

Abstract
Pseudomonas aeruginosa is an opportunistic pathogen and causes a wide range of acute and chronic infections. P. aeruginosa infections are kept in check by an effective immune surveillance in the healthy host, while any imbalance or defect in the normal immune response can manifest in disease. Invasive acute infection in the immunocompromised patients is mediated by potent extracellular and cell bound bacterial virulence factors. Life-threatening chronic infection in cystic fibrosis patients is maintained by pathogenic variants that contribute to evade detection and clearance by the immune system. Here, we reviewed the molecular basis of receptor-mediated recognition of P. aeruginosa and their role in initiating inflammation and the colonization. In addition, the consequence of the P. aeruginosa genetic adaptation for the antibacterial defence and the maintaining of chronic infection are discussed.
1. Pathogenesis of P. aeruginosa Pneumonia
Pseudomonas aeruginosa rarely causes infection in healthy host although it is one of the most important agents of nosocomial infections in diverse clinical setting. The immunocompetent host usually offers effective immune surveillance against infection by P. aeruginosa; however, any imbalance or defect in the normal immune response to this opportunistic pathogen can lead to infection and manifest in disease. The spectrum of clinical diseases caused by P. aeruginosa in humans ranges from invasive acute infections as in patients who are mechanically ventilated, individuals who are immunocompromised, and patients with malignancies or HIV infection, to life-threatening chronic infections as in cystic fibrosis (CF) patients. P. aeruginosa adaptability to environments that are inhospitable to most other microorganisms, minimal nutritional requirements, and high resistance to antibiotics allow it to survive in different hosts [1].
The first step in mounting a protective immune response is the recognition of the bacterial pathogen by cell surface receptors, which are located on professional phagocytes (granulocytes and monocytes/macrophages) and dendritic cells as well as nonimmune cells (Figure 1(a)) [2]. This is followed by the activation of intracellular signalling pathways and stimulation of inflammatory mediators. Subsequently, effector immune mechanisms are triggered such as neutrophil and macrophage activation as well as initiation of adaptive immunity through T helper cell (Th)1 or Th2 responses. In most cases, the disease process begins with some alteration or circumvention of normal host defenses. In patients with damaged mucosal barriers from mechanical ventilation, trauma or antecedent viral infection, P. aeruginosa colonization of the respiratory tract is often followed by acute pneumonia, sepsis, and death. Loss of defence mechanism and an inappropriate immune-response in patients who are immucompromised, particularly transplant recipients, burn patients, patients with cancer, neutropenia, and with HIV are important risk factors for P. aeruginosa infection.
Figure 1.
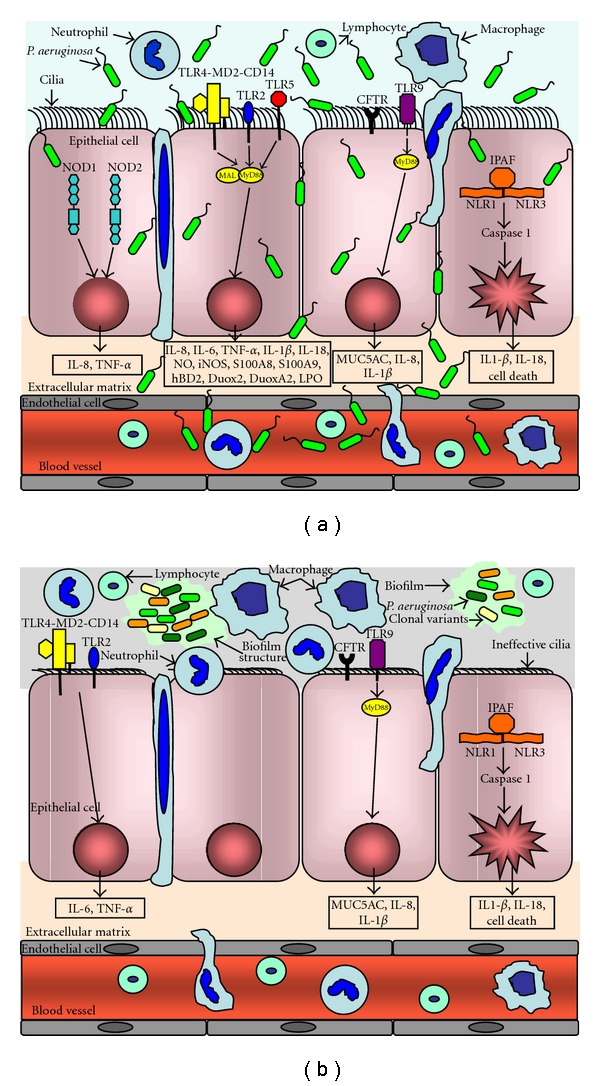
Pseudomonas aeruginosa recognition by PRRs during acute and chronic lung infection. (a) During acute lung infection, P. aeruginosa can invade, disseminate and lead to extensive tissue damage by means of potent array of extracellular and cell bound virulence factors. However the immunocompetent host mounts an effective immune response characterized of bacterial recognition by cell surface receptors, which are located on immune cells as well as epithelial cells. Plasma membrane-bound TLRs (TLR2, TLR4-MD2-CD14, TLR5, and TLR9) and cytosolic NLRs (NOD1, NOD2, and IPAF) recognise P. aeruginosa PAMPs and recruit adaptors to induce downstream signalling cascades, which result in transcription of pro-inflammatory mediators and mucins. These pro-inflammatory mediators, including chemokines, recruit immune cells to the lung in order to clear P. aeruginosa and resolve the infection. (b) During chronic lung infection, P. aeruginosa, enmeshed in biofilm structures, does not have direct contact to the airways epithelium and probably only immunogenic bacterial components can access to airway epithelium and immune cells in the lung. In addition, during long-term colonization, bacteria undergo a number of genetic changes and gain the ability to evade detection and clearance by the immune system, thus surviving in the host. The loss or modification of several PAMPs (flagellin, LPS, and PGN) lead to reduced recognition by TLRs and NLRs although components of the alginate capsule can still be recognized both by TLR2 and TLR4. The inadequate immune response may explain the chronic colonization of P. aeruginosa strains. NOD, Nucleotide-binding oligomerization domain; TLR, Toll-like receptor; NLR, Nod-like receptor; IPAF, ICE-protease activating factor; MyD88, myeloid differentiation primary response protein; CFTR, cystic fibrosis transmembrane conductance regulator.
In CF, generalized immune deficiency and specific abnormalities in acquired immunity are highly unlikely, since systemic infection is not characteristic of this disease (Figure 1(b)). In fact, sepsis due to P. aeruginosa, even after decades of lung infection, is rare, presumably due to effective humoral immunity. More likely, the disease represents a failure of local airway defense. It has been suggested that bacteria adhere more readily to CF airway epithelial cells due to enhanced expression of the cell surface ganglioside asialoGM1, promoting infection [3]. Paradoxically, it has also been proposed that cystic fibrosis transmembrane conductance regulator (CFTR) serves as a bacterial receptor and that its absence leads to failure to internalize and kill bacteria [4, 5]. Furthermore, an abnormal accumulation of ceramide in the lungs of CF mice and in epithelial cells from CF patients has been shown to result in an increased death rate of respiratory epithelial cells and DNA deposits on the respiratory epithelium, which facilitate bacterial adherence [6].
Defects in antimicrobial activity of airway fluid [7], in neutrophil phagocytosis [8] or excessive neutrophil extracellular traps (NETs) formation [9] likely occur in the inflamed CF airway environment but are unlikely to be the primary defect. The respiratory tract pathophysiology in CF principally results from the inability to secrete Cl− and regulate Na+ absorption, which causes relative dehydration of the airway surface, depleting the periciliary layer and causing accumulation of hyperviscous mucus that cannot be cleared by mucociliary clearance or cough [10]. Mucus plaques and plugs serve as a nidus for intra-luminal infection [11]. Bacteria resident within the thickened luminal mucus may evade chemical antimicrobial factors and phagocytes [12]. Complex bacterial evolution and host adaptation occur in the chronically infected airway, which is likely unique in CF due to the constant and severe degree of mucus dehydration and impaired mucus clearance. Bacterial colonies exhibiting biofilm-like properties may develop, which are difficult or impossible to eradicate. The continuous presence of bacteria and the accompanying intense inflammation ultimately remodel the airway wall, causing the ubiquitous mucous secretory cell hyperplasia and metaplasia, submucosal gland enlargement, hypertrophy of the bronchial circulation, ectasis of bronchi and bronchioles, and variable parenchymal cyst formation, sometimes progressing to cavitary disease, with adjacent fibrosis and pleural involvement [13].
The pathogenesis of P. aeruginosa infections appears to be complex and multifactorial. The conditions present in acute infections force pathogens to injure or kill the host, with multiorgan failure sometimes occurring in hours or days. In contrast, chronic infections occur without acute injury and in the presence of biofilm structures—a population of microorganisms that aggregates on a matrix—that develop over days or weeks, and bacterial genetic variants may grow in the biofilms. A potent array of P. aeruginosa extracellular and cell bound virulence factors are critical for the initial colonization phase of infection and then invasion, dissemination, and extensive tissue damage [14]. Flagella and pili, the motile surface appendages of P. aeruginosa, are responsible for bacterial motility, progression towards epithelial contact and dissemination throughout the host organism. These appendages also act as initial tethers in facilitating bacteria to epithelial cell contact by binding to the cell surface receptors. Additionally, lipopolysaccharide (LPS) plays a similar role in bacterial adhesion [15]. Upon cell contact, the type III secretion system (T3SS), a major virulence determinant, is activated [16, 17]. The T3SS allows P. aeruginosa to inject secreted toxins through a syringe-like apparatus directly into the eukaryotic cytoplasm. Four effector proteins are known: ExoY, ExoS, ExoT, and ExoU and all participate, at varying levels, in the cytotoxicity of P. aeruginosa leading to invasion and dissemination [18]. Other virulence factors secreted via type II secretion system into the extracellular space such as elastase, alkaline phosphatase, exotoxin A, and phospholipase C are also liable for invasion and dissemination by destroying the protective glycocalix of the respiratory epithelium and exposing epithelial ligands to P. aeruginosa and participate in cytotoxicity [19]. A similar role has also been reported for pyoverdine and pyocyanin. Most of these P. aeruginosa invasive functions are selected against in CF chronic infection leading to less virulent but more persistent phenotypes [20–22]. The steric shielding or modification of these exposed molecules is the most effective strategy for avoiding host's innate recognition and establishing persistent chronic infection [23]. In addition, in the lung of CF patients, P. aeruginosa forms microcolonies encapsulated by mucoid exopolysaccharide. The emergence of mucoid variants is believed to mark the transition to the fatal, chronic stage of the infection [24].
2. Sensing and Defence against Pseudomonas aeruginosa
The innate immune system is the first line of defence against pathogens. Innate immune responses depend on a vast array of nonclonally expressed receptors, named pattern recognition receptors (PRRs), aimed at pathogen recognition. PRR bind to highly conserved invariant molecular complexes called pathogens (microbe-) associated molecular Patterns (PAMP or MAMPs), which are widespread and conserved among microorganisms [36]. PRR binding with PAMPs elicits a signalling response which results in a rapid response against any encountered microorganisms, including potential pathogens. This complexity of bindings allows the immune system not only to tailor its response to a specific pathogen but also to discriminate the site of infection or the microbial burden.
The best studied PRRs are Toll-like receptors (TLRs), which are transmembrane proteins present at the cell surface or on the membrane of endocytic vesicles or other intracellular organelles [37]. The extracellular domain of TLRs is characterized by leucine-rich repeats (LRRs) that are involved in ligand binding. Ligand recognition induces homodimerization or heterodimerization of the ectodomains, allowing the intracellular domains to initiate signalling. The cytoplasmic domain contains the highly conserved Toll/interleukin-1 (IL-1) receptor (TIR) domain, which interacts with various adaptor molecules such as myeloid differentiation primary response protein (MyD88) to elicit signalling. There are currently 12 known mammalian TLRs; they recognize and bind a wide variety of bacterial PAMPs, including LPS (typically recognized by TLR4 although some LPS species can be recognized by TLR2), lipopeptides (TLR1, TLR2, TLR6), lipoarabinomannan and lipoteichoic acid (TLR2 and other TLRs), flagellin (TLR5), and bacterial DNA (TLR9) [38].
Although the TLRs is the family of PRRs best studied another PRR family identified through homology to plant R proteins has emerged as playing a crucial role in host response. The PRR family of nucleotide-binding oligomerization nomain-(NOD-) like receptor (NLR) proteins includes 23 members in humans, and it is divided in 5 subfamilies according to their effector domains. Several studies highlighted that NLR NOD1 and NOD2 are key cytoplasmic PRR [39, 40]. These proteins are characterized by a N-terminal effector domain, a centrally located nucleotide-binding domain and multiple leucin-rich repeats in their C-terminal end. While Nod1 is ubiquitous, Nod2 seems to be more restricted to myelomonocytic and epithelial cells. Both Nod1 and Nod2 sense peptidoglycan (PGN) motifs. Nod1 exhibits specificity for a diaminopimelate containing GlcNAc-MurNAc tripeptide (GM-TriDap) fragment that is almost exclusively found in Gram-negative bacterial PGN, while Nod2 binds muramyl dipeptide (MDP) motif that is common to Gram-positive and Gram-negative bacteria [41, 42]. After recognition of bacterial ligands through the LRRs domain Nod1 and Nod2 activate the Nuclear Factor κB (NF-κB) and elicit the production of pro-inflammatory cytokines [43]. Other NLR members, such as NLR1, NLR3 and Ipaf, are involved in building a multisubunit protein complex called inflammasome following PAMP and DAMP (danger-associated molecular Patterns) detection [44]. This is a tightly controlled process leading to the proteolytic processing of procaspase-1, which in turns activate the interleukin-1β. Inflammasome activation also accounts for a peculiar type of cell death called pyroptosis characterized by membrane cell lesions leading to the release of mature IL-1β [45].
P. aeruginosa stimulates pro-inflammatory cytokine production [46]. P. aeruginosa expresses numerous PAMPs (Table 1), among which LPS and flagellin have been reported to play a special role in signalling bacterial presence in host. LPS is a glycolipid that constitutes the major portion of the outermost membrane of Gram-negative bacteria. LPS consists of three distinct regions: O-antigen, core, and lipid A. Both O-antigen and core consist of polysaccharide chains, whereas lipid A consists of fatty acid and phosphate moieties bonded to a central glucosamine dimer [47]. This last portion of LPS molecule is recognized by TLR4, in association with CD14 and the adaptor molecule MD2 [48, 49]. It has been reported that P. aeruginosa can vary LPS acylation (number and structure of fatty acids) during its biosynthesis [50, 51]. This variation results on a modified sensing of this PAMP from the cognate receptor TLR4, which classically recognizes hexa-acylated LPS, and/or recognition from TLR2, which can bind tetra- and penta-acylated pseudomonal LPS. The role of TLR2 in P. aeruginosa recognition is widely treated in several studies. TLR2 is a “promiscuous” member of the TLR family as it recognizes several ligands. TLR2 has been reported to participate in recognition of multiple P. aeruginosa components, including lipoproteins [52], alginate [27], flagellin [53], and exoenzyme S [25].
Table 1.
PRRs sensing P. aeruginosa-associated molecular patterns.
| Receptors | P. aeruginosa PAMP | References |
|---|---|---|
| Toll-like receptors | ||
| TLR2 | PGN | [25] |
| LPS | [26] | |
| ExoS | [25] | |
| mannuronic acid polymers | [27] | |
| flagellin | [28] | |
| Slime-GLP | [29] | |
| TLR4-MD2-CD14 | LPS | [30] |
| ExoS | [25] | |
| mannuronic acid polymers | [27] | |
| TLR5 | Flagellin | [31] |
| TLR9 | DNA | [32] [33] |
| NLR receptors | ||
| Nod 1 | PGN | [30] |
| NLRC4/Ipaf | flagellin | [34] |
| T3SS | [34] | |
| Other receptors | ||
| CFTR | LPS | [35] |
In addition to direct TLR2 activation it has been proposed that P. aeruginosa triggers tumor necrosis factor TNF-α production by human monocytes through slime glycolipoprotein (slime-GLP) [29], an extracellular glycolipid component, which is present at the bacterial surface and produced during in vivo infection by mucoid and nonmucoid strains. P. aeruginosa slime-GLP is rich in mannose, a sugar which is absent from the homologous LPS. Slime-GLP can activate innate immune responses through interaction with MR (Mannose receptors) which synergize and complement the activity of TLRs. Following interaction with P. aeruginosa MR activation elicits proinflammatory cytokine production by stimulating NF-κB and MAPK. P. aeruginosa MR activation synergizes TLR2 activity to trigger maximum NF-κB stimulation and proinflammatory cytokine production [54]. Likewise, studies in TLR9 deficient mice demonstrated that signalling induced by this TLR plays a role in cornea inflammation experimentally provoked by P. aeruginosa [55]. However, a more recent report identified a new-based CpG motif (which is the natural agonist of TLR9), CpG-ODNc41, in P. aeruginosa genome [56]. In human and murine monocytes CpG-ODNc41 was nonstimulatory and noncytotoxic and acted as an antagonist by inhibiting the stimulatory activity of conventional CpG-DNAs.
Modification in host sensing may switch the immune response from an appropriate reaction to the presence of the pathogen to a vicious circle in which inapt signalling leads to excessive inflammation which exacerbates the effect of P. aeruginosa presence in the organism. Flagellin is a protein that arranges itself in a hollow cylinder to form the filament bacterial flagellum. Indeed, P. aeruginosa is motile via a single polar flagellum that has structural properties very similar to those of enteric Gram-negative bacteria, with the added structural feature of being glycosylated [57]. A specific motif [58–60] in flagellin monomer is a ligand for TLR5 and induces the expression of proinflammatory mediators in monocytes, macrophages, intestinal, airway, and corneal epithelial cells, resulting from the activation of the transcription factor NF-kB and production of pro-inflammatory cytokines [61]. The majority of studies focused on the interaction of TLR activity and P. aeruginosa have been carried out in vitro, on cellular models. However, up to now no single TLR deficiency in transgenic mice seems to affect the host response to P. aeruginosa in lung infection, which is one of the more common and serious aspect of P. aeruginosa pathogenesis. Though controversial results have been obtained by different groups, common opinion emerged that mice lacking individual TLR4, TLR2, or TLR5 are equally sensitive to lung P. aeruginosa infection [62–64]. Likewise, the combination of TLR4 and TLR2 absence does not impair the ability of P. aeruginosa to establish infection albeit the defects in cytokine responses have been recorded. Multitransgenic mice lacking TLR5 and TLR4 appear to be more sensitive to P. aeruginosa [62]. Most of TLR signalling following bacterial sensing depends on the MyD88 adaptor protein that leads the activation of NF-kB and the production of pro-inflammatory cytokines. Therefore, mice lacking MyD88 infected intranasally with P. aeruginosa displayed elevated bacterial counts in lung [65] along with reduced levels of cytokines such as TNF-α and IL-1β.
Moreover, several studies report that P. aeruginosa is sensed by the NLR family members. Notably, P. aeruginosa is recognized by the Nod1 PRR following infection in HEK293 cells overexpressing this receptor [66]. Likewise by exploiting various cell model systems, it was demonstrated that Nod1, but not Nod2, recognizes in a different way P. aeruginosa PGN of clonal clinical strains isolated by airways of a patient of CF at the initial and chronic stages of infection [30].
In contrast to TLR5, which senses extracellular flagellin, the Ipaf inflammasome is generally activated by cytosolic flagellin, which is sufficient for IPAF-dependent caspase-1 activation [67, 68]. In accordance with this issue, during infection, P. aeruginosa is recognized by Ipaf through flagellin sensing [34, 69]. However, parallel studies reported that Ipaf-mediated inflammasome is also mounted by the presence of a T3SS carried by several pathogens, including P. aeruginosa [70]. More recently, a basal body rod component of the T3SS apparatus has been identified as the T3SS-dependent responsible for Ipaf activation. This component has been found in Salmonella typhimurium (PrgJ), Burkholderia pseudomallei (BsaK), Escherichia coli (EprJ and EscI), Shigella flexneri (MxiI), and P. aeruginosa (PscI), inducing caspase-1 activation [71]. These rod proteins share a sequence motif that is essential for detection by Ipaf, and that is similar to the flagellin motif recognized by the same receptor.
3. Avoiding Host Recognition in Pseudomonas aeruginosaf and Establishing Chronic Infection
Although the process of recognition of PAMPs is rapid and efficient, it is now clear that bacteria are able to alter their structures in order to avoid or modulate this immune recognition. This bacterial behaviour is classed as an “immune evasion strategy”. Most CF patients acquire chronic P. aeruginosa infections by their teenage years and these respiratory infections are responsible for much of the morbidity and mortality. It has been established that the majority of P. aeruginosa strains infecting the lungs of CF patients are acquired independently, presumably from diverse environmental reservoirs. However, the long-term colonization of the CF host is maintained by P. aeruginosa pathoadaptive lineages, which are clonal with the initially acquired strain and carried phenotypic variants [77]. A variety of host-derived inflammatory product and environmental factors contribute to select clonal variants in P. aeruginosa. As a result of inflammation, P. aeruginosa is exposed to high levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which are generated primarily by neutrophils as part of the host's innate immune response. ROS and RNS contribute to mutations that confer an adaptive advantage to P. aeruginosa in the airway [78, 79]. Furthermore, within the highly viscous mucus, a microaerobic/anaerobic milieu prevails due to oxygen consumption by bacterial pathogens or invading neutrophils. P. aeruginosa growth in oxygen restricted environments leads to changes in the bacterial phenotypes that facilitate chronic infection [11, 80].
Pathogenicity-adaptive mutations represent a genetic mechanism for enhancing bacterial virulence without horizontal transfer of specific virulence factors [81]. Common mutations are consistently acquired by most CF patients as those in regulators of alginate biosynthesis (mucA and algT) [74] and virulence genes including in the LPS (pagL) and muropeptide modifications [30], motility (rpoN) [72], in the quorum-sensing regulator (lasR) [75, 76], in the T3SS [82], in the multidrug-efflux pump (mexA) and in mutator phenotypes (mutS) [83] (Table 2). Whole-genome comparison between an isogenic early and late P. aeruginosa pair recovered from one patient 90 months apart showed that the late isolate accumulated 68 mutations [20]. Interestingly, virulence factors required for the initiation of acute infections were selected against during chronic infection. This indicates reduced virulence of the late strains with regard to their ability to provoke acute infection and host recognition [23, 84]. High adaptive genetic diversification underlines the immune evasion strategies, resulting in increasing chances of bacterial survival in their niche, that is, in the airway environment of CF patients. This evolutionary scenario is similar to that of the genomes of pathogens which establish chronic infection as Escherichia coli [85], Haemophilus influenzae [86], or Helicobacter pylori [87].
Table 2.
Frequent mutations in P. aeruginosa virulence factors of CF airways isolates.
| Virulence factors | Mutation* | References |
|---|---|---|
| LPS | pagL | [30] |
| Flagella | rpoN, fleQ | [72] [20] |
| Alginate | mucA | [73] [74] |
| Quorum sensing | LasR | [75] [76] |
| T3SS | exsA | [20] |
*Only most common mutations in P. aeruginosa clinical strains are reported.
Recent reports have showed that P. aeruginosa exploits PAMPs modification as a strategy to hijack genes involved in innate immune responses and to favor survival in CF patients [28, 30]. Loss of flagellar expression enables immune evasion by the bacteria due to loss of engagement by phagocytic receptors that recognize flagellar components and loss of immune activation through flagellin-mediated TLR signaling. Using a variety of in vitro, ex vivo, and in vivo infection models, Amiel et al. [28], showed that loss of P. aeruginosa motility dramatically alters immune responses to these bacteria compared to those for motile isogenic bacterial strains and that it is the loss of flagellum-mediated motility, but not flagellum expression itself, that results in dramatic bacterial resistance to phagocytosis by murine and human phagocytes. Likewise, studies in the agar beads murine model by using the P. aeruginosa isolates from patients with CF demonstrated that the risk of chronic infection is increased by the absence of pili and flagella [21]. These studies provide an explanation for the clinical observation that P. aeruginosa isolates obtained from CF hosts often exhibit a nonmotile phenotype [88] and explain how this phenotype can confer a survival advantage for bacteria that modulate or lose their motility during chronic infection.
Chemical structure of LPS and PGN were determined for three P. aeruginosa clones isolated from airways of a CF patient during a period of 7.5 years [30]. Lipid A, that is variably penta-, hexa-, or hepta-acylated, was temporally associated with different stages of CF infection. Among the three strains LPS lipid A diversity was observed in the number and location of fatty-acid side chains. Early and late mucoid P. aeruginosa strains synthesized a LPS blend essentially composed by tetra-, penta-, and hexa-acylated species. In contrast, the late nonmucoid strain was constituted by homologue lipid A species which carried hexa-acylated and hepta-acylated moieties. These findings are in accordance with previous observations [50, 89, 90], that P. aeruginosa synthesizes more highly acylated (hexa- and hepta-acylated) LPS structures during adaptation to the CF airways. Characterization of the bacterial genes that modify lipid A revealed that the pagL gene was mutated in the strain obtained at the later stage of CF. As for the PGN, diversity in early and late P. aeruginosa strains consisted in different distribution of canonical monomeric and dimeric species. When tested in human cells including those of CF origin, the strong inflammatory response induced by P. aeruginosa LPS and PGN isolated at early stage of infection was attenuated at late stage. Significantly higher NF-κB activation, IL-8 expression and production were detected after direct activation of TLR4/MD2-CD14 by LPS and Nod1 by PGN of early strain when compared to late strains [30].
Lipid A structures of P. aeruginosa affected also the inflammatory response in mice [30]. Leukocyte recruitment in the bronchoalveolar lavage fluid (BALF) of mice exposed to different LPS structures of clinical strains showed striking differences in total differential cell counts. Significant higher recruitment of neutrophils was observed in mice exposed to LPS from early strain in comparison to those treated with late strains. Cytokine levels, tested in murine lung homogenates, showed higher MIP-2 levels for mice treated with early LPS than late LPS. Similar trends were obtained with KC and IL-1β. The impaired ability of the host to mount an adequate immune response could explain the ineffective eradication of the infection and the resulting persistent infection of late strains in comparison to the clonal early strain in the agar beads mouse model [21].
A prominent feature of P. aeruginosa strains infecting CF patients is the conversion to a mucoid, exopolysaccharide alginate-overproducing phenotype. Mannuronic acid polymers, the main components of the alginate capsule, induce immunostimulation via TLR2 and TLR4 pathways [27]. However, the overproduction of alginate by P. aeruginosa may be advantageous for the bacteria by impeding phagocytosis, and providing protection against reactive oxygen species and antibiotics [91–93]. The response of airway epithelia to the stimuli presented by mucoid P. aeruginosa is not pro-inflammatory and, hence, may not be conducive to the effective elimination of the pathogen [94]. Indeed, in vivo studies suggest that clearance of mucoid strains from murine lungs is diminished compared with nonmucoid strains, indicating improved survival of alginate-producing strains in the respiratory tract [95, 96]. Alginate enhances mucin secretion by tracheal epithelial cells and may inhibit neutrophil migration to the sites of infection. Interestingly, the production of flagellin and alginate by P. aeruginosa are inversely regulated by the alternative sigma factor AlgT, which is a positive regulator of mucoidy and a negative regulator of flagella mediated motility.
Among the four effector proteins secreted by the T3SS, exogenous ExoS has been demonstrated to activate TLR2 and TLR4 [25], thus contributing to inflammatory responses. However, it has been proposed that, following the infection of CF patient airways, P. aeruginosa strains evolve to reduce T3SS expression [18], or that populations of cells gradually change from a type III protein secretion-positive phenotype to a secretion-negative phenotype [97]. In addition, QS-related and T3SS genes were downregulated in a modified artificial-sputum medium, more closely resembling CF sputum [98]. It has also been reported that the cyclic AMP/Vfr-dependent signaling (CVS) pathway, responsible for the regulation of the expression of multiple invasive virulence factors, including T3SS, exotoxin A, protease IV, and TFP, is defective in mucA mutants [99]. All these findings suggest that mucoid conversion and inhibition of invasive virulence determinants may both confer a selective advantage to P. aeruginosa strains in the CF lung.
4. Conclusions
In conclusion, the last decade has witnessed an increase in our understanding of the molecular mechanisms involved in the recognition of P. aeruginosa as invading and standing pathogen. It is expected that future efforts will try to elucidate in much more detail and precision the role of genetic variability in the PRRs for the susceptibility to P. aeruginosa infection and the mechanism of P. aeruginosa adaptation to host. Research in the field might lead to novel discoveries and new therapies for patients at risk factors of P. aeruginosa infection and might have an important impact on the quality of life for patients with CF.
Acknowledgments
Funded by European Commission (NABATIVI-223670, EU-FP7-HEALTH-2007-B) and Italian Cystic Fibrosis Research Foundation, Grants FFC#17/2009 and FFC#8/2007, with the contribution of Antonio Guadagnin e Figlio s.r.l., Delegazione FFC Roma 2 “Vorrei 2009” Rome and Potenza, Associazione Amici di Cortina, Furla Spa, GVS Spa Zola Predosa (BO), and Delegazione FFC Bologna.
References
- 1.Stover CK, Pham XQ, Erwin AL, et al. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature. 2000;406(6799):959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 2.Sadikot RT, Blackwell TS, Christman JW, Prince AS. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. American Journal of Respiratory & Critical Care Medicine. 2005;171(11):1209–1223. doi: 10.1164/rccm.200408-1044SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saiman L, Prince A. Pseudomonas aeruginosa pili bind to asialoGM1 which is increased on the surface of cystic fibrosis epithelial cells. Journal of Clinical Investigation. 1993;92(4):1875–1880. doi: 10.1172/JCI116779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pier GB, Grout M, Zaidi TS, et al. Role of mutant CFTR in hypersusceptibility of cystic fibrosis patients to lung infections. Science. 1996;271(5245):64–67. doi: 10.1126/science.271.5245.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pier GB, Grout M, Zaidi TS. Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(22):12088–12093. doi: 10.1073/pnas.94.22.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teichgräber V, Ulrich M, Endlich N, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nature Medicine. 2008;14(4):382–391. doi: 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- 7.Ganz T. Antimicrobial polypeptides in host defense of the respiratory tract. Journal of Clinical Investigation. 2002;109(6):693–697. doi: 10.1172/JCI15218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berger M, Sorensen RU, Tosi MF, Dearborn DG, Döring G. Complement receptor expression on neutrophils at an inflammatory site, the Pseudomonas-infected lung in cystic fibrosis. Journal of Clinical Investigation. 1989;84(4):1302–1313. doi: 10.1172/JCI114298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marcos V, Zhou Z, Yildirim AO, et al. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nature Medicine. 2010;16(9):1018–1023. doi: 10.1038/nm.2209. [DOI] [PubMed] [Google Scholar]
- 10.Matsui H, Grubb BR, Tarran R, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95(7):1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- 11.Worlitzsch D, Tarran R, Ulrich M, et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. Journal of Clinical Investigation. 2002;109(3):317–325. doi: 10.1172/JCI13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsui H, Verghese MW, Kesimer M, et al. Reduced three-dimensional motility in dehydrated airway mucus prevents neutrophil capture and killing bacteria on airway epithelial surfaces. Journal of Immunology. 2005;175(2):1090–1099. doi: 10.4049/jimmunol.175.2.1090. [DOI] [PubMed] [Google Scholar]
- 13.Livraghi A, Randell SH. Cystic fibrosis and other respiratory diseases of impaired mucus clearance. Toxicologic Pathology. 2007;35(1):116–129. doi: 10.1080/01926230601060025. [DOI] [PubMed] [Google Scholar]
- 14.Tang HB, Dimango E, Bryan R, et al. Contribution of specific Pseudomonas aeruginosa virulence factors to pathogenesis of pneumonia in a neonatal mouse model of infection. Infection & Immunity. 1996;64(1):37–43. doi: 10.1128/iai.64.1.37-43.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madjdpour C, Oertli B, Ziegler U, Bonvini JM, Pasch T, Beck-Schimmer B. Lipopolysaccharide induces functional ICAM-1 expression in rat alveolar epithelial cells in vitro. American Journal of Physiology. 2000;278(3):L572–L579. doi: 10.1152/ajplung.2000.278.3.L572. [DOI] [PubMed] [Google Scholar]
- 16.Filloux A, Bally M, Ball G, Akrim M, Tommassen J, Lazdunski A. Protein secretion in gram-negative bacteria: transport across the outer membrane involves common mechanisms in different bacteria. EMBO Journal. 1990;9(13):4323–4329. doi: 10.1002/j.1460-2075.1990.tb07881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yahr TL, Goranson J, Frank DW. Exoenzyme of S of Pseudomonas aeruginosa is secreted by a type III pathway. Molecular Microbiology. 1996;22(5):991–1003. doi: 10.1046/j.1365-2958.1996.01554.x. [DOI] [PubMed] [Google Scholar]
- 18.Lee VT, Smith RS, Tümmler B, Lory S. Activities of Pseudomonas aeruginosa effectors secreted by the type III secretion system in vitro and during infection. Infection & Immunity. 2005;73(3):1695–1705. doi: 10.1128/IAI.73.3.1695-1705.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ball G, Durand É, Lazdunski A, Filloux A. A novel type II secretion system in Pseudomonas aeruginosa. Molecular Microbiology. 2002;43(2):475–485. doi: 10.1046/j.1365-2958.2002.02759.x. [DOI] [PubMed] [Google Scholar]
- 20.Smith EE, Buckley DG, Wu Z, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(22):8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bragonzi A, Paroni M, Nonis A, et al. Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. American Journal of Respiratory & Critical Care Medicine. 2009;180(2):138–145. doi: 10.1164/rccm.200812-1943OC. [DOI] [PubMed] [Google Scholar]
- 22.Bianconi I, Milani A, Cigana C, et al. Positive signature-tagged mutagenesis in Pseudomonas aeruginosa: tracking patho-adaptive mutations promoting airways chronic infection. PLoS Pathogens. 2011;7(2) doi: 10.1371/journal.ppat.1001270. Article ID e1001270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen D, Singh PK. Evolving stealth: genetic adaptation of Pseudomonas aeruginosa during cystic fibrosis infections. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(22):8305–8306. doi: 10.1073/pnas.0602526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoiby N. Pseudomonas aeruginosa infection in cystic fibrosis. Diagnostic and prognostic significance of Pseudomonas aeruginosa precipitins determined by means of crossed immunoelectrophoresis. A survey. Acta Pathologica et Microbiologica Scandinavica Supplement. 1977;(262):1–96. [PubMed] [Google Scholar]
- 25.Epelman S, Stack D, Bell C, et al. Different domains of Pseudomonas aeruginosa exoenzyme S activate distinct TLRs. Journal of Immunology. 2004;173(3):2031–2040. doi: 10.4049/jimmunol.173.3.2031. [DOI] [PubMed] [Google Scholar]
- 26.Erridge C, Pridmore A, Eley A, Stewart J, Poxton IR. Lipopolysaccharides of Bacteroides fragilis, Chlamydia trachomatis and Pseudomonas aeruginosa signal via toll-like receptor 2. Journal of Medical Microbiology. 2004;53(8):735–740. doi: 10.1099/jmm.0.45598-0. [DOI] [PubMed] [Google Scholar]
- 27.Flo T, Ryan L, Latz E, et al. Involvement of Toll-like receptor (TLR) 2 and TLR4 in cell activation by mannuronic acid polymers. The Journal of Biological Chemistry. 2002;277(38):35489–35495. doi: 10.1074/jbc.M201366200. [DOI] [PubMed] [Google Scholar]
- 28.Amiel E, Lovewell RR, O’Toole GA, Hogan DA, Berwin B. Pseudomonas aeruginosa evasion of phagocytosis is mediated by loss of swimming motility and is independent of flagellum expression. Infection & Immunity. 2010;78(7):2937–2945. doi: 10.1128/IAI.00144-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagoumintzis G, Christofidou M, Dimitracopoulos G, Paliogianni F. Pseudomonas aeruginosa slime glycolipoprotein is a potent stimulant of tumor necrosis factor alpha gene expression and activation of transcription activators nuclear factor kappa B and activator protein 1 in human monocytes. Infection & Immunity . 2003;71(8):4614–4622. doi: 10.1128/IAI.71.8.4614-4622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cigana C, Curcurù L, Leone MR, et al. Pseudomonas aeruginosa exploits lipid A and muropeptides modification as a strategy to lower innate immunity during cystic fibrosis lung infection. PloS one. 2009;4(12):p. e8439. doi: 10.1371/journal.pone.0008439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z, Louboutin JP, Weiner DJ, Goldberg JB, Wilson JM. Human airway epithelial cells sense Pseudomonas aeruginosa infection via recognition of flagellin by toll-like receptor 5. Infection & Immunity. 2005;73(11):7151–7160. doi: 10.1128/IAI.73.11.7151-7160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magnusson M, Tobes R, Sancho J, Pareja E. Cutting edge: natural DNA repetitive extragenic sequences from gram-negative pathogens strongly stimulate TLR9. Journal of Immunology. 2007;179(1):31–35. doi: 10.4049/jimmunol.179.1.31. [DOI] [PubMed] [Google Scholar]
- 33.Hemmi H, Takeuchi O, Kawai T, et al. A toll-like receptor recognizes bacterial DNA. Nature. 2000;408(6813):740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 34.Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, Núñez G. Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. European Journal of Immunology. 2007;37(11):3030–3039. doi: 10.1002/eji.200737532. [DOI] [PubMed] [Google Scholar]
- 35.Schroeder TH, Lee MM, Yacono PW, et al. CFTR is a pattern recognition molecule that extracts Pseudomonas aeruginosa LPS from the outer membrane into epithelial cells and activates NF-kappa B translocation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(10):6907–6912. doi: 10.1073/pnas.092160899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Janeway CA., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harbor Symposia on Quantitative Biology. 1989;54(1):1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 37.Akira S, Takeda K. Toll-like receptor signalling. Nature Reviews Immunology. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 38.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 39.Inohara N, Koseki T, Del Peso L, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. The Journal of Biological Chemistry. 1999;274(21):14560–14567. doi: 10.1074/jbc.274.21.14560. [DOI] [PubMed] [Google Scholar]
- 40.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. The Journal of Biological Chemistry. 2001;276(7):4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 41.Girardin SE, Boneca IG, Carneiro LA, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300(5625):1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- 42.Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2: implications for crohn’s disease. The Journal of Biological Chemistry. 2003;278(8):5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 43.Inohara N, Chamaillard M, McDonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annual Review of Biochemistry. 2005;74:355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 44.Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nature Immunology. 2006;7(12):1250–1257. doi: 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- 45.Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends in Microbiology. 2001;9(3):113–114. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 46.Lyczak JB, Cannon CL, Pier GB. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes and Infection. 2000;2(9):1051–1060. doi: 10.1016/s1286-4579(00)01259-4. [DOI] [PubMed] [Google Scholar]
- 47.Alexander C, Rietschel ET. Bacterial lipopolysaccharides and innate immunity. Journal of Endotoxin Research. 2001;7(3):167–202. [PubMed] [Google Scholar]
- 48.Lien E, Means TK, Heine H, et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. Journal of Clinical Investigation. 2000;105(4):497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitchens R. Role of CD14 in cellular recognition of bacterial lipopolysaccharides. Chemical Immunology. 1999;74:61–82. doi: 10.1159/000058750. [DOI] [PubMed] [Google Scholar]
- 50.Ernst RK, Yi EC, Guo L, et al. Specific lipopolysaccharide found in cystic fibrosis airway Pseudomonas aeruginosa. Science. 1999;286(5444):1561–1565. doi: 10.1126/science.286.5444.1561. [DOI] [PubMed] [Google Scholar]
- 51.Ernst R, Hajjar AM, Tsai JH, Moskowitz SM, Wilson CB, Miller SI. Pseudomonas aeruginosa lipid A diversity and its recognition by Toll-like receptor 4. Journal of Endotoxin Research. 2003;9(6):395–400. doi: 10.1179/096805103225002764. [DOI] [PubMed] [Google Scholar]
- 52.Firoved AM, Ornatowski W, Deretic V. Microarray analysis reveals induction of lipoprotein genes in mucoid Pseudomonas aeruginosa: implications for inflammation in cystic fibrosis. Infection & Immunity. 2004;72(9):5012–5018. doi: 10.1128/IAI.72.9.5012-5018.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adamo R, Sokol S, Soong G, Gomez MI, Prince A. Pseudomonas aeruginosa flagella activate airway epithelial cells through asialoGM1 and toll-like receptor 2 as well as toll-like receptor 5. American Journal of Respiratory Cell & Molecular Biology. 2004;30(5):627–634. doi: 10.1165/rcmb.2003-0260OC. [DOI] [PubMed] [Google Scholar]
- 54.Xaplanteri P, Lagoumintzis G, Dimitracopoulos G, Paliogianni F. Synergistic regulation of Pseudomonas aeruginosa-induced cytokine production in human monocytes by mannose receptor and TLR2. European Journal of Immunology. 2009;39(3):730–740. doi: 10.1002/eji.200838872. [DOI] [PubMed] [Google Scholar]
- 55.Huang X, Barrett RP, McClellan SA, Hazlett LD. Silencing toll-like receptor-9 in Pseudomonas aeruginosa keratitis. Investigative Ophthalmology & Visual Science. 2005;46(11):4209–4216. doi: 10.1167/iovs.05-0185. [DOI] [PubMed] [Google Scholar]
- 56.Sun X, Sui H, Fisher JT, et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. The Journal of Clinical Investigation. 2010;120(9):3149–3160. doi: 10.1172/JCI43052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schirm M, Arora SK, Verma A, et al. Structural and genetic characterization of glycosylation of type a flagellin in Pseudomonas aeruginosa. Journal of Bacteriology. 2004;186(9):2523–2531. doi: 10.1128/JB.186.9.2523-2531.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Donnelly MA, Steiner TS. Two nonadjacent regions in enteroaggregative Escherichia coli flagellin are required for activation of toll-like receptor 5. The Journal of Biological Chemistry. 2002;277(43):40456–40461. doi: 10.1074/jbc.M206851200. [DOI] [PubMed] [Google Scholar]
- 59.Murthy KG, Deb A, Goonesekera S, Szabó C, Salzman AL. Identification of conserved domains in Salmonella muenchen flagellin that are essential for Its ability to activate TLR5 and to induce an inflammatory response in vitro. The Journal of Biological Chemistry. 2004;279(7):5667–5675. doi: 10.1074/jbc.M307759200. [DOI] [PubMed] [Google Scholar]
- 60.Verma A, Arora SK, Kuravi SK, Ramphal R. Roles of specific amino acids in the N terminus of Pseudomonas aeruginosa flagellin and of flagellin glycosylation in the innate immune response. Infection & Immunity. 2005;73(12):8237–8246. doi: 10.1128/IAI.73.12.8237-8246.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eaves-Pyles T, Murthy K, Liaudet L. Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: I kappa B alpha degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. The Journal of Immunology. 2001;166(2):1248–1260. doi: 10.4049/jimmunol.166.2.1248. [DOI] [PubMed] [Google Scholar]
- 62.Feuillet V, Medjane S, Mondor I, et al. Involvement of toll-like receptor 5 in the recognition of flagellated bacteria. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(33):12487–12492. doi: 10.1073/pnas.0605200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Power MR, Peng Y, Maydanski E, Marshall JS, Lin TJ. The development of early host response to Pseudomonas aeruginosa lung infection is critically dependent on myeloid differentiation factor 88 in mice. The Journal of Biological Chemistry. 2004;279(47):49315–49322. doi: 10.1074/jbc.M402111200. [DOI] [PubMed] [Google Scholar]
- 64.Ramphal R, Balloy V, Huerre M, Si-Tahar M, Chignard M. TLRs 2 and 4 are not involved in hypersusceptibility to acute Pseudomonas aeruginosa lung infections. Journal of Immunology. 2005;175(6):3927–3934. doi: 10.4049/jimmunol.175.6.3927. [DOI] [PubMed] [Google Scholar]
- 65.Skerrett SJ, Liggitt HD, Hajjar AM, Wilson CB. Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. Journal of Immunology. 2004;172(6):3377–3381. doi: 10.4049/jimmunol.172.6.3377. [DOI] [PubMed] [Google Scholar]
- 66.Travassos LH, Carneiro LA, Girardin SE, et al. Nod1 participates in the innate immune response to Pseudomonas aeruginosa. The Journal of Biological Chemistry. 2005;280(44):36714–36718. doi: 10.1074/jbc.M501649200. [DOI] [PubMed] [Google Scholar]
- 67.Franchi L, Amer A, Body-Malapel M, et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in Salmonella-infected macrophages. Nature Immunology. 2006;7(6):576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 68.Miao EA, Andersen-Nissen E, Warren SE, Aderem A. TLR5 and Ipaf: dual sensors of bacterial flagellin in the innate immune system. Seminars in Immunopathology. 2007;29(3):275–288. doi: 10.1007/s00281-007-0078-z. [DOI] [PubMed] [Google Scholar]
- 69.Miao EA, Ernst RK, Dors M, Mao DP, Aderem A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(7):2562–2567. doi: 10.1073/pnas.0712183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. Journal of Experimental Medicine. 2007;204(13):3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miao EA, Mao DP, Yudkovsky N, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mahenthiralingam E, Campbell ME, Speert DP. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infection and Immunity. 1994;62(2):596–605. doi: 10.1128/iai.62.2.596-605.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin DW, Schurr MJ, Mudd MH, Govan JRW, Holloway BW, Deretic V. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(18):8377–8381. doi: 10.1073/pnas.90.18.8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bragonzi A, Wiehlmann L, Klockgether J, et al. Sequence diversity of the mucABD locus in Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Microbiology. 2006;152(11):3261–3269. doi: 10.1099/mic.0.29175-0. [DOI] [PubMed] [Google Scholar]
- 75.D’Argenio DA, Wu M, Hoffman LR, et al. Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Molecular Microbiology. 2007;64(2):512–533. doi: 10.1111/j.1365-2958.2007.05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hoffman L, Kulasekara HD, Emerson J, et al. Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. Journal of Cystic Fibrosis. 2009;8(1):66–70. doi: 10.1016/j.jcf.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Burns JL, Gibson RL, McNamara S, et al. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. Journal of Infectious Diseases. 2001;183(3):444–452. doi: 10.1086/318075. [DOI] [PubMed] [Google Scholar]
- 78.Ciofu O, Riis B, Pressler T, Poulsen HE, Høiby N. Occurrence of hypermutable Pseudomonas aeruginosa in cystic fibrosis patients is associated with the oxidative stress caused by chronic lung inflammation. Antimicrobial Agents & Chemotherapy. 2005;49(6):2276–2282. doi: 10.1128/AAC.49.6.2276-2282.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mathee K, Ciofu O, Sternberg C, et al. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology. 1999;145(6):1349–1357. doi: 10.1099/13500872-145-6-1349. [DOI] [PubMed] [Google Scholar]
- 80.Bragonzi A, Worlitzsch D, Pier GB, et al. Nonmucoid Pseudomonas aeruginosa expresses alginate in the lungs of patients with cystic fibrosis and in a mouse model. Journal of Infectious Diseases. 2005;192(3):410–419. doi: 10.1086/431516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sokurenko EV, Hasty DL, Dykhuizen DE. Pathoadaptive mutations: gene loss and variation in bacterial pathogens. Trends in Microbiology. 1999;7(5):191–195. doi: 10.1016/s0966-842x(99)01493-6. [DOI] [PubMed] [Google Scholar]
- 82.Jain M, Ramirez D, Seshadri R, et al. Type III secretion phenotypes of Pseudomonas aeruginosa strains change during infection of individuals with cystic fibrosis. Journal of Clinical Microbiology. 2004;42(11):5229–5237. doi: 10.1128/JCM.42.11.5229-5237.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oliver A, Cantón R, Campo P, Baquero F, Blàzquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288(5469):1251–1253. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- 84.Young D, Hussell T, Dougan G. Chronic bacterial infections: living with unwanted guests. Nature Immunology. 2002;3(11):1026–1032. doi: 10.1038/ni1102-1026. [DOI] [PubMed] [Google Scholar]
- 85.Sokurenko EV, Chesnokova V, Dykhuizen DE, et al. Pathogenic adaptation of Escherichia coli by natural variation of the FimH adhesin. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(15):8922–8926. doi: 10.1073/pnas.95.15.8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moxon ER, Murphy PA. Haemophilus influenzae bacteremia and meningitis resulting from survival of a single organism. Proceedings of the National Academy of Sciences of the United States of America. 1978;75(3):1534–1536. doi: 10.1073/pnas.75.3.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Akopyants NS, Eaton KA, Berg DE. Adaptive mutation and cocolonization during Helicobacter pylori infection of gnotobiotic piglets. Infection & Immunity. 1995;63(1):116–121. doi: 10.1128/iai.63.1.116-121.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Luzar MA, Thomassen MJ, Montie TC. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: relationship to patient clinical condition. Infection & Immunity. 1985;50(2):577–582. doi: 10.1128/iai.50.2.577-582.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ernst RK, Moskowitz SM, Emerson JC, et al. Unique lipid A modifications in Pseudomonas aeruginosa isolated from the airways of patients with cystic fibrosis. Journal of Infectious Diseases. 2007;196(7):1088–1092. doi: 10.1086/521367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hajjar AM, Ernst RK, Tsai JH, Wilson CB, Miller SI. Human toll-like receptor 4 recognizes host-specific LPS modifications. Nature Immunology. 2002;3(4):354–359. doi: 10.1038/ni777. [DOI] [PubMed] [Google Scholar]
- 91.Simpson JA, Smith SE, Dean RT. Alginate inhibition of the uptake of Pseudomonas aeruginosa by macrophages. Journal of General Microbiology. 1988;134(1):29–36. doi: 10.1099/00221287-134-1-29. [DOI] [PubMed] [Google Scholar]
- 92.Simpson JA, Smith SE, Dean RT. Alginate may accumulate in cystic fibrosis lung because the enzymatic and free radical capacities of phagocytic cells are inadequate for its degradation. Biochemistry & Molecular Biology International. 1993;30(6):1021–1034. [PubMed] [Google Scholar]
- 93.Høiby N, Krogh Johansen H, Moser C, Song Z, Ciofu O, Kharazmi A. Pseudomonas aeruginosa and the in vitro and in vivo biofilm mode of growth. Microbes & Infection. 2001;3(1):23–35. doi: 10.1016/s1286-4579(00)01349-6. [DOI] [PubMed] [Google Scholar]
- 94.Cobb LM, Mychaleckyj JC, Wozniak DJ, López-Boado YS. Pseudomonas aeruginosa flagellin and alginate elicit very distinct gene expression patterns in airway epithelial cells: implications for cystic fibrosis disease. Journal of Immunology. 2004;173(9):5659–5670. doi: 10.4049/jimmunol.173.9.5659. [DOI] [PubMed] [Google Scholar]
- 95.Govan JR, Fyfe JA, Baker NR. Heterogeneity and reduction in pulmonary clearance of mucoid Pseudomonas aeruginosa. Reviews of Infectious Diseases. 1983;5:S874–S879. doi: 10.1093/clinids/5.supplement_5.s874. [DOI] [PubMed] [Google Scholar]
- 96.Boucher JC, Yu H, Mudd MH, Deretic V. Mucoid Pseudomonas aeruginosa in cystic fibrosis: characterization of muc mutations in clinical isolates and analysis of clearance in a mouse model of respiratory infection. Infection & Immunity. 1997;65(9):3838–3846. doi: 10.1128/iai.65.9.3838-3846.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jain M, Ramirez D, Seshadri R, et al. Type III secretion phenotypes of Pseudomonas aeruginosa strains change during infection of individuals with cystic fibrosis. Journal of Clinical Microbiology. 2004;42(11):5229–5237. doi: 10.1128/JCM.42.11.5229-5237.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fung C, Naughton S, Turnbull L, et al. Gene expression of Pseudomonas aeruginosa in a mucin-containing synthetic growth medium mimicking cystic fibrosis lung sputum. Journal of Medical Microbiology. 2010;59(9):1089–1100. doi: 10.1099/jmm.0.019984-0. [DOI] [PubMed] [Google Scholar]
- 99.Jones AK, Fulcher NB, Balzer GJ. Activation of the Pseudomonas aeruginosa AlgU regulon through mucA mutation inhibits cyclic AMP/Vfr signaling. The Journal of Bacteriology. 2010;192(21):5709–5717. doi: 10.1128/JB.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]