, 61]. However, a clear-cut understanding of the mechanisms of Nrf2 upstream activation remains unclear to date.
It has been shown that Nrf2 is constitutively expressed and localized in the cytosolic compartment and maintains a repressed state by complexing with the actin-associating protein, Keap1. This heterodimerization limits most of Nrf2 to the cytoskeleton and away from the nucleus. Because of a cysteine-rich surface, Keap1 has vulnerability to oxidation during, escalation of intracellular oxidative and nitrosative stress ultimately resulting in global conformational changes to Keap1, thereby, leading to the liberation of Nrf2. After such a reaction, the monomeric Nrf2 becomes available to translocate to the nucleus (Figure 2). In this manner, Keap1 acts as a redox sensor that upregulates ARE antioxidant responses through Nrf2 [16, 56, 62]. Nrf2 activation also has been shown to be mediated through phosphorylation of Nrf2 by mitogen-activated protein kinases (MAPKs), protein kinase C (atypical isoform), and phosphoinositol-3-kinase (PI3K) [54, 63].
In response to oxidative stress, Nrf2 normally translocates from the cytoplasm into the nucleus and transactivates expression of genes with antioxidant activity. Despite this cellular mechanism, severe oxidative damage is not uncommon in Alzheimer (AD) and Parkinson disease (PD). Intense mechanistic investigations in this arena have revealed that Nrf2 expression is abundant in both the nucleus and the cytoplasm of neurons in normal hippocampi with predominant expression in the nucleus. However, in Alzheimer's, Nrf2 was predominantly cytoplasmic rather than nuclear in hippocampal neurons and was not a major component of beta amyloid plaques or neurofibrillary tangles. In contrast, the magnitude of expression of nuclear Nrf2 was much stronger in PD nigral neurons, but it was cytoplasm centric in substantia nigra of normal Alzheimer's. Such observations suggest that Nrf2-mediated transcription is not robust in neurons in AD despite the presence of oxidative stress. But in PD, despite a stronger nuclear localization of Nrf2, the impact of Nrf2 may be inadequate to protect neurodegeneration [53]. Because of this differential Nrf2 expression, it can be considered as a potential therapeutic target for conditions that are sensitive to free radical damage. Unfortunately, these observations do not account for additional contributory roles played by microglia and astrocytes in the overall neuronal system. Future studies may unravel their alleviating or aggravating role during oxidative stress. Nevertheless, mitochondrial dysfunction and buildup of reactive oxygen species are so common in most neurodegenerative disorders that targeting Nrf2 may be a novel way of combating conditions with variable causes and etiologies [64, 65].
2. Therapeutic Advances in Alzheimer's Disease and Parkinson's Disease
There are a variety of antioxidants that can attenuate the effects of oxidative stress through multiple mechanisms, and the importance of antioxidants maintaining redox balance is well known. However, no single antioxidant or combination of antioxidants has been discovered to completely halt the progression or cure the diseases that are associated with the destructive properties of oxidative stress. In AD, oxidative stress is a factor throughout the entire brain, making it difficult to find treatment that is specific to the symptoms of the disease. On the other hand, PD is a localized disease, where dopaminergic neurons in the substantia nigra can be monitored, and treatment can be more streamlined and targeted.
Recent studies for the treatment of PD and AD have been directed at agents that target and stabilize the mitochondria [28]. The most promising treatment for AD is the administration of methylene blue, a potential mitochondrial stabilizer at complex I and IV (Table 1). However, to date there is limited amount of published data [66].
Table 1.
Therapeutic modalities undergoing preclinical/clinical trials for neurodegenerative diseases.
| Compound/Chemical | Disease | Effect |
|---|---|---|
| CoQ10 | PD | Reduces the loss of DA neurons in the SNpc (Cleren et al., 2008, [12], Somayajulu-Niţu et al., 2009, [13]) |
|
| ||
| Trehalose | PD | Autophagy-mediated, short-term reduction of phosphorylated tau and β-amyloid plaques in parkin (PK−/−/TauVLW) mouse model (Rodríguez-Navarro et al., 2010, [14]) |
|
| ||
| SR-3306 (JNK inhibitor) | PD | Reduces the loss of dopaminergic cell bodies in the SNpc and their terminals in the striatum (Crocker et al., 2011, [15]) |
|
| ||
| Curcumin | PD | Reduces synuclein toxicity, intracellular ROS, and apoptosis in neuroblastoma cells (Dinkova-Kostova et al., 2010, [16]) |
|
| ||
| AD | Blockes Aβ aggregation (Guo et al., 2010, [17]) | |
| Inhibites Aβ insult (Ono et al., 2004, [18]) | ||
| Protects Sprague-Dawley rats from Aβ-induced damage (Frautschy et al., 2001, [19]) | ||
| Inhibits neuroglial cell proliferation (Ambegaokar et al., 2003, [20]) | ||
| Inhibits Aβ-induced cytochemokine gene expression and CCR5-mediated chemotaxis of THP-1 monocytes by modulating EGR-1 (Giri et al., 2004, [21]) | ||
| Inhibits α-synuclein aggregation (Pandey et al., 2008, [22]) | ||
|
| ||
| Methylene blue | AD | Inhibits cGMP pathway |
| Attenuates amyloid plaque formation and neurofibrillary tangles (Wischik et al., 2008, [23] Oz et al., 2009, [24]) | ||
|
| ||
| Viral vector Aβ cDNA (oral vaccination) | AD | Alleviates progressive cognitive impairment with decreased Aβ deposition, insoluble Aβ, soluble Aβ oligomer, microglial attraction, and synaptic degeneration induced in brain regions (Mouri et al., 2007, [25]) |
|
| ||
| AL-108 | AD | Stabilizes microtubules and blocks Aβ aggregation (Masters and Beyreuther, 2006, [26]) |
|
| ||
| Curcumin derivative | Stroke | Enhances memory, contributes to neurotrophic activity, and prevents cell death (Lapchak et al., 2011, [27]) |
Antioxidant and anti-inflammatory drugs are the focus of research in combating oxidative stress aimed at stabilizing the mitochondria by quenching ROS generated from the ETC. Common antioxidants such as flavonoids, vitamin C, beta hydroxy acid (BHA), butylated hydroxytoluene (BHT), and nordihydroguaiaretic acid may be unable to access the ubiquinone-binding sites at complex I and II due to their hydrophobicity. These antioxidants are better at combating ROS at the flavin mononucleotide site of complex I [67]. This insight demonstrates that new therapeutic agents need to be specific to the pathophysiological conditions of the site where ROS are generated. Coenzyme Q10 has been shown to have some neuroprotective effects and is under clinical trial for Parkinson's disease (Table 1). However, due to its highly hydrophobic nature, the oil-soluble formulation of CoQ10 could not be studied in cell culture models and its efficacy in in vivo studies is found only at very high doses. A new water-soluble formulation of CoQ10, developed by combining it with tocopherol and poly ethylene glycol, has shown very promising results. The water-soluble CoQ10 (WS-CoQ10) protected human neuronal cells against oxidative stress-induced cell death in several in vitro culture models [31, 32, 39]. Furthermore, it has been shown that WS-CoQ10 not only decreased the oxidative stress, but stabilized mitochondria and prevented Bax-induced mitochondrial permeabilization [33]. Most importantly, in a recent study with a paraquat-induced Parkinson's disease rat model, WS-CoQ10 was shown to be very effective in preventing neuronal loss and amelioration of PD-related symptoms [13]. As shown in Figure 2, levels of oxidative stress induced by paraquat was decreased in WS-CoQ10-treated rats.
Recently a disaccharide, Trehalose, has been shown to protect SNpc neurons by the induction of autophagy, short-term reduction of phosphorylated tau and β-amyloid plaques in parkin (PK−/−/TauVLW) mouse model [14].
Exercise has been shown to combat oxidative stress in PD by inducing the production of antioxidants and neurotrophic factors [68] and has also been shown to clear Aβ peptides in AD [68, 69].
There are numerous antioxidants on the market that are extremely useful at combating oxidative stress. By assessing and evaluating these antioxidants, it is hoped that they may be used therapeutically for PD, AD, or stroke-related injury.
3. Implications of Oxidative Stress in Stroke and Ischemic Related Brain Injury
In ischemic related brain injuries, one of the main perpetrators of cellular damage is oxidative stress. Many studies have indicated that the increase in oxidative stress contributes to lipid damage, protein alterations, and DNA damage. Ironically, the return of blood flow to the infarcted area of the brain causes harm along with its benefits due to the increase in oxygen availability and the increase in oxidative stress that reperfusion causes. In these situations, lactic acid accumulates in the affected neurons promoting prooxidant effects by increasing the H+ concentration within the cells and generating more ROS [70]. The primary source of ROS is the superoxide anion radical (O2 ∙−), which is generated by leakage from complex III of the electron transport chain of malfunctioning mitochondria [71].
While oxygen may be the main culprit associated with damage due to oxidative stress, it does not act without its accomplices. Hydrogen peroxide is converted to the hydroxyl radical (•OH), and the nitric oxide (NO) species that are produced can have extensive implications in neuronal signaling. During the ensuing inflammatory response, O2 ∙− can undergo a lethal reaction with NO to produce the highly detrimental peroxynitrite anion (ONOO−), which in turn leads to DNA fragmentation and lipid peroxidation [72].
Since the brain makes up only 2% of the total body weight of a human, yet consumes approximately 20% of the available oxygen, it is an excellent environment for the occurrence of oxidative stress [73]. The brain also contains high levels of lipids while possessing low amounts of antioxidants, thus further increasing its susceptibility to damage as the result of ROS and oxidative stress [74].
Stroke is a leading cause of death and long-term disability in industrialized nations [75, 76] and is a condition that regularly leaves its victims in a state of impaired cognition and motor deficits, with nearly 40% of patients not expected to make a full recovery [70]. The damage and detrimental effects of stroke are heavily influenced by oxidative stress and the production of free radicals.
Two types of stroke can occur, hemorrhagic stroke, and the more common, ischemic stroke. In hemorrhagic stroke, rupture of an artery results in uncontrolled bleeding to the affected area of the brain. In ischemic stroke, there is a blockage of blood flow to the brain due to the formation of a blood clot. This deprivation of oxygenated blood results in the formation of the ischemic core where cells die rather quickly and irreversibly due to necrosis. The onset of lipolysis, protein degradation, and the breakdown of ion homeostasis are some of the events responsible for the rapid death of these cells [77].
In the area between the unaffected brain and the ischemic core lies a region where the struggle between the life and death of neurons ensues. This region of the brain is known as the penumbra. It is here that the brain is composed of damaged and malfunctioning, yet salvageable tissue. Cells in this region are susceptible to a programmed form of cell death known as apoptosis. These cells can remain viable and for several days following the onset of stroke [78].
Here in the penumbra region is where a host of events related to oxidative stress take place. Ironically enough, reperfusion acts as a double-edged sword. While reperfusion is essential to save the cells affected by ischemia, it also brings along with it its own threat. When reperfusion occurs, there is a large and rapid influx of oxygenated blood to the infarct region. While this delivers the necessary blood, it also brings with it the elements necessary for producing ROS that contribute to the oxidative stress placed upon the already damaged brain tissue.
When platelets are exposed to conditions of reperfusion, they generate additional ROS in the form of O2 ∙− and •OH. Furthermore, the ROS that are produced during reperfusion are responsible for the activation and transcription of many proteins. For example, ROS stimulate the production of JNK and mitogen-activated protein kinase phosphorylation (p38 MAPK) in the affected neurons of the penumbra. JNK-1 is favored in the nucleus of neurons during reperfusion, and activator protein-1 (AP-1) binding is also enhanced [78]. The activation of AP-1 is necessary for the induction of apoptosis to occur [79]. This action, along with the activation of caspase-3, are several examples of how reperfusion is responsible for initiating cell death within the neurons of the penumbra by controlling the expression of certain genes.
Along with their role in effecting the transcription of various proteins, ROS generated by reperfusion can itself cause direct cellular stress. Reperfusion causes such a large influx of oxygen that all of it cannot be used by the mitochondria, and normal radical scavenging mechanisms such as superoxide dismutase (SOD), glutathione peroxidase, and catalase are overwhelmed and cannot adequately quench the multitude of free radicals that are produced and leak from the system [80]. The cells of the penumbra are already vulnerable to damage, and the generation of ROS exacerbates the damage that may have already occurred to these cells by lipid peroxidation. In particular, phospholipid membrane degradation is a major concern. The brain is especially susceptible to such damage due to the large amount of lipids that compose its structure. Lipid peroxidation targets the polyunsaturated fatty acids (PUFAs) in the brain, thus, decreasing the membrane integrity. The decrease in membrane stability is especially important because the membrane contains receptor proteins and ion channel entities. Along with its own deleterious effects, lipid peroxidation is also responsible for the inhibition of lipid repair enzymes such as lysophosphatidylcholine acyltransferase and fatty acyl CoA-synthase [81].
While cells in the infarct region die instantly via necrosis, cells of the penumbra are likely to die by means of apoptosis. Apoptosis is a programmed form of cell death where the cell expends energy towards its own demise. It is controlled by a complex interconnection of proteins and is often triggered by oxidative stress and the release of cytochrome c from the mitochondria [82]. The increased level of ROS is involved in generating the signal that causes permeabilization of the mitochondrial membrane, and, thus, the release of cytochrome c into the cytosol. Once this occurs, the initiation of the cascade of caspases occurs. Activation of caspases 3, 8, and 9 will eventually lead to the death of the cell and other surrounding cells [83].
3.1. Stroke-Induced Inflammation
Inflammation is a biological response to harmful stimuli and often occurs as a result of stroke. One of the key contributors to the inflammatory response are glial cells, more specifically microglia, that secrete proinflammatory cytokines and chemokines that contribute to the damage done to the penumbra. The most common contributors to the damage due to inflammation are tumour necrosis factor alpha (TNF-alpha) and interlenukin-B (IL-Beta), among others [84]. These cytokines are responsible for the increased expression of cellular adhesion molecules (CAMs) that in combination with platelets, adhere to vessel walls causing a “no-flow” constriction [85] and the release of more proinflammatory molecules. Ultimately, the inflammatory response results in decreased blood brain barrier function, increased cerebral edema, and cell death [84].
3.2. The Role of Proapoptotic Proteins in Stroke
As previously mentioned, apoptosis is controlled by a wide range of proteins. Oxidative stress can cause the activation of p53-tumor suppressor gene which in turn is responsible for the increased transcription of Bcl-2 associated X protein (Bax) and the p53 upregulated modulator of apoptosis (PUMA) [86]. PUMA has been shown to be able to interact with the Bcl-2 family of proteins to assist in initiating apoptosis. It has been shown that PUMA is able to associate with the mitochondrial membrane along with Bax to promote the release of cytochrome c [87]. Studies involving the PD-associated DJ-1 gene indicate that this gene protects the cells against excitotoxicity and the effects of ischemia. DJ-1 was found to decrease the presence of oxidative stress markers, and, thus, protect the cell due to the alleviation of the effects of oxidative stress [88].
A large majority of the proteins responsible for the balance between death and survival belong to the Bcl-2 family of proteins that protect cells from apoptosis induced by a wide variety of stimuli. One of these proteins is the proapoptotic protein Bax which exists in the cytosol as a harmless 24 kDa monomer. However, in cases of increased oxidative stress, the protein product of the p53 tumor suppressor gene causes increased transcription of Bax to occur [89]. In response to this increased amount of Bax due to oxidative stress, Bax undergoes dimerization either with itself or other members of the Bcl-2 family (e.g., tBID) through interactions of alpha helix 2 with the BH3 domain [90]. This dimerized form of Bax then begins its migration towards the mitochondria. Once in range of the mitochondria, the dimerized form of Bax may associate with the protein transition pore (PTP) of the voltage-dependent anion channel (VDAC) of the mitochondrial membrane. This action allows for the uncontrolled flow of cytotoxic factors, such as cytochrome c, to be released from the mitochondria into the cytosol, and inevitably, the demise of the mitochondria [91].
Studies have demonstrated that Bax channel activity is necessary for apoptosis to occur since cell death was halted with the use of Bax channel blockers [92]. Since Bax is an essential protein in the regulation of apoptosis, it is an excellent target candidate for therapeutic approaches. Not only does its extensive involvement in the process of cell death make Bax a good therapeutic target, its position in the apoptosis cascade does as well. While focusing on antioxidants may be a valid point of investigation, bolstering of the antioxidant defense machinery still results in permeabilized mitochondria. Blocking the initiation of apoptosis further up the chain by inhibiting Bax function may save the mitochondria and halt apoptosis.
3.3. Experimental Models of Stroke
In the cellular model of stroke, an excellent way to mimic ischemic assault is by inducing hypoxia. Hypoxia is the deprivation of an adequate amount of oxygen to tissues, usually accompanied by detrimental effects. To accomplish this in a cellular model, cells can be placed in an oxygen-free chamber for a period of time before being removed [93]. It was found that when conditions of hypoxia exist, hypoxia-inducible factor alpha (HIF-1α) expression increases [94] and, therefore, is implicated in hypoxia-induced apoptosis. HIF-1α participates by stabilizing the structure of the tumor suppressor gene p53, which leads to the expression of apoptosis-related genes [94, 95].
HIF-1α has also been shown to have antiapoptotic effects because those cells with increased levels of HIF-1α show resistance to hypoxia-induced apoptosis [96]. The deciding factor of whether HIF-1α is protective or harmful to a cell seems to depend on the level of hypoxia. If conditions of mild hypoxia exist, HIF-1α is phosphorylated and associated with HIF-1β, and the transcription of p53 is low with anti-apoptotic genes being activated [97, 98]. However, in high hypoxic conditions, the reverse is the case. HIF-1α is dephosphorylated and p53 levels are unregulated, eventually leading to the activation of pro-apoptotic proteins such as Bax [97, 99, 100]. This is similar to conditions of ischemic stroke where up to 24 hours after ischemia, most pro-apoptotic genes are upregulated, whereas 48 hours to 8 days after ischemia anti-apoptotic genes are the majority of those induced [94].
There are a variety of different in vivo models of stroke ranging from middle cerebral artery occlusion (MCAO) [101] and four vessel carotid artery occlusion [102]. One model that our laboratory has employed to investigate the effects of stroke in a rat model is the bilateral carotid artery occlusion and two vessel occlusion hypovolemic hypotension (2VO/HT) model [103]. In this model, global forebrain ischemia is induced by occluding the 2 carotid arteries and removing a certain volume of blood from the animal to maintain a mean arterial pressure of 50 mm Hg. This results in an interruption of blood flow to the brain, successfully creating an infarct region similar to stroke and the surrounding penumbra. This model can be used to accurately investigate the effects of various therapeutic reagents and their abilities to protect neurons under conditions similar to stroke.
3.4. Therapeutic Approaches for Stroke
At this time, the only known treatment for victims of stroke is the use of thrombolytics, most commonly, tissue plasminogen activator. The downfall to this avenue of treatment is that it must be administered within 3 hours of the onset of stroke. This is relatively hard to accomplish, as most stroke victims do not arrive at hospital to receive treatment within this timeframe. Also, thrombolytics can lead to an increased likelihood of hemorrhages occurring within the brain [85]. Hypothermia has also been investigated as a possible treatment for stroke. It has been found that lowering the body temperature of a stroke victim may improve the neurological outcome [104]. However this technique remains highly experimental as the temperature, duration, and onset of cooling still remains to be accurately determined.
An emerging field of study for treatment of ischemia includes the use of bone marrow stromal cells (BMSC). These cells can differentiate into neural and glial cells, both in vivo and in vitro, after being transplanted into animal model brains following neurological insult such as intracerebral hemorrhage (ICH) [105]. These neural stem cells migrate to the area of the brain that is injured in order to replace the neuron deficit that was lost due to hemorrhagic stroke. Recent studies have found that these BMSCs are able to generate functional recovery in Wistar rats following ICH [106].
Another interesting avenue of exploration into potential therapeutics for stroke is the use of curcumin derivatives. Curcumin has been shown to prevent Alzheimer's markers in animal models of the disease [107] and has also been shown to be effective in reducing the deficits of middle cerebral artery occlusion in a rat [108, 109]. Recent studies have shown that when used as a treatment in a model of stroke, a pyrazole derivative of curcumin was able to enhance memory, contribute to neurotrophic activity, and, most importantly, prevent cell death [110].
Research has focused on blocking pro-apoptotic proteins that are responsible for causing cell death. Recently, advances in treatments for stoke have been made by the use of low molecular weight compounds that inhibit proteins (such as Bax) that are critical in the apoptosis cascade. This is a critical stage for the inhibition of apoptosis due to the fact that Bax channel formation is required for the destabilization of the mitochondria, and subsequent release of cytotoxic factors [92]. These inhibitory compounds were modeled after single-domain antibodies that were able to bind specifically to Bax [93]. They are small enough to have the potential to cross the blood brain barrier and are not susceptible to proteolysis. Recent research completed in our laboratory indicates that these compounds show a high specificity towards the pro-apoptotic protein Bax and are able to block its function and save the neurons of the penumbra from apoptosis [111]. These compounds are able to competitively bind to Bax even when in the presence of single-domain antibodies that are specific to Bax. By binding in some manner to Bax, these compounds prevent the association of Bax with the mitochondria and prevent mitochondrial destabilization, thus, limiting the influx of cytotoxic factors into the cytosol. It is hoped that these compounds will not need-to be administered in such a short-time frame following stroke as is the case for thrombolytics, nor pose the risk of hemorrhage that thrombolytics do. With more investigation, it is likely that the use of low molecular weight compounds will become valid treatment options for stroke patients.
4. Conclusion
It is now well established that oxidative stress and mitochondrial dysfunction are the early and key biochemical mechanisms leading to Alzheimer's disease, Parkinson's disease, and stroke-related pathologies. Mitochondria are greatly involved in neuronal cell death due to the vicious cycle of oxidative toxicity, which causes mitochondrial dysfunction that leads to more ROS and the potential collapse of the mitochondrial membrane. Environmental toxins, amyloid-beta toxicity, and ischemia/hyper-perfusion-related toxicity all lead to oxidative toxicity directly or indirectly by mitochondrial destabilization. Significant progress has been made to inhibit neuronal cell death by using anti-oxidants or blockers of pro-apoptotic proteins, but a combinatorial treatment to reduce oxidative stress and stabilize mitochondria to halt neuronal loss needs to be explored.
Figure 3.
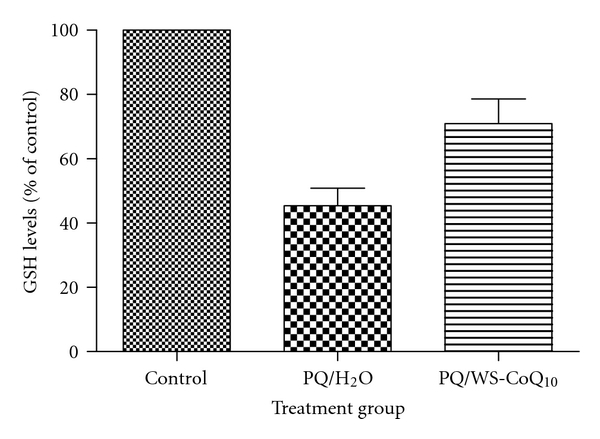
GSH assay. Measuring oxidative stress levels in brain tissue from rats challenged with paraquat and treated with WS-CoQ10. GSH levels decrease in the presence of toxin-inducted oxidative stress and are brought back up to control levels in the presence of the antioxidant (WS-CoQ10).
Acknowledgments
The authors gratefully acknowledge the funding from NSERC and CIHR for this research project. K. Facecchia and L.-A. Fochesato contributed equally to this paper.
References
- 1.Moura MB, Dos Santos LS, Van Houten B. Mitochondrial dysfunction in neurodegenerative diseases and cancer. Environmental and Molecular Mutagenesis. 2010;51(5):391–405. doi: 10.1002/em.20575. [DOI] [PubMed] [Google Scholar]
- 2.Bittar PG, Charnay Y, Pellerin L, Bouras C, Magistretti PJ. Selective distribution of lactate dehydrogenase isoenzymes in neurons and astrocytes of human brain. Journal of Cerebral Blood Flow and Metabolism. 1996;16(6):1079–1089. doi: 10.1097/00004647-199611000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Tholey G, Roth-Schechter BF, Mandel P. Activity and isoenzyme pattern of lactate dehydrogenase in neurons and astroblasts cultured from brains of chick embryos. Journal of Neurochemistry. 1981;36(1):77–81. doi: 10.1111/j.1471-4159.1981.tb02379.x. [DOI] [PubMed] [Google Scholar]
- 4.Harrison JF, Hollensworth SB, Spitz DR, Copeland WC, Wilson GL, LeDoux SP. Oxidative stress-induced apoptosis in neurons correlates with mitochondrial DNA base excision repair pathway imbalance. Nucleic Acids Research. 2005;33(14):4660–4671. doi: 10.1093/nar/gki759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nobre Jr. HV, Fonteles MMDF, De Freitas RMD. Acute seizure activity promotes lipid peroxidation, increased nitrite levels and adaptive pathways against oxidative stress in the frontal cortex and striatum. Oxidative Medicine and Cellular Longevity. 2009;2(3):130–137. doi: 10.4161/oxim.2.3.8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jomova K, Vondrakova D, Lawson M, Valko M. Metals, oxidative stress and neurodegenerative disorders. Molecular and Cellular Biochemistry. 2010;345(1-2):91–104. doi: 10.1007/s11010-010-0563-x. [DOI] [PubMed] [Google Scholar]
- 7.Cantu D, Schaack J, Patel M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS ONE. 2009;4(9, article e7095) doi: 10.1371/journal.pone.0007095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu W-C, Hu D-N, Gao H-X, et al. Subtoxic levels hydrogen peroxide-induced production of interleukin-6 by retinal pigment epithelial cells. Molecular Vision. 2010;16:1864–1873. [PMC free article] [PubMed] [Google Scholar]
- 9.Chadwick W, Zhou Y, Park S-S, et al. Minimal peroxide exposure of neuronal cells induces multifaceted adaptive responses. PLoS ONE. 2010;5(12, article e14352) doi: 10.1371/journal.pone.0014352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sen P, Chakraborty PK, Raha S. p38 Mitogen-activated protein kinase (p38MAPK) upregulates catalase levels in response to low dose H2O2 treatment through enhancement of mRNA stability. FEBS Letters. 2005;579(20):4402–4406. doi: 10.1016/j.febslet.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 11.Gomez-Cabrera MC, Domenech E, Viña J. Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radical Biology and Medicine. 2008;44(2):126–131. doi: 10.1016/j.freeradbiomed.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Cleren C, Yang L, Lorenzo B, et al. Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of Parkinsonism. Journal of Neurochemistry. 2008;104(6):1613–1621. doi: 10.1111/j.1471-4159.2007.05097.x. [DOI] [PubMed] [Google Scholar]
- 13.Somayajulu-Niţu M, Sandhu JK, Cohen J, et al. Paraquat induces oxidative stress, neuronal loss in substantia nigra region and Parkinsonism in adult rats: neuroprotection and amelioration of symptoms by water-soluble formulation of coenzyme Q10 . BMC Neuroscience. 2009;10, article88 doi: 10.1186/1471-2202-10-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodríguez-Navarro JA, Rodríguez L, Casarejos MJ, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiology of Disease. 2010;39(3):423–438. doi: 10.1016/j.nbd.2010.05.014. [DOI] [PubMed] [Google Scholar]
- 15.Crocker CE, Khan S, Cameron MD, Robertson HA, Robertson GS, Lograsso P. JNK inhibition protects dopamine neurons and provides behavioral improvement in a rat 6-hydroxydopamine model of Parkinson's disease. ACS Chemical Neuroscience. 2011;2(4):207–212. doi: 10.1021/cn1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dinkova-Kostova AT, Talalay P, Sharkey J, et al. An exceptionally potent inducer of cytoprotective enzymes: elucidation of the structural features that determine inducer potency and reactivity with Keap1. Journal of Biological Chemistry. 2010;285(44):33747–33755. doi: 10.1074/jbc.M110.163485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo J-P, Yu S, McGeer PL. Simple in vitro assays to identify amyloid-β aggregation blockers for Alzheimer's disease therapy. Journal of Alzheimer's Disease. 2010;19(4):1359–1370. doi: 10.3233/JAD-2010-1331. [DOI] [PubMed] [Google Scholar]
- 18.Ono K, Hasegawa K, Naiki H, Yamada M. Curcumin has potent anti-amyloidogenic effects for Alzheimer's β-amyloid fibrils in vitro. Journal of Neuroscience Research. 2004;75(6):742–750. doi: 10.1002/jnr.20025. [DOI] [PubMed] [Google Scholar]
- 19.Frautschy SA, Hu W, Kim P, et al. Phenolic anti-inflammatory antioxidant reversal of Aβ-induced cognitive deficits and neuropathology. Neurobiology of Aging. 2001;22(6):993–1005. doi: 10.1016/s0197-4580(01)00300-1. [DOI] [PubMed] [Google Scholar]
- 20.Ambegaokar SS, Wu L, Alamshahi K, et al. Curcumin inhibits dose-dependently and time-dependently neuroglial cell proliferation and growth. Neuroendocrinology Letters. 2003;24(6):469–473. [PubMed] [Google Scholar]
- 21.Giri RK, Rajagopal V, Kalra VK. Curcumin, the active constituent of turmeric, inhibits amyloid peptide-induced cytochemokine gene expression and CCR5-mediated chemotaxis of THP-1 monocytes by modulating early growth response-1 transcription factor. Journal of Neurochemistry. 2004;91(5):1199–1210. doi: 10.1111/j.1471-4159.2004.02800.x. [DOI] [PubMed] [Google Scholar]
- 22.Pandey N, Strider J, Nolan WC, Yan SX, Galvin JE. Curcumin inhibits aggregation of alpha-synuclein. Acta Neuropathologica. 2008;115(4):479–489. doi: 10.1007/s00401-007-0332-4. [DOI] [PubMed] [Google Scholar]
- 23.Wischik CM, Bentham P, Wischik DJ, Seng KM. Tau aggregation inhibitor (TAI) therapy with rememberTM arrests disease progression in mild and moderate Alzheimer’s disease over 50 weeks. Alzheimer’s Dementia. 2008;4:p. T167. [Google Scholar]
- 24.Oz M, Lorke DE, Petroianu GA. Methylene blue and Alzheimer’s disease. Biochemical Pharmacology. 2009;78(8):927–932. doi: 10.1016/j.bcp.2009.04.034. [DOI] [PubMed] [Google Scholar]
- 25.Mouri A, Noda Y, Hara H, Mizoguchi H, Tabira T, Nabeshima T. Oral vaccination with a viral vector containing Aβ cDNA attenuates age-related Aβ accumulation and memory deficits without causing inflammation in a mouse Alzheimer model. FASEB Journal. 2007;21(9):2135–2148. doi: 10.1096/fj.06-7685com. [DOI] [PubMed] [Google Scholar]
- 26.Masters CL, Beyreuther K. Alzheimer’s centennial legacy: prospects for rational therapeutic intervention targeting the Aβ amyloid pathway. Brain. 2006;129(11):2823–2839. doi: 10.1093/brain/awl251. [DOI] [PubMed] [Google Scholar]
- 27.Lapchak PA, Schubert DR, Maher PA. Delayed treatment with a novel neurotrophic compound reduces behavioral deficits in rabbit ischemic stroke. Journal of Neurochemistry. 2011;116(1):122–131. doi: 10.1111/j.1471-4159.2010.07090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Müller WE, Eckert A, Kurz C, Eckert GP, Leuner K. Mitochondrial dysfunction: common final pathway in brain aging and Alzheimer's disease-therapeutic aspects. Molecular Neurobiology. 2010;41(2-3):159–171. doi: 10.1007/s12035-010-8141-5. [DOI] [PubMed] [Google Scholar]
- 29.Atlante A, Bobba A, Petragallo VA, Marra E. Alzheimer's proteins, oxidative stress, and mitochondrial dysfunction interplay in a neuronal model of Alzheimer's disease. International Journal of Alzheimer's Disease. 2010;2010:11 pages. doi: 10.4061/2010/621870. Article ID 621870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown TP, Rumsby PC, Capleton AC, Rushton L, Levy LS. Pesticides and Parkinson's disease—is there a link? Environmental Health Perspectives. 2006;114(2):156–164. doi: 10.1289/ehp.8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandhu JK, Pandey S, Ribecco-Lutkiewicz M, et al. Molecular mechanisms of glutamate neurotoxicity in mixed cultures of NT2-derived neurons and astrocytes: protective effects of coenzyme Q10 . Journal of Neuroscience Research. 2003;72(6):691–703. doi: 10.1002/jnr.10579. [DOI] [PubMed] [Google Scholar]
- 32.Somayajulu M, McCarthy S, Hung M, Sikorska M, Borowy-Borowski H, Pandey S. Role of mitochondria in neuronal cell death induced by oxidative stress; neuroprotection by Coenzyme Q10 . Neurobiology of Disease. 2005;18(3):618–627. doi: 10.1016/j.nbd.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 33.Naderi J, Somayajulu-Nitu M, Mukerji A, et al. Water-soluble formulation of Coenzyme QM10 inhibits Bax-induced destabilization of mitochondria in mammalian cells. Apoptosis. 2006;11(8):1359–1369. doi: 10.1007/s10495-006-8417-4. [DOI] [PubMed] [Google Scholar]
- 34.Smeyne RJ, Jackson-Lewis V. The MPTP model of Parkinson's disease. Molecular Brain Research. 2005;134(1):57–66. doi: 10.1016/j.molbrainres.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 35.Kim RH, Smith PD, Aleyasin H, et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6- tetrahydropyrindine (MPTP) and oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(14):5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liou HH, Tsai MC, Chen CJ, et al. Environmental risk factors and Parkinson’s disease: a case-control study in Taiwan. Neurology. 1997;48(6):1583–1588. doi: 10.1212/wnl.48.6.1583. [DOI] [PubMed] [Google Scholar]
- 37.Ritz B, Yu F. Parkinson's disease mortality and pesticide exposure in California 1984–1994. International Journal of Epidemiology. 2000;29(2):323–329. doi: 10.1093/ije/29.2.323. [DOI] [PubMed] [Google Scholar]
- 38.Dhillon AS, Tarbutton GL, Levin JL, et al. Pesticide/environmental exposures and Parkinson’s disease in East Texas. Journal of Agromedicine. 2008;13(1):37–48. doi: 10.1080/10599240801986215. [DOI] [PubMed] [Google Scholar]
- 39.McCarthy S, Somayajulu M, Sikorska M, Borowy-Borowski H, Pandey S. Paraquat induces oxidative stress and neuronal cell death; neuroprotection by water-soluble Coenzyme Q10 . Toxicology and Applied Pharmacology. 2004;201(1):21–31. doi: 10.1016/j.taap.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 40.Somayajulu-Nitu M, Domazet-Damjanov D, Matei A, Schwartzenberger E, Cohen J, Pandey S. Role of environmental and inflammatory toxicity in neuronal cell death. The Open Toxicology Journal. 2008;2:26–42. [Google Scholar]
- 41.Neves FF, Sousa RB, Pazin-Filho A, Cupo P, Elias J, Jr., Nogueira-Barbosa MH. Severe paraquat poisoning: clinical and radiological findings in a survivor. Jornal Brasileiro de Pneumologia. 2010;36(4):513–516. doi: 10.1590/s1806-37132010000400019. [DOI] [PubMed] [Google Scholar]
- 42.Reuters T. EU court reimposes ban on paraquat weedkiller. 2007 , http://uk.reuters.com/article/idUKL1166680020070711.
- 43.Syngenta Crop Protection AG. Paraquat Information Center. 2011, http://www.paraquat.com/english/paraquat-information-center.
- 44.McCormack AL, Thiruchelvam M, Manning-Bog AB, et al. Environmental risk factors and Parkinson’s disease: selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiology of Disease. 2002;10(2):119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- 45.McCormack AL, Atienza JG, Johnston LC, Andersen JK, Vu S, Di Monte DA. Role of oxidative stress in paraquat-induced dopaminergic cell degeneration. Journal of Neurochemistry. 2005;93(4):1030–1037. doi: 10.1111/j.1471-4159.2005.03088.x. [DOI] [PubMed] [Google Scholar]
- 46.Choi WS, Abel G, Klintworth H, Flavell RA, Xia Z. JNK3 mediates paraquat-and rotenone-induced dopaminergic neuron death. Journal of Neuropathology and Experimental Neurology. 2010;69(5):511–520. doi: 10.1097/NEN.0b013e3181db8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng J, Mao XO, Stevenson FF, Hsu M, Andersen JK. The herbicide paraquat induces dopaminergic nigral apoptosis through sustained activation of the JNK pathway. Journal of Biological Chemistry. 2004;279(31):32626–32632. doi: 10.1074/jbc.M404596200. [DOI] [PubMed] [Google Scholar]
- 48.Ramachandiran S, Hansen JM, Jones DP, Richardson JR, Miller GW. Divergent mechanisms of paraquat, MPP+, and rotenone toxicity: oxidation of thioredoxin and caspase-3 activation. Toxicological Sciences. 2007;95(1):163–171. doi: 10.1093/toxsci/kfl125. [DOI] [PubMed] [Google Scholar]
- 49.Cristóvão AC, Choi DH, Baltazar G, Beal MF, Kim YS. The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxidants and Redox Signaling. 2009;11(9):2105–2118. doi: 10.1089/ars.2009.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anantharam V, Kaul S, Song C, Kanthasamy A, Kanthasamy AG. Pharmacological inhibition of neuronal NADPH oxidase protects against 1-methyl-4-phenylpyridinium (MPP+)-induced oxidative stress and apoptosis in mesencephalic dopaminergic neuronal cells. NeuroToxicology. 2007;28(5):988–997. doi: 10.1016/j.neuro.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao HM, Liu B, Hong JS. Critical role for microglial NADPH oxidase in rotenone-induced degeneration of dopaminergic neurons. Journal of Neuroscience. 2003;23(15):6181–6187. doi: 10.1523/JNEUROSCI.23-15-06181.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shaikh SB, Nicholson LFB. Effects of chronic low dose rotenone treatment on human microglial cells. Molecular Neurodegeneration. 2009;4(1, article 55) doi: 10.1186/1750-1326-4-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramsey CP, Glass CA, Montgomery MB, et al. Expression of Nrf2 in neurodegenerative diseases. Journal of Neuropathology and Experimental Neurology. 2007;66(1):75–85. doi: 10.1097/nen.0b013e31802d6da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Calkins MJ, Johnson DA, Townsend JA, et al. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxidants and Redox Signaling. 2009;11(3):497–508. doi: 10.1089/ars.2008.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. Journal of Neuroscience. 2008;28(50):13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radical Biology and Medicine. 2004;36(10):1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 57.Lee JM, Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. Journal of Biochemistry and Molecular Biology. 2004;37(2):139–143. doi: 10.5483/bmbrep.2004.37.2.139. [DOI] [PubMed] [Google Scholar]
- 58.Numazawa S, Yoshida T. Nrf2-dependent gene expressions: a molecular toxicological aspect. Journal of Toxicological Sciences. 2004;29(2):81–89. doi: 10.2131/jts.29.81. [DOI] [PubMed] [Google Scholar]
- 59.Ho HK, White CC, Fernandez C, et al. Nrf2 activation involves an oxidative-stress independent pathway in tetrafluoroethylcysteine-induced cytotoxicity. Toxicological Sciences. 2005;86(2):354–364. doi: 10.1093/toxsci/kfi205. [DOI] [PubMed] [Google Scholar]
- 60.Katsuoka F, Motohashi H, Engel JD, Yamamoto M. Nrf2 transcriptionally activates the mafG gene through an antioxidant response element. Journal of Biological Chemistry. 2005;280(6):4483–4490. doi: 10.1074/jbc.M411451200. [DOI] [PubMed] [Google Scholar]
- 61.Sakurai A, Nishimoto M, Himeno S, et al. Transcriptional regulation of thioredoxin reductase 1 expression by cadmium in vascular endothelial cells: role of NF-E2-related factor-2. Journal of Cellular Physiology. 2005;203(3):529–537. doi: 10.1002/jcp.20246. [DOI] [PubMed] [Google Scholar]
- 62.Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):2046–2051. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakaso K, Yano H, Fukuhara Y, Takeshima T, Wada-Isoe K, Nakashima K. PI3K is a key molecule in the Nrf2-mediated regulation of antioxidative proteins by hemin in human neuroblastoma cells. FEBS Letters. 2003;546(2-3):181–184. doi: 10.1016/s0014-5793(03)00517-9. [DOI] [PubMed] [Google Scholar]
- 64.Johnson JA, Johnson DA, Kraft AD, et al. The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Annals of the New York Academy of Sciences. 2008;1147:61–69. doi: 10.1196/annals.1427.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Greco T, Fiskum G. Neuroprotection through stimulation of mitochondrial antioxidant protein expression. Journal of Alzheimer's Disease. 2010;20(supplement 2):S427–S437. doi: 10.3233/JAD-2010-100519. [DOI] [PubMed] [Google Scholar]
- 66.Gura T. Hope in Alzheimer’s fight emerges from unexpected places. Nature Medicine. 2008;14(9):p. 894. doi: 10.1038/nm0908-894. [DOI] [PubMed] [Google Scholar]
- 67.Liu Y, Schubert DR. The specificity of neuroprotection by antioxidants. Journal of Biomedical Science. 2009;16(1, article 98) doi: 10.1186/1423-0127-16-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bloomer RJ, Schilling BK, Karlage RE, Ledoux MS, Pfeiffer RF, Callegari J. Effect of resistance training on blood oxidative stress in Parkinson disease. Medicine and Science in Sports and Exercise. 2008;40(8):1385–1389. doi: 10.1249/MSS.0b013e31816f1550. [DOI] [PubMed] [Google Scholar]
- 69.Ang ET, Tai YK, Lo SQ, Seet R, Soong TW. Neurodegerative diseases: exercising toward neurogensis and neuoregeneration. Frontiers in Aging Neuroscience. 2010;25:p. 2. doi: 10.3389/fnagi.2010.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. International journal of stroke. 2009;4(6):461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 71.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Archives of Biochemistry and Biophysics. 1985;237(2):408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 72.Bergendi L, Benes L, Duracková Z, Ferencik M. Chemistry, physiology and pathology of free radicals. Life Sciences. 1999;65(18-19):1865–1874. doi: 10.1016/s0024-3205(99)00439-7. [DOI] [PubMed] [Google Scholar]
- 73.Clarke DD, Sokoloff L. Baisic Neurochemistry: Molecular, Cellular and Medical Aspects. Philadelphia, Pa, USA: Lippincott-Raven; 1999. Circulation and energy metabolism of the brain; pp. 637–669. [Google Scholar]
- 74.Saeed SA, Shad KF, Saleem T, Javed F, Khan MU. Some new prospects in the understanding of the molecular basis of the pathogenesis of stroke. Experimental Brain Research. 2007;182(1):1–10. doi: 10.1007/s00221-007-1050-9. [DOI] [PubMed] [Google Scholar]
- 75.Alexandrova ML, Bochev PG. Oxidative stress during the chronic phase after stroke. Free Radical Biology and Medicine. 2005;39(3):297–316. doi: 10.1016/j.freeradbiomed.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 76.Moro MA, Almeida A, Bolaños JP, Lizasoain I. Mitochondrial respiratory chain and free radical generation in stroke. Free Radical Biology and Medicine. 2005;39(10):1291–1304. doi: 10.1016/j.freeradbiomed.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 77.Brouns R, De Deyn PP. The complexity of neurobiological processes in acute ischemic stroke. Clinical Neurology and Neurosurgery. 2009;111(6):483–495. doi: 10.1016/j.clineuro.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 78.Schaller B, Graf R. Cerebral ischemia and reperfusion: the pathophysiologic concept as a basis for clinical therapy. Journal of Cerebral Blood Flow and Metabolism. 2004;24(4):351–371. doi: 10.1097/00004647-200404000-00001. [DOI] [PubMed] [Google Scholar]
- 79.Chen YC, Tsai SH, Lin-Shiau SY, Lin JK. Elevation of apoptotic potential by anoxia hyperoxia shift in NIH3T3 cells. Molecular and Cellular Biochemistry. 1999;197(1-2):147–159. doi: 10.1023/a:1006941630901. [DOI] [PubMed] [Google Scholar]
- 80.Li C, Wright M, Jackson R. Reactive species-mediated lung epithelia cells death after hypoxia and reoxygenation. Experimental Lung Research. 2002;28:373–389. doi: 10.1080/01902140290092001. [DOI] [PubMed] [Google Scholar]
- 81.Schaller B, Graf R. Cerebral ischemia and reperfusion: the pathophysiologic concept as a basis for clinical therapy. Journal of Cerebral Blood Flow and Metabolism. 2004;24(4):351–371. doi: 10.1097/00004647-200404000-00001. [DOI] [PubMed] [Google Scholar]
- 82.Soriano ME, Scorrano L. The interplay between BCL-2 family proteins and mitochondrial morphology in the regulation of apoptosis. Advances in Experimental Medicine and Biology. 2010;687:97–114. doi: 10.1007/978-1-4419-6706-0_6. [DOI] [PubMed] [Google Scholar]
- 83.Moffitt KL, Martin SL, Walker B. From sentencing to execution—the processes of apoptosis. Journal of Pharmacy and Pharmacology. 2010;62(5):547–562. doi: 10.1211/jpp.62.05.0001. [DOI] [PubMed] [Google Scholar]
- 84.Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. Journal of Translational Medicine. 2009;7, article 97 doi: 10.1186/1479-5876-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.del Zoppo GJ. Acute anti-inflammatory approaches to ischemic stroke. Annals of the New York Academy of Sciences. 2010;1207:143–148. doi: 10.1111/j.1749-6632.2010.05761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Molecular Cell. 2001;7(3):683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 87.Zinkel S, Gross A, Yang E. BCL2 family in DNA damage and cell cycle control. Cell Death and Differentiation. 2006;13(8):1351–1359. doi: 10.1038/sj.cdd.4401987. [DOI] [PubMed] [Google Scholar]
- 88.Aleyasin H, Rousseaux MWC, Phillips M, et al. The Parkinson’s disease gene DJ-1 is also a key regulator of stroke-induced damage. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(47):18748–18753. doi: 10.1073/pnas.0709379104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Culmsee C, Mattson MP. p53 in neuronal apoptosis. Biochemical and Biophysical Research Communications. 2005;331(3):761–777. doi: 10.1016/j.bbrc.2005.03.149. [DOI] [PubMed] [Google Scholar]
- 90.Yin XM, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369(6478):321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- 91.Scharstuhl A, Mutsaers HAM, Pennings SWC, Russel FGM, Wagener FADTG. Involvement of VDAC, Bax and ceramides in the efflux of AIF from mitochondria during curcumin-induced apoptosis. PLoS ONE. 2009;4(8, article e6688) doi: 10.1371/journal.pone.0006688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hetz C, Vitte PA, Bombrun A, et al. Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. Journal of Biological Chemistry. 2005;280(52):42960–42970. doi: 10.1074/jbc.M505843200. [DOI] [PubMed] [Google Scholar]
- 93.Gueorguieva D, Li S, Walsh N, Mukerji A, Tanha J, Pandey S. Identification of single-domain, Bax-specific intrabodies that confer resistance to mammalian cells against oxidative-stress-induced apoptosis. The FASEB Journal. 2006;20(14):2636–2638. doi: 10.1096/fj.06-6306fje. [DOI] [PubMed] [Google Scholar]
- 94.Li L, Qu YI, Li J, Xiong Y, Mao M, Mu D. Relationship between HIF-1α expression and neuronal apoptosis in neonatal rats with hypoxia-ischemia brain injury. Brain Research. 2007;1180(1):133–139. doi: 10.1016/j.brainres.2007.08.059. [DOI] [PubMed] [Google Scholar]
- 95.Fan X, Heijnen CJ, van der Kooij MA, Groenendaal F, van Bel F. The role and regulation of hypoxia-inducible factor-1α expression in brain development and neonatal hypoxic-ischemic brain injury. Brain Research Reviews. 2009;62(1):99–108. doi: 10.1016/j.brainresrev.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 96.Chen D, Li M, Luo J, Gu W. Direct interactions between HIF-1α and Mdm2 modulate p53 function. Journal of Biological Chemistry. 2003;278(16):13595–13598. doi: 10.1074/jbc.C200694200. [DOI] [PubMed] [Google Scholar]
- 97.Greijer AE, van der Groep P, Kemming D, et al. Up-regualtion of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor I (HIF-I) Journal of Pathology. 2005;206(3):291–304. doi: 10.1002/path.1778. [DOI] [PubMed] [Google Scholar]
- 98.Halterman MW, Miller CC, Federoff HJ. Hypoxia-inducible factor-1α mediates hypoxia-induced delayed neuronal death that involves p53. Journal of Neuroscience. 1999;19(16):6818–6824. doi: 10.1523/JNEUROSCI.19-16-06818.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Suzuki H, Tomida A, Tsuruo T. Dephosphorylated hypoxia-inducible factor 1α as a mediator of p53-dependent apoptosis during hypoxia. Oncogene. 2001;20(41):5779–5788. doi: 10.1038/sj.onc.1204742. [DOI] [PubMed] [Google Scholar]
- 100.Piret JP, Mottet D, Raes M, Michiels C. Is HIF-1α a pro- or an anti-apoptotic protein? Biochemical Pharmacology. 2002;64(5-6):889–892. doi: 10.1016/s0006-2952(02)01155-3. [DOI] [PubMed] [Google Scholar]
- 101.Calvert JW, Cahill J, Yamaguchi-Okada M, Zhang JH. Oxygen treatment after experimental hypoxia-ischemia in neonatal rats alters the expression of HIF-1α and its downstream target genes. Journal of Applied Physiology. 2006;101(3):853–865. doi: 10.1152/japplphysiol.00268.2006. [DOI] [PubMed] [Google Scholar]
- 102.Koizumi J, Yoshida Y, Nakazawa T, Ooneda G. Experimental studies of ischemic brain edema. 1. A new experimental model of cerebral embolism in rats in which recirculation can be introduced in the ischemic area. Japanese Journal of Stroke. 1986;8:1–8. [Google Scholar]
- 103.Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10(3):267–272. doi: 10.1161/01.str.10.3.267. [DOI] [PubMed] [Google Scholar]
- 104.Roberts GG, Di Loreto MJ, Marshall M, Wang J, DeGracia DJ. Hippocampal cellular stress responses after global brain ischemia and reperfusion. Antioxidants and Redox Signaling. 2007;9(12):2265–2275. doi: 10.1089/ars.2007.1786. [DOI] [PubMed] [Google Scholar]
- 105.Yenari MA, Hemmen TM. Therapeutic hypothermia for brain ischemia: where have we come and where do we go? Stroke. 2010;41(10 SUPPL. 1):S72–S74. doi: 10.1161/STROKEAHA.110.595371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Woodbury D, Schwarz EJ, Prockop DJ, Black IB. Adult rat and human bone marrow stromal cells differentiate into neurons. Journal of Neuroscience Research. 2000;61(4):364–370. doi: 10.1002/1097-4547(20000815)61:4<364::AID-JNR2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 107.Otero L, Zurita M, Bonilla C, et al. Late transplantation of allogeneic bone marrow stromal cells improves neurologic deficits subsequent to intracerebral hemorrhage. Cytotherapy. 2011;13(5):562–571. doi: 10.3109/14653249.2010.544720. [DOI] [PubMed] [Google Scholar]
- 108.Frautschy SA, Hu W, Kim P, et al. Phenolic anti-inflammatory antioxidant reversal of Aβ-induced cognitive deficits and neuropathology. Neurobiology of Aging. 2001;22(6):993–1005. doi: 10.1016/s0197-4580(01)00300-1. [DOI] [PubMed] [Google Scholar]
- 109.Thiyagarajan M, Sharma SS. Neuroprotective effect of curcumin in middle cerebral artery occlusion induced focal cerebral ischemia in rats. Life Sciences. 2004;74(8):969–985. doi: 10.1016/j.lfs.2003.06.042. [DOI] [PubMed] [Google Scholar]
- 110.Dohare P, Garg P, Jain V, Nath C, Ray M. Dose dependence and therapeutic window for the neuroprotective effects of curcumin in thromboembolic model of rat. Behavioural Brain Research. 2008;193(2):289–297. doi: 10.1016/j.bbr.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 111.McGonigal K, Tanha J, Palazov E, Li S, Gueorguieva-Owens D, Pandey S. Isolation and functional characterization of single domain antibody modulators of caspase-3 and apoptosis. Applied Biochemistry and Biotechnology. 2009;157(2):226–236. doi: 10.1007/s12010-008-8266-4. [DOI] [PubMed] [Google Scholar]