

Abstract
Accumulating evidence suggests an important role for cyclooxygenase-2 (COX-2) in the pathogenesis of a wide range of malignancies. The protumorigenic properties of COX-2 are generally thought to be mediated by its product, PGE2, which is shown to promote tumor spread and growth by multiple mechanisms but most importantly through modulation of the local immune response in the tumor. Pancreatic tumor cells produce various amounts of PGE2, some of them being even deficient in COX enzymes or other PGE2 synthases. Here we describe that, beside pancreatic tumor cells or stromal fibroblasts, human peripheral blood mononuclear cells can also produce PGE2 upon coculture with pancreatic cancer cells. Stimulating of cellular cPLA2 within PBMCs by secreted factors, presumably sPLA2, from tumor cells appeared crucial, while the direct contact between PBMCs and PDACs seemed to be dispensable for this effect. Our data is emphasizing the complex interactions participating in the formation of the tolerogenic immune milieu within pancreatic tumors.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal gastrointestinal malignancies. It is the fourth most frequent cause of cancer-related deaths in North America, the sixth in Europe, and the fifth in the UK [1].
Beside surgery and chemotherapy, cyclooxygenases (COX)—the constitutive (COX-1) and the inducible (COX-2)—have been investigated as targets for treatment and prevention of pancreatic cancer since their expression seems to correlate with poor prognosis in PDAC patients [2, 3]. The two forms of COX catalyze the conversion of arachidonic acid (produced by the activity of phospholipase A2 and other enzymes) into prostaglandins [4]. One of the main representatives of prostaglandins is PGE2, characterized as a mediator with suppressive activity at several levels of the immune response [5]. It selectively impairs the production of interleukin-2 (IL-2) and interferon-γ (IFNγ) in T cells, blocks the production of dendritic-cell- (DC-) produced proinflammatory cytokines, including IL-12p70, and induces IL-10 production [6–8]. Earlier results suggest an involvement of COX-2 in the control of tumor-dependent angiogenesis and growth in certain pancreatic cancers and provide the rationale for inhibition of the COX pathway as an effective therapeutic approach for pancreatic tumors [3, 9]. In addition, cyclooxygenase-2 (COX-2) overexpression in transgenic mice induces a sequence of metaplasia-dysplasia events leading to neoplastic transformation in the exocrine pancreas [10].
Several works have focused on the production of PGE2 and the activity of enzymes involved in the earlier steps of prostaglandin production in pancreatic cancer [11, 12]. Diagnostically, PGE2 levels have been shown to be helpful to distinguish early intraductal papillary neoplasms (high PGE2) from cystic neoplasms (low PGE2) in patients with known mucinous lesions in the pancreas [13]. Investigations report that either COX-1 or both COX-1 and COX-2 genes can be silenced in pancreatic cancers. Furthermore, recent evidence supports that pancreatic cancers lacking COX enzymes can exploit stromal fibroblasts as an exogenous source of prostaglandins and PGE2 in particular [11, 14]. Such fibroblasts named also pancreatic stellate cells (PSCs) promote the progression of pancreatic cancer by increasing cancer cells' proliferation and invasion, as well as by protecting them from radiation- and gemcitabine-induced apoptosis. However, it has to be noted that, beside PSCs, other cells types such as macrophages, dendritic cells, and lymphocytes that infiltrate pancreatic carcinoma tissue actively participate in the formation of the cellular and cytokine milieu within the tumor [15]. These cells are considered to be quickly suppressed by the tolerogenic microenvironment of the PDAC tumor, for which PGE2 seems to play an important role [16]. In addition, recent work showed that PGE2 present in the PDAC patients' plasma was involved in the induction of semimature DC phenotype that does not allow these cells to function as professional antigen-presenting cells [17].
In our work, we describe that the culture of human pancreatic cancer cells (or their conditioned media) with peripheral blood mononuclear cells induces the release of PGE2 irrespective of the COX status and PGE2 producing capacity of the PDAC cell line used in the assay.
2. Materials and Methods
2.1. Cells and Treatment
Human pancreatic carcinoma cell lines were obtained from ATCC (Manassas, VA) and grown in RPMI 1640 (BxPC-3, T3M-4, AsPC-1) or DMEM (MiaPaCa-2) supplemented with 10% fetal calf serum (FCS), penicillin (100 U/mL), and streptomycin (100 μg/mL).
Buffy coats of randomly selected healthy donors were obtained from the blood bank of the Heidelberg University, and peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation over Histopaque 1077 gradient (Sigma, Hamburg, Germany), at 400 g, no brake, for 45 min at room temperature. Cells were cultured in RPMI supplemented with 10% FCS and penicillin (100 U/mL)/streptomycin (100 μg/mL) at 37°C, in a 5% CO2 incubator. For corresponding experiments cells were simultaneously stimulated using LPS and ConA (Sigma) both at final concentration of 5 μg/mL.
The supernatants for either PDAC cells or PBMCs were obtained after centrifugation at 10000 rpm for 5 minutes before application onto the cells or use for PGE2 ELISA experiments.
The PLA2 inhibitor N-{(2S,4R)-4-(Biphenyl-2-ylmethyl-isobutyl-amino)-1-[2-(2,4-difluorobenzoyl)-benzoyl]-pyrrolidin-2-ylmethyl}-3-[4-(2,4-dioxothiazolidin-5 ylidenemethyl)-phenyl]acrylamide (Pyrrolidin 1) was obtained from Merck Biosciences, Germany and used at a final concentration of 1 μM. The sPLA2 inhibitor 4-[(1-oxo-7-phenylheptyl)amino]-(4R)-octanoic acid (CAY10590) was used at final concentration of 91 μM. The COX-1 inhibitor SC-560, was used at a final concentration of 10 nM. The COX-2 inhibitor NS-398 was used at a final concentration of 2 μM. CAY10590, SC-560, and NS-398 were obtained from Cayman chemical, USA.
2.2. PGE2 Detection
The PGE2 measurement in culture media was performed using a PGE2 ELISA kit obtained from Enzo Life Sciences GmbH (Lörrach, Germany) following manufacturer's instructions. Experiments were repeated in triplicate with at least three different blood donors, and statistical significance was calculated using paired Student's t-test.
3. Results and Discussion
3.1. Pancreatic Cancer Cells Induce the Production of PGE2 from Human Peripheral Blood Mononuclear Cells
In our study we used several human pancreatic cancer cell lines from primary (MiaPaCa-2, BxPC-3) and metastatic origin (AsPC-1, T3M-4) that have been previously characterized to have various expression of the enzymes involved in PGE2 production. BxPC-3 have been formerly reported to intrinsically produce PGE2, since they are endowed with several active elements of the PGE2 enzymatic cascade, namely, cPLA2, COX-1, and COX-2 [12]. On the other hand in the same study MiaPaCa-2 and AsPC-1 were shown to be deficient in the expression of these enzymes and therefore unable to produce PGE2. The cell line T3M-4 derives from a metastatic tumor and has not been previously characterized in terms of its PGE2 production status.
We tested the expression of PGE2 from these cells and found that AsPC-1 and MiaPaCa-2 did not produce any PGE2 after 48 hours of cultivation (Figure 1(a)). In contrast both BxPC-3 and T3M-4 could release significant amounts of this prostaglandin. Nonstimulated mononuclear cells from peripheral blood did not secrete PGE2 under normal culture conditions but could be strongly induced to release it upon treatment with LPS and ConA (Figure 1(b)). Plating of unstimulated PBMCs together with pancreatic cancer cells caused a burst of PGE2 activity in the medium 48 hours after coculturing the cells. The levels were comparable in all four cocultures reaching the amounts obtained after LPS/ConA stimulation (Figure 1(b)). While in the case of BxPC-3 and T3M-4, pancreatic cells could have actively contributed to the increase in PGE2 concentration, for AsPC-1 and MiaPaCa-2 this was rather unlikely, since they were reported as completely devoid of PGE2 synthases and did not produce any PGE2 in our experiments. Therefore, PBMCs seemed to be the only possible source of the prostaglandin within these cocultures.
Figure 1.
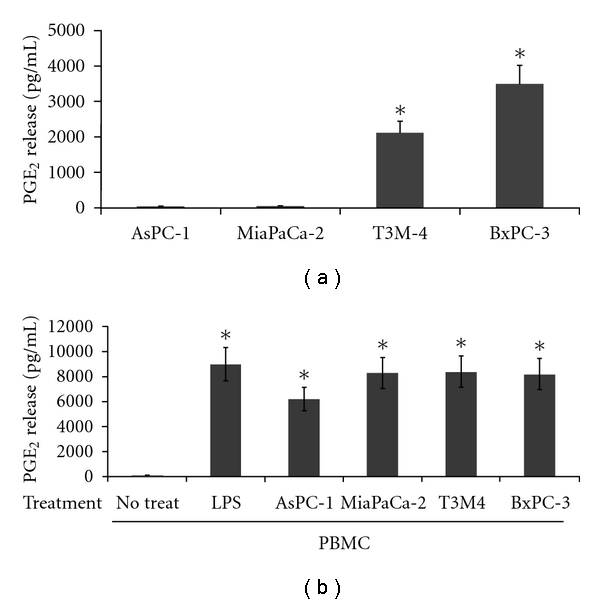
Release of PGE2 from pancreatic carcinoma cells and peripheral blood mononuclear cells. (a) PGE2 production from PDAC monocultures of cells. Cells (AsPC-1, MiaPaCa-2, T3M-4, BxPC-3) were plated in 48-well plates at a density of 2.5 × 104 cells/well in 500 μL of medium/well, and supernatants were collected at 48 hrs for PGE2 ELISA measurement. (b) PGE2 production from PBMCs. After purification over a Histopaque gradient mononuclear cells were plated in a 48-well plate (5 × 105 cells/well) either alone or onto preseeded PDAC cultures (2.5 × 104 cells/well). 48 hours after coculturing the cells, supernatants were collected and analysed for PGE2 expression. PBMCs treated with LPS and ConA served as controls. All experiments were repeated in triplicate with three different healthy blood donors. *Standard deviations were calculated, and results were considered significant with P values from Student's t-test below 0.05.
3.2. Production of PGE2 from PBMCs Is Induced by Factor(s) Secreted by Pancreatic Cancer Cells
In our next step we aimed at establishing whether PGE2 production may be induced by a factor secreted by tumor cells or whether it requires direct cell to cell contact. Supernatants from cultured pancreatic cancer cells were applied onto preseeded PBMCs. We selected to use conditioned media from AsPC-1 and MiaPaCa-2 cells since, as already determined above, these cells were unable to produce PGE2 on their own. As shown in Figure 2, LPS/ConA treatment induced a very potent PGE2 response from PBMCs. The supernatants of AsPC-1 and MiaPaCa-2 could also provoke the release of significant amount of the prostaglandin from PBMCs, although to a lesser extent (Figure 2). We reconfirmed that the media of AsPC-1 and MiaPaCa-2 were devoid of any PGE2 before being applied onto the PBMCs (data not shown). In conclusion, these data suggest that direct cell contact between PBMCs and PDACs is dispensable for PGE2 induction from immune cells.
Figure 2.
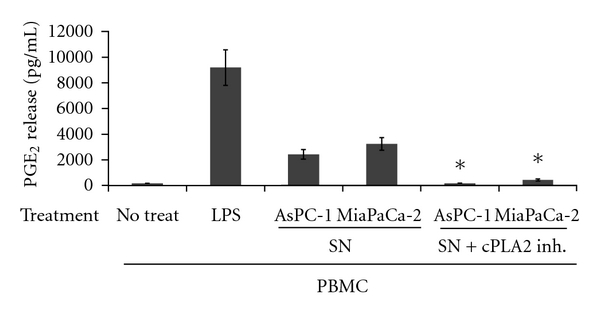
Release of PGE2 from peripheral blood mononuclear cells treated with PDAC supernatants. Supernatants (SN) from AsPC-1 and MiaPaCa-2 cells, derived 24 hrs after plating of the cells, were treated (SN + cPLA2 Inh.) or not (SN) with Pyrolidin 1 and applied onto cultured PBMCs (1 × 106 cells/well) in a 24 well plate. Two parts of PDAC medium (400 μL) were added onto one part medium overlaying the PBMCs (200 μL) and 48 hrs later samples were collected and analysed for PGE2 expression. The experiment was repeated in duplicate with three different healthy blood donors. *Standard deviations were calculated and results were considered significant with P values from Student's t-test below 0.05.
The first enzymatic activity required for prostaglandin production is phospholipase A2, which among other enzymes catalyzes the release of arachidonic acid from cell membrane lipids [4]. This enzyme has several forms, the most potent being the cytoplasmic phospholipase A2 (cPLA2) [18] that can potentially be activated in PBMCs by PDACs or their secreted factors to induce PGE2 release. For that purpose we treated the immune cell cultures with a selective cPLA2 inhibitor (pyrolidin 1) [19] immediately after application of the conditioned AsPC-1 and MiaPaCa-2 media. The presence of the inhibitor could drastically reduce PGE2 production induced by pancreatic cancer supernatants (Figure 2), suggesting indeed that the inherent cPLA2 activity in PBMCs is triggered by factors secreted by pancreatic cancer cells. Given that sPLA2 may trigger PGE2 production, although to a lesser extent than cPLA2, and taking into account their known ability to stimulate the cPLA2 enzyme [20, 21], we evaluated the effect of different inhibitors of the PGE2 production cascade in cultures of human PBMCs treated with AsPC-1 and MiaPaCa-2 media. As shown in Table 1 the COX-1 enzyme was not a major player contributing to PGE2 production form PBMCs, while COX-2 seemed to be strongly involved in the latter process. Interestingly, both the cPLA2 and sPLA2 inhibitors seemed to have a strong inhibitory effect on PGE2 production, suggesting that sPLA2 enzymes most likely released from AsPC-1 and MiaPaCa-2 could represent a potential candidate to indirectly trigger PGE2 expression.
Table 1.
PGE2 production from PBMCs in presence of supernatants from PDAC cell lines and specific inhibitors.
| Treatment | Inhibitors | ||||
|---|---|---|---|---|---|
| No inh. | cPLA2 | sPLA2 | COX-1 | COX-2 | |
| PBMC + AsPC-1 SN1 | 100% ± 0 | 8.84% ± 5.64* | 3.50% ± 2.94* | 100% ± 81.16 | 1.99% ± 1.82* |
| PBMC + MiaPaCa-2 SN1 | 100% ± 0 | 14.40% ± 2.5* | 6.02% ± 2.06* | 68.69% ± 29.58 | 2.89% ± 1.58* |
PGE2 production was measured in the coculture supernatants 48 hours posttreatment. Data are represented as percentages of PGE2 production, compared to that induced by PDAC supernatants on PBMCs (taken as 100%) plus. The names and concentrations of respective inhibitors are indicated in Materials and Methods. The experiment was repeated with four independent donors. Standard error was calculated using SigmaPlot 10.1 program, and the statistical significance (*) of differences was assessed by a two-tailed Student's paired t-test. P values below 0.05 were considered significant.
1SN-supernatant.
Very recent study by Omura et al. [11] as well as earlier reports [14] has proposed that cancer-associated fibroblasts, representing the highest percentage of pancreatic cancer stromal cells, may serve as exogenous sources of prostaglandins for cyclooxygenase-deficient pancreatic cancers. In this study we could confirm our assumption that the immune component, which is also widely represented in PDAC stromal reaction, may represent an important alternative source of prostaglandin production in the presence of tumor cells. For instance, macrophages and dendritic cells could become such a source, since they are well known to produce high levels of PGE2 upon stimulation [22, 23].
Omura et al. [11] have also demonstrated that, in some of the clinical cancer samples and cell lines testing negative for COX-1 mRNA, certain level of expression could still be induced by epigenetic treatment with methyltransferase inhibitor 5-aza-dC or histone deacetylase inhibitor TSA. However, it is rather unlikely that in our coculture experiments PBMCs could induce such a high level of PGE-2 expression from PDACs, following mechanisms similar to chemical stimulation.
The levels of PGE2 induced by supernatants of AsPC-1 and MiaPaCa-2 cells applied on the PBMCs were lower than the ones achieved in the corresponding cocultures, but one has to take into account that the concentrations of the probable secreted stimulators should be much higher in the immediate proximity of the cells than in the overlaying medium that we used for the stimulation.
4. Conclusion
In our work we describe a previously unknown, feature of pancreatic cancer stromal interaction, pointing that pancreatic cancer cells can induce PGE2 production from immune cells and further modulate host cell functions in their favor through a mechanism that may also be characteristic of other tumor entities.
Abbreviations
- PDAC:
Pancreatic ductal adenocarcinoma
- COX:
Cyclooxygenase
- PBMCs:
Peripheral blood mononuclear cells
- PSC:
Pancreatic stellate cells
- PLA2:
Phospholipase A2.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer Journal for Clinicians. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Juuti A, Louhimo J, Nordling S, Ristimäki A, Haglund C. Cyclooxygenase-2 expression correlates with poor prognosis in pancreatic cancer. Journal of Clinical Pathology. 2006;59(4):382–386. doi: 10.1136/jcp.2005.026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mukherjee P, Basu GD, Tinder TL, et al. Progression of pancreatic adenocarcinoma is significantly impeded with a combination of vaccine and COX-2 inhibition. Journal of Immunology. 2009;182(1):216–224. [PMC free article] [PubMed] [Google Scholar]
- 4.Scott KF, Sajinovic M, Hein J, et al. Emerging roles for phospholipase A2 enzymes in cancer. Biochimie. 2010;92(6):601–610. doi: 10.1016/j.biochi.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 5.Phipps RP, Stein SH, Roper RL. A new view of prostaglandin E regulation of the immune response. Immunology Today. 1991;12(10):349–352. doi: 10.1016/0167-5699(91)90064-Z. [DOI] [PubMed] [Google Scholar]
- 6.Edwards JP, Emens LA. The multikinase inhibitor Sorafenib reverses the suppression of IL-12 and enhancement of IL-10 by PGE2 in murine macrophages. International Immunopharmacology. 2010;10(10):1220–1228. doi: 10.1016/j.intimp.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snijdewint FG, Kalinski P, Wierenga EA, Bos JD, Kapsenberg ML. Prostaglandin E2 differentially modulates cytokine secretion profiles of human T helper lymphocytes. Journal of Immunology. 1993;150(12):5321–5329. [PubMed] [Google Scholar]
- 8.Hilkens CM, Snijders A, Vermeulen H, Van Der Meide PH, Wierenga EA, Kapsenberg ML. Accessory cell-derived IL-12 and prostaglandin E2 determine the IFN-γ level of activated human CD4+ T cells. Journal of Immunology. 1996;156(5):1722–1727. [PubMed] [Google Scholar]
- 9.Chu J, Lloyd FL, Trifan OC, Knapp B, Rizzo MT. Potential involvement of the cyclooxygenase-2 pathway in the regulation of tumor-associated angiogenesis and growth in pancreatic cancer. Molecular Cancer Therapeutics. 2003;2(1):1–7. [PubMed] [Google Scholar]
- 10.Colby JK, Klein RD, McArthur MJ, et al. Progressive metaplastic and dysplastic changes in mouse pancreas induced by cyclooxygenase-2 overexpression. Neoplasia. 2008;10(8):782–796. doi: 10.1593/neo.08330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Omura N, Griffith M, Vincent A, et al. Cyclooxygenase-deficient pancreatic cancer cells use exogenous sources of prostaglandins. Molecular Cancer Research. 2010;8(6):821–832. doi: 10.1158/1541-7786.MCR-09-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasan S, Satake M, Dawson DW, et al. Expression analysis of the prostaglandin E2 production pathway in human pancreatic cancers. Pancreas. 2008;37(2):121–127. doi: 10.1097/MPA.0b013e31816618ba. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt CM, Yip-Schneider MT, Ralstin MC, et al. PGE(2) in pancreatic cyst fluid helps differentiate IPMN from MCN and predict IPMN dysplasia. Journal of Gastrointestinal Surgery. 2008;12(2):243–249. doi: 10.1007/s11605-007-0404-8. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida S, Ujiki M, Ding XZ, et al. Pancreatic stellate cells (PSCs) express cyclooxygenase-2 (COX-2) and pancreatic cancer stimulates COX-2 in PSCs. Molecular Cancer. 2005;4:p. 27. doi: 10.1186/1476-4598-4-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Research. 2007;67(19):9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 16.Tjomsland V, Sandström P, Spångeus A, et al. Pancreatic adenocarcinoma exerts systemic effects on the peripheral blood myeloid and plasmacytoid dendritic cells: an indicator of disease severity? BMC Cancer. 2010;10:p. 87. doi: 10.1186/1471-2407-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tjomsland V, Spångeus A, Sandström P, Borch K, Messmer D, Larsson M. Semi mature blood dendritic cells exist in patients with ductal pancreatic adenocarcinoma owing to inflammatory factors released from the tumor. PLoS One. 2010;5(10, article e13441) doi: 10.1371/journal.pone.0013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leslie CC, Gangelhoff TA, Gelb MH. Localization and function of cytosolic phospholipase A2alpha at the Golgi. Biochimie. 2010;92(6):620–626. doi: 10.1016/j.biochi.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghomashchi F, Stewart A, Hefner Y, et al. A pyrrolidine-based specific inhibitor of cytosolic phospholipase A(2)alpha blocks arachidonic acid release in a variety of mammalian cells. Biochimica et Biophysica Acta. 2010;1513(2):160–166. doi: 10.1016/s0005-2736(01)00349-2. [DOI] [PubMed] [Google Scholar]
- 20.Gorovetz M, Schwob O, Krimsky M, Yedgar S, Reich R. MMP production in human fibrosarcoma cells and their invasiveness are regulated by group IB secretory phospholipase A2 receptor-mediated activation of cytosolic phospholipase A2. Frontiers in Bioscience. 2008;13(5):1917–1925. doi: 10.2741/2811. [DOI] [PubMed] [Google Scholar]
- 21.Fonteh AN, Atsumi GI, LaPorte T, Chilton FH. Secretory phospholipase A2 receptor-mediated activation of cytosolic phospholipase A2 in murine bone marrow-derived mast cells. Journal of Immunology. 2000;165(5):2773–2782. doi: 10.4049/jimmunol.165.5.2773. [DOI] [PubMed] [Google Scholar]
- 22.Fogel-Petrovic M, Long JA, Knight DA, Thompson PJ, Upham JW. Activated human dendritic cells express inducible cyclo-oxygenase and synthesize prostaglandin E2 but not prostaglandin D2. Immunology and Cell Biology. 2004;82(1):47–54. doi: 10.1111/j.1440-1711.2004.01213.x. [DOI] [PubMed] [Google Scholar]
- 23.Harizi H, Gualde N. Dendritic cells produce eicosanoids, which modulate generation and functions of antigen-presenting cells. Prostaglandins Leukotrienes and Essential Fatty Acids. 2002;66(5-6):459–466. doi: 10.1054/plef.2002.0383. [DOI] [PubMed] [Google Scholar]