

Abstract
Tn7 transposition has been hypothesized to require a heteromeric transposase formed by two Tn7-encoded proteins, TnsA and TnsB, and accessory proteins that activate the transposase when they are associated with an appropriate target DNA. This study investigates the mechanism of Tn7 transposase activation by isolation and analysis of transposase gain-of-function mutants that are active in the absence of these accessory proteins. This work shows directly that TnsA and TnsB are essential and sufficient components of the Tn7 transposase and also provides insight into the signals that activate the transposase. We also describe a protein–protein interaction between TnsA and TnsC, a regulatory accessory protein, that is likely to be critical for transposase activation.
Keywords: ATP/conformational change/enzyme activation/protein–protein interaction/transposition
Introduction
Many cellular processes such as transcription, replication and recombination involve the execution of an enzymatic activity at a particular site on a DNA substrate. Controlling the initiation of these processes is important since random events may have deleterious effects. We are interested in the control of transposition, a recombination reaction in which a discrete DNA segment translocates from a donor DNA to a non-homologous target DNA.
We study the bacterial transposon Tn7 (Barth et al., 1976). This element is distinguished among transposons in that its transposition requires multiple transposon-encoded proteins, TnsA, B, C, D and E (Craig, 1995). The different combinations of Tns proteins provide regulated and differential signals for transposition. TnsABC+D mediate Tn7 insertion into a unique site attTn7 on the Escherichia coli chromosome at high frequency, while TnsABC+E direct the transposon into many non-attTn7 sites at a relatively low frequency (Rogers et al., 1986; Waddell and Craig, 1988; Kubo and Craig, 1990).
Although no transposition is observed in the presence of just TnsA and TnsB, several observations suggest that these proteins together form the Tn7 transposase. Based on sequence similarities, TnsB is a member of a superfamily that includes bacterial transposases, retrotransposon transposases and retroviral integrases (Kulkosky et al., 1992; Rezsohazy et al., 1993; Radstrom et al., 1994; Polard and Chandler, 1995; Sarnovsky et al., 1996). Transposases of this class are characterized by a conserved D, D(35)E amino acid motif that is required for catalytic activity (Baker and Luo, 1994; Dyda et al., 1994; Kim et al., 1995; Rice and Mizuuchi, 1995; Bujacz et al., 1996). We have shown that alterations of these amino acids in TnsB result in catalytic defects in transposition, in particular blocking the breakage and joining events at the 3′ ends of Tn7 (Sarnovsky et al., 1996). We have also found that mutations in TnsA result in a catalytic block to cleavage at the 5′ ends of Tn7 (May and Craig, 1996; Sarnovsky et al., 1996). Structural analyses of TnsA (Hickman et al., 2000) have recently revealed that this protein most resembles a type II restriction enzyme, which is consistent with the proposed role of TnsA in catalysis (May and Craig, 1996; Sarnovsky et al., 1996). We have also found that under unusual reaction conditions, i.e. in the presence of Mn2+ and glycerol, TnsAB alone can mediate breakage events at the transposon ends and unusual intramolecular joining events in which one broken transposon end joins to the other end of the transposon rather than to an intermolecular target DNA (Biery et al., 2000a).
Based on these observations, we have proposed that TnsA and TnsB together form a heteromeric transposase, with each protein having its own distinct function: TnsB binds specifically to the ends of the transposon (Arciszewska and Craig, 1991; Arciszewska et al., 1991) and carries out breakage and joining reactions at the 3′ ends of the transposon while TnsA executes breakage at the 5′ end of the transposon. The heteromeric state of Tn7 transposase is in contrast to Tn10, which also transposes via a cut-and-paste mechanism but uses a single gene product to carry out both 5′ and 3′ reactions (Haniford et al., 1989; Chalmers and Kleckner, 1994; Bolland and Kleckner, 1996). Other cut-and-paste mobile elements such as the Tc1 and Tc3 elements of Caenorhabditis elegans also use a single gene product to mediate their translocation (van Luenen et al., 1994; Vos et al., 1996).
We have argued elsewhere that the remaining Tns proteins—TnsC, TnsD and TnsE—control the activity of the TnsAB transposase, proposing in particular that TnsC controls the TnsAB transposase, and that TnsD and TnsE are responsible for selecting the proper site for Tn7 insertions by activating TnsC in the presence of particular target sites (Bainton et al., 1993; Wolkow et al., 1996). However, we have been unable to exclude the possibility that Tns proteins other than TnsA and TnsB also contribute residues to form an active transposase.
Little is known about how TnsC activates the TnsAB transposase. There are several possibilities: TnsC may facilitate TnsA and TnsB assembly into an active complex on the transposon ends; the interaction between TnsC and TnsAB may introduce conformational changes in the catalytic sites of the transposase; it remains possible that TnsC may serve as part of Tn7 transposase. Here, we identify transposase gain-of-function mutants in either TnsA or TnsB that are able to promote transposition with only TnsABC and, in some cases, with only TnsAB. The genetic and biochemical characterization of these mutants provides valuable information on the mechanism of Tn7 transposase activation.
Results
Isolation of transposase gain-of-function mutants
Our working model is that activation of the TnsAB transposase is modulated directly by TnsC, whose activity is in turn controlled by signals from target DNA. We have proposed that the ability of TnsC to modulate TnsAB is controlled through its ATP state and that the TnsD-bound target complex TnsD+attTn7 can switch TnsC into an ATP-bound active form by forming a TnsCATP+TnsD+attTn7 complex (Craig, 1995; Stellwagen and Craig, 1998). However, we do observe a low level of recombination in the presence of TnsABC and ATP, and we postulate that a small fraction of TnsC does exist in the TnsCATP conformation, even in the absence of the TnsD+attTn7 target signal. Based on this hypothesis, we set out to isolate TnsAB mutants that promote transposition with TnsC present in limiting amounts, i.e. in the presence of TnsC but in the absence of TnsD+attTn7. Such mutants could result from intrinsic changes in transposase activity that are independent of TnsC or from their more efficient interaction with a limiting amount of ‘active’ TnsC.
To screen for transposase ‘gain-of-function’ mutations, we employed a papillation assay, which enabled us to examine visually the transposition frequency in individual colonies (Stellwagen and Craig, 1997). This assay utilizes a miniTn7lac element carrying a promoterless lacZYA operon; the lac genes are not expressed unless the miniTn7lac element inserts downstream of an actively transcribed promoter. When a strain carrying the miniTn7lac element located in a transcriptionally silent donor site is plated on MacConkey lactose indicator plates, it forms white colonies. If the strain is supplied with Tn7 transposition proteins, it initially forms white colonies, but, over time, Tn7 transposition in individual cells generates Lac+ progeny, which form red papillae within the white colony. The number of these papillae is a measure of the frequency of transposition events. After 3 days of growth, few Lac+ red papillae are visible in colonies with only TnsABC whereas colonies containing TnsABC+E are saturated with papillae.
In one screen, a chemically mutagenized tnsA plasmid was introduced into cells containing a miniTn7lac donor plasmid and a tnsBC plasmid; transformants that gave rise to an increased number of red Lac+ papillae compared with the colonies with wild-type tnsABC were selected as the candidates for transposase gain-of-function mutants (see Materials and methods). Among 2000 transformants screened, 13 candidates from individual transformations were isolated and by subcloning and sequencing, seven different mutations were identified in the tnsA gene (Table I).
Table I. Tn7 transposase gain-of-function mutations.
| Screen | Allele | Class | No. of independent isolates | Papillation level |
|---|---|---|---|---|
| tnsA mutants in TnsA+B+C transposition | ||||
| AS69N | I | 2 | ++++++ | |
| AE73K | I | 2 | ++++ | |
| AA65V | I | 2 | +++ | |
| AE185K | II | 1 | ++++++ | |
| AQ261Z | II | 1 | ++++++ | |
| AG239S | III | 3 | +++++ | |
| AG239D | III | 2 | ++++ | |
| AWT | – | – | + | |
| tnsA and tnsB mutants in TnsA+B+C transposition | AE185K | II | 2 | ++++++ |
| AQ261Z | II | 2 | ++++++ | |
| BM366I | II | 1 | ++++++ | |
| BA325T | II | 4 | ++++++ | |
| BA325V | I | 2 | ++++++ | |
| AWTBWT | – | – | + | |
| TnsABC+E | – | – | ++++++ (day 2) |
Plasmid configurations in two screens were different as explained in Materials and methods. TnsAQ261Z contains a nonsense mutation (see Materials and methods). Cells were patched on MacConkey agar plates and the papillation level was recorded after 3 days of incubation at 30°C. TnsABC+E transposition events appeared earlier than all the mutants and were recorded after 2 days; after a 3-day incubation, the papillae were too numerous to be counted. The relative differences in papillation are indicated by +/–. In a patch (∼1 inch long): –, <5 papillae; +, 5–15 papillae; ++, 15–30 papillae; +++, 30–50 papillae; ++++, 50–100 papillae; +++++, 100–150 papillae; and ++++++ corresponds to the amount of papillae that saturated the entire patch.
In another screen, we looked for transposase gain-of-function mutants by introducing a chemically mutagenized tnsAB plasmid into cells containing a tnsC plasmid. Among 5000 transformants screened, 11 candidates from individual transformations were isolated and by subcloning and sequencing, five different mutations were identified (Table I); three were in the tnsB gene while two were in the tnsA gene. The positions of all the mutations are shown in Figure 1.

Fig. 1. Location of the transposase gain-of-function mutations. Schematic illustration of the functional domains of TnsA and TnsB and various point mutations collected in this study. The shaded boxes represent the DNA binding domain of TnsB (R.Sarnovsky and N.L.Craig, unpublished data), the catalytic domain of TnsB (Sarnovsky et al., 1996), and the catalytic domain of TnsA (Sarnovsky et al., 1996; Hickman et al., 2000). The characters ‘D’, ‘E’, ‘K’ and ‘Q’ indicate the relative positions of amino acids implicated in catalysis: D273, D361 and E396 in TnsB; E63, D114, Q130, K132 and E149 in TnsA. The arrows indicate the relative positions of amino acid changes in the transposase gain-of-function mutants. The mutant classes are explained in the text.
In these screens, all the mutations except TnsBM366I have been isolated multiple times. The transposition levels of each mutant based on the papillation assay are shown in Table I, and compared with wild-type TnsABC and TnsABC+E levels. The transposition frequencies of all the mutants are lower than the TnsABC+E level.
The papillation assay does not differentiate intermolecular from intramolecular events. To evaluate the nature of transposition events generated by the mutants, we examined whether the original Lac– donor F plasmid was converted to Lac+, representing intramolecular transposition. For each mutant, 20 red papillae were purified and the linkage of their Lac+ phenotype to the donor plasmid was determined. For all the mutants, >90% of the papillae examined contained Lac– plasmids, suggesting that intermolecular transposition of miniTn7lac into the E.coli chromosome occurred. Chromosomal DNA containing Lac+ insertions was also analyzed by Southern blotting using a probe specific to miniTn7; no apparent insertion hot spots in the chromosome for the miniTn7lac element were observed (data not shown). These analyses thus demonstrate that the transposase gain-of-function mutants promote intermolecular recombination.
As described below, the mutants represent three distinct classes based on their phenotypes (Table II). Compared with the wild-type transposase, whose activity is latent and can only be turned on by TnsC in combination with the target selection proteins, class I mutants (TnsAA65V, TnsAS69N and TnsAE73K) can be activated by TnsC alone, and can be further activated by TnsC in the presence of TnsD and TnsE. The class II mutants (TnsAE185K, TnsAQ261Z, TnsBM366I and TnsBA325T) have similar phenotypes as class I mutants in their response to TnsC and the targeting proteins. Furthermore, they have increased intrinsic transposase activity, i.e. they can promote transposition independent of TnsC. The class III mutants (TnsAG239D and TnsAG239S) can also be activated by TnsC alone in vivo but their ability to respond to the target selecting proteins TnsD and TnsE is impaired.
Table II. Three distinct classes of Tn7 transposase gain-of-function mutants.
| Class | TnsA*+B* | TnsA*BC or TnsAB*C | TnsA*BC+D/E or TnsAB*C+D/E |
|---|---|---|---|
| I | – | ++ | +++++ |
| II | + | ++ | +++++ |
| III | – | ++ | +++ |
| Wild type | – | – | +++++ |
The relative transposition frequencies of wild type and transposase mutants (see text) are indicated by the +/–, which are independent of the signs in Table I.
Transposition activity in vitro
Tn7 transposes via a cut-and-paste mechanism, as demonstrated in the reconstituted in vitro Tn7 transposition system (Figure 2A) (Bainton et al., 1991, 1993). Tn7 is excised from a donor DNA through a pair of double-strand breaks (DSB) at the junction of the transposon ends and flanking donor DNA to give rise to an excised linear transposon (ELT) intermediate. The DSB are not necessarily concerted, which leads to recombination intermediates that contain breaks at either the left or right end of the transposon, named as DSB.L or DSB.R. The ELT is joined to the target to give the end product, a simple insertion (SI).

Fig. 2. Recombination activity of the mutants in TnsABC reaction and the effect of ATP cofactor. (A) The Tn7 transposition pathway is illustrated on the left. Donor DNA (solid line) containing the miniTn7 transposon (rectangle); target DNA (dashed line). The cis-acting recombination sequences at the left and right ends of the transposon are indicated by open and filled triangles, respectively. miniTn7 is excised from the donor DNA by double-strand breaks, through the intermediates DSB.L (double-strand break at the left end), DSB.R (double-strand break at the right end) and ELT (excised linear transposon). ELT then can be inserted into the target DNA to give a simple insertion product (SI). The positions of the NdeI sites used to linearize the plasmid substrates are indicated by slashed lines. A Southern blot of the in vitro transposition reactions is shown on the right. The proteins included in each reaction are indicated above each lane. (B) TnsA*BC reactions: TnsAE185K from class II (lanes 2 and 6), TnsAS69N from class I (lanes 3 and 7) and TnsAG239D from class III (lanes 4 and 8). As indicated, either ADP or ATP was used in the assembly reaction mixture. (C) TnsAB*C reactions: TnsBM366I (lanes 2 and 5) and TnsBA325T (lanes 3 and 6) from class II.
As shown in Figure 2A, no recombination activity can be observed with wild-type TnsAB, reflecting the dormant state of the transposase. The successful completion of transposition requires the presence of all four proteins, TnsABC+D. Although TnsABC does not support any simple insertion formation in vitro, some DSB recombination intermediates are observed. One explanation for this result is that TnsC may exist in a mixed population in which a small fraction of the TnsC is bound to ATP even in the absence of the usual target cofactors TnsD+attTn7, the remaining TnsC being inactive, i.e. ADP bound or lacking nucleotide.
Transposase gain-of-function mutants were expressed and purified as recombinant proteins (see Materials and methods) and their catalytic activities evaluated in the in vitro transposition system. Representatives were chosen from each category of mutants: TnsAS69N from class I; TnsAE185K, TnsBM366I and TnsBA325T from class II; and TnsAG239D from class III.
In TnsABC reactions (Figure 2B), both class I mutant TnsAS69N and class II mutant TnsAE185K are more efficient than TnsAwt at promoting strand breakage in the presence of TnsC, generating more DSB and ELT intermediates. For the TnsB mutants (Figure 2C), two class II mutants (TnsBM366I and TnsBA325T) also show increased strand breakage activity compared with TnsBwt. The finding that recombination by these mutants is inhibited by the addition of ADP, as is the wild type, reveals that the gain-of-function phenotypes of these transposase mutants are likely to result from alteration of their interaction with TnsCATP.
Taken together, compared with the wild-type transposase, both class I and class II mutants show increased substrate breakage activity, indicating their more efficient use of TnsC. The class III mutant appears to have less breakage activity than wild-type protein in TnsABC reactions in vitro (Figure 2B, lane 4). The finding that the class III mutant does not respond to TnsC in vitro as well as in vivo will be discussed later.
Transposition activity in the absence of TnsC
Can any of the mutants we have isolated promote transposition in the absence of the regulator TnsC? To test this, we used the papillation assay with cells containing the miniTn7lac element with different combinations of the mutant TnsA and TnsB proteins. The class II mutants differentiate themselves from the rest of the mutants by their ability to promote transposition without TnsC. Virtually no papillae were seen in cells with TnsAwt+TnsBwt, reflecting the fact that TnsA and TnsB by themselves can not promote transposition (Table III). Class II TnsB mutants (TnsBM366I and TnsBA325T) when paired with class II TnsA mutants (TnsAE185K and TnsAQ261Z) were able to give rise to red Lac+ papillae, indicating that these transposase mutants promote transposition in the absence of TnsC. This is the first time that we have observed any transposition activity with only TnsAB in vivo. For all the strains, >85% of Lac+ papillae represented intermolecular insertions of the miniTn7lac element into different positions in the E.coli chromosome (data not shown). These results with the class II mutants indicate that TnsA and TnsB contain all the essential activities to complete an intermolecular transposition event.
Table III. TnsA and TnsB complementation using the papillation assay.
| BWT | BM366I | BA325T | BA325V | |
|---|---|---|---|---|
| AWT | – | – | – | – |
| AE185K | – | + | ++ | – |
| AQ261Z | – | – | + | – |
| Other TnsA* | – | – | – | – |
The papillation assay was carried out as described in Materials and methods by supplying TnsA and TnsB on separate but compatible plasmids. Cells were patched on MacConkey agar plates and the plates scored after 3 days of incubation at 30°C. The –/+ code is the same as in Table I. Other TnsA* stands for class I mutants (TnsAA65V, TnsAS69N, TnsAE73K) and class III mutants (TnsAG239D and TnsAG239S), which all gave the same results.
We also examined the transposase activity of the gain-of-function mutants in vitro in the absence of TnsC (Figure 3). The class II mutants when paired with each other, i.e. TnsA*+TnsB* (TnsAE185K + TnsBM366I or TnsAE185K + TnsBA325T), do support recombination, resulting in the accumulation of DSB intermediates. Also visible are unusual recombination products in which one transposon end has broken away from the donor backbone and, instead of joining to an intermolecular target DNA, has joined to the other end of the element in an intramolecular reaction to form a circularized transposon species. We have heretofore observed these joining products under only aberrant recombination conditions of Mn2+ and high glycerol (Biery et al., 2000a). The class II TnsB mutants, in combination with TnsAwt or the other classes of TnsA mutants, showed a detectable but low level of activity. In these reactions, additional glycerol (20%) was included in the reaction mixture. At low glycerol concentrations (<2%), we do not see any activity with any combination of TnsA and TnsB mutants (data not shown). Glycerol has been shown to stimulate enzyme activity in other in vitro recombination systems (Craigie et al., 1990). Glycerol might act as a nucleophile in the strand cleavage reactions (Engelman et al., 1991; Vink et al., 1991) or as a volume excluder, increasing protein–protein and protein–DNA interactions.

Fig. 3. Transposase gain-of-function mutants can support breakage and joining reaction with only TnsA and TnsB. TnsAB reactions with mutant proteins. The assembly step was carried out without either TnsC or TnsD under the conditions specified in Materials and methods except for the additional 20% glycerol. ‘intra-mol joining’ indicates intramolecular joining products where one end of the transposon has been cleaved and subsequently joined within the same donor in the region near to the other end of the transposon (Biery et al., 2000b). The low-level cross-hybridization to the target DNA occasionally observed with this probe is indicated.
The presence of recombination products reveals that class II mutants contain increased intrinsic transposase activity compared with the wild-type TnsAB, while class I and class III mutants have no apparent changes. The modification in the transposase intrinsic activity could result from increased interaction between TnsA and TnsB, or from conformational changes in the catalytic domains of TnsA or TnsB.
The response of transposase mutants to the target signal
We have shown that the gain-of-function mutants are altered in the way they respond to TnsC. Do these mutational changes have any impact on the response of the transposase to the conventional activation signal from TnsC+D or TnsC+E? To answer this question, we compared the transposition properties of the mutant transposases in TnsABC+D and TnsABC+E reactions using an in vivo λ hop assay. In the λ hop assay, transposition frequency is evaluated by scoring the insertion events of a miniTn7KmR element from an integration- and replication-defective phage into the bacterial chromosome, by selecting for the kanamycin resistance marker. The transposition frequency was determined by the number of KmR colonies per plaque forming unit of phage particles (p.f.u.).
As shown in Table IV, wild-type TnsABC+D promotes transposition at a frequency of 4 × 10–3 transposition events/p.f.u. For class I and class II mutants, their transposition frequencies are similar to the wild type, i.e. these mutants are highly activated by the TnsC+D signal. The response of the transposase mutants to targeting signals from TnsE was also evaluated by using the λ hop assay, and again we found that class I and class II mutants responded like wild type to the TnsC+E signal. In contrast, class III mutants did not respond to the TnsC+D and TnsC+E signal as well as wild type; thus, these mutations in the transposase result in deficiencies in both TnsABC+D and TnsABC+E transposition. The levels of transposition promoted by the gain-of-function mutants alone or with TnsC is below the threshold of sensitivity of the λ hop assay (<2 × 10–8 transposition events/p.f.u.) and can only be measured by the papillation assay.
Table IV. Effects of the target selection protein TnsD/E.
| Mutations | Allele | TnsABC+D | TnsABC+E |
|---|---|---|---|
| TnsAa | wild type | 4.0 × 10–3 | 4.3 × 10–6 |
| Class I | S69N | 4.0 × 10–3 | 9.3 × 10–6 |
| E73K | 5.1 × 10–3 | 4.3 × 10–6 | |
| A65V | 2.1 × 10–3 | 5.8 × 10–6 | |
| Class II | E185K | 5.2 × 10–3 | 9.5 × 10–6 |
| Q261Z | 7.8 × 10–3 | 1.0 × 10–5 | |
| Class III | G239D | 3.6 × 10–5 | 1.0 × 10–7 |
| G239S | 2.7 × 10–4 | 5.4 × 10–7 | |
| No TnsA | – | 2.6 × 10–8 | 2.1 × 10–8 |
| TnsBb | wild type | 9.7 × 10–6 | ND |
| Class II | M366I | 1.3 × 10–5 | ND |
| A325T | 1.4 × 10–5 | ND | |
| No TnsB | – | 4.1 × 10–8 | ND |
Transposition frequency of a miniTn7KmR element from an integration-defective λ phage into the E.coli chromosome was measured in strains containing the gain-of-function transposase mutants. The results are the average of five trials and shown as number of transposition events/p.f.u.
aTnsA mutants were tested in the top panel.
bTnsB mutants were tested in the bottom panel. Owing to the difference in their plasmid configuration (see Materials and methods), the transposition frequency of the TnsABC+D reaction of the wild-type proteins is ∼400-fold lower than that found above while the transposition frequency of the TnsABC+E reaction was below the level of detection.
We also examined the activity of the transposase mutants in the in vitro TnsABC+D transposition reaction (Figure 4). Compared with the wild-type transposase, class I and class II mutants show similar levels of simple insertions while the class III mutant (TnsAG239D) showed much lower activity. These in vitro results are consistent with the phenotype of the gain-of-function mutants shown above in the λ hop assay. Taken together, both class I and class II mutants responded to the conventional target signals from TnsD and TnsE as do wild-type TnsA+TnsB. However, the class III mutants can only be partially activated by the target signals, suggesting that these mutations may prevent the transposase from being converted into the fully active state.

Fig. 4. Effect of the target selection protein TnsD in vitro. (A) TnsA*BC+D reactions: TnsAS69N from class I (lane 2), TnsAE185K from class II (lane 3) and TnsAG239D from class III (lane 4). The three bands above the donor substrate, indicated with a bracket, are intermediate products resulting from the joining between a partially cleaved donor substrate and the target DNA. (B) TnsAB*C+D reactions: TnsBM366I (lane 2) and TnsBA325T (lane 3) from class I.
Identification of loss-of-function TnsA mutations that are defective in TnsC-mediated transposase activation
We noticed that the class I mutants (TnsAA65V, TnsAS69N and TnsAE73K) that show more efficient use of TnsC are clustered in a 9 amino acid region at the N-terminal region of TnsA. One reasonable hypothesis is that this region of TnsA may be functionally important in channeling a TnsC-mediated activation signal.
To probe further the region of TnsA where the class I gain-of-function mutations lie, we constructed alanine substitutions in the 70–81 region of TnsA and analyzed the effect of these mutations on transposase’s ability to respond to a variety of activation signals (Table V). The mutations at L70A/E71A/W72A and D78A/E81A partially or completely blocked transposition in any pathway as shown by the λ hop assay. The transposition promoted by TnsAwtBC+D occurs at a frequency of 3 × 10–3 transposition events/p.f.u. while the transposition frequencies of TnsA*BC+D were substantially decreased. Compared with the TnsAwtBC+E level, the transposition events mediated by TnsA*BC+E were also reduced substantially. Therefore, TnsA mutants containing changes at particular residues (L70A/E71A/W72A and D78A/E81A) have lost the ability to respond to the conventional targeting signals from TnsD or TnsE.
Table V. Phenotypes of the site-directed mutations in the region of TnsA that are implicated in interaction with TnsC.
| A*BCa | aA*BCA225V | A*BC+Db | A*BC+Eb | A*BC+ABCDc | |
|---|---|---|---|---|---|
| Wild type | – | ++++ | 3.1 × 10–3 | 6.2 × 10–6 | 6.4 × 10–5 |
| L70A/E71A/W72A | – | – | <1.6 × 10–8 | <1.6 × 10–8 | 1.5 × 10–6 |
| D78A/E81A | – | – | 1.7 × 10–6 | <1.6 × 10–8 | 8.3 × 10–7 |
aPapillation phenotype of the transposase mutants in TnsABC reactions with either wild-type TnsC or a gain-of-function mutant TnsCA225V. Data were collected after cells had gone through 3 days incubation at 30°C on MacConkey agar plates.
bTransposition frequencies of the mutants in TnsABC+D or TnsABC+E reactions were based on the λ hop assay.
cTransposition frequencies of the TnsABC+D reactions when the TnsA mutant was co-expressed with wild-type TnsA were based on the λ hop assay. Owing to the difference in their strain background (see Materials and methods), the transposition frequency of the TnsABC+D reaction of the wild-type proteins is different from that found in the assay above.
We have also found that these alanine substitution mutants could not be activated by TnsC alone (Table V). Based on the papillation assay, the transposase activities of these mutants can not be stimulated by a gain-of-function TnsC mutant (TnsCA225V), which was originally isolated based on its ability to enable TnsABC to promote transposition in the absence of TnsD or TnsE (Stellwagen and Craig, 1997). Therefore, the mutations L70A/E71A/W72A and D78A/E81A abolished the transposase’s ability to respond to TnsC-mediated activation signals.
To exclude the possibility that the loss-of-function phenotype of these mutants is due to improper protein folding, we also examined the effects of the mutant proteins on the TnsAwtBC+D transposition when they are co-expressed with TnsAwt in the same cell (Table V). These mutants showed dominant negative phenotypes, inhibiting the TnsAwtBC+D reactions as evaluated by the λ hop assay. This dominant negative phenotype indicates that these TnsA mutants retain their ability to communicate with TnsB and/or TnsC in the functional transposition complexes. Therefore, mutations at L70A/E71A/W72A and D78A/E81A affect TnsA specifically with respect to transposase activation while still retaining the ability to interact with other transposition proteins.
Evidence for a direct interaction between the transposase and the activator TnsC
Previous pull-down experiments with GST–TnsC derivatives revealed that a physical interaction can be detected between TnsA and TnsC under in vitro recombination conditions (A.Stellwagen and N.L.Craig, unpublished observation). To localize the site(s) where TnsA interacts with TnsC, we employed protease footprinting assays. Purified TnsA was subjected to proteolysis in the absence and presence of TnsC. In the absence of TnsC, tryspin cleaved preferentially at four sites to give a ladder of TnsA fragments as detected by an anti-TnsA polyclonal antibody. Sequencing of the individual cleavage products revealed that trypsin cleaved after amino acid residues R21, R43/46, R91 and R105 of TnsA (Figure 5A and D). The species that was truncated at R105 was largely resistant to digestion by trypsin even with the addition of 10-fold excess over the level sufficient to cleave all the input TnsA (data not shown); thus, the carboxyl portion of TnsA appeared to be folded into a protease resistant structural domain. Similar results were obtained using a chymotrypsin digestion of TnsA (Figure 5B and D). Chymotrypsin cleaves TnsA predominantly after amino acid residues W32, Y48 and F67.

Fig. 5. Localization of a specific region in TnsA that is involved in direct interaction with TnsC. (A) Protease footprinting of TnsA using trypsin. Lane 1, untreated TnsA; lanes 2–4, TnsA treated with increasing amounts of trypsin (10, 33 and 100 ng); lanes 5–7, TnsA was preincubated with TnsC and then treated with increasing amounts of trypsin (33, 100 and 333 ng). The first N-terminal amino acid of the proteolytic products is labeled. (B) Protease footprinting of TnsA using chymotrypsin. Lane 1, untreated TnsA; lanes 2–4, TnsA treated with chymotrypsin (3.3, 10 and 33 ng); lanes 5–7, TnsA was preincubated with TnsC and then treated with chymotrypsin (10, 33 and 100 ng). (C) Protease footprinting of TnsA mutants. Lane 1, untreated TnsA; lanes 2–4, TnsA treated with increasing amounts of trypsin (10, 33 and 100 ng); lanes 5–7, TnsA preincubated with TnsC and then treated with increasing amounts of trypsin (10, 33 and 100 ng). (D) The protease cleavage sites on TnsA are indicated by arrows (top: trypsin; bottom: chymotrypsin). The region protected by TnsC is marked between amino acids 43 and 105. The shaded box represents the catalytic domain of TnsA (Sarnovsky et al., 1996; Hickman et al., 2000). The characters ‘D’ and ‘E’ indicate the D114 and E149 amino acids implicated in catalysis; other positions—E63, Q130 and K132—are not indicated in this view.
We also performed proteolysis of TnsA in the presence of TnsC. As shown in Figure 5, in the presence of TnsC, a substantial fraction of TnsA was resistant to the protease, especially at sites R43/46, R91 and R105 for the trypsin digest and Y48 and F67 for the chymotrypsin digest. The resistance to cleavage of TnsA extends from amino acid 43 to 105, which contains the region identified by both gain-of-function and loss-of-function mutants that have altered response to TnsC-mediated activation. Moreover, the loss-of-function TnsA proteins are accessible to protease attack even in the presence of TnsC (Figure 5C). A reasonable hypothesis is that we have identified a specific region in TnsA that is involved in a direct interaction with TnsC; alternatively, the conformational change in the 43–105 region may result from a global structural change in TnsA induced by TnsC interaction with TnsA at another site.
Formation of a TnsA+TnsC+TnsD+attTn7 complex
In the presence of ATP, TnsC and TnsD can form a distinct complex on attTn7 as evaluated by a gel mobility shift assay (Figure 6, lane 1) (Bainton et al., 1993). When the TnsC+TnsD+attTn7 complex was further incubated with increasing amounts of TnsA, an even slower migrating species accumulated, which is believed to be a TnsA+TnsC+TnsD+attTn7 complex (Figure 6, lanes 2–4) (K.Kubo, P.Kuduvalli and N.L.Craig, unpublished data). The formation of this complex is likely to occur through a TnsA–TnsC interaction. Using a gel mobility shift assay and judging the appearance of the TnsA+TnsC+ TnsD+attTn7 complex, we evaluated the interaction between different TnsA mutants and TnsC. The TnsA loss-of-function mutants, L70A/E71A/W72A and D78A/E81A, which are defective in TnsC-mediated transposase activation, were unable to form the putative TnsA+TnsC+ TnsD+attTn7 complex (Figure 6, lanes 4–9).
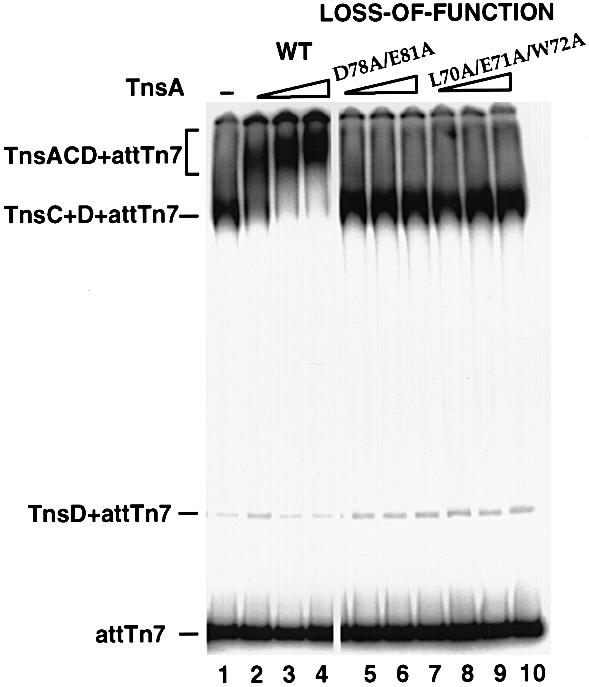
Fig. 6. Effect of TnsA loss-of-function mutations on the TnsA–TnsC interaction analyzed by gel mobility shift assay. The DNA substrate contains attTn7 (–52 to +64). TnsC and TnsD were present in all the reactions. Lane 1, no TnsA was added. Lanes 2–4, wild-type TnsA (13, 26 and 40 ng). Lanes 5–10, loss-of-function mutants were added: lanes 5–7, mutant D78A/E81A (13, 26 and 40 ng); lanes 8–10, mutant L70A/E71A/W72A (13, 26 and 40 ng).
These observations support the hypothesis that the formation of TnsA+TnsC+TnsD+attTn7 complexes is directly dependent on the TnsA–TnsC interaction. They also support the view that the interaction between this 65–81 amino acid region of TnsA and TnsC, identified by both gain-of-function and loss-of-function mutations as well as protease footprinting assays, is likely to be critical in TnsC-mediated transposase activation.
Discussion
Multiple transposon-encoded proteins are involved in executing the strand breakage and joining activities that underlie Tn7 transposition. In order to investigate further the roles and interactions between different Tns proteins, we have isolated and characterized Tn7 transposase gain-of-function mutants that have altered intrinsic transposase activity or are altered in their ability to respond to other proteins that regulate Tn7 transposition. This work supports and extends our hypothesis that TnsA and TnsB form the transposase. This work also identifies an important functional interaction between the transposition regulatory ATP-utilizing protein TnsC and TnsA that is likely to be critical to the activation of the transposase.
TnsA and TnsB form the Tn7 transposase
Our hypothesis that TnsAB collaborate in an interdependent fashion to mediate transposition is derived from two lines of evidence. Mutational studies suggest the existence of catalytic sites in both the TnsA and TnsB proteins. Mutations in TnsB lead to the loss of cleavage and joining activities at the 3′ ends of the element and mutations in TnsA result in the loss of cleavage at the 5′ ends (May and Craig, 1996; Sarnovsky et al., 1996). Furthermore, TnsAB-dependent breakage and joining events are observed in vitro under aberrant reaction conditions (Biery et al., 2000a). The work reported here shows that TnsA+TnsB are sufficient to form the transposase. We have isolated class II gain-of-function TnsA and TnsB mutants that can execute transposition in the absence of any other Tns proteins, providing strong evidence that these proteins alone form the actual transposase that executes all the breakage and joining activities which underlie transposition. It should be noted that TnsAB transposition was observed only with certain combinations of TnsA and TnsB mutants. This finding could be interpreted to show some sort of allele-specific interaction between mutants, or to show that mutations are required in both proteins because of low levels of transposition with just a single mutant.
The recent crystallographic analysis of TnsA (Hickman et al., 2000) allows us to consider the disposition of the TnsA mutations with respect to other features of TnsA (Figure 7). Several amino acids, E53, D114, Q130, K132 and E149, are known to form part of the TnsA active site. Very interestingly, the gain-of-function class I TnsA mutations that alter the activation of TnsAB in the presence of TnsC (A65V, S69N and E73K), and the loss-of-function TnsA alleles (L70A/E71A/W72A and D78A/E81A) are located on the α-helix that contains the active site residue E63. The back of this helix is part of a large hydrophobic patch that we speculate may be an interaction site for TnsC. Thus, alteration of amino acids in these regions ultimately may change interaction of TnsC with TnsA and consequently the disposition of E63 in the active site, changing the catalytic activity of TnsA. The other gain-of-function mutations in TnsA, the class II alleles E185K and Q261Z and the class III mutants at G239, map in the C-terminal region of TnsA that forms a globular domain of TnsA that is distinct from the N-terminal region. Further analysis will be required to dissect how the class II and III mutations affect TnsA structure and function.

Fig. 7. Location of TnsA mutations on the structure of TnsA. (A) Ribbons (Carson, 1997) representation of the three-dimensional structure of TnsA. The N-terminal catalytic domain is in blue, the C-terminal helix–turn–helix containing domain is in orange. Class I residues on helix α2 are marked in red, class II residues are in green. The location of the class III mutations is marked on the main chain just upstream of α10. Residues that are essential for catalytic activity (E63, D114 and K132) and the two bound catalytic Mg2+ residues are also shown. (B) The molecular surface of TnsA overlaid to its ribbons trace. Exposed hydrophobic areas are painted green on the surface. Class I residues are marked in red; they are located on helix α2, which forms one of the boundaries that is part of a large hydrophobic patch on the surface. The surface calculations were performed with SPOCK (Christopher, 1998), the ribbon was drawn with Molscript (Kraulis, 1991) and the final figure was rendered with Povray 3.1 (persistence of vision at http://www.povray.org).
The two TnsB mutations, A325T and M366I, that result in an increased transposase activity in the absence of TnsC lie in the catalytic domain, near the second D of the D, D(35)E motif (Sarnovsky et al., 1996). An attractive view is that these particular mutations induce a conformational change in TnsB, usually executed in response to TnsC and an appropriate target signal, that facilitates the transition of the catalytic pocket into an active state.
We have not observed recombination in the absence of either component of the TnsAB transposase, suggesting that both of these proteins are necessary for recombination and that each of these proteins can influence the activity of the other. Interestingly, when the 5′ cleavage activity of TnsA is not required, the presence of TnsA is still essential for promoting TnsB catalytic activity (May and Craig, 1996; Sarnovsky et al., 1996). TnsA can therefore affect the catalytic activity of TnsB. The fact that we have been able to isolate mutations in both TnsA and TnsB that activate the intrinsic activities of the TnsAB transposase further supports the notion that TnsA and TnsB impose modulatory effects on each other (May and Craig, 1996; Sarnovsky et al., 1996).
There are examples of other recombination reactions involving multiple recombinase proteins. The recombinase for Xer site-specific recombination is composed of two proteins, XerC and XerD (Blakely et al., 1993). Both proteins contain specific DNA-binding activities as well as the active sites for catalysis. Like TnsA and TnsB, neither XerC nor XerD on its own is active in DNA strand cleavage or strand exchange, implying that the interaction between two proteins is necessary to induce an active configuration that is ready for catalysis (Subramanya et al., 1997; Spiers and Sherratt, 1999). Another example of the collaboration of two proteins is the RAG1/RAG2 recombinase active in VDJ recombination. Both RAG1 and RAG2 are essential for the breakage and joining reactions at the junction between the recombination signal sequences and coding sequences (McBlane et al., 1995; Melek et al., 1998; Roth and Craig, 1998).
TnsC activates the TnsAB transposase
No transposition can be observed in the presence of TnsAwtBwt alone; other proteins are required to activate the transposase to execute recombination. We have previously argued that TnsC interacts with TnsAB to activate recombination (Craig, 1995; Stellwagen and Craig, 1998). As shown in Figure 8, we have proposed that TnsC, an ATPase and ATP-dependent non-specific DNA binding protein, exists in two states (‘on’ and ‘off’) differing in ability to activate the TnsAB transposase. The ‘on’ conformation is TnsCATP. The ‘off’ conformation of TnsC is bound to ADP or lacking nucleotide; TnsC does not activate TnsAB in the presence of ADP or in the absence of nucleotide cofactor. We believe that the control of the TnsD-mediated target signal over Tn7 transposition is executed by affecting the equilibrium of TnsC between ATP- and ADP-bound forms. ATP binding assay has revealed that the presence of TnsD and attTn7 can increase the ATP binding efficiency of TnsC (A.Stellwagen and N.L.Craig, unpublished observation). Therefore, TnsD may communicate with TnsC through modifying its nucleotide binding status. We note that a low amount of TnsABC-mediated recombination is observed in vitro when ATP is present; a reasonable explanation is that there is a small population of TnsCATP present that can activate TnsAB.

Fig. 8. Model for Tn7 transposase activation. TnsC is present as two forms: TnsCon (circle) and TnsCoff (rhombus). The equilibrium between the two forms is determined by ATP and targeting cofactors TnsD and attTn7. The wild-type TnsAB transposase by itself is inactive (clear) and is unable to promote transposition (Tnp–). TnsAB can be activated (gray) by active TnsC in TnsCATP+TnsD+attTn7 complex and mediate transposition (Tnp+). The impact of mutational changes on the transposase is shown at the bottom and elucidated in the text.
Both class I and class II mutants of TnsA and TnsB exhibit gain-of-function phenotypes in the in vitro TnsABC reaction, which is consistent with their increased transposition levels detected in vivo in the papillation assay, suggesting they are better activated by TnsC than the wild-type transposase. The class I mutations are likely to enable the transposase to establish a more effective interaction with TnsC, which allows more transposase molecules to be activated when a limited amount of TnsCon is available in TnsABC reactions. In the presence of TnsD or TnsE, TnsC is driven into the TnsCon state. Now there are sufficient numbers of TnsCon available to interact with and activate all the TnsAB molecules and the efficiency of the transposase–TnsC interaction is no longer a limiting factor in Tn7 transposition. Therefore, the class I mutants promote transposition at a level similar to the wild-type transposase in both TnsABC+D and TnsABC+E reactions.
While both class I and class II mutants show increased activation by TnsC, only class II mutants have increased intrinsic transposase activity. The dual effects of class II mutations on transposase activity imply that the mechanism of TnsC-mediated activation and the self-activation of the transposase may not be mutually exclusive. The class II mutations may allow the transposase to assume a constitutively active conformation that consequently leads to a better interaction with TnsC. As shown in Figure 8, we propose that Tn7 transposase activation involves two interconnected steps: the interaction of TnsAB with the regulator TnsC and the transformation of the transposase from the dormant to an active state.
Class III mutants: different target signals, a common activation pathway
Compared with the class I and II mutants, the class III mutants are very different with respect to TnsC-mediated activation in the presence and absence of target signals. In vivo, class III mutants exhibit a gain-of-function phenotype in TnsABC transposition, but they are defective in both TnsABC+D and TnsABC+E transposition. Class III mutants are also defective in the TnsABC reaction in vitro. We propose that these mutants have gained a better interaction with TnsC at the expense of losing their capacity to become fully activated after interacting with TnsC. In TnsABC transposition in vivo when a limited number of TnsCon molecules exist, the better interaction with TnsC allows more transposase molecules to be activated even though the mutant transposase can not be fully activated to the extent of wild type. The combination of two effects resulted in gain-of-function phenotypes. In the presence of target activation signals, the efficiency of the transposase–TnsC interaction is no longer a limiting factor for transposition and consequently the defect of class III mutants in their ability to be fully activated by TnsC becomes visible.
Both the TnsABC+D and the TnsABC+E transposition pathways utilize the TnsABC core recombination machinery, which implies that two pathways may share a similar transposase activation mechanism (Craig, 1995). Consistent with this view is that the class III mutants responded similarly to the target signals from both TnsD and TnsE. Furthermore, the TnsA loss-of-function mutants generated by site-directed mutagenesis that are insensitive to TnsC stimulation have also lost their ability to be activated by TnsD and TnsE targeting signals. These findings suggest that a common transposase activation pathway mediated through the regulator TnsC ensures the maintenance of Tn7 transposon in a diversified background, which reflects the regulatory simplicity of a complicated recombination system.
Interactions between TnsA and TnsC
How does TnsC interact with the TnsAB transposase? Our screen for transposase gain-of-function mutants was performed in the presence of TnsC. We hypothesized that one type of mutant would allow the transposase to interact more efficiently with this (presumably) limiting TnsCon population. We isolated several gain-of-function mutants (class I) in a small segment (65–73) of TnsA that have the properties expected of altered usage of TnsC. Their increase in recombination activity was observed in vivo by increased transposition frequency and in vitro by increased accumulation of recombination products with TnsCATP. These mutants also responded well with the activation signals from TnsD and TnsE. One attractive explanation is that these mutations fall in a region involved in TnsA–TnsC interaction. That TnsA and TnsC could interact was previously demonstrated by Stellwagen et al. in pull-down assays (A.Stellwagen and N.L.Craig, unpublished observation). We have shown here that both gain-of-function mutations identified by screening for a particular transposition phenotype and loss-of-function mutations generated by site-directed mutagenesis in the 65–81 amino acid region of TnsA appear to affect TnsA–TnsC signals. The fact that this segment is included in a region of TnsA that is protected from protease attack supports the view that the 65–81 amino acid region is key to TnsA–TnsC interaction. Strikingly, the 65–81 segment is included on an α-helix that supports an active site reside (E63) on one region of TnsA and which is also exposed to a large hydrophobic surface on another face of TnsA (Hickman et al., 2000). An attractive hypothesis is that a specific interaction between TnsA and TnsC in this hydrophobic region induces a structural change in TnsA that brings all residues participating in catalysis into the required proximity and precise orientation. Alternatively, the TnsA–TnsC interaction may facilitate assembly of the functional transposition complex. Future analysis will be required to investigate these interesting possibilities.
Materials and methods
Bacterial strains, plasmids and phage
NLC51 is E.coli F–araD139 Δ (argF-lac) U169 rpsl150 relA1 flbB5301 deoC1 ptsF25 rbsR valR recA56 (McKown et al., 1987); CW51 is E.coli F– ara– arg– lac proXIII recA56 NalR RifR (Waddell and Craig, 1988). Several conjugable derivatives of F plasmid were used. pOX-G carries a gentamicin resistance gene (Johnson and Reznikoff, 1984). pOX-miniTn7lac is pOX-G containing a kanamycin resistance gene and a promoterless lacZY operon flanked by the essential transposon end sequences, 166 bp from the left end of Tn7(Tn7L) and 90 bp from the right end of (Tn7R) (R.Deboy and N.L.Craig, unpublished observation). Phage λKK1 is a derivative of λ780 (b2::hisOGD b522 cI857 Pam80 nin5) with hisG9424::Tn10 del16 del17::attTn7(–342 to +165):: miniTn7KmR (McKown et al., 1988).
Mutagenesis of tnsAB and papillation assay
pCW43 (tnsA) (Waddell and Craig, 1988) was treated with 1 M hydroxylamine hydrochloride (in NaOH) at 37°C for 24 h. The mutagen was dialyzed away from the DNA in TE buffer. The DNA was recovered by ethanol precipitation and transformed by electroporation into CW51(pOX-miniTn7lac) with wild-type tnsBC on a compatible plasmid pFLU290. pFLU290 (tnsBC) was constructed by deleting the BglII fragment from pCW15 (Waddell and Craig, 1988). The transformants were plated on MacConkey plates containing appropriate antibiotics. The plates were incubated at 30°C for 3 days and transformants were screened for the papillation phenotype.
Another screen was carried out for the gain-of-function mutants for both tnsA and tnsB genes. The mutagenized plasmid pFLU211(tnsAB) was transformed into CW51(pOX-miniTn7lac) while the wild-type tnsC was supplied on a compatible plasmid pKAO53 (Orle and Craig, 1991). pFLU211(tnsAB) was constructed by deleting the BamHI fragment from pCW15 (Waddell and Craig, 1988); ∼5000 transformants were screened. The transformants giving rise to red papillae were selected as candidates for the transposase gain-of-function mutants. Plasmid DNA was extracted from these potential mutants and retransformed into the same strain background to verify their phenotypes. For the nonsense mutant TnsAQ261Z, its genotype was confirmed by generating a truncated tnsA gene deleting the last 36 bp.
Complementation between TnsA and TnsB mutants was examined using the papillation assay by co-transforming pCW43(tnsA/A*) and pFLU250(tnsB/B*) into CW51(pOX-miniTn7lac). pFLU250 was constructed by deleting the BglII fragment of pFLU211 containing wild-type or mutant TnsB.
λ hop assay
The transposition frequency of a miniTn7KmR element from the integration- and replication-defective phage λKK1 into the chromosome of E.coli strain NLC51 was evaluated when Tns proteins were supplied. The protocol was adapted from McKown et al. (1988). The transposition frequency was determined by the number of KmR colonies/p.f.u. For TnsA mutants, tnsABC was provided by pCW15 while tnsD and tnsE were provided by pCW23 and pCW30, respectively (Waddell and Craig, 1988). pCW15(tnsA*BC) was constructed by subcloning the NcoI–BglII fragment of pCW43(tnsA*) into pCW15. For TnsB mutants, tnsAB was provided by pFLU211 containing wild-type or mutant TnsB while tnsCD was provided by pFLU280. pFLU280 was constructed by inserting the PstI–EcoRI fragment from pCW53 (Waddell and Craig, 1988) into pGB2 (Churchward et al., 1984).
Tns proteins
GST–TnsA expression vector pFLU221 was constructed by inserting a PCR-generated fragment that encodes TnsA into pGEX-1λT between the BamHI and EcoRI sites. The TnsA mutations were cloned into pFLU221 by replacing the NcoI–EcoRI fragment with one from pCW43(tnsA*). GST–TnsA was expressed and purified as described previously (Bainton et al., 1993). TnsA was freed from the GST moiety through a limited proteolytic treatment with thrombin. TnsB was expressed as a fusion protein TnsB-HSV-His6, which contains amino acids 1–694 of TnsB (702 amino acids) fused to a 15 amino acid linker containing an HSV epitope followed by a His6 tag (Sarnovsky et al., 1996). The TnsB mutations were subcloned into the TnsB-HSV-His6 expression vector pBTAG by replacing the AflII–NcoI fragment with one from pFLU211(tnsAB*) and expressed and purified as described previously (Sarnovsky et al., 1996). TnsC was fraction III (Gamas and Craig, 1992). TnsD was TnsD-His, a derivative containing a C-terminal polyhistidine tag (Sarnovsky et al., 1996; Sharpe and Craig, 1998).
In vitro transposition reactions
Reactions were performed essentially as described previously (Bainton et al., 1993). The donor plasmid pEMΔ (5.9 kb) contains a 1.6 kb miniTn7KmR element. The 3.2 kb target plasmid pRM2 (McKown et al., 1988) contains a 555 bp attTn7 segment (–342 to +165) in the AccI site of pUC18. Reaction mixtures (100 µl final volume) contained 0.25 nM pEMΔ donor DNA, 2.5 nM pRM2 target plasmid, 28 mM HEPES pH 8.0, 2.2 mM diothiothreitol (DTT), 4.4 mM Tris pH 7.5, 100 µg/ml tRNA, 50 µg/ml bovine serum albumin (BSA), 0.16 mM EDTA, 0.1 mM MgCl2, 0.1 mM CHAPS, 30 mM NaCl, 21 mM KCl, 1.8% glycerol, 2.0 mM ATP and 15 mM MgAc unless indicated otherwise. Tns proteins were added as follows: 40 ng of TnsA, 25 ng of TnsB, 30 ng of TnsC, 22 ng of TnsD. Reaction mixtures containing all components except donor DNA, TnsA, TnsB and MgAc were assembled on ice. In Figure 2B and C, TnsD was omitted from the assembly reaction. In Figure 3, both TnsC and TnsD were omitted from the assembly reaction and 20% of glycerol was included in the mixtures. The assembly reaction mixtures were incubated for 30 min at 30°C; donor DNA, TnsA, TnsB, and MgAc were then added and the incubation was continued for an additional 20 min (Figure 4), or for 80 min (Figures 2 and 3). Reactions were stopped by adjusting to 25 mM EDTA and 300 mM NaAc, followed by extraction with phenol:chloroform (1:1); the DNA was ethanol precipitated, digested with NdeI and analyzed on 0.8% agarose gel or 1.2% agarose gel for Figure 3. Electrophoresis was carried out at 50 V for 16 h. The DNAs were transferred to Gene Screen Plus and analyzed by hybridization with a probe specific to miniTn7KmR. The probe was labeled by random priming with [α-32P]dCTP and the Klenow fragment of DNA polymerase I (BMB). All blots were analyzed using a Molecular Dynamics PhosphorImager.
Peptide sequencing and protease footprinting assay
TnsA was subjected to limited proteolysis by chymotryspin or trypsin. Proteolysis was stopped by addition of phenylmethylsulfonyl fluoride (PMSF) and fragments were resolved by Tricine–PAGE. Polypeptides were transferred to Immobolin P membrane, stained with Coomassie Blue and subjected to automated sequencing. For the protease footprinting assay, 180 ng of TnsA were incubated without or with 250 ng of TnsC for 30 min at 30°C in 30 µl of the buffer containing 25 mM HEPES pH 8.0, 1.25 mM DTT, 290 mM NaCl, 1 mM EDTA, 5.8% glycerol, 10 mM Tris pH 8.0, 1 mM CaCl2, and components derived from TnsC buffer, 0.16 mM ATP, 1.6 mM MgCl2, 1.6 mM CHAPS (final concentrations). The mixture was then treated with protease in the presence of 50 mM (NH4)2CO3. The amounts of chymotrypsin and trypsin added are indicated in the figure legends. After 15 min incubation at 37°C, the reactions were terminated by addition of PMSF to 0.5 mM and resolved by 10% SDS–PAGE. Protein samples were then transferred to Immobolin P membrane. Western blot analyses were performed by using polyclonal antiserum to TnsA and a chemiluminescent kit (Pierce).
Genetic analysis of site-directed TnsA mutations
Site-directed mutations in TnsA were generated by PCR with oligonucleotides containing appropriately altered tnsA sequence, and then introduced back into pCW43 plasmid by cloning and verified by direct DNA sequencing. The transposition frequencies of the mutants in TnsABC and TnsABCA225V reactions were determined based on papillation assay. pCW43(tnsA/A*) was transformed into CW51(pOX-miniTn7lac) with a compatible plasmid pFLU290 containing either tnsBCWT or tnsBCA225V. The TnsABC+D and TnsABC+E transposition frequencies of the mutants were determined based on λ hop assay in strain NLC51. tnsABC came from pCW15. pCW15(tnsA*BC) was constructed by subcloning the NcoI–BglII fragment of pCW43(tnsA*) into pCW15. tnsD and tnsE were provided by pCW23 and pCW30, respectively (Waddell and Craig, 1988). The dominant negative assay used a strain NLC51(pOX::TpRattTn7::tnsABCD) (Z.S.Skelding and N.L.Craig, unpublished data) containing pCW15(tnsA*BC). The effects of TnsA mutants on the transposition frequency of TnsAWTBC+D were determined by the λ hop assay.
Gel mobility shift assay
Protein–DNA complexes were examined using a gel mobility shift assay. The DNA substrate is a 250 bp fragment derived from pPK13 (K.Kubo, P.Kuduvalli and N.L.Craig, unpublished data) by XbaI–HindIII digestion, which contains attTn7 (–52 to +64). Reaction mixtures (20 µl final volume) contained 36 mM HEPES pH 8.0, 4.2 mM DTT, 5.2 mM Tris pH 7.5, 50 µg/ml BSA, 0.48 mM EDTA, 0.3 mM MgCl2, 0.3 mM CHAPS, 94 mM NaCl, 27 mM KCl, 5% glycerol, 7 mM ATP, 40 µg/ml sheared salmon sperm DNA, and ∼0.01 pmol of 5′ α-32P-labeled DNA substrate. Tns proteins were added as follows: 30 ng of TnsC, 22 ng of TnsD and various amounts of TnsA as indicated. Preincubation reaction mixtures containing all components except TnsA were assembled on ice and incubated for 20 min at 30°C; TnsA was then added and the incubation was continued for an additional 20 min. The reactions were electrophoresed through 4.8% polyacrylamide gels (37.5:1 acrylamide: N, N′-methylene-biacrylamide) in Tris–borate–EDTA buffer at 10 V/cm for 5 h. Gels were vacuum dried and exposed to a PhosphorImager screen.
Acknowledgments
Acknowledgements
We are grateful to Alison Hickman and Fred Dyda for their information about the crystal structure of TnsA. We also thank Jef Boeke, Brendan Cormack, and members of the Craig laboratory for comments on the manuscript and Patti Eckhoff for her assistance in its preparation. N.L.C. is an Investigator of the Howard Hughes Medical Institute.
References
- Arciszewska L.K. and Craig,N.L. (1991) Interaction of the Tn7-encoded transposition protein TnsB with the ends of the transposon. Nucleic Acids Res., 19, 5021–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arciszewska L.K., McKown,R.L. and Craig,N.L. (1991) Purification of TnsB, a transposition protein that binds to the ends of Tn7. J. Biol. Chem., 266, 21736–21744. [PubMed] [Google Scholar]
- Bainton R., Gamas,P. and Craig,N.L. (1991) Tn7 transposition in vitro proceeds through an excised transposon intermediate generated by staggered breaks in DNA. Cell, 65, 805–816. [DOI] [PubMed] [Google Scholar]
- Bainton R.J., Kubo,K.M., Feng,J.-N. and Craig,N.L. (1993) Tn7 transposition: target DNA recognition is mediated by multiple Tn7-encoded proteins in a purified in vitro system. Cell, 72, 931–943. [DOI] [PubMed] [Google Scholar]
- Baker T.A. and Luo,L. (1994) Identification of residues in the Mu transposase essential for catalysis. Proc. Natl Acad. Sci. USA, 91, 6654–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth P.T., Datta,N., Hedges,R.W. and Grinter,N.J. (1976) Transposition of a deoxyribonucleic acid sequence encoding trimethoprim and streptomycin resistances from R483 to other replicons. J. Bacteriol., 125, 800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biery M., Lopata,M. and Craig,N.L. (2000a) A minimal system for Tn7 transposition: the transposon-encoded proteins TnsA and TnsB can execute DNA breakage and joining reactions that generate circularized Tn7 species. J. Mol. Biol., 29, 25–37. [DOI] [PubMed] [Google Scholar]
- Biery M.C., Steward,F., Stellwagen,A.E., Raleigh,E.A. and Craig,N.L. (2000b) A simple in vitro Tn7-based transposition system with low target site selectivity for genome and gene analysis. Nucleic Acids Res., 28, 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely G., May,G., McCulloch,R., Arciszewska,L.K., Burke,M., Lovett,S.T. and Sherratt,D.J. (1993) Two related recombinases are required for site-specific recombination at dif and cer in E.coli K12. Cell, 75, 351–361. [DOI] [PubMed] [Google Scholar]
- Bolland S. and Kleckner,N. (1996) The three chemical steps of Tn10/IS10 transposition involve repeated utilization of a single active site. Cell, 84, 223–233. [DOI] [PubMed] [Google Scholar]
- Bujacz G., Jaskólski,M., Alexandratos,J., Wlodawer,A., Merkel,G., Katz,R.A. and Skalka,A.M. (1996) The catalytic domain of avian sarcoma virus integrase: conformation of the active-site residues in the presence of divalent cations. Structure, 4, 89–96. [DOI] [PubMed] [Google Scholar]
- Carson M. (1997) ‘Ribbons’. Methods Enzymol., 277, 493–505. [PubMed] [Google Scholar]
- Chalmers R.M. and Kleckner,N. (1994) Tn10/IS10 transposase purification, activation and in vitro reaction. J. Biol. Chem., 269, 8029–8035. [PubMed] [Google Scholar]
- Christopher J.A. (1998) SPOCK: The Structural Properties Observation and Calculation Kit (Program Manual). Texas A&M University, College Station, TX. [Google Scholar]
- Churchward G., Beline,D. and Nagamine,Y. (1984) A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene, 31, 165–171. [DOI] [PubMed] [Google Scholar]
- Craig N.L. (1995) Transposon Tn7. Curr. Top. Microbiol. Immunol., 204, 27–48. [DOI] [PubMed] [Google Scholar]
- Craigie R., Fujiwara,T. and Bushman,F.D. (1990) The IN protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro. Cell, 62, 829–837. [DOI] [PubMed] [Google Scholar]
- Dyda F., Hickman,A.B., Jenkins,T.M., Engelman,A., Craigie,R. and Davies,D.R. (1994) Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science, 266, 1981–1986. [DOI] [PubMed] [Google Scholar]
- Engelman A., Mizuuchi,K. and Craigie,R. (1991) HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell, 67, 1211–1221. [DOI] [PubMed] [Google Scholar]
- Gamas P. and Craig,N.L. (1992) Purification and characterization of TnsC, a Tn7 transposition protein that binds ATP and DNA. Nucleic Acids Res., 20, 2525–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haniford D.B., Chelouche,A.R. and Kleckner,N. (1989) A specific class of IS10 transposase mutants are blocked for target site interactions and promote formation of an excised transposon fragment. Cell, 59, 385–394. [DOI] [PubMed] [Google Scholar]
- Hickman A.B., Li,L., Mathew,S.V., May,E.W., Craig,N.L. and Dyda,F. (2000) Unexpected structural diversity in DNA recombination: the restriction endonuclease connection. Mol. Cell, in press. [DOI] [PubMed] [Google Scholar]
- Johnson R.C. and Reznikoff,W.S. (1984) Copy number control of Tn5 transposition. Genetics, 107, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K., Namgoong,S.-Y., Jayaram,M. and Harshey,R.M. (1995) Step-arrest mutants of phage Mu transposase. J. Biol. Chem., 270, 1472–1479. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Kubo K.M. and Craig,N.L. (1990) Bacterial transposon Tn7 utilizes two classes of target sites. J. Bacteriol., 172, 2774–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkosky J., Jones,K.S., Katz,R.S., Mack,J.P.G. and Skalka,A.M. (1992) Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol. Cell. Biol., 12, 2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May E.W. and Craig,N.L. (1996) Switching from cut-and-paste to replicative Tn7 transposition. Science, 272, 401–404. [DOI] [PubMed] [Google Scholar]
- McBlane J.F., van Gent,D.C., Ramsden,D.A., Romeo,C., Cuomo,C.A., Gellert,M. and Oettinger,M.A. (1995) Cleavage at a V(D)J recombination signal requires only RAG1 and RAG2 proteins and occurs in two steps. Cell, 83, 387–395. [DOI] [PubMed] [Google Scholar]
- McKown R.L., Waddell,C.S., Arciszewska,L.A. and Craig,N.L. (1987) Identification of a transposon Tn7-dependent DNA-binding activity that recognizes the ends of Tn7. Proc. Natl Acad. Sci. USA, 84, 7807–7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKown R.L., Orle,K.A., Chen,T. and Craig,N.L. (1988) Sequence requirements of Escherichia coli attTn7, a specific site of transposon Tn7 insertion. J. Bacteriol., 170, 352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melek M., Gellert,M. and van Gent,D. (1998) Rejoining of DNA by the RAG1 and RAG2 proteins. Science, 280, 301–303. [DOI] [PubMed] [Google Scholar]
- Orle K.A. and Craig,N.L. (1991) Identification of transposition proteins encoded by the bacterial transposon Tn7. Gene, 104, 125–131. [DOI] [PubMed] [Google Scholar]
- Polard P. and Chandler,M. (1995) Bacterial transposases and retroviral integrases. Mol. Microbiol., 15, 1–23. [DOI] [PubMed] [Google Scholar]
- Radstrom P., Skold,O., Swedberg,G., Flensburg,F., Roy,P.H. and Sundstrom,L. (1994) Transposon Tn5090 of plasmid R751, which carries an integron, is related to Tn7, Mu and the retroelements. J. Bacteriol., 176, 3257–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezsohazy R., Hallet,B., Delcour,J. and Mahillon,J. (1993) The IS4 family of insertion sequences: evidence for a conserved transposase motif. Mol. Microbiol., 9, 1283–1295. [DOI] [PubMed] [Google Scholar]
- Rice P. and Mizuuchi,K. (1995) Structure of the bacteriophage mu transposase core: a common structural motif for DNA transposition and retroviral integration. Cell, 82, 209–220. [DOI] [PubMed] [Google Scholar]
- Rogers M., Ekaterinaki,N., Nimmo,E. and Sherratt,D. (1986) Analysis of Tn7 transposition. Mol. Gen. Genet., 205, 550–556. [DOI] [PubMed] [Google Scholar]
- Roth D.B. and Craig,N.L. (1998) VDJ recombination: a transposase goes to work. Cell, 94, 1–20. [DOI] [PubMed] [Google Scholar]
- Sarnovsky R., May,E.W. and Craig,N.L. (1996) The Tn7 transposase is a heteromeric complex in which DNA breakage and joining activities are distributed between different gene products. EMBO J., 15, 6348–6361. [PMC free article] [PubMed] [Google Scholar]
- Sharpe P. and Craig,N.L. (1998) Host proteins can stimulate Tn7 transposition: a novel role for the ribosomal protein L29 and the acyl carrier protein. EMBO J., 17, 5822–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiers A.J. and Sherratt,D.J. (1999) C-terminal interactions between the XerC and XerD site-specific recombinases. Mol. Microbiol., 32, 1031–1042. [DOI] [PubMed] [Google Scholar]
- Stellwagen A. and Craig,N.L. (1997) Gain-of-function mutations in TnsC, an ATP-dependent transposition protein which activates the bacterial transposon Tn7. Genetics, 145, 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen A. and Craig,N.L. (1998) Mobile DNA elements: controlling transposition with ATP-dependent molecular switches. Trends Biol. Sci., 23, 486–490. [DOI] [PubMed] [Google Scholar]
- Subramanya H.S., Arciszewska,L.K., Baker,R.A., Bird,L.E., Sherratt,D.J. and Wigley,D.B. (1997) Crystal structure of the site-specific recombinase, XerD. EMBO J., 16, 5178–5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Luenen H.G., Colloms,S.D. and Plasterk,R.H.A. (1994) The mechanism of transposition of Tc3 in C.elegans. Cell, 79, 293–301. [DOI] [PubMed] [Google Scholar]
- Vink C., Yeheskiely,E., van der Marel,G.A., van Boom,J.H. and Plasterk,R.H. (1991) Site-specific hydrolysis and alcoholysis of human immunodeficiency virus DNA termini mediated by the viral integrase protein. Nucleic Acids Res., 19, 6691–6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos J.C., De Baere,I. and Plasterk,R.H.A. (1996) Transposase is the only nematode protein required for in vitro transposition of Tc1. Genes Dev., 10, 755–761. [DOI] [PubMed] [Google Scholar]
- Waddell C.S. and Craig,N.L. (1988) Tn7 transposition: two transposition pathways directed by five Tn7-encoded genes. Genes Dev., 2, 137–149. [DOI] [PubMed] [Google Scholar]
- Wolkow C.A., DeBoy,R.T. and Craig,N.L. (1996) Conjugating plasmids are preferred targets for Tn7. Genes Dev., 10, 2145–2157. [DOI] [PubMed] [Google Scholar]