

Abstract
The molecular circadian clock consists of a feedback loop in which canonical clock proteins negatively regulate transcription of their own genes. Timed nuclear entry of these proteins is critical, but regulation of this event is poorly understood. In Drosophila melanogaster, the idea that nuclear entry of PERIOD (PER) is controlled by its partner protein TIMELESS (TIM) has been challenged by several studies. We identify here a novel mutation in the tim gene that eliminates behavioral rhythms while allowing robust expression of TIM and PER. Mutant TIM can bind to and stabilize PER. However, neither protein is expressed cyclically, and phosphorylation of both is reduced. In addition, TIM and PER are localized in the cytoplasm at all times of day, and mutant TIM attenuates transcriptional feedback by PER in cultured cells, suggesting that it holds PER in the cytoplasm. In fact, much of the reduced phosphorylation of PER in the new tim mutant appears to result from the cytoplasmic localization of PER. Interestingly, mutating a threonine near the original mutation produces similar phenotypes, raising the possibility that defective phosphorylation is the basis of TIM dysfunction in the novel tim mutant. We also show that a stable form of PER is cytoplasmic in tim-null flies. These studies establish an essential role of TIM in the timed nuclear entry of PER.
Introduction
The basic circadian clock mechanism consists of molecular feedback loops in which clock proteins negatively regulate expression of their own genes in a circadian fashion. Despite the wide acceptance of this model, major questions about clock mechanisms remain unanswered (Zheng and Sehgal, 2008). For instance, we still do not know how these loops are sustained and, additionally, how they generate a period of ∼24 h.
Maintenance of these feedback loops must require tight control over levels/activity of relevant proteins and also accurate timing of events within the cycle. In Drosophila, levels/activity of the period protein (PER) are critical for the determination of circadian period, and nuclear entry of PER occurs at a specific time of day (Smith and Konopka, 1982; Curtin et al., 1995; Ashmore et al., 2003). Within the nucleus, PER represses the activity of the transcriptional activators, CLOCK (CLK) and CYCLE (CYC) (Hardin, 2005). The expression of Drosophila per is coregulated with that of the timeless (tim) gene, and TIM is required for PER stability and, thereby, its cyclic expression (Zheng and Sehgal, 2008). TIM was also thought to be required for the nuclear localization of PER (Vosshall et al., 1994), but several studies have challenged this notion.
Since PER is unstable in the absence of TIM (Price et al., 1995), effects of a tim-null mutant on the localization of PER were based upon analysis of a PER–β-galactosidase fusion protein, which was nuclear in wild-type flies and cytoplasmic in tim mutants (Vosshall et al., 1994). A subsequent study using transfected cells supported the idea that the two proteins require each other for nuclear entry (Saez and Young, 1996). However, other experiments using the same cultured cells demonstrated that PER can repress transcription by itself, indicating that it can enter the nucleus independently (Ashmore et al., 2003; Chang and Reppert, 2003; Nawathean and Rosbash, 2004). In addition, PER enters the nucleus before TIM in Drosophila clock cells (Shafer et al., 2002). Moreover, when PER was stabilized in tim-null flies, through reduction of the destabilizing PER kinase DOUBLE-TIME (DBT), it was in the nucleus (Cyran et al., 2005). While this may reflect TIM relieving the cytoplasm-anchoring effects of DBT, other studies do not support such an effect of DBT (Bao et al., 2001; Nawathean and Rosbash, 2004).
We report here a new allele of tim, which renders flies arrhythmic and yet expresses high levels of TIM protein. PER is also expressed at high levels and is hypophosphorylated. Robust expression of both proteins allowed us to address the role of TIM in the regulation of PER. Through cell culture experiments and in vivo fly data, we show that TIM is required for the nuclear entry of PER. The effects of the new tim mutation are recapitulated by mutating a nearby threonine, raising the intriguing possibility that disruption of TIM phosphorylation underlies the mutant phenotypes. Based upon these findings, we propose a modified model for the regulation of the nuclear entry of PER.
Materials and Methods
Drosophila genetics.
We identified an arrhythmic fly line (2611) from the ethyl methanesulfonate-mutagenized Zuker collection (Koundakjian et al., 2004), as previously described (Wu et al., 2008). The new mutant did not complement tim01 for arrhythmicity, suggesting that the mutation lies in the tim locus. Sequencing of the tim coding region revealed a single-base mutation that results in a proline to leucine (PL) at position 115. Therefore, we named the new allele timPL. We outcrossed the mutant line to a w1118 background (iso31) for five generations.
Analysis of circadian behavior.
Male flies were entrained to 12 h light/dark cycles at 25°C for at least 3 d. Locomotor activity of 1- to 7-d-old adult males was monitored and analyzed as described previously (Williams et al., 2001).
Antibodies and reagents.
PA1139 (rabbit) and PA1140 (guinea pig) antibodies to PER were made against amino acids 1–240 of PER. Antibodies to TIM (UPR8) (Hunter-Ensor et al., 1996), PER (UPR34), and JETLAG (JET; UP971) (Koh et al., 2006) were described previously. Antibodies to MYC (Roche) and MAPK (Sigma) were purchased. MG132, AR-A014418 (AR), okadaic acid (OA), and tautomycin (TTM) were purchased from Calbiochem. Leptomycin B (LMB) was purchased from Sigma-Aldrich.
Expression constructs.
The cDNA encoding short-tim (s-tim) was cloned into the expression vector pIZ/V5-His (Invitrogen). s-timPL or s-timT113A (s-timTA) was generated by site-directed mutagenesis with the Quick Change mutagenesis kit (Stratagene). pCaspeR-hs-per-CFP and pIZ-tim-YFP were gifts from Dr. Michael Young (Rockefeller University, New York, NY) (Meyer et al., 2006). PCR-amplified tim-yellow fluorescent protein (YFP) was cloned into the NotI site of pCaspeR-hs vector.
Transient transfection and immunoprecipitation assays.
Drosophila S2R+ cells were cultured in a standard Schneider medium. Drosophila S2 cells were cultured in M3 media. Cells were transfected with the Effectene kit (Qiagen) according to the manufacturer's protocol. For immunoprecipitation (IP) assays, 48 h after transfection cells were collected and lysed in lysis buffer (50 mm Tris-HCl at pH 7.5, 150 mm NaCl, 1 mm EDTA, 0.5% Triton X-100, and protease inhibitors; Roche). Cell lysates were incubated with guinea pig anti-PER antibody (PA1140) and Protein G Dynabeads (Invitrogen) in lysis buffer for 2 h at 4°C.
Western blot analysis.
Flies were entrained to a 12 h light/dark cycle for at least 3 d and collected on dry ice at different time points. For examining light-dependent TIM degradation, flies were exposed to a 60 min light pulse, and S2 cells were exposed to a 30 min light pulse. Western blots of fly head extracts and cell lysates were performed as described previously (Sathyanarayanan et al., 2004). Antibodies to TIM (UPR8), PER (PA1139), and JET (UP971) were used at 1:1000, and those to MAPK at 1:5000.
Generation of transgenic strains.
The UAS–tim constructs were generated by inserting PCR-amplified wild-type s-tim or s-timTA coding region into the pUAST-attB vector. The UAS–tim constructs were targeted to the identical 86F8 insertion site via the phiC31–integrase system (Venken et al., 2006) using a standard embryo injection technique (Rainbow Transgenic Flies).
Immunohistochemistry.
Three- to seven-day-old flies entrained to 12 h light/dark cycles for at least 3 d were collected at different time points, and immunostaining of whole-mount brain samples was performed as described previously (Koh et al., 2006). Samples were incubated with antibodies to PER (UPR34) or TIM (UPR8) at 1: 2000 or 1:1500, respectively. Samples were costained with antibody to Pigment Dispensing Factor (PDF; HH74) at 1:1000 to facilitate identification of ventral lateral neurons (LNvs). FITC- or Cy3-conjugated secondary antibodies (Jackson ImmunoResearch) were used at 1:1500. Six to ten fly brains were examined per condition. Immunostained samples were imaged with a Leica TCS-SP5 confocal microscope.
Scoring of subcellular localization.
Subcellular localization of PER and TIM in fly brains as well as in S2 cells was scored without knowledge of the identity of the samples. Large LNvs were scored as predominantly nuclear, predominantly cytoplasmic, or both nuclear and cytoplasmic, using PDF as a cytoplasmic marker. For scoring of S2 cells, only cells with detectable PER and TIM expression were included. S2 cells were scored nuclear if stronger nuclear expression was detected surrounded by weaker cytoplasmic expression, and cytoplasmic if the opposite was the case. Cells where expression was essentially uniform except in the nucleolus were scored as both nuclear and cytoplasmic.
Cell-based transcription assays.
Plasmid DNA (10 ng of pAc-dClk, 50 ng of per-E-box-luciferease (luc), 10 ng of pIZ-tim, 10 ng of pAc-per, and 50 ng of pAc-Rluc) was transfected into S2R+ cells in 96 well plates. Thirty-six hours after transfection, cells were harvested, and luciferase activity was measured in a Victor 3 plate reader (PerkinElmer) by using Stop & Glow reagents (Promega). Luc/R-luc readings were normalized to the full activation seen in the dClk-alone sample (set as 100).
Results
Identification of a new allele of tim
In the course of a genetic screen for mutants affecting sleep, we identified a number of novel circadian mutations (Wu et al., 2008). Although the sleep screen was conducted in the presence of light/dark cycles, which is not how a typical circadian screen would be conducted, we were able to detect circadian defects in several lines based upon their lack of anticipation of light/dark transitions. One such line turned out to be arrhythmic in constant darkness (DD) and carried a mutation, a proline-to-leucine substitution at position 115, in the tim gene. We outcrossed the mutation, termed timPL, several times into an isogenized w118 (Iso31) background and then rechecked the phenotype. As initially observed, timPL homozygotes are arrhythmic in DD (Fig. 1A; Table 1), and the timPL allele does not complement a null allele of tim (tim01). Surprisingly, given that the tim-null mutation does not have a dominant phenotype, the timPL heterozygotes show a longer period (Fig. 1A; Table 1). Thus, this tim allele may interfere, or compete, with the activity of wild-type TIM.
Figure 1.
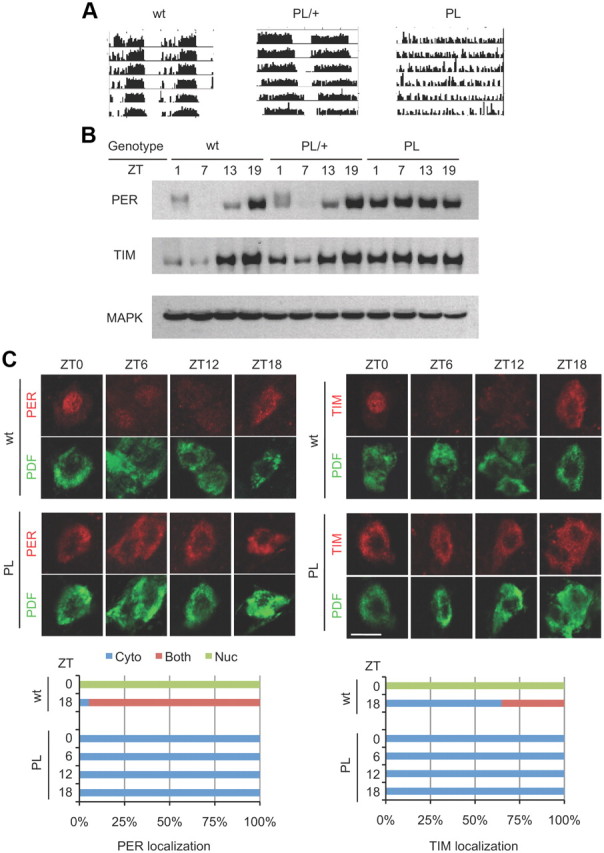
The timPL mutation has dramatic effects on behavioral rhythms and on the post-translational regulation of TIM and PER. A, Wild-type flies, timPL mutants, and flies heterozygous for the PL mutation were assayed for rest/activity rhythms in constant darkness. Representative activity records of the different genotypes are shown. Quantification of the free-running rhythm phenotypes is presented in Table 1. B, Western blots of adult fly heads of wild-type, heterozygous, and homozygous timPL mutants were probed for the expression of PER and TIM. The blots were also probed with antibody to MAPK as a loading control. Similar results were obtained in three independent experiments. C, Cytoplasmic expression of PER and TIM in large LNvs of timPL flies. Wild-type and timPL mutants were collected at the times shown, and whole-mount brains were stained with anti-PER and anti-TIM antibodies. Scale bar, 10 μm. PDF signal was used to mark the cytoplasm of large LNvs. Quantification shows PER and TIM subcellular localization, scored as nuclear (green), cytoplasmic (blue), or both nuclear and cytoplasmic (red). Control ZT6 and ZT12 were not scored because the low signal intensity made it difficult to determine subcellular localization. At least 15 cells were counted for each genotype and time point.
Table 1.
Free-running circadian locomotor rhythm phenotypes of tim mutants
| Genotype | % Rhythmic | n | Period ± SEM (h) |
|---|---|---|---|
| tim+/+ | 100 | 32 | 23.56 ± 0.05 |
| timPL/+ | 98.4 | 62 | 25.01 ± 0.04 |
| timPL/PL | 3.2 | 62 | 31.75 ± 0.75 |
| tim01/+ | 100 | 32 | 23.51 ± 0.03 |
| tim01/PL | 0 | 32 | NA |
NA, Not applicable.
Molecular oscillations and nuclear expression of PER and TIM are abrogated in timPL flies
The previously identified arrhythmic allele of tim (tim01) contains a small deletion in the coding region (Sehgal et al., 1994). Thus, it does not produce full-length TIM and is a functional null. In tim01 flies, PER is expressed at very low levels, which led to the conclusion that TIM stabilizes PER (Price et al., 1995). We found that timPL flies express high levels of TIM and PER (Fig. 1B). However, neither is expressed cyclically, and phosphorylation-induced mobility shifts of PER at specific times of day are not observed (Fig. 1B). Since low-mobility forms are conspicuously lacking, we infer that PER is hypophosphorylated in timPL flies.
As mentioned earlier, an effect of TIM on nuclear localization of PER was based upon the expression of a PER-βGAL fusion protein in tim01 flies (Vosshall et al., 1994). However, this fusion protein contains only the N-terminal half of PER and is nonfunctional. Stable expression of TIM and PER in timPL flies allowed us to ask whether the nuclear expression of the two proteins is affected. As shown in Figure 1C, in wild-type flies PER is nuclear at zeitgeber time 0 (ZT0) and is cytoplasmic at ZT18. At ZT6 and ZT12, levels are low, but nuclear expression is discernable at ZT6. In the timPL mutant, it is cytoplasmic at all times. Likewise, TIM in wild-type flies is nuclear at ZT0 and cytoplasmic at other times (TIM disappears from the nucleus earlier than PER in the early morning), but in timPL flies it is detected in the cytoplasm at all times (Fig. 1C).
timPL and a mutation in a neighboring threonine block TIM import into the nucleus and light-induced TIM degradation
As noted above, the PL mutation affects a proline residue, and so it could affect secondary structure of the protein or disrupt a specific post-translational modification. Since phosphorylation is the dominant, and perhaps most important, post-translational modification undergone by clock proteins, we examined the sequence around the PL mutation for possible phosphorylation sites. A candidate was a threonine at position 113, which is highly conserved in insects. We mutated this threonine to alanine and compared properties of the resulting protein, TIM-TA, with those of the original mutant, TIM-PL.
In Drosophila cultured S2 cells, TIM is usually detected in the cytoplasm. However, it shuttles in and out of the nucleus and so can be held in the nucleus if nuclear export is inhibited by leptomycin (Ashmore et al., 2003). We found that neither TIM-TA nor TIM-PL showed significant nuclear expression when cells were treated with leptomycin (Fig. 2A). Thus, both proteins are defective in nuclear expression and the defect is in nuclear import rather than accelerated nuclear export.
Figure 2.

The timPL mutation and a mutation in a neighboring threonine prevent nuclear localization and light-induced degradation of TIM. A, S2R+ cells were transiently transfected with wild-type tim (wt), timPL (PL), or tim carrying a mutation in a neighboring threonine tim (TA). All tim constructs were tagged with YFP. Cells were treated with 10 nm LMB for 2 h, and then fixed and examined by fluorescence confocal microscopy. Scale bar, 25 μm. The distribution of cells showing mostly nuclear, mostly cytoplasmic, or both nuclear and cytoplasmic TIM is indicated. Two additional independent experiments showed similar results. B, Light-induced degradation of TIM is reduced in timPL mutants. Flies were collected at ZT20 with or without a 1 h light exposure, and fly head extracts were examined by Western blot analysis. C, Light-induced TIM degradation is inhibited by the PL (left) and TA (right) mutations. S2 cells were transiently transfected with pAc-tim along with MYC epitope-tagged pIZ-cry and FLAG epitope-tagged pIZ-jet, with or without 10 nm LMB. MAPK was probed to control for loading. Two other identical independent experiments showed similar results.
In response to light, wild-type TIM is degraded and mediates resetting of the circadian clock (Ashmore and Sehgal, 2003). The circadian expression profile of TIM-PL in flies suggested that light-induced degradation is impaired because the protein remains high during daylight hours. Indeed, examination of fly head extracts after a 1 h light pulse during the dark period showed that TIM was not degraded by light in timPL mutants (Fig. 2B). Light-induced degradation can be reconstituted in cultured cells if TIM is cotransfected with the circadian photoreceptor cryptochrome (CRY) and the E3 ligase JET (Koh et al., 2006). As shown in Figure 2C, TIM-PL and TIM-TA fail to be degraded in response to light in S2 cells. To address whether the defect in light-induced degradation is a consequence of the altered subcellular localization, we also determined whether TIM is preferentially degraded in the nucleus. Thus, we assayed TIM degradation by light in cells treated with leptomycin. Wild-type TIM degraded equally well in the presence and absence of leptomycin, and neither of the two mutant TIM proteins was degraded in either condition, indicating that subcellular localization does not affect the efficacy of light-induced degradation (Fig. 2C). Notably, CRY was degraded in the presence of TIM mutants, indicating that CRY degradation in response to light does not require TIM degradation. This is consistent with previous work showing that CRY can be degraded in the absence of TIM (Koh et al., 2006).
Together, these experiments demonstrate that both TIM-TA and TIM-PL are impaired in nuclear import and light-induced degradation, although these two effects are not necessarily linked. Given that the major defects observed with TIM-PL are recapitulated by mutating a nearby threonine, we speculate that disruption of a specific post-translational modification accounts for the timPL phenotype. However, Thr113 is not a known consensus site for any kinase, and it has yet to be determined whether the residue is phosphorylated.
The timTA mutation introduced into flies mimics the timPL mutation
To further examine the role of the putatively phosphorylated threonine at position 113 in producing the behavioral phenotypes of timPL mutants, we introduced the TIM-TA protein into flies by generating a UAS transgene. As previously observed (Yang and Sehgal, 2001), overexpression of wild-type TIM in a wild-type background by a clock cell GAL4 driver, tim-UAS-Gal4 (TUG), produced arrhythmia (Table 2). In contrast, overexpression of TIM-TA in a wild-type background lengthened circadian period to ∼26 h (Fig. 3; Table 2), similar to the semidominant phenotype seen in the timPL heterozygous flies (Fig. 1A). Since both wild-type and mutant TIM proteins are expressed in flies overexpressing TIM-TA as in heterozygous mutants, this finding suggests that TIM-TA can recapitulate a timPL behavioral phenotype. We infer that arrhythmia results only when the overexpressed TIM is fully functional, and that this is not the case for TIM-TA. Accordingly, expression of TIM-TA in tim01 flies produced little to no rescue of rhythms (2–11%; Table 2). In contrast, we obtained up to ∼50% rescue with a wild-type UAS–tim construct (24–48%). For the rescue experiments, we used the pan-neuronal driver, elav, since TUG-Gal4 was ineffective, perhaps because it overexpresses TIM. The relatively weak rescue by the wild-type tim transgene could, in part, be a function of the construct; we used the more light-sensitive form of TIM, s-TIM (Sandrelli et al., 2007), which has not been used before for rescue. In addition, we note that rescue of tim01 flies by a UAS–tim construct is typically not very efficient, with the maximal rescue being 70% in a previous study (Yang and Sehgal, 2001). This may be due to the difficulty of controlling TIM levels with the UAS–GAL4 system (see Discussion).
Table 2.
Free-running circadian locomotor rhythm phenotypes of flies expressing wild-type tim or timTA transgenes
| Genotype | % Rhythmic | n | Period ± SEM (h) |
|---|---|---|---|
| elavGal4/Y; tim01; UAS-tim(wt)#2/+ | 24.21 | 95 | 24.10 ± 0.121 |
| elavGal4/Y; tim01; UAS-tim(wt)#3/+ | 25.81 | 31 | 23.39 ± 0.218 |
| elavGal4/Y; tim01; UAS-tim(wt)#5/+ | 48.44 | 64 | 24.07 ± 0.135 |
| elavGal4/Y; tim01; UAS-tim(TA)#1/+ | 7.81 | 64 | 24.53 ± 0.123 |
| elavGal4/Y; tim01; UAS-tim(TA)#2/+ | 11.11 | 63 | 24.36 ± 0.186 |
| elavGal4/Y; tim01; UAS-tim(TA)#7/+ | 1.56 | 64 | 24.50 |
| elavGal4/Y; tim01 | 0 | 32 | NA |
| tim01; UAS-tim(wt)#2/+ | 3.13 | 32 | 23.92 |
| tim01; UAS-tim(wt)#3/+ | 0 | 16 | NA |
| tim01; UAS-tim(wt)#5/+ | 9.38 | 32 | 23.67 ± 0.125 |
| tim01; UAS-tim(TA)#1/+ | 0 | 32 | NA |
| tim01; UAS-tim(TA)#2/+ | 0 | 32 | NA |
| tim01; UAS-tim(TA)#7/+ | 0 | 32 | NA |
| UAS-tim(wt)#2 | 100 | 15 | 24.02 ± 0.078 |
| UAS-tim(wt)#3 | 92 | 13 | 24.2 ± 0.114 |
| UAS-tim(wt)#5 | 100 | 8 | 24.14 ± 0.08 |
| TUG;UAS-tim(wt)#2 | 0 | 15 | NA |
| TUG;UAS-tim(wt)#3 | 0 | 15 | NA |
| TUG;UAS-tim(wt)#5 | 0 | 8 | NA |
| UAS-tim(TA)#1 | 100 | 16 | 24.24 ± 0.120 |
| UAS-tim(TA)#7 | 100 | 16 | 23.96 ± 0.079 |
| TUG;UAS-tim(TA)#1 | 94 | 16 | 25.9 ± 0.167 |
| TUG;UAS-tim(TA)#7 | 93 | 15 | 26.12 ± 0.107 |
| tim01,TUG/tim01; UAS-tim(wt)#2/+ | 0 | 10 | NA |
| tim01,TUG/tim01; UAS-tim(wt)#3/+ | 0 | 15 | NA |
| tim01,TUG/tim01; UAS-tim(TA)#1/+ | 0 | 23 | NA |
| tim01,TUG/tim01; UAS-tim(TA)#7/+ | 0 | 23 | NA |
NA, Not applicable.
Figure 3.

Effects of TIM-TA on activity rhythms in wild-type and tim mutant flies. Top, Activity rhythms of wild-type flies overexpressing either a wt or a TIM-TA transgene (TA) under control of the TUG-Gal4 driver. Bottom, Activity records of tim01 mutant flies carrying UAS-tim (wt) or UAS-tim (TA) under control of an elav-Gal4 driver.
Mutant TIM proteins affect nuclear entry of and negative feedback by PER
Like TIM, PER is detected only in the cytoplasm when expressed by itself in S2 cells. When the two proteins are expressed together, they are found in the nucleus after a delay of several hours (Meyer et al., 2006). To determine whether TIM-PL affects the nuclear entry of PER in S2 cells, as it does in flies, we expressed both proteins together in S2 cells. Both genes were tagged with fluorescent reporters (TIM-YFP and PER-GFP) under the control of a heat-shock promoter. Eight hours after expression was induced, we detected wild-type TIM and PER in the nuclei of many cells (Fig. 4A). However, TIM-PL and PER remained cytoplasmic. Similarly, when PER is coexpressed with TIM-TA, both proteins remain cytoplasmic (Fig. 4A). Thus, neither TIM-PL nor TIM-TA promotes nuclear expression of PER, as does wild-type TIM. We note that previously we did not observe nuclear expression of PER and wild-type TIM when they were coexpressed in S2 cells (Ashmore et al., 2003). The longer induction time (8 h) in the present study compared with the shorter induction time (2 h) in the previous study may account for the difference. This interpretation is supported by the work of Meyer et al. (2006), who reported that TIM and PER enter the nucleus ∼6 h after induction.
Figure 4.
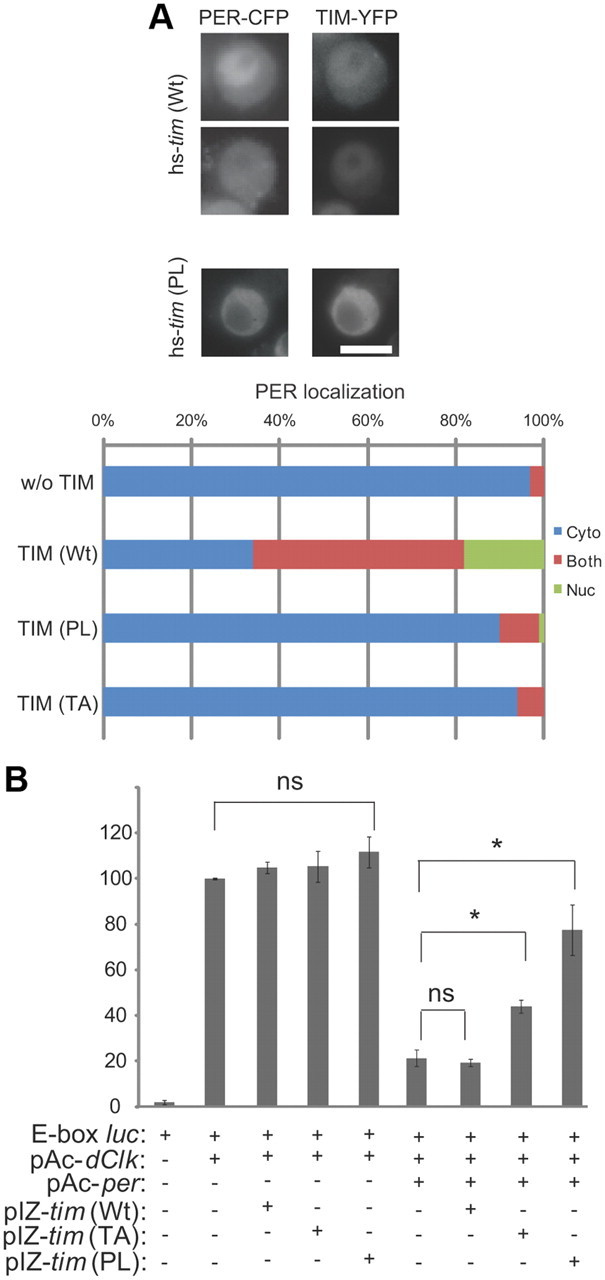
TIM mutants affect nuclear expression of and feedback repression by PER. A, Mutant TIM proteins anchor PER in the cytoplasm. S2 cells transfected with hs-YFP tagged wild-type tim (wt) or mutant tim (PL) and hs-CFP tagged per were fixed 8 h after heat shock induction of TIM and PER. Scale bar, 10 μm. Quantification shows PER localization in TIM-expressing cells, scored as mostly nuclear (green), mostly cytoplasmic (blue), or both nuclear and cytoplasmic (red). The top and bottom panels of the hs-tim(WT) images show nuclear PER and PER in both the nucleus and cytoplasm, respectively. At least 100 cells were counted for each condition in three independent experiments. B, TIM mutants inhibit feedback repression by PER on CLK-CYC-mediated transcription. Drosophila S2 cells were transiently transfected with two reporter genes (E-box-luc and actin-Rluc), and various combinations of pAc-dClk, wt, or mutant (TA or PL) pIZ-s-tim, and pAc-per, as indicated. After 48 h, cells were harvested and assayed for luciferase. Data were normalized to Rluc expression to control for transfection efficiency. Results are presented as the percentage of full activation seen in the dClk alone condition and expressed as averages of three independent experiments. Error bars indicate SEM. *p < 0.05 by t test.
Although PER is not detected in the nucleus when expressed by itself in S2 cells, significant amounts are clearly transported in because it can repress CLK-mediated transcription of a per promoter-luciferase reporter (Ashmore et al., 2003; Chang and Reppert, 2003; Nawathean and Rosbash, 2004). We found that the addition of TIM carrying either the proline or the threonine mutation significantly suppressed feedback by PER (Fig. 4B). We speculate that this is because the mutant TIM proteins hold PER in the cytoplasm. Co-IP experiments confirmed that TIM-PL and TIM-TA bind robustly to PER (Fig. 5). However, the co-IPs did not permit quantification of the PER–TIM interaction with enough precision to determine whether the mutant TIM proteins bind PER more tightly than the wild-type TIM protein does.
Figure 5.

Mutant TIM proteins bind effectively to PER. S2 cells were transiently transfected with pAc-tim (wt), tim (PL), or tim (TA) in the presence (+) or absence (−) of pAc-per-HA. After 48 h, cells were subjected to IP using antibodies against PER. Two other identical independent experiments showed similar results.
timPL and timTA affect the phosphorylation of sites recognized by protein phosphatases
Since the TA mutation suggested a defect in TIM phosphorylation, we examined the effects of proline and threonine mutations on phosphorylation. Both PER and TIM display a 24 h cycle in their phosphorylation profiles, which appears to be regulated, at least in part, through rhythmic dephosphorylation (Sathyanarayanan et al., 2004; Fang et al., 2007). Whereas dephosphorylation by protein phosphatase 1 (PP1) largely determines protein stability, PP2A also has an effect on nuclear entry. We used TTM to block activity of PP1 and OA to block phosphatase activity in general (Dombradi et al., 2004), and we examined the phosphorylated states of wild-type and mutant proteins.
As reported previously (Sathyanarayanan et al., 2004), treatment with OA in cultured cells produced a substantial shift in the mobility of wild-type TIM (Fig. 6A). It also shifted the mobility of TIM-PL and TIM-TA proteins, but to a much lesser extent, suggesting that these proteins are resistant to one or more phosphorylation events regulated by PP2A and other protein phosphatases. TTM did not produce any shift in the mutant TIM proteins, but since the shift in wild-type TIM is also very small (Fang et al., 2007), differences were barely discernable (Fig. 6B). TTM did, however, reduce expression of mutant TIM proteins as it did of wild-type TIM, indicating that mutant TIM is sensitive to the destabilizing effects of PP1 inhibition.
Figure 6.
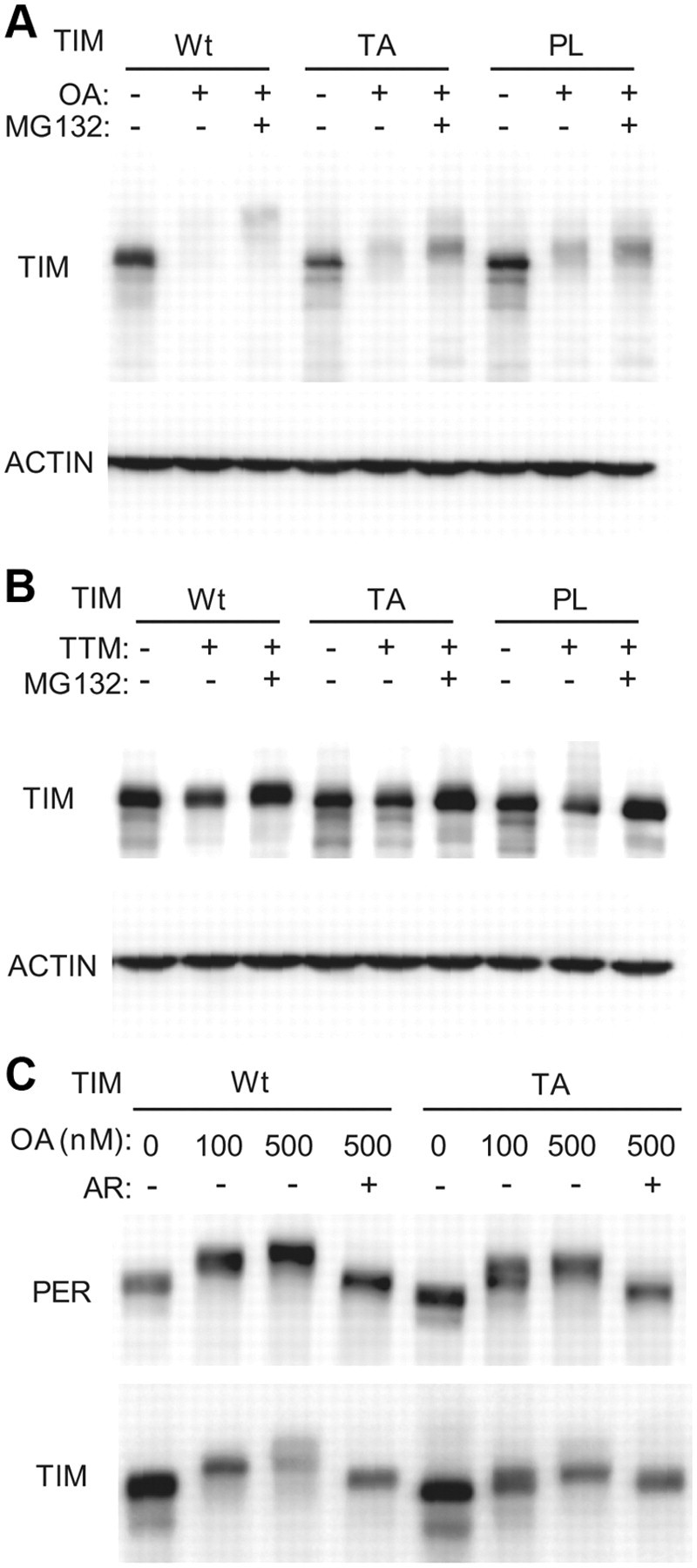
Mutant TIM proteins affect phosphorylation of TIM and PER. A, Mutant TIMs affect phosphorylation of sites recognized by PP2A. S2 cells were transiently transfected with wild-type tim or mutant tim constructs. Transfected cells were treated with PP2A inhibitor OA (500 nm), with or without proteasome inhibitor MG132 (10 μm) for 4 h. The ACTIN band is shown as a loading control. Similar results were obtained in three independent experiments. B, Effects of PP1 inhibition on phosphorylation of wild-type and mutant TIMs. S2 cells were transiently transfected with wild-type tim or mutant tim constructs. Transfected cells were treated with PP1 inhibitor TTM (4 mm), with or without proteasome inhibitor MG132 (10 μm). The ACTIN band is shown as a loading control. C, TIM mutants affect PER phosphorylation by GSK-3β. S2 cells were transfected with both per and wild-type tim (wt) or mutant tim (TA). Transfected cells were treated with varying concentrations of OA with proteasome inhibitor MG132 (10 μm), in some cases along with the GSK-3β inhibitor AR (10 μm) as indicated.
We also examined the effect of wild-type and the TIM-TA mutant on PER phosphorylation. As in the case of TIM, phosphorylation of PER through PP1 inhibition does not produce a detectable shift in protein mobility (Fang et al., 2007), and so we only looked at effects of general phosphatase inhibition. We showed previously that PER mobility in response to OA treatment in S2 cells is independent of TIM (Fang et al., 2007). We found, however, that the TA mutant did have a small effect on PER mobility such that low concentrations of OA (100 nm) resulted in a second PER band under the major shifted form. We infer that the TA mutant does not permit maximal phosphorylation of PER upon a weak block of phosphatase activity (with 100 nm OA). A stronger block of phosphatase activity (with 500 nm OA) produced a complete shift of PER mobility (Fig. 6C). These mobility shifts were abolished when a glycogen synthase kinase-3β (GSK-3β) inhibitor was added, suggesting that most of the phosphorylation is dependent on GSK-3β, although we cannot rule out the possibility that the drug has nonspecific effects on other kinases.
Hyperphosphorylated forms of PER are detectable predominantly in the nucleus
Western blots of fly head extracts indicated that PER is hypophosphorylated in timPL mutants (Fig. 1B). TIM-PL may have direct effects on PER phosphorylation in flies or its effects on PER could be indirect; namely, PER may be hypophosphorylated because it is not transported to the nucleus in timPL flies. To determine whether PER is phosphorylated preferentially in the nucleus, we added a strong nuclear localization signal (NLS), derived from the SV40 T antigen, to the per gene and expressed it in S2 cells. The NLS-tagged PER was expressed in the nucleus, while untagged PER was cytoplasmic (Fig. 7A). Western blot analysis of these PER proteins revealed low-mobility forms of the tagged PER that were not seen with untagged PER (Fig. 7B). To confirm that the low-mobility forms were due to phosphorylation and not to the addition of the small tag, we treated the lysates with phosphatase. Phosphatase treatment reduced the molecular weight of all forms, confirming that the mobility shifts were due to phosphorylation. Leptomycin did not have any effect on PER mobility, consistent with previous data indicating that it does not have a significant effect on the subcellular localization of PER (Ashmore et al., 2003). These data suggest that hyperphosphorylated forms of PER are detectable mostly in the nucleus, and so the gross PER phosphorylation phenotype of timPL mutants could be secondary to the defect in nuclear localization.
Figure 7.
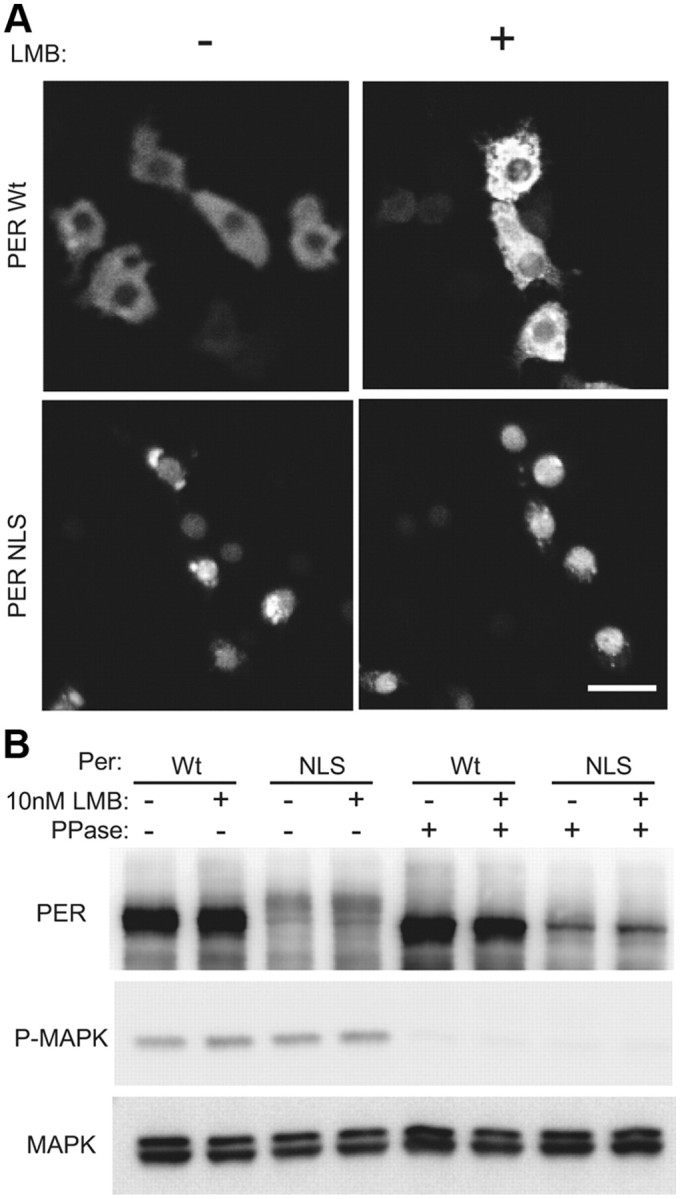
PER is phosphorylated preferentially in the nucleus. A, NLS-tagged PER (PER-NLS) is predominantly localized in the nucleus. S2R+ cells were transiently transfected with wild-type per or per tagged with NLS. Cells were fixed and stained with antibody to PER, and the localization of PER was examined by confocal microscopy. Scale bar, 20 μm. B, PER-NLS is hyperphosphorylated. S2R+ cells were transiently transfected with wild-type per or per tagged with NLS. Transfected cells were lysed with or without phosphatase. The resulting lysates were examined by immunoblot analysis using indicated antibodies. Some lysates were incubated with λ-phosphatase (New England Biolabs) for 30 min at 37°C, before gel analysis.
A stable form of PER is expressed in the cytoplasm in tim01 flies
The experiments above indicated that the major effect of the timPL mutation on PER is on its nuclear entry. At this point, the only experiments indicating that TIM actually promotes nuclear entry of PER were the coexpression studies in S2 cells (Saez and Young, 1996). However, S2 cells are not clock cells. Thus, we decided to use a recently described stable form of PER to address the subcellular localization of PER in tim01 flies. The PERS47A blocks a phosphorylation site and renders PER resistant to degradation via the E3 ligase, SLIMB (Grima et al., 2002; Ko et al., 2002; Chiu et al., 2008). We introduced a PERS47A construct into tim01 flies and examined its localization. In wild-type flies, this protein was found in the nucleus and the cytoplasm at both ZT1 and ZT20 (Fig. 8A). In tim01 mutants, however, it was clearly cytoplasmic at both times (Fig. 8A). These data appear to be in conflict with previous results showing that in cultured cells, expression of PER by itself is sufficient to produce repression of CLK-mediated transcription. The apparent discrepancy could be due to excessive overexpression of PER in S2 cells or different sensitivities of immunostaining versus luciferase assays. Regardless, our data show definitively that TIM is required for robust nuclear expression of PER in vivo.
Figure 8.
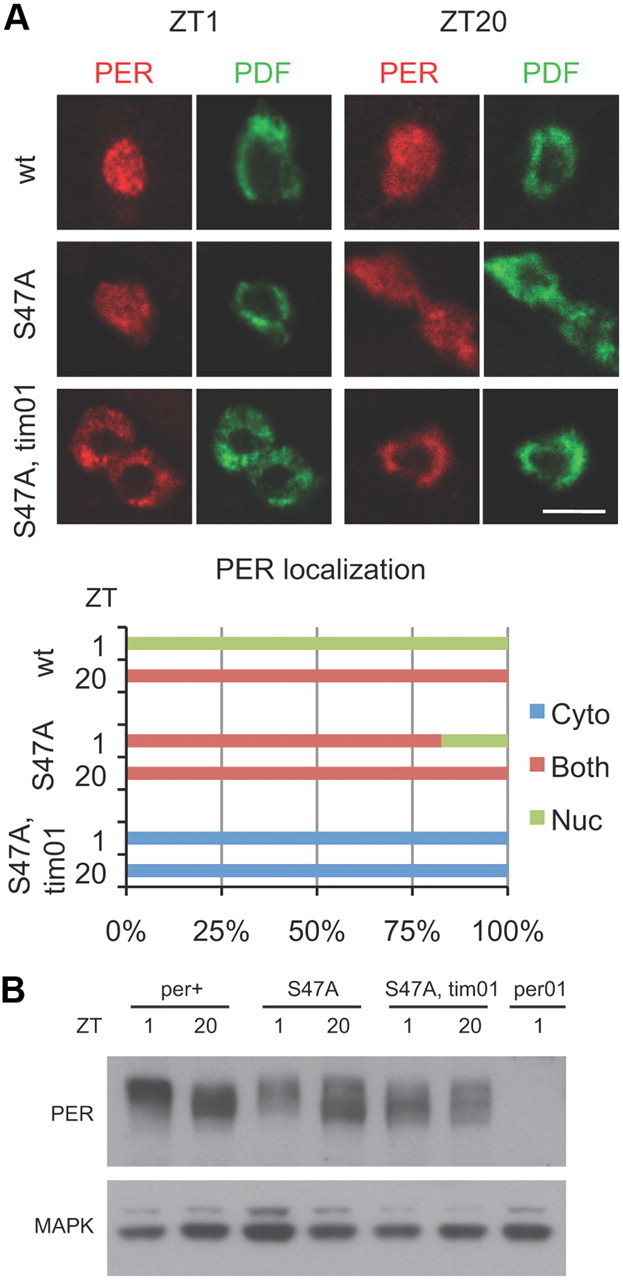
A stable form of PER is expressed in the cytoplasm in tim01 flies. A, Expression of PER in central clock neurons (large LNvs) of wild-type (top), perS47A (middle), or perS47A; tim01 (bottom) flies. Flies were collected at indicated times, and whole-mount brains were stained with antibodies to PER and PDF. PDF signal was used to identify the cytoplasm of large LNvs. Scale bar, 10 μm. Quantification shows PER subcellular localization, scored as nuclear (Nuc; green), cytoplasmic (Cyto; blue), or both nuclear and cytoplasmic (Both; red). At least 19 neurons were counted for each genotype and time point. B, Western blot analysis of head extracts from per0, wild-type, perS47A; tim+, and perS47A; tim01 flies. PERS47A is expressed at equivalent levels in tim+ and tim01 backgrounds, and it is hyperphosphorylated in tim01 flies. The MAPK band is shown as a loading control. Similar results were obtained in three independent experiments.
Our cell culture experiments using NLS-tagged PER showed that hyperphosphorylated PER is detectable mostly when PER is nuclear. This could be because most phosphorylation events occur in the nucleus or because hyperphosphorylated PER is stable in the nucleus, whereas it is degraded rapidly in the cytoplasm. To address this issue, we determined the phosphorylation status of PERS47A, which is cytoplasmic and stable in tim01 flies. Low-mobility forms of PERS47A were found in tim01 flies (Fig. 8B), which suggests that hyperphosphorylated wild-type PER is typically associated with nuclear expression because it is not stable in the cytoplasm. When PER is made stable via the S47A mutation, highly phosphorylated forms can be detected in the cytoplasm.
Discussion
The identification of an arrhythmic mutant of tim that expresses high levels of TIM and PER has provided us with a valuable tool to address outstanding questions regarding the regulation of these two critical clock molecules. Cell culture experiments indicate that the original proline mutation affects phosphorylation of TIM. This is supported by the similar phenotypes obtained when a neighboring threonine is mutated. A mass spectrometry analysis of TIM was uninformative because the region including the threonine was not covered (data not shown). Mutation of the threonine blocks nuclear entry of TIM and PER, affects phosphorylation of TIM in response to OA, inhibits transcriptional feedback by PER, lengthens the period when coexpressed with wild-type TIM, and is ineffective in rescuing rhythms in tim01 flies. Rescue by the wild-type tim construct was also relatively weak in these experiments. In part, this could be a result of using the s-TIM isoform (Sandrelli et al., 2007), which has not been used previously for rescue experiments. We note, however, that the UAS–GAL4 system has never produced full rescue of tim01 flies. We believe that the difficulty lies in the control of protein levels. Overexpression of TIM very readily produces arrhythmia, more so than does overexpression of PER (Yang and Sehgal, 2001). Given the stability of GAL4 and the amplification achieved with this system, TIM protein may easily increase to levels that disrupt the clock. The lack of rescue by the TUG driver may be caused by this same effect.
In a wild-type genetic background, the wild-type UAS–tim construct caused arrhythmia, as previously reported (Yang and Sehgal, 2001), but the threonine mutant produced long periods. Overexpression of wild-type TIM probably increases expression and negative feedback by PER, thereby jamming the system and preventing progression to the next point in the cycle. In contrast, TIM-TA probably competes with endogenous TIM for binding to PER, and the PER bound to TIM-TA is likely held in the cytoplasm. This would reduce the amount of PER available for feedback, and might even delay nuclear entry, resulting in a longer period.
The new PL allele affects PER on multiple levels. Its effects on nuclear entry and on phosphorylation prompted us to investigate the relationship between the subcellular localization and phosphorylation status of PER. We find that the bulk of highly phosphorylated PER is present in the nucleus. We also find that PERS47A is stably expressed in the cytoplasm in tim01 flies and some of it is highly phosphorylated. This suggests that although PER can be hyperphosphorylated in the cytoplasm, it is very unstable. It appears mobility-shifting phosphorylation events are not as destabilizing in the nucleus as in the cytoplasm. Consistent with this interpretation, highly phosphorylated nuclear PER stays around for several hours even after TIM has been degraded (Rothenfluh et al., 2000). In case of the timPL mutant, PER is expressed stably in the cytoplasm and yet hyperphosphorylated forms of it are not detected. This could be because either phosphorylation is a primary defect or, even though it can be highly phosphorylated in the timPL mutant, PER gets degraded due to its cytoplasmic localization.
Interestingly, we find that a GSK-3β inhibitor completely blocks OA-dependent mobility shifts of PER. Given the importance of DBT for PER phosphorylation (Kloss et al., 1998), our finding suggests that DBT and GSK-3β cooperate to produce the mobility shifts. This view is supported by recent work demonstrating that multiple kinases, including GSK-3β, are involved in PER phosphorylation and regulation of its nuclear entry (Ko et al., 2010) as well as by our earlier work indicating that GSK-3β-dependent phosphorylation of PER produces mobility shifts (Fang et al., 2007).
The stability of PER in timPL is almost certainly due to its interaction with TIM, which may prevent phosphorylation at specific sites. Typically, this effect of TIM occurs later in the cycle. In wild-type flies, before the appearance of TIM, phosphorylation events likely destabilize PER in the cytoplasm and produce a delay in peak expression of protein. After it is stabilized by TIM, PER accumulates and then moves into the nucleus where its progressive phosphorylation determines its eventual decline. We suggest that in timPL mutants PER is arrested in the state in which it normally occurs in the early part of the night, when it just starts to accumulate in the cytoplasm.
We report here the first direct evidence that TIM plays a critical role in the timed nuclear entry of PER. The timPL mutant reduces negative feedback, presumably by holding PER in the cytoplasm. This effect of the TIM-PL protein, which would explain the semidominant phenotype of the mutant, could reflect a new property of the mutant protein. However, we do not believe this is the case; a stable form of PER is nuclear in wild-type flies and cytoplasmic in tim01 mutants. Whether TIM transports PER into the nucleus or regulates it in some other way is not known. A previous study that assayed interactions between TIM and PER in S2 cells through FRET analysis raised the possibility that TIM and PER dissociate before nuclear entry (Meyer et al., 2006). In addition, in fly clock neurons, PER is detected in the nucleus before TIM (Shafer et al., 2002). Our present findings are consistent with, but do not prove, the hypothesis that PER needs to be dissociated from TIM to enter the nucleus.
We propose a modified model for the regulation of nuclear entry of PER and TIM. TIM is present in excess, and some of it shuttles in and out of the nucleus constantly. However, much of TIM stays bound to PER in the cytoplasm until the appropriate time of day, at which point it mediates a specific modification of PER. Since TIM is not required for nuclear entry in the absence of DBT, this modification probably counteracts some effect of DBT (Cyran et al., 2005). Thus, it could be the removal of a phosphate residue added by DBT, or a secondary phosphorylation event that negates the effect of a DBT-phospho-site. Candidate molecules for the relevant phosphatase and kinase would be PP2A and Shaggy, both of which are implicated in nuclear entry (Martinek et al., 2001; Sathyanarayanan et al., 2004). Once PER is modified, it is imported into the nucleus. It then retains TIM, which would otherwise be exported out, in the nucleus.
Together, these findings shed new light on a process of long-standing interest. They also highlight the importance of genetic mutants for addressing biological mechanisms. The general belief is that forward genetic screens for circadian clock mutants in Drosophila have been saturated, and so all nonessential, nonredundant components have already been identified. While this may be true, it is clear that different types of mutants in the same components can be very useful for addressing mechanistic questions. Along these lines, we recently clarified the role of Pdp1 in the clock by isolating a novel allele through a genetic screen (Zheng et al., 2009).
Footnotes
T.H. was supported by the Japan Society for the Promotion of Science Research Fellowships for Young Scientists. The work was supported by Grants 2R01NS048471 and 2R56NS048471 from the National Institute of Neurological Diseases and Stroke. We thank Lino Saez and Michael Young for constructs and cells; Isaac Edery for providing perS47A flies; Corinne Smith, Drew Peterson, and Huihui Pan for technical assistance; James Jepson for help with immunostaining and confocal imaging; and members of the Sehgal laboratory for helpful discussions.
References
- Ashmore LJ, Sehgal A. A fly's eye view of circadian entrainment. J Biol Rhythms. 2003;18:206–216. doi: 10.1177/0748730403018003003. [DOI] [PubMed] [Google Scholar]
- Ashmore LJ, Sathyanarayanan S, Silvestre DW, Emerson MM, Schotland P, Sehgal A. Novel insights into the regulation of the timeless protein. J Neurosci. 2003;23:7810–7819. doi: 10.1523/JNEUROSCI.23-21-07810.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Rihel J, Bjes E, Fan JY, Price JL. The Drosophila double-timeS mutation delays the nuclear accumulation of period protein and affects the feedback regulation of period mRNA. J Neurosci. 2001;21:7117–7126. doi: 10.1523/JNEUROSCI.21-18-07117.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DC, Reppert SM. A novel C-terminal domain of Drosophila PERIOD inhibits dCLOCK:CYCLE-mediated transcription. Curr Biol. 2003;13:758–762. doi: 10.1016/s0960-9822(03)00286-0. [DOI] [PubMed] [Google Scholar]
- Chiu JC, Vanselow JT, Kramer A, Edery I. The phospho-occupancy of an atypical SLIMB-binding site on PERIOD that is phosphorylated by DOUBLETIME controls the pace of the clock. Genes Dev. 2008;22:1758–1772. doi: 10.1101/gad.1682708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin KD, Huang ZJ, Rosbash M. Temporally regulated nuclear entry of the Drosophila period protein contributes to the circadian clock. Neuron. 1995;14:365–372. doi: 10.1016/0896-6273(95)90292-9. [DOI] [PubMed] [Google Scholar]
- Cyran SA, Yiannoulos G, Buchsbaum AM, Saez L, Young MW, Blau J. The double-time protein kinase regulates the subcellular localization of the Drosophila clock protein period. J Neurosci. 2005;25:5430–5437. doi: 10.1523/JNEUROSCI.0263-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombradi V, Kokai E, Farkas I. Protein phosphatase 1. In: Arino J, Alexander DR, editors. Protein phosphatases: topics in current genetics. Vol 5. Berlin, Heidelberg: Springer; 2004. pp. 21–44. [Google Scholar]
- Fang Y, Sathyanarayanan S, Sehgal A. Post-translational regulation of the Drosophila circadian clock requires protein phosphatase 1 (PP1) Genes Dev. 2007;21:1506–1518. doi: 10.1101/gad.1541607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grima B, Lamouroux A, Chélot E, Papin C, Limbourg-Bouchon B, Rouyer F. The F-box protein slimb controls the levels of clock proteins period and timeless. Nature. 2002;420:178–182. doi: 10.1038/nature01122. [DOI] [PubMed] [Google Scholar]
- Hardin PE. The circadian timekeeping system of Drosophila. Curr Biol. 2005;15:R714–R722. doi: 10.1016/j.cub.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Hunter-Ensor M, Ousley A, Sehgal A. Regulation of the Drosophila protein timeless suggests a mechanism for resetting the circadian clock by light. Cell. 1996;84:677–685. doi: 10.1016/s0092-8674(00)81046-6. [DOI] [PubMed] [Google Scholar]
- Kloss B, Price JL, Saez L, Blau J, Rothenfluh A, Wesley CS, Young MW. The Drosophila clock gene double-time encodes a protein closely related to human casein kinase I epsilon. Cell. 1998;94:97–107. doi: 10.1016/s0092-8674(00)81225-8. [DOI] [PubMed] [Google Scholar]
- Ko HW, Jiang J, Edery I. Role for Slimb in the degradation of Drosophila Period protein phosphorylated by Doubletime. Nature. 2002;420:673–678. doi: 10.1038/nature01272. [DOI] [PubMed] [Google Scholar]
- Ko HW, Kim EY, Chiu J, Vanselow JT, Kramer A, Edery I. A hierarchical phosphorylation cascade that regulates the timing of PERIOD nuclear entry reveals novel roles for proline-directed kinases and GSK-3β/SGG in circadian clocks. J Neurosci. 2010;30:12664–12675. doi: 10.1523/JNEUROSCI.1586-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh K, Zheng X, Sehgal A. JETLAG resets the Drosophila circadian clock by promoting light-induced degradation of TIMELESS. Science. 2006;312:1809–1812. doi: 10.1126/science.1124951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koundakjian EJ, Cowan DM, Hardy RW, Becker AH. The Zuker collection: a resource for the analysis of autosomal gene function in Drosophila melanogaster. Genetics. 2004;167:203–206. doi: 10.1534/genetics.167.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinek S, Inonog S, Manoukian AS, Young MW. A role for the segment polarity gene shaggy/GSK-3 in the Drosophila circadian clock. Cell. 2001;105:769–779. doi: 10.1016/s0092-8674(01)00383-x. [DOI] [PubMed] [Google Scholar]
- Meyer P, Saez L, Young MW. PER-TIM interactions in living Drosophila cells: an interval timer for the circadian clock. Science. 2006;311:226–229. doi: 10.1126/science.1118126. [DOI] [PubMed] [Google Scholar]
- Nawathean P, Rosbash M. The doubletime and CKII kinases collaborate to potentiate Drosophila PER transcriptional repressor activity. Mol Cell. 2004;13:213–223. doi: 10.1016/s1097-2765(03)00503-3. [DOI] [PubMed] [Google Scholar]
- Price JL, Dembinska ME, Young MW, Rosbash M. Suppression of PERIOD protein abundance and circadian cycling by the Drosophila clock mutation timeless. EMBO J. 1995;14:4044–4049. doi: 10.1002/j.1460-2075.1995.tb00075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfluh A, Young MW, Saez L. A TIMELESS-independent function for PERIOD proteins in the Drosophila clock. Neuron. 2000;26:505–514. doi: 10.1016/s0896-6273(00)81182-4. [DOI] [PubMed] [Google Scholar]
- Saez L, Young MW. Regulation of nuclear entry of the Drosophila clock proteins period and timeless. Neuron. 1996;17:911–920. doi: 10.1016/s0896-6273(00)80222-6. [DOI] [PubMed] [Google Scholar]
- Sandrelli F, Tauber E, Pegoraro M, Mazzotta G, Cisotto P, Landskron J, Stanewsky R, Piccin A, Rosato E, Zordan M, Costa R, Kyriacou CP. A molecular basis for natural selection at the timeless locus in Drosophila melanogaster. Science. 2007;316:1898–1900. doi: 10.1126/science.1138426. [DOI] [PubMed] [Google Scholar]
- Sathyanarayanan S, Zheng X, Xiao R, Sehgal A. Post-translational modification of Drosophila PERIOD protein by protein phosphatase 2A. Cell. 2004;116:603–615. doi: 10.1016/s0092-8674(04)00128-x. [DOI] [PubMed] [Google Scholar]
- Sehgal A, Price JL, Man B, Young MW. Loss of circadian behavioral rhythms and per RNA oscillations in the Drosophila mutant timeless. Science. 1994;263:1603–1606. doi: 10.1126/science.8128246. [DOI] [PubMed] [Google Scholar]
- Shafer OT, Rosbash M, Truman JW. Sequential nuclear accumulation of the clock proteins period and timeless in the pacemaker neurons of Drosophila melanogaster. J Neurosci. 2002;22:5946–5954. doi: 10.1523/JNEUROSCI.22-14-05946.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Konopka R. Effects of dosage alterations at the per locus on the period of the circadian clock of Drosophila. Mol Gen Genet. 1982;185:30–36. [Google Scholar]
- Venken KJ, He Y, Hoskins RA, Bellen HJ. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314:1747–1751. doi: 10.1126/science.1134426. [DOI] [PubMed] [Google Scholar]
- Vosshall LB, Price JL, Sehgal A, Saez L, Young MW. Block in nuclear localization of period protein by a second clock mutation, timeless. Science. 1994;263:1606–1609. doi: 10.1126/science.8128247. [DOI] [PubMed] [Google Scholar]
- Williams JA, Su HS, Bernards A, Field J, Sehgal A. A circadian output in Drosophila mediated by neurofibromatosis-1 and Ras/MAPK. Science. 2001;293:2251–2256. doi: 10.1126/science.1063097. [DOI] [PubMed] [Google Scholar]
- Wu MN, Koh K, Yue Z, Joiner WJ, Sehgal A. A genetic screen for sleep and circadian mutants reveals mechanisms underlying regulation of sleep in Drosophila. Sleep. 2008;31:465–472. doi: 10.1093/sleep/31.4.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Sehgal A. Role of molecular oscillations in generating behavioral rhythms in Drosophila. Neuron. 2001;29:453–467. doi: 10.1016/s0896-6273(01)00218-5. [DOI] [PubMed] [Google Scholar]
- Zheng X, Sehgal A. Probing the relative importance of molecular oscillations in the circadian clock. Genetics. 2008;178:1147–1155. doi: 10.1534/genetics.107.088658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Koh K, Sowcik M, Smith CJ, Chen D, Wu MN, Sehgal A. An isoform-specific mutant reveals a role of PDP1epsilon in the circadian oscillator. J Neurosci. 2009;29:10920–10927. doi: 10.1523/JNEUROSCI.2133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]