

Abstract
Introduction. Gastroesophageal reflux has been associated with chronic inflammatory diseases and may be a cause of airway remodelling. Aspiration of gastric fluids may cause damage to airway epithelial cells, not only because acidity is toxic to bronchial epithelial cells, but also since it contains digestive enzymes, such as pepsin. Aim. To study whether pepsin enhances cytotoxicity and inflammation in airway epithelial cells, and whether this is pH-dependent. Methods. Human bronchial epithelial cells were exposed to increasing pepsin concentrations in varying acidic milieus, and cell proliferation and cytokine release were assessed. Results. Cell survival was decreased by pepsin exposure depending on its concentration (F = 17.4) and pH level of the medium (F = 6.5) (both P < 0.01). Pepsin-induced interleukin-8 release was greater at lower pH (F = 5.1; P < 0.01). Interleukin-6 induction by pepsin was greater at pH 1.5 compared to pH 2.5 (mean difference 434%; P = 0.03). Conclusion. Pepsin is cytotoxic to bronchial epithelial cells and induces inflammation in addition to acid alone, dependent on the level of acidity. Future studies should assess whether chronic aspiration causes airway remodelling in chronic inflammatory lung diseases.
1. Introduction
Aspiration of gastric fluids damages airway epithelial cells [1] due to the toxicity of its low pH [2]. Several in vivo and in vitro models have assessed the effect of acid aspiration on lung injury and inflammation, using a hydrochloric acid solution with a pH ranging from 1 to 1.5 [2–5]. In addition, gastric particles have been found to contribute to lung injury [6]. Previous in vivo and in vitro studies have shown that acid aspiration causes an IL-6 and IL-8 mediated neutrophil influx into the lungs [2, 3, 7, 8]. A correlation between acid aspiration, increased IL-8 levels, and airway neutrophil counts has been found in asthma patients [9]. However, the acidity of gastric fluids might not be the only cause of damage and inflammatory response. Digestive enzymes such as pepsin might be an important factor as well.
Pepsin is stored as inactive pepsinogen in the chief cells of the gastric mucosa. It is a protease involved in the digestion of food, and its activity is acid-dependent. The conversion of pepsinogen to pepsin in the stomach starts slowly at pH 6 and reaches optimal activity between pH 1.5 to 2.5. Above pH 6.8, pepsin becomes inactive and above pH 7.5 it is fully inactive and irreversibly denatured [10]. In human gastric fluid, the pH varies from 1.5 to 3, which agrees with pepsin's activity optimum, and the concentration of pepsin varies from 0.5 to 1 mg/mL [11].
In recent years, pepsin has become an important and reliable biomarker for gastric aspiration [12–14], and has been associated with gastroesophageal reflux disease, lung rejection after transplantation, and bronchopulmonary dysplasia in children [15, 16]. Experimental studies are needed to investigate whether pepsin is solely a marker of aspiration, or that these associations are partly caused by the pathological actions of pepsin. The combination of protein breakdown by pepsin and acid damage might result in an amplification of cell and tissue damage. Pepsin in acid has been shown to damage the esophageal tissue of rabbits, more so than acid alone [17]. The cytotoxicity and inflammation caused by a combination of acid and pepsin on airway epithelium has, until now, not been investigated.
To assess the additional effect of pepsin in inducing inflammation and cytotoxicity to airway cells, we briefly exposed bronchial epithelial cells to various pH levels with and without pepsin. We hypothesised that pepsin concentrations at a lower pH would result in increased cytotoxicity and induce proinflammatory cytokine release.
2. Methods
All reagents were obtained from Sigma-Aldrich, United Kingdom unless otherwise stated.
2.1. Cell Culture
Human bronchial epithelial cells (16-HBE, an SV-40 transformed cell-line) were maintained in continuous culture in Dulbecco's Modified Eagle's Medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM glutamate, and penicillin (100 IU/mL)/streptomycin (100 μg/mL). Cells were grown to confluency at 37°C in a humidified atmosphere containing 5% CO2, washed with Ca2+/Mg2+-free phosphate-buffered saline (PBS-CMF), harvested with Trypsin-EDTA, and passaged.
The cells were seeded at a density of 60,000 per well in 24-well culture plates, and grown until approximately 80% confluent in complete media.
2.2. Experimental Model of Gastric Fluid Aspiration
0.01 M hydrochloric acid (26 mL, 13 mL, and 10 mL) was added to cell culture medium (25 mL) until pH was 1.5, 2, and 2.5, respectively. Pepsin concentrations of 40, 20, 10, 5, 2.5, and 1.25 mg/mL in 0.9% sodium chloride were prepared. Cell media was discarded and the wells were washed with PBS-CMF. One hundred μL of pepsin solution was added to 900 μL of acidified culture medium in the wells, and cells were exposed for 5 minutes to pepsin in each of the combinations of acidified culture medium, resulting in final pepsin concentrations of 4, 2, 1, 0.5, 0.25, and 0.125 mg/mL. Exposure medium was removed and cells were incubated for 20 hours in culture medium. Medium was centrifuged; cytotoxicity and cytokines were assessed in the supernatant. Each experiment was performed 3 times in duplicate.
2.3. Assessment of Cytotoxicity and Cytokine Release
Viability of cells was assessed by lactate dehydrogenase (LDH) (Roche Molecular Biochemicals, West Sussex, United Kingdom) release using pyruvic acid as a substrate and cell proliferation by Methylene Blue staining [18] by spectrometry. Interleukin-1β (IL-1β), IL-5, IL-6, IL-8, IL-10, tumor necrosis factor-α (TNF-α), and vascular endothelial growth factor (VEGF) were measured by a cytometric bead array (BD Biosciences, Oxford, United Kingdom).
2.4. Statistics
Cell proliferation was expressed as an absorption percentage of the media control without pepsin with corresponding pH; cytokine levels were corrected for cell proliferation and expressed as a percentage of the level of the corresponding pH without pepsin. Cytotoxicity measured by LDH was expressed as an absorption percentage of exposure to 100% lysis (by Triton X). Cell proliferation, LDH cytotoxicity, and cytokine levels were analysed using a 2-way ANOVA with pepsin concentration with pH as a fixed factor, and Tukey post-hoc testing. We considered P < 0.05 as statistically significant. All data were analysed using the SPSS statistical package for Windows, version 12.0.1 (SPSS inc, Chicago, Illinois).
3. Results
3.1. Cytoxicity
Acidification of the cell culture induced cytotoxicity as measured by LDH significantly more at pH 2.0 (mean (SEM) 57% ± 5.77%) compared to both pH 1.5 and pH 2.5 (22% ± 2.17%, and 28% ± 2.81%, resp., both P < 0.01, Figure 1). LDH-release therefore was pH-dependent (P < 0.01). There was a trend for a pepsin effect (P = 0.078), with lower LDH release with pepsin at all concentrations compared to no pepsin, independent of pH levels.
Figure 1.
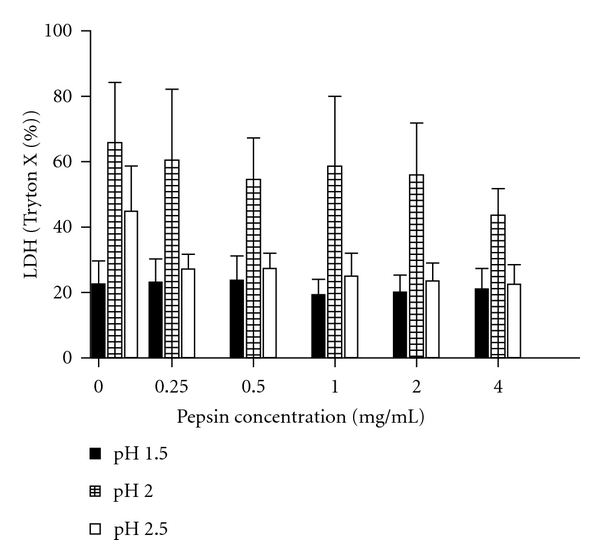
Exposure of cells for 5 minutes induced more cytotoxicity as measured by LDH after 20 hours incubation at pH 2.0 (mean (SEM) 57% ± 5.77%) compared to both pH 1.5 and pH 2.5 (22% ± 2.17%, and 28% ± 2.81%, resp., both P < 0.01). LDH release was pH-dependent (P < 0.01). There was a trend for a pepsin effect (P = 0.078), with lower LDH release with pepsin at all concentrations compared to no pepsin, independent of pH levels.
The exposure of cells to media with pH 1.5 for 5 minutes resulted in significantly less cell survival after 20 hours of incubation compared to pH 2.5 (mean absorption 0.22 versus 0.57; P = 0.03, Figure 2). The addition of pepsin caused a decrease in cell survival, which was dependant on both pepsin and pH (both P < 0.01, Figure 3 and supplemental Figure 1 in Supplementary Material online at doi: 10.4061/2011/569416). The effects of pepsin and acidified medium on cell survival were interactive (P < 0.01). Pepsin, resulted in lower cell survival compared to no pepsin at pH 1.5 at all concentrations tested (P < 0.01). At pH 2 and pH 2.5, there were trends towards lower cell survival with higher pepsin concentrations.
Figure 2.
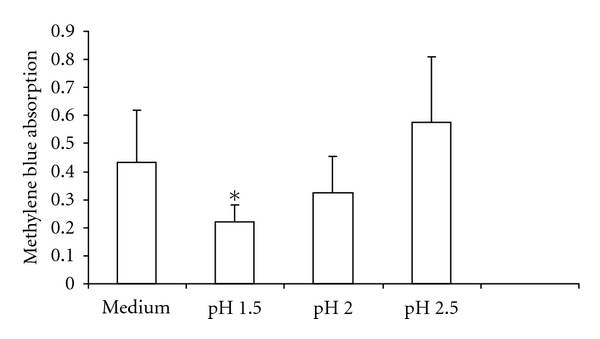
pH dependence of 16-HBE cell survival, as measured by methylene blue absorption. *Exposure of cells to medium with pH 1.5 for 5 minutes resulted in significantly less cell proliferation after 20 hours of incubation compared to exposure to pH 2.5 (mean absorption 0.22 versus 0.57; P = 0.03, n = 3). pH of control medium was 7.9. Histograms represent the means and the bars the SEM.
Figure 3.
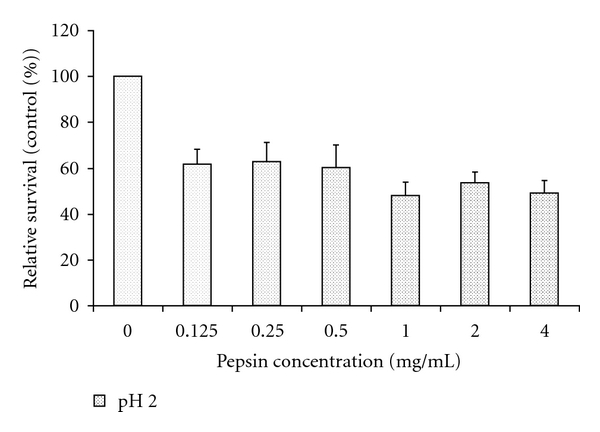
Effects of pepsin on cell survival, as measured by methylene blue absorption, and expressed as a % of absorbance of treatment with the corresponding pH without pepsin (control). Pepsin induced a significant decrease in cell survival independent of pH (F = 6.5; P < 0.01). Data are expressed as means (histograms) and SEM (bars) for 3 replicates. See supplement Figure 1 for the effect of pepsin on cell survival of all the pH levels used in the statistical analysis.
3.2. Cytokine Release
Figure 4 shows the effects of pepsin, dependent on dose and pH level, on IL-6 release. There was a trend towards more IL-6 release induced by pepsin after exposure to media with a lower pH (P = 0.09). Posthoc analysis showed that exposure at a pH of 1.5 resulted in a greater induction of IL-6 release by pepsin compared to pH 2.5 for all pepsin concentrations (mean difference 283%; P = 0.03) (see Figure 4 and supplement Figure 2).
Figure 4.
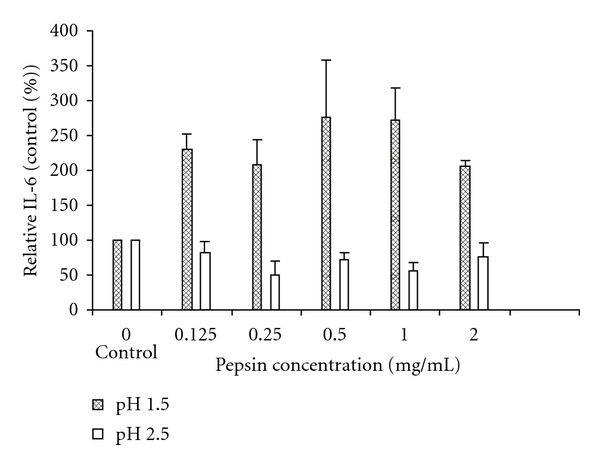
Interleukin-6 production by 16-HBE cells, corrected for cell proliferation and expressed as a % of exposure with the corresponding pH without pepsin (control). Interleukin-6 release induced by pepsin is higher at pH 1.5 compared to pH 2.5 (mean difference 283%; P = 0.03). Data are expressed as means (histograms) and SEM (bars) for 3 replicates. See supplement Figure 2 for induction of IL-6 at all pepsin concentrations and pH levels used in the statistical analysis.
The induction of IL-8 release by pepsin was greater at lower pH levels (P < 0.01). Pepsin at a pH of 1.5 induced significantly more IL-8 compared to pH 2.5 for all pepsin concentrations (mean difference 221%; P < 0.01, Figure 5, and supplement Figure 3). We were unable to detect VEGF-B with pH 1.5 and pH 2. With pH 2.5, there was no effect of pepsin on VEGF-B release. IL-1β, IL-5, and TNF-α were not detected.
Figure 5.
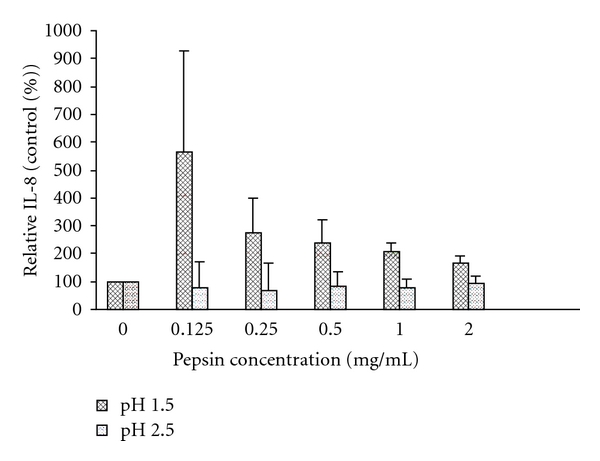
Interleukin-8 production by 16-HBE cells, corrected for cell proliferation and expressed as a % of exposure with the corresponding pH without pepsin (control). Interleukin-8 release induced by pepsin is higher at pH 1.5 compared to pH 2.5 (mean difference 221%; P < 0.01). Data expressed as mean (histograms) and SEM (bars) for 3 replicates. See supplement Figure 3 for induction of IL-8 at all pepsin concentrations and pH levels used in the statistical analysis.
4. Discussion
We found that pepsin is cytotoxic and induces inflammation in bronchial epithelial cells. Pepsin-induced cytotoxicity to 16HBE cells is pH-dependent, with the greatest effect seen at the lowest acidity. Likewise, pepsin-mediated IL-6 and IL-8 release is greatest in media with a lower pH.
We assessed cell damage by both lactate dehydrogenase (LDH) release from airway epithelial cells that indicated disruption of the cell membrane and Methylene Blue staining of, predominantly, nucleic acids. Methylene Blue is generally used to measure cell proliferation, and also gives an indication of cell damage due to detachment. The LDH assay showed lower levels with more toxic concentrations of pH and pepsin. This is probably due to the removal of medium used during cell exposure before incubation, which resulted in removal of the LDH released in the first 5 minutes. Therefore, our study does not report an acute toxic effect at least as measured by LDH release.
A disadvantage of assessing cell number with Methylene Blue might be the loss of living cells when the exposure medium is exchanged for incubation medium: pepsin may destroy adhesion molecules, resulting in free floating live cells, which could be lost during the removal of the medium. Acid exposure makes the cells more susceptible, facilitating greater cell detachment by pepsin. To minimise loss of cells due to detachment, we did not wash the wells after exposure, but carefully removed the exposure medium. Nonetheless, it is quite possible that we lost live cells during this procedure. In vivo studies have found that destruction of junctional molecules by pepsin causes erosive lesions in the oesophageal epithelium, and similar lesions have been found in the airways due to gastric fluid aspiration [1, 17]. We did not determine, in our model, the number of cells that were lost due to detachment or due to cell death.
We used an experimental protocol that is very cytotoxic: we had to expose cells to low pH levels, since pepsin requires a pH of 3 or under to become activated [17]. Therefore, we corrected for cell number to analyse cytokine release. We assessed exposure times of 5 minutes and 1 hour, but found the 1 hour exposure time to be markedly toxic to the cells due to extended exposure to low pH. Likewise, the cells were exposed in full media, containing physiologically relevant 10% serum, to offer some protection, since preliminary experiments (not shown) showed that the low pH and pepsin were very toxic. However, it should be noted that acidifying the medium with HCl to the various pH levels would have resulted in dilution of the medium causing changes in the osmolarity, as well as changes in the medium due to reaction of HCl with media components. Thus, we expressed the results of the effects of pepsin as a ratio to the medium of the corresponding pH without pepsin to correct for this bias.
Chronic aspiration has been associated with chronic inflammatory lung diseases. There are few data on the consequences of chronic aspiration. It is tempting to hypothesise that chronic aspiration leads to airway remodelling and the development of COPD, since recent reports suggest strong associations between COPD and gastroesophageal reflux disease [19–21]. Whether aspiration is the cause of this association is unknown. Our in vitro model shows that aspiration could lead to direct epithelial damage and the release of inflammatory mediators, involving at least the cytokines IL-6 and IL-8. These cytokines are involved in recruitment of neutrophils, and induction of acute phase reactants, and are increased in sputum during COPD exacerbations [22]. Models of chronic aspiration are required to determine which consequences our acute observations have in the development of COPD. Since we found cytotoxic and pro-inflammatory effects of pepsin in addition to those caused by acids alone, future studies investigating the consequences of chronic aspiration should include pepsin in aspiration models. Future human studies should assess whether chronic aspiration causes airway remodelling.
Supplementary Material
In our experiments, human bronchial epithelial cells were exposed to increasing pepsin concentrations in varying acidic milieus, and cell proliferation and cytokine release were assessed. In the article, we selected concentrations best representing the effects of pepsin to show in the figures. In the supplementary material, we present the figures of the effects of pepsin on cell survival and induction of cytokine release of all the pepsin concentrations and pH levels used in the statistical analysis.
Acknowledgment
The authors would like to thank Shonna Johnston for performing the bead flow-cytometry, Dr Rodger Duffin for the 16-HBE cells, Huib Kerstjens for critical appraisal and support, and the European Respiratory Society, and Stichting Astma Bestrijding for their financial support. This paper was funded by Stichting Asthma Bestrijding, the Netherlands. Erik Bathoorn is the recipient of a European Respiratory Society Fellowship (no. 382).
Abbreviations
- COPD:
Chronic obstructive pulmonary disease
- GER:
Gastroesophageal reflux
- HBE:
Human bronchial epithelial cells
- IL:
Interleukin
- PBS-CMF:
Ca2+/Mg2+-free phosphate-buffered saline
- TNF-α:
Tumor necrosis factor-α
- VEGF:
Vascular endothelial growth factor
- LDH:
Lactate dehydrogenase.
References
- 1.Wynne JW, Ramphal R, Hood CI. Tracheal mucosal damage after aspiration. A scanning electron microscope study. American Review of Respiratory Disease. 1981;124(6):728–732. doi: 10.1164/arrd.1981.124.6.728. [DOI] [PubMed] [Google Scholar]
- 2.Bonnans C, Fukunaga K, Levy MA, Levy BD. Lipoxin A4 regulates bronchial epithelial cell responses to acid injury. American Journal of Pathology. 2006;168(4):1064–1072. doi: 10.2353/ajpath.2006.051056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck-Schimmer B, Rosenberger DS, Neff SB, et al. Pulmonary aspiration: new therapeutic approaches in the experimental model. Anesthesiology. 2005;103(3):556–566. doi: 10.1097/00000542-200509000-00019. [DOI] [PubMed] [Google Scholar]
- 4.Nader ND, Davidson BA, Tait AR, Holm BA, Knight PR. Serine antiproteinase administration preserves innate superoxide dismutase levels after acid aspiration and hyperoxia but does not decrease lung injury. Anesthesia and Analgesia. 2005;101(1):213–219. doi: 10.1213/01.ANE.0000152188.65226.FE. [DOI] [PubMed] [Google Scholar]
- 5.Nagase T, Uozumi N, Ishii S, et al. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nature Immunology. 2000;1(1):42–45. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
- 6.Davidson BA, Knight PR, Wang Z, et al. Surfactant alterations in acute inflammatory lung injury from aspiration of acid and gastric particulates. American Journal of Physiology. 2005;288(4):L699–L708. doi: 10.1152/ajplung.00229.2004. [DOI] [PubMed] [Google Scholar]
- 7.Kennedy TP, Johnson KJ, Kunkel RG, Ward PA, Knight PR, Finch JS. Acute acid aspiration lung injury in the rat: biphasic pathogenesis. Anesthesia and Analgesia. 1989;69(1):87–92. [PubMed] [Google Scholar]
- 8.Folkesson HG, Matthay MA, Hebert CA, Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin- 8-dependent mechanisms. Journal of Clinical Investigation. 1995;96(1):107–116. doi: 10.1172/JCI118009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sacco O, Silvestri M, Sabatini F, et al. IL-8 and airway neutrophilia in children with gastroesophageal reflux and asthma-like symptoms. Respiratory medicine. 2006;100(2):307–315. doi: 10.1016/j.rmed.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 10.Piper DW, Fenton BH. pH stability and activity curves of pepsin with special reference to their clinical importance. Gut. 1965;6(5):506–508. doi: 10.1136/gut.6.5.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balan KK, Jones AT, Roberts NB, Pearson JP, Critchley M, Jenkins SA. The effects of Helicobacter pylori colonization on gastric function and the incidence of portal hypertensive gastropathy in patients with cirrhosis of the liver. American Journal of Gastroenterology. 1996;91(7):1400–1406. [PubMed] [Google Scholar]
- 12.Metheny NA, Chang YH, Ye JS, et al. Pepsin as a marker for pulmonary aspiration. American Journal of Critical Care. 2002;11(2):150–154. [PMC free article] [PubMed] [Google Scholar]
- 13.Farrell S, McMaster C, Gibson D, Shields MD, McCallion WA. Pepsin in bronchoalveolar lavage fluid: a specific and sensitive method of diagnosing gastro-oesophageal reflux-related pulmonary aspiration. Journal of Pediatric Surgery. 2006;41(2):289–293. doi: 10.1016/j.jpedsurg.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Farhath S, Aghai ZH, Nakhla T, et al. Pepsin, a reliable marker of gastric aspiration, is frequently detected in tracheal aspirates from premature ventilated neonates: relationship with feeding and methylxanthine therapy. Journal of Pediatric Gastroenterology and Nutrition. 2006;43(3):336–341. doi: 10.1097/01.mpg.0000232015.56155.03. [DOI] [PubMed] [Google Scholar]
- 15.Stovold R, Forrest IA, Corris PA, et al. Pepsin, a biomarker of gastric aspiration in lung allografts: a putative association with rejection. American Journal of Respiratory and Critical Care Medicine. 2007;175(12):1298–1303. doi: 10.1164/rccm.200610-1485OC. [DOI] [PubMed] [Google Scholar]
- 16.Farhath S, He Z, Nakhla T, et al. Pepsin, a marker of gastric contents, is increased in tracheal aspirates from preterm infants who develop bronchopulmonary dysplasia. Pediatrics. 2008;121(2):e253–e259. doi: 10.1542/peds.2007-0056. [DOI] [PubMed] [Google Scholar]
- 17.Tobey NA, Hosseini SS, Caymaz-Bor C, Wyatt HR, Orlando GS, Orlando RC. The role of pepsin in acid injury to esophageal epithelium. American Journal of Gastroenterology. 2001;96(11):3062–3070. doi: 10.1111/j.1572-0241.2001.05260.x. [DOI] [PubMed] [Google Scholar]
- 18.Oliver MH, Harrison NK, Bishop JE, Cole PJ, Laurent GJ. A rapid and convenient assay for counting cells cultured in microwell plates: application for assessment of growth factors. Journal of Cell Science. 1989;92(3):513–518. doi: 10.1242/jcs.92.3.513. [DOI] [PubMed] [Google Scholar]
- 19.Casanova C, Baudet JS, del Valle Velasco M, et al. Increased gastro-oesophageal reflux disease in patients with severe COPD. European Respiratory Journal. 2004;23(6):841–845. doi: 10.1183/09031936.04.00107004. [DOI] [PubMed] [Google Scholar]
- 20.Mokhlesi B, Morris AL, Huang CF, Curcio AJ, Barrett TA, Kamp DW. Increased prevalence of gastroesophageal reflux symptoms in patients with COPD. Chest. 2001;119(4):1043–1048. doi: 10.1378/chest.119.4.1043. [DOI] [PubMed] [Google Scholar]
- 21.Ducolone A, Vandevenne A, Jouin H, et al. Gastroesophageal reflux in patients with asthma and chronic bronchitis. Allergie et Immunologie. 1988;20(6):218–225. [PubMed] [Google Scholar]
- 22.Bathoorn E, Liesker JJW, Postma DS, et al. Change in inflammation in out-patient COPD patients from stable phase to a subsequent exacerbation. International Journal of COPD. 2009;4(1):101–109. doi: 10.2147/copd.s4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In our experiments, human bronchial epithelial cells were exposed to increasing pepsin concentrations in varying acidic milieus, and cell proliferation and cytokine release were assessed. In the article, we selected concentrations best representing the effects of pepsin to show in the figures. In the supplementary material, we present the figures of the effects of pepsin on cell survival and induction of cytokine release of all the pepsin concentrations and pH levels used in the statistical analysis.