

Abstract
The innate immune system uses Toll family receptors to signal for the presence of microbes and initiate host defense. Bacterial lipoproteins (BLPs), which are expressed by all bacteria, are potent activators of Toll-like receptor-2 (TLR2). Here we show that the adaptor molecule, myeloid differentiation factor 88 (MyD88), mediates both apoptosis and nuclear factor-κB (NF-κB) activation by BLP-stimulated TLR2. Inhibition of the NF-κB pathway downstream of MyD88 potentiates apoptosis, indicating that these two pathways bifurcate at the level of MyD88. TLR2 signals for apoptosis through MyD88 via a pathway involving Fas-associated death domain protein (FADD) and caspase 8. Moreover, MyD88 binds FADD and is sufficient to induce apoptosis. These data indicate that TLR2 is a novel ‘death receptor’ that engages the apoptotic machinery without a conventional cytoplasmic death domain. Through TLR2, BLP induces the synthesis of the precursor of the pro-inflammatory cytokine interleukin-1β (IL-1β). Interestingly, BLP also activates caspase 1 through TLR2, resulting in proteolysis and secretion of mature IL-1β. These results indicate that caspase activation is an innate immune response to microbial pathogens, culminating in apoptosis and cytokine production.
Keywords: apoptosis/bacterial lipoprotein/MyD88/NF-κB/TLR2
Introduction
Bacterial infections are a major cause of morbidity and mortality worldwide (Hinman, 1998). The ability of the innate immune system to respond to pathogens is central to host defense. Unlike adaptive immunity, which takes weeks to generate, innate immune responses are mobilized within hours of exposure to a pathogen. This is accomplished by germline-encoded receptors on the surface of cells of the innate immune system, which identify molecules produced exclusively by bacteria, termed pathogen-associated molecular patterns (PAMPs) (Medzhitov and Janeway, 1997b). Examples of PAMPs are lipopolysaccharide (LPS), bacterial lipoprotein (BLP), peptidoglycans and lipoteichoic acids (Henderson et al., 1996). Subsequent to recognition, numerous host defense mechanisms are enabled, including the production of pro-inflammatory cytokines, antimicrobial compounds (Medzhitov and Janeway, 1997b) and co-stimulatory molecules (Medzhitov and Janeway, 1997a).
Originally identified as components of developmental pathways in Drosophila, members of the Toll family have emerged as signaling receptors for PAMPs (Kopp and Medzhitov, 1999). Human Toll-like receptor-2 (TLR2) transmits intracellular signals in response to BLP. The characteristic N-terminal lipo-amino acid, N-acyl-S- diacylglyceryl cysteine, common to all BLPs, is necessary to activate TLR2 signaling. Synthetic BLP analogs consisting of a palmitylated version of N-acyl-S-diacylglyceryl cysteine (Pam3Cys) and a few C-terminal amino acids can mimic the immunomodulatory effects of BLP, including TLR2 activation (Aliprantis et al., 1999; Brightbill et al., 1999; Hirschfeld et al., 1999). BLPs are particularly attractive targets for innate immune recognition since they are produced by all bacterial pathogens. TLR2 has also been implicated in signaling induced by Gram-positive cell walls, peptidoglycan and mycobacterial factors (Means et al., 1999; Schwandner et al., 1999; Takeuchi et al., 1999; Yoshimura et al., 1999). TLR4 is required for cellular responses to LPS, an outer membrane glycolipid found in Gram-negative bacteria (Poltorak et al., 1998; Hoshino et al., 1999; Shimazu et al., 1999; Takeuchi et al., 1999).
Toll receptors have an extracellular leucine-rich repeat (LRR) domain and an interleukin-1 receptor (IL-1R) type 1-like intracellular signaling domain (Rock et al., 1998), called the Toll/IL-1R homology region (TIR). Through their TIR domains, these receptors recruit the adaptor molecule, myeloid differentiation factor 88 (MyD88). This interaction is mediated through a homotypic interaction with a TIR domain in the C-terminus of MyD88 (Medzhitov et al., 1998; Muzio et al., 1998a; O’Neill and Greene, 1998). MyD88 is necessary for Toll receptor and IL-1R signaling since MyD88-deficient mice are hyporesponsive to LPS and IL-1 (Adachi et al., 1998; Kawai et al., 1999). MyD88 initiates a signaling pathway which sequentially involves IL-1R-associated kinase 1 (IRAK1), tumor necrosis factor-α receptor (TNFR)-associated factor 6 (TRAF6) and nuclear factor (NF)-κB-inducing kinase (NIK) (Medzhitov et al., 1998; Muzio et al., 1998a; O’Neill and Greene, 1998; Kopp and Medzhitov, 1999; Yang et al., 1999; Zhang et al., 1999). The NF-κB/Rel family of transcription factors are maintained in the cytoplasm as inactive complexes with inhibitory proteins, called IκBs (Baeuerle, 1998). NIK activates the IκB kinase complex (IKKα and IKKβ) (Ling et al., 1998), which phosphorylates the IκBs, targeting them for ubiquitylation and degradation by the proteasome (DiDonato et al., 1997; Zandi et al., 1997). Degradation of IκB liberates NF-κB–Rel dimers, which translocate to the nucleus and augment expression of the immune response and anti-apoptotic genes (for reviews see Baeuerle and Baichwal, 1997; Baichwal and Baeuerle, 1997).
We have demonstrated recently that in addition to stimulating NF-κB, TLR2 transmits a pro-apoptotic signal (Aliprantis et al., 1999). Members of the TNFR superfamily, also known as death receptors (DRs), activate these two pathways as well. DRs induce apoptosis through adaptor molecules, which couple the receptor to the apoptotic machinery (Ashkenazi and Dixit, 1998). Fas-associated death domain protein (FADD) is a central adaptor. FADD contains a C-terminal death domain (DD) that tethers it to the receptor complex, and an N-terminal death effector domain (DED) that binds a homologous region in the pro-domain of caspase 8 (Baker and Reddy, 1998). Caspases are cytoplasmic cysteine proteases that initiate and propagate apoptotic signals (Thornberry and Lazebnik, 1998). Caspases are synthesized as zymogens and mature into active enzymes by limited proteolysis. Oligomerization of caspase 8 at the receptor complex by FADD results in maturation of the enzyme (Martin et al., 1998; Muzio et al., 1998b). Mature caspase 8 cleaves downstream effector caspases, which execute the apoptotic program.
Here, we show that MyD88, which contains an N-terminal DD (Hardiman et al., 1996), transmits the signals for NF-κB activation and apoptosis downstream of TLR2 activated by BLP. Inhibition of NF-κB activation downstream of MyD88 promotes the induction of apoptosis in both 293 cells and monocytes, suggesting that NF-κB up-regulates survival signals. MyD88 engages FADD, which can bind to and activate caspase 8. These interactions couple TLR2 to the apoptotic machinery, culminating in cell death. Concomitant with BLP-induced apoptosis, caspase 1 is activated. Although the enzymatic activity of caspase 1 is not necessary for cell death to proceed, this enzyme cleaves pro-IL-1β to its mature, pro-inflammatory form, which is released from the dying cells.
Results
Identification of MyD88 as a common mediator of TLR2-induced apoptosis and NF-κB activation
293 cells transfected with exogenous TLR2 activate NF-κB and undergo apoptosis in response to BLP (Aliprantis et al., 1999). We investigated how TLR2 activates NF-κB and apoptosis after stimulation by BLP. 293 cells were transiently co-transfected with a gD epitope-tagged version of human TLR2 (gD-TLR2) and dominant-negative (DN) versions of one of the following signaling proteins: MyD88, TRAF2, TRAF6, NIK or IKKβ. After transfection, the cells were exposed to the synthetic BLP, Pam3CysSerLys4 (sBLP), and tested for their ability to activate an NF-κB-regulated reporter gene (Figure 1A) or undergo apoptosis (Figure 1B). Both pathways were only activated in the presence of sBLP. Cells transfected with a gD epitope-tagged 141 amino acid C-terminal deletion mutant of TLR2 (TLR2Δ2), which lacks a large portion of the intracellular TIR domain, activated neither apoptosis nor the NF-κB-regulated reporter gene in response to sBLP (Figure 1A and B). MyD88-DN, which lacks the N-terminal DD, inhibited both NF-κB activation and apoptosis after BLP stimulation (Figure 1A and B). Consistent with the previously described signaling pathway downstream of the TLRs (Medzhitov et al., 1998; Muzio et al., 1998a; Yang et al., 1999), TRAF6-DN, NIK-DN and IKKβ-DN all attenuated NF-κB activation (Figure 1A). TRAF6-DN consistently inhibited TLR2-mediated NF-κB signaling to a lesser extent than MyD88-DN or NIK-DN, suggesting that TRAF6-independent pathways may exist downstream of MyD88, which converge at the level of NIK. In contrast to MyD88-DN, the dominant-negative versions of TRAF6, NIK and IKKβ failed to prevent apoptosis (Figure 1B). TRAF2-DN inhibited neither pathway, indicating that co-transfection alone is not responsible for the observed inhibitory effects. This is consistent with prior studies, which demonstrated that TRAF2-DN blocks NF-κB activation by TNFR, but not by IL-1R or TLR2 (Cao et al., 1996; Yang et al., 1999), although TRAF2 is not absolutely required for TNFR activation of NF-κB (Yeh et al., 1997). As a control, we verified that expression of gD-TLR2 was equal in this co-transfection assay (Figure 1C). Taken together, these data indicate that in response to BLP, the TIR domain of TLR2 relays signals for NF-κB activation and apoptosis through the adaptor molecule MyD88.
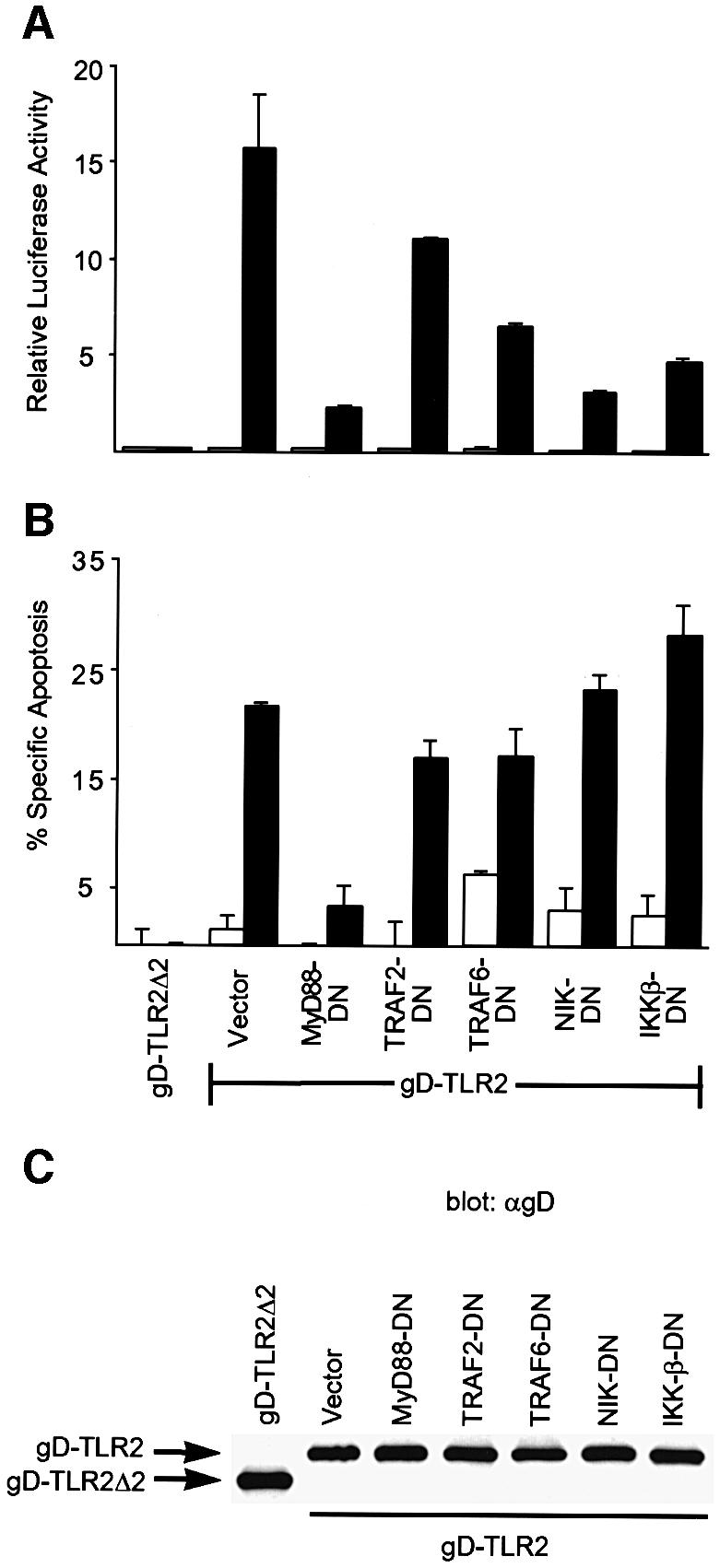
Fig. 1. MyD88 is a component of the NF-κB and apoptotic pathways initiated by BLP through hTLR2. (A and B) 293 cells were transiently co-transfected with equivalent amounts (0.25 µg) of expression plasmids encoding gD-TLR2 or gD-TLR2Δ2 and the indicated dominant negatives. An NF-κB-regulated luciferase reporter plasmid was also transfected. At 24 h post-transfection, the cells were incubated in medium alone (white bars) or in medium with 1 µg/ml sBLP (black bars). The induction of NF-κB-dependent transcription (A) was determined. Apoptosis (B) was measured by TUNEL analysis. Data in (B) are reported as the percentage specific apoptosis relative to cells transfected with gD-TLR2Δ2 without sBLP. Results are the average ± SD of two independent samples. (C) Anti-gD western blot of lysates prepared from cells transfected as in (A) and (B).
Inhibition of NF-κB activation downstream of TLR2 augments the apoptotic response
Both NIK-DN and IKKβ-DN slightly enhanced the cytotoxic effect of sBLP on cells transfected with TLR2 (Figure 1B), suggesting that the NF-κB pathway protects cells from apoptosis. To explore this further, we transiently co-transfected 293 cells with decreasing amounts of gD-TLR2 and a constant amount of NIK-DN, exposed the cells to sBLP and measured the apoptotic response (Figure 2A). As little as 0.01 µg of the gD-TLR2 vector was sufficient to induce a maximal sBLP-dependent apoptotic response when co-transfected with NIK-DN. In the absence of NIK-DN, 10-fold more gD-TLR2 vector (0.1 µg) was required to measure appreciable sBLP-induced cell death. Transfection of cells with NIK-DN alone, with or without sBLP, did not induce cell death, indicating that gD-TLR2 is necessary to mediate apoptosis in this system. Finally, we confirmed that the expression of gD-TLR2 was proportional to the amount of transfected gD-TLR2 vector and independent of co-transfection with NIK-DN (Figure 2B).

Fig. 2. Inhibition of the NF-κB pathway facilitates TLR2-mediated apoptosis. (A) 293 cells were transiently transfected with 0.25, 0.1, 0.04, 0.01 and, in addition for co-transfections with NIK-DN, 0.002, 0.0004 and 0.00008 µg of an expression plasmid encoding gD-TLR2 indicated in a gray gradient from black (0.25 µg) to white (0.00008 µg). Where indicated, the cells were also transfected with 0.25 µg of an expression plasmid encoding NIK-DN. Control cells transfected with vector DNA only are indicated with hatched bars. At 24 h post-transfection, the cells were incubated with or without 1 µg/ml sBLP as indicated and apoptosis was determined by TUNEL staining. (B) Anti-gD western blot of lysates prepared from cells transfected as in (A). Cells transfected with 0.002 µg or less had undetectable levels of gD-TLR2.
The human monocytic cell line THP-1 expresses TLR2 endogenously (Kirschning et al., 1998; Zhang et al., 1999). It was reported that THP-1 cells activate NF-κB in response to BLP (Norgard et al., 1996). This was confirmed by following the degradation of the NF-κB inhibitor, IκB-α, by western blot analysis after treating the cells with sBLP. As early as 10 (100 ng/ml sBLP) or 30 min (5 ng/ml sBLP) after exposure to sBLP, IκB-α was degraded in THP-1 cells (Figure 3A). By 120 min, IκB-α was re-synthesized, consistent with the observation that the IκB-α gene contains a κB enhancer element and is up-regulated by NF-κB (Chiao et al., 1994). As a control, we showed that the levels of expression of phospholipase C γ (PLCγ) do not change in response to sBLP. Cells incubated with an inactive, monoacylated derivative of sBLP (msBLP), generated by base hydrolysis (Aliprantis et al., 1999), did not degrade IκB-α.

Fig. 3. Inhibition of IκB-α degradation augments TLR2-mediated cell death in THP-1 cells. (A) sBLP induces the degradation of IκB-α in THP-1 cells. THP-1 cells were incubated with medium, msBLP (100 ng/ml) or sBLP (5 or 100 ng/ml) for the indicated times and analyzed by western blotting for IκB-α and for PLCγ, as a loading control. (B) A proteasome inhibitor and an anti-TLR2 blocking mAb inhibit IκB-α degradation in sBLP-treated THP-1 cells. THP-1 cells were pre-incubated with either medium alone, the indicated concentrations of lactacystin β-lactone, 25 µg/ml anti-TLR2 mAb 2392 or an isotype control mAb. sBLP was added to a final concentration of 5 ng/ml. After 30 min, the cells were analyzed as in (A). (C) Lactacystin β-lactone facilitates sBLP-mediated apoptosis in THP-1 cells. THP-1 cells were pre-incubated with the indicated concentrations of lactacystin β-lactone. Either medium alone (circles) or medium with sBLP was added to yield final sBLP concentrations of 2.5 ng/ml (diamonds) or 0.25 ng/ml (squares). (D) sBLP and MLP, but not Pam3Cys, msBLP or LPS, kill lactacystin β-lactone-treated THP-1 cells. THP-1 cells were pre-incubated for 1 h with 5 µM lactacystin β-lactone. Medium only (cross) or medium with either sBLP (diamonds), MLP (squares), Pam3Cys (circles), msBLP (triangles) or LPS (stars) was added to yield the indicated final concentrations. (E) sBLP-induced death of THP-1 cells pre-treated with lactacystin β-lactone is dependent on TLR2. THP-1 cells were pre-incubated for 1 h with 10 µM lactacystin β-lactone in either medium alone (diamonds) or medium with 25 µg/ml anti-TLR2 mAb 2392 (circles) or an isotype control mAb (squares). sBLP was added to yield the indicated final concentrations. Some standard deviations in (C), (D) and (E) are within the limits of the data points.
Transfection of THP-1 cells is very inefficient. Therefore, to assess the role of NF-κB activation on BLP-mediated apoptosis in THP-1 cells, we used the selective proteasome inhibitor, lactacystin β-lactone (Fenteany and Schreiber, 1998). Inhibition of the proteasome precludes IκB-α degradation and thereby prevents NF-κB activation (Palombella et al., 1994; Delic et al., 1998). Pre-treatment of THP-1 cells with 10 µM lactacystin β-lactone inhibited IκB-α degradation and augmented sBLP-mediated cell death in a dose-dependent manner (Figure 3B and C). Doses of lactacystin β-lactone that partially inhibited IκB-α degradation (i.e. 2.5 µM) only marginally increased sBLP killing. The cell death response was proportional to the dose of sBLP employed (Figure 3C, D and E). Purified Escherichia coli murein lipoprotein (MLP) also killed lactacystin β-lactone-treated THP-1 cells. However, neither msBLP nor the lipo-amino acid Pam3Cys were active in this assay (Figure 3D). This is consistent with previous observations that both the acyl groups and peptide moieties of sBLP are critical for biological activity. Shigella flexneri 1A LPS was also non-toxic, confirming that the cytotoxic response is specific for BLP. Moreover, BLP-mediated degradation of IκB-α and apoptosis were inhibited by pre-treatment of the cells with the anti-TLR2 monoclonal antibody (mAb) 2392 (Aliprantis et al., 1999) but not an isotype-matched control mAb (Figure 3B and E). These data indicate that endogenously expressed TLR2 activates NF-κB as well as apoptosis in response to BLP. In addition, these experiments complement the studies with NIK-DN (Figure 2) to demonstrate that the NF-κB pathway protects cells from apoptotic signals generated by TLR2.
TLR2 induces apoptosis through an FADD/caspase 8 pathway
Members of the TNFR superfamily induce apoptosis through DDs located in their cytoplasmic regions (Ashkenazi and Dixit, 1998). The receptor DD binds DD-containing adaptor molecules, through homotypic interactions.
FADD, which contains a C-terminal DD, is critical for the induction of apoptosis by DRs. TLRs do not contain sequences with homology to conventional DDs. However, MyD88, which mediates TLR2-induced apoptosis (Figure 1), has a DD (Hardiman et al., 1996). To test whether FADD is involved in TLR2-mediated apoptosis, we transiently co-transfected 293 cells with gD-TLR2 and FADD-DN and exposed them to sBLP. FADD-DN consists solely of its DD, lacking the N-terminal DED that binds a homologous region in caspase 8. FADD-DN inhibited sBLP-induced apoptosis through gD-TLR2, similarly to MyD88-DN (Figure 4A). The Bcl-2 family of proteins does not block apoptosis initiated by the Fas receptor (Strasser et al., 1995; Huang et al., 1999) and, as shown in Figure 4A, Bcl-xl was also unable to block TLR2-mediated apoptosis, even though it was strongly expressed. Inhibition could not be attributed to transfection variation, as gD-TLR2 was expressed similarly among all transfections (Figure 4B).

Fig. 4. TLR2-induced apoptosis proceeds through a FADD/caspase 8 pathway. (A) TLR2-mediated apoptosis is inhibited by FADD-DN but not by Bcl-xl. 293 cells were transiently co-transfected with 0.25 µg of an expression plasmid encoding gD-TLR2 and 0.3 µg of expression plasmids encoding the indicated dominant negatives or Bcl-xl. At 24 h post-transfection, the cells were incubated with or without 1 µg/ml sBLP as indicated and apoptosis was determined by TUNEL staining. (B) Anti-gD western blot of lysates prepared from cells transfected as in (A) indicating equivalent expression of gD-TLR2 among transfections. (C) TLR2-mediated apoptosis is inhibited by a catalytically inactive mutant of caspase 8. 293 cells were transiently co-transfected with 0.15 µg of an expression plasmid encoding gD-TLR2 and either 0.05 (+), 0.15 (++) or 0.45 µg (+++) of expression plasmids encoding gD-caspase 8-DN or Flag-caspase 1-DN. Apoptosis was induced and quantified as in (A). Data in (A) and (C) are reported as the percentage specific apoptosis relative to cells transfected with gD-TLR2 without sBLP. (D) Anti-gD (upper panel) and anti-Flag (lower panel) western blots of lysates prepared from cells transfected as in (C). (E) sBLP-mediated cell death of THP-1 cells is attenuated by a peptide inhibitor of caspase 8. THP-1 cells were pre-incubated with medium containing 75 µg/ml cycloheximide without (star) or with the indicated concentrations of z-FA-fmk (squares), Ac-YVAD-cmk (circles), z-IETD-fmk (triangles) or z-VAD-fmk (diamonds). sBLP was added to a final concentration of 2 ng/ml. Some standard deviations are within the limits of the data points. (F) Anti-casapse-8 western blots of lysates prepared from 293 cells co-transfected with gD-TLR2 and NIK-DN and incubated for the indicated times (h) with sBLP (1 µg/ml). Pro-caspase 8 (p55) and the p28 proteolytic intermediate are shown. (G) sBLP-mediated cell death is not mediated through TNF or Fas. THP-1 cells were pre-incubated for 4.5 h with medium only (1) or with 5 µg/ml anti-TLR2 mAb 2392 (2), an isotype-matched control mAb (3), anti-Fas (4) or anti-TNF mAb (5) and then with 100 pg/ml sBLP. Cytotoxicity was assayed by LDH release.
Initiator caspases, such as caspase 8, harboring a point mutation of the active site cysteine, can act as dominant-negative inhibitors of TNFR1- and Fas-mediated apoptosis (Boldin et al., 1996; Muzio et al., 1996; Hu et al., 1997). To test whether TLR2 uses caspase 8 to induce apoptosis, we transiently co-transfected 293 cells with gD-TLR2 and increasing amounts of a gD epitope-tagged mutant of caspase 8 with the active site cysteine exchanged for a serine residue (C360S) (gD-caspase 8-DN). gD-caspase 8-DN inhibited sBLP-dependent cell death in a dose-dependent manner (Figure 4C). As a negative control, we co-transfected a Flag epitope-tagged mutant of caspase 1, which harbors a cysteine to alanine exchange at the active site (C285A) (Flag-caspase 1-DN). Caspase 1 is considered to be a non-apoptotic caspase but is critical for the proteolytic maturation of two cytokines: pro-IL-1β and pro-IL-18 (Thornberry and Lazebnik, 1998; Fantuzzi and Dinarello, 1999). Flag-caspase 1-DN did not affect gD-TLR2-mediated apoptosis. Inhibition of cell death by gD-caspase 8-DN was not due to differences in gD-TLR2 expression, as similar levels of receptor were expressed among all samples (Figure 4D).
To investigate which caspases are involved in sBLP-mediated cell death in THP-1 cells, irreversible, specific, peptide inhibitors of caspases were used. Apoptosis was inhibited by 65% in cells pre-incubated with 75 µM of the caspase 8 inhibitor, z-IETD-fluoromethylketone (z-IETD-fmk) (Figure 4E). Previous reports have documented that this peptide inhibits caspase 8 in vitro (Garcia-Calvo et al., 1998) and attenuates Fas- and caspase 8-mediated apoptosis in vivo (Martin et al., 1998; Bortner and Cidlowski, 1999). Moreover, a broad spectrum caspase inhibitor, z-VAD-fmk, completely prevented sBLP-induced apoptosis. A caspase 1 inhibitor, acetyl-YVAD-chlomethylketone (Ac-YVAD-cmk) (Garcia-Calvo et al., 1998), only slightly attenuated cell death. The cathepsin-B peptide inhibitor z-FA-fmk (Esser et al., 1993), which lacks the critical aspartate residue at position P1 necessary for caspase inhibition, was used as a control in these experiments and did not affect cell death. These data indicate that in cells expressing endogenous TLR2, sBLP initiates a cell death pathway through caspase 8. Furthermore, the proteolytic maturation of pro-caspase 8 was activated in response to sBLP (Figure 4F).
Here, we report that FADD and caspase 8 execute TLR2-mediated apoptosis. These molecules are also involved in the cell death pathways downstream of TNFR and Fas (Ashkenazi and Dixit, 1998). We excluded the possibility that TLR2 was activating apoptosis through either of these receptors. Specific blocking mAb to either TNF or Fas did not inhibit sBLP-mediated cytotoxicity in THP-1 cells (Figure 4G). Neither the anti-TNF nor the anti-Fas mAb were cytotoxic by themselves (data not shown). The specific anti-TLR2 mAb 2392 was included as a positive control for inhibition. Taken together, these data indicate that BLP mediates apoptosis specifically through TLR2, and not by activating DR family members.
Overexpression of MyD88 induces apoptosis
We tested whether MyD88 is sufficient to induce apoptosis by overexpressing the protein in 293 cells. Cells transfected with AU1 epitope-tagged full-length MyD88 (AU1-MyD88-FL) exhibited morphological changes characteristic of apoptosis, including cell shrinkage, vacuolation and membrane blebbing (Figure 5A). In addition, MyD88-transfected cells became annexin V positive (Figure 5E), indicating the presence of phosphatidylserine on the outer leaflet of the plasma membrane, a biochemical consequence of apoptotic death. Cells co-expressing AU1-MyD88-FL and NIK-DN underwent more cell death than those transfected with AU1-MyD88-FL alone (Figure 5B and E). In addition, NIK-DN sensitized cells to apoptosis at lower concentrations of AU1-MyD88-FL (Figure 5E). The DD of MyD88 was required to induce cell death since cells transfected with the TIR domain of MyD88 (AU1-MyD88-DN) did not undergo apoptosis (Figure 5C–E). The relative levels of expression of AU1-MyD88-FL and AU1-MyD88-DN were similar, proportional to the amount of transfected DNA and independent of co-transfection with NIK-DN (Figure 5F). These data indicate that MyD88 is sufficient to induce cell death. Similarly to TLR2-mediated apoptosis, suppression of NF-κB activation promotes apoptotic signals generated by MyD88.

Fig. 5. MyD88-FL, but not MyD88-DN, induces apoptosis in 293 cells. (A–D) 293 cells were transiently transfected with 0.25 µg of expression plasmids encoding AU1-MyD88-FL (A and B) or AU1-MyD88-DN (C and D). In (B) and (D), 0.25 µg of an expression plasmid encoding NIK-DN was co-transfected. A plasmid encoding GFP (0.025 µg) was included in the transfection to label transfected cells. GFP-positive cells were photographed 48 h post-transfection. Arrowheads indicate cells with apoptotic morphology. (E) Inhibition of the NF-κB pathway facilitates MyD88-mediated apoptosis. 293 cells were transiently transfected with 0.25 (white bars), 0.1 (gray), 0.04 (black) or 0 µg (diagonal lines) of expression plasmids encoding AU1-MyD88-FL or AU1-MyD88-DN. Where indicated, 0.25 µg of an expression plasmid encoding NIK-DN was co-transfected. A plasmid encoding GFP (0.025 µg) was included to label transfected cells. At 60 h post-transfection, apoptosis was determined by annexin V staining. The data are reported as the percentage specific apoptosis in the GFP-positive (transfected) population relative to cells transfected with vector DNA. A specific increase in apoptosis was only detected in the GFP-positive population. (F) Anti-AU1 western blot of lysates prepared from cells transfected as in (E).
MyD88 binds the cell death adaptor molecule, FADD
To investigate whether AU1-MyD88-FL and FADD interact in vivo, these proteins were overexpressed in 293 cells and a co-immunoprecipitation was performed with an anti-AU1 mAb. The immunoprecipitates were resolved by SDS–PAGE and blotted for FADD (Figure 6A). FADD immunoprecipitated with AU1-MyD88-FL, but not AU1-MyD88-DN. Furthermore, endogenous FADD was detected in the immunoprecipitates of cells transfected with AU1-MyD88-FL alone, indicating that binding is not an artifact of overexpression of both proteins. Western blot analysis of cell lysates and immunoprecipitates for AU1-MyD88-FL and AU1-MyD88-DN indicated similar levels of expression and immunoprecipitation (data not shown). The DD of MyD88 (MyD88-DD) was sufficient to immunoprecipitate FADD (Figure 6B), suggesting that MyD88 binds FADD through a DD–DD interaction.

Fig. 6. MyD88 associates with FADD through its DD–DD interaction. (A) 293 cells were transiently transfected with expression plasmids (1 µg each) encoding FADD, AU1-MyD88-FL or AU1-MyD88-DN alone, or in combination as indicated. At 24 h post-transfection, cell lysates were prepared, and co-immunoprecipitation (IP) and western blotting were performed using antisera as indicated. (B) 293 cells were transiently transfected with expression plasmids (1 µg each) encoding FADD and/or HA epitope-tagged MyD88-DD. The experiment was performed as in (A) except that an anti-HA mAb was used for the IP. (C) 293 cells were transiently transfected with expression plasmids (1 µg each) encoding the death domains (DDs) of MyD88, FADD or RIP alone or in combination. At 24 h post-transfection, cell lysates were prepared, and immunoprecipitation (IP) and western blotting (WB) were performed using the indicated antibodies.
We also showed that the DD–DD interaction between MyD88 and FADD is specific. Hemagglutinin (HA)-MyD88-DD was expressed in 293 cells with AU1-FADD-DD or Myc-RIP-DD (receptor-interacting protein). Immunoprecipitation with an anti-AU1 mAb, which immunoprecipitates AU1-FADD-DD, also pulled down HA-MyD88-DD (Figure 6C). In contrast, immunoprecipitation with an anti-Myc mAb, which precipitates RIP, does not co-immunoprecipitate HA-MyD88-DD. Taken together, these data demonstrate the specificity of the interaction between the DDs of MyD88 and FADD.
BLPs activate caspase 1 through TLR2
To investigate whether TLR2-mediated caspase activation contributes to inflammation, we measured the production of mature IL-1β by THP-1 cells treated with sBLP. IL-1β is synthesized as a 31 kDa, inactive, cytoplasmic precursor lacking a secretion signal sequence. A single proteolytic cleavage of pro-IL-1β, by caspase 1, produces a 17 kDa mature cytokine, which is released from cells by an unknown mechanism (Dinarello, 1997). An increase in the expression of pro-IL-1β was detected in the lysates of THP-1 cells treated for 3 h with sBLP (Figure 7). Mature IL-1β was also released into the culture supernatants, indicating that sBLP activates caspase 1 (Figure 7). Mature IL-1β was not detected in cell lysates (not shown). Pre-treating the cells with the caspase 1-specific inhibitor, Ac-YVAD-cmk, inhibited the maturation of IL-1β after sBLP stimulation (Figure 7), but did not affect pro-IL-1β synthesis (Figure 7). Furthermore, the anti-TLR2 mAb 2392, but not an isotype control mAb, inhibited the production of both pro- and mature IL-1β. No increase in the production of either molecule was detected when cells were treated with the negative control compound msBLP. Taken together, these data show that in monocytic cells, BLP stimulates the synthesis of the cytokine pro-IL-1β through TLR2. TLR2 also activates caspase 1, which processes pro-IL-1β into a mature, pro-inflammatory cytokine that is released from the cell.
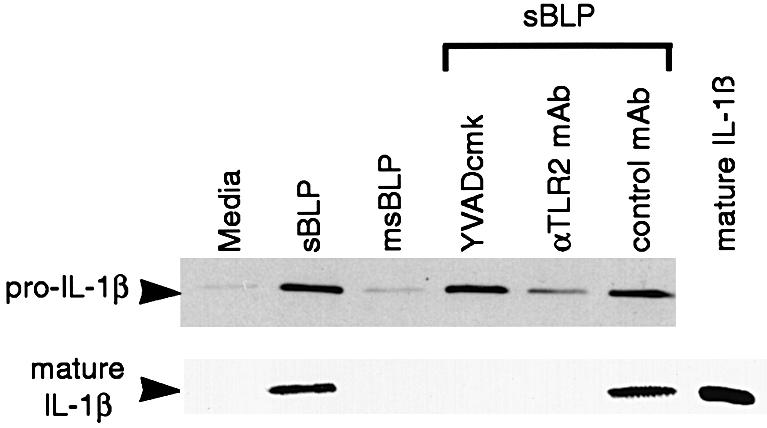
Fig. 7. TLR2 mediates pro-IL-1β synthesis and caspase 1 activation in THP-1 cells treated with sBLP. THP-1 cells were pre-treated for 6 h with 32 nM PMA in the indicated conditions. Where shown, cells were pre-incubated with 10 µM Ac-YVAD-cmk, 25 µg/ml anti-TLR2 mAb 2392 or isotype control mAb and then treated with sBLP at 4 ng/ml. After 3 h, cell lysates were resolved by SDS–PAGE and blotted for pro-IL-1β. Culture supernatants were collected, resolved by SDS–PAGE and blotted for mature IL-1β. As a control for the molecular weight of mature IL-1β, 1 ng of recombinant mature IL-1β was resolved and blotted in parallel.
Discussion
TLR2 signaling: divergence at the level of MyD88
BLP can mediate two responses through TLR2: apoptosis and activation of NF-κB (Aliprantis et al., 1999). Here we describe how TLR2 signals for these two end points. The signal transduction pathway to activate NF-κB downstream of TLR4 and TLR2 was described previously (Medzhitov et al., 1998; Muzio et al., 1998a; Yang et al., 1999). Our results are in accordance with these previous findings and indicate that in response to BLP, the pathway proceeds from the intracellular TIR domain of TLR2 through MyD88, TRAF6, NIK and the IKK complex (Figure 1A).
The pathway from TLR2 to apoptosis also requires the intracellular TIR domain of the receptor and MyD88 (Figure 1B). However, dominant-negative versions of TRAF6, NIK and IKKβ do not inhibit cell death. In fact, inhibition of the NF-κB pathway by NIK-DN promotes BLP-mediated apoptosis in 293 cells expressing TLR2 (Figure 2). Moreover, inhibition of IκB-α degradation by a proteasome inhibitor augments BLP-induced cell death in THP-1 cells (Figure 3), which express endogenous TLR2. Therefore, survival signals regulated by NF-κB are important to suppress TLR2-mediated cell death.
Our data indicate that MyD88 couples TLR2 to the FADD/caspase 8 apoptotic machinery. This conclusion has been reached on the basis of five independent lines of investigation: (i) BLP-induced cell death is inhibited specifically by dominant-negative versions of MyD88, FADD and caspase 8 in 293 cells expressing TLR2 (Figures 1B, 4A and C); (ii) a specific peptide inhibitor of caspase 8 inhibits BLP-induced cell death in THP-1 cells (Figure 4E); (iii) caspase 8 is activated in response to sBLP in cells expressing TLR2 (Figure 4F); (iv) the DD of MyD88 specifically co-immunoprecipitates FADD (Figure 6); and (v) overexpression of MyD88, but not a truncation mutant lacking the DD, is sufficient to induce apoptosis (Figure 5; Jaunin et al., 1998). It is important to note that this last conclusion is based on overexpression studies and in the future the role of MyD88 in apoptosis should be defined in other systems.
TLR2 cell death signaling pathway: parallels to the TNFR superfamily
The proposed signaling pathways to activate NF-κB via TLR and TNFR1 are analogous (Kopp and Medzhitov, 1999). In both cases, the receptors recruit adaptor molecules [MyD88 or TNFR-receptor associated death domain protein (TRADD)] that bind to a serine/threonine kinase (IRAK1 or RIP), which stimulates TRAF proteins (TRAF6 or TRAF2) to activate NIK. NF-κB is generally considered an anti-apoptotic transcription factor, as it up-regulates the expression of cell death inhibitors (Wang et al., 1998). However, NF-κB has been implicated in cell death induction in some cell types (Baichwal and Baeuerle, 1997). Interestingly, and with similarity to TNFR1-induced apoptosis, inhibition of NF-κB activation augments TLR2-mediated cell death (Figures 2 and 3). Using neutralizing monoclonal antibodies to both TNF and Fas, we excluded the possibility that sBLP induces apoptosis through either of these two proteins (Figure 4).
In addition to the NF-κB pathway, TRADD couples TNFR1 to the apoptotic machinery (Yuan, 1997). TRADD recruits FADD to the receptor complex through homotypic interactions mediated by DDs in each protein. Through its DED, FADD binds pro-caspase 8, resulting in oligomerization and enzyme maturation (Martin et al., 1998; Muzio et al., 1998b). Similarly to TRADD, the DD of MyD88 co-immunoprecipitates FADD. Whether this interaction is direct or mediated by other adaptor molecules awaits further experimentation. Unlike the DD of TRADD, the DD of MyD88 does not interact with RIP (Figure 6).
As shown for Fas-mediated apoptosis (Strasser et al., 1995; Huang et al., 1999), Bcl-xl (Figure 4A) and Bcl-2 (our unpublished data) do not inhibit TLR2 activation of cell death. This observation further illustrates the parallels between TLR2 and conventional DR-induced apoptosis. Interestingly, FADD also plays a role in cell proliferation (Newton et al., 1998; Zhang et al., 1998). Whether TLR2 engages cell proliferation pathways through FADD remains to be determined. In light of the role of Toll receptors in Drosophila development, this possibility is quite attractive.
Do other TLR family members engage the cell death pathway?
Multiple groups have reported that LPS can induce cell death. In endothelial cells, LPS-induced apoptosis is augmented by cycloheximide, an inhibitor of protein synthesis (Choi et al., 1998). In macrophages, inhibition of NF-κB activation, by either a proteasome inhibitor (Ruckdeschel et al., 1998) or an IκB-α super-repressor mutant (Kitamura, 1999), promotes LPS-induced cell death. As we have observed for BLP-mediated apoptosis through TLR2 (Figures 2 and 3), it appears that NF-κB suppresses pro-apoptotic signals generated by LPS. It is likely that LPS-mediated cell death proceeds through TLR4 and a MyD88/FADD pathway since: (i) TLR4 is required for all tested responses to LPS (Hoshino et al., 1999; Takeuchi et al., 1999); (ii) MyD88-deficient mice are hyporesponsive to LPS (Kawai et al., 1999); and (iii) LPS-induced apoptosis in endothelial cells, which express TLR4 (Zhang et al., 1999), is blocked by FADD-DN (Choi et al., 1998). We have also reported LPS-mediated cell death in THP-1 cells (Aliprantis et al., 1999). However, in our experimental system, concentrations of LPS 1000-fold higher than BLP are required to kill THP-1 cells. Although these cells express TLR4 (Zhang et al., 1999), they might lack upstream molecules necessary to mediate sensitive TLR4/LPS signals, such as CD14 or MD-2 (Shimazu et al., 1999). Alternatively, LPS might engage a cell death inhibitory pathway in THP-1 cells, which is not activated by BLP.
The induction of cell death by Toll-related receptors might be an evolutionarily conserved mechanism for innate immune defense. Numerous plant disease resistance genes (R genes) have been identified with homology to the TLRs, including N protein of tobacco, L6 protein from flax and RPP5 from Arabidopsis (Yang et al., 1997; Medzhitov and Janeway, 1998). Each of these proteins contains LRR and TIR homology domains. R gene products mediate a hypersensitive response (HR) upon encounter with a pathogen. The HR is characterized by localized cell death at the site of pathogen invasion, which limits the spread of microbes (Whitham et al., 1994; Staskawicz, 1995). The HR displays similarity to animal programmed cell death (Richberg et al., 1998) and can be attenuated by caspase inhibitors (Pozo, 1998). In analogy to TLR2-mediated apoptosis, it is tempting to speculate that R proteins induce cell death through their TIR domains by activating a caspase pathway.
Relevance of TLR-mediated apoptosis in vivo
Pathogenic bacteria may use TLRs to kill host macrophages. Yersinia outer protein J (YopJ) is necessary for Yersinia sp. to induce apoptosis in macrophages (Monack et al., 1997). YopJ is secreted by the bacterium into the cytoplasm of macrophages (Schesser et al., 1998), where it binds multiple MAP kinase kinase family members, including IKKβ, and inhibits NF-κB activation (Orth et al., 1999). The pathogens Shigella dysenteriae, Corynebacterium diphtheriae and Pseudomonas aeruginosa kill macrophages through toxins that inhibit protein synthesis (Moss et al., 1999). It is possible that BLP, LPS or other PAMPs from these bacteria are signaling for cell death through a TLR. Inhibition of protein synthesis or NF-κB activation (e.g. YopJ) could prevent the synthesis of anti-apoptotic molecules necessary to rescue the macrophage from death signals generated by TLRs.
In addition to the TLRs, the IL-1R also has an intracellular TIR domain. IL-1 has been implicated in the pathogenesis of two disorders of inappropriate apoptosis: diabetes mellitus (Sjoholm, 1998) and acute neurodegeneration (Rothwell et al., 1997). In vitro, IL-1 is cytotoxic to insulin-producing pancreatic β cells and potentiates excitotoxic neuronal cell apoptosis. In vivo, IL-1R antagonist, a cytokine that binds the IL-1R and inhibits signaling, protects laboratory animals from diabetogenic processes (Sandberg et al., 1997) and ischemic neural injury (Relton and Rothwell, 1992). Future work with appropriate cell types (i.e. pancreatic β cells and neurons) will determine whether IL-1 mediates cell death in these systems through an MyD88/FADD/caspase 8 pathway.
Although many bacterial pathogens induce apoptosis in host cells (Moss et al., 1999), the implications of this phenomenon remain elusive. Activation of TLR2 by BLP stimulates the synthesis of the precursor form of IL-1β, a potent pro-inflammatory cytokine (Figure 7A). In addition, TLR2 activates caspase 1 during BLP-mediated apoptosis, resulting in the cleavage and release of mature IL-1β (Figure 7B). These signaling events could be crucial for generating inflammatory signals during infection. To resolve inflammation, immune effector cells, such as neutrophils, must be eliminated by apoptosis (Savill, 1997). Activation of TLRs by PAMPs could limit the lifespan of inflammatory cells and thus control the duration of acute responses to bacteria. Finally, TLR-mediated cell death could facilitate long-term immunity. The recent hypothesis that intracellular contents released from dying cells stimulate adaptive immune responses (Matzinger, 1998) and the observation that synthetic bacterial lipopeptides are excellent adjuvants support this idea (Bessler et al., 1997). Thus, the apoptotic pathway initiated by TLRs potentially fulfills multiple roles in the genesis and progression of innate and adaptive immune responses to bacteria.
Materials and methods
Biological reagents
Stocks of sBLP (Boehringer Mannheim Biochemica, USA) and Pam3Cys (Calbiochem-Novabiochem Corp., San Diego, CA) were prepared at 1 mg/ml in endotoxin-free water with 0.05% horse serum albumin (HSA) (Grifols, Miami, FL). msBLP was described previously (Aliprantis et al., 1999). Purified E.coli MLP (Zhang et al., 1997) was a gift of Dr Klimpel (University of Texas at Galveston, TX). Stocks of S.flexneri type 1A LPS (Sigma, St Louis, MO) were prepared in endotoxin-free phosphate-buffered saline (PBS). Stocks of Ac-YVAD-cmk (BACHEM, Torrance, CA), z-VAD-fmk, z-IETD-fmk and z-FA-fmk (Enzyme Systems Products, Livermore, CA) were prepared in dimethylsulfoxide (DMSO) at 10 mM. Stocks of clasto-lactacystin β-lactone (Boston Biochem, Cambridge, MA) were prepared in DMSO at 5 mM. Recombinant mature IL-1β was a gift of the Vilcek Laboratory (NYU Medical Center, NY). Anti-TNF mAb 210 was from R and D Systems and anti-Fas ZB4 was obtained from Upstate Biotechnology. Unless indicated otherwise, other reagents were obtained from Sigma Biochemicals.
Cell culture
Human kidney epithelial 293 cells and human monocytic THP-1 cells were maintained as described previously (Aliprantis et al., 1999).
Expression vectors
Expression plasmids encoding the following proteins were described previously: (i) gD epitope-tagged human TLR2 and a 141 amino acid C-terminal deletion mutant (TLR2Δ2) (Yang et al., 1998); (ii) AU1 epitope-tagged MyD88-FL, AU1-epitope tagged MyD88-DN (152–296) and HA-epitope tagged MyD88-DD (1–151) (Muzio et al., 1997); (iii) TRAF2-DN (87–501) (Cao et al., 1996); (iv) TRAF6-DN (289–522) (Cao et al., 1996); (v) NIK-DN (KK429–430AA) (Malinin et al., 1997); (vi) IKKβ-DN (K44A) (Zandi et al., 1997); (vii) FADD and FADD-DN (80–208) (Chinnaiyan et al., 1995); (viii) RIP-DD (Ting et al., 1996); and (ix) green fluorescent protein (GFP) (Aliprantis et al., 1999). Expression vectors pSFFV-neo-bcl-2 and pSFFV-mu-FLAG-bcl-xl were kindly provided by Dr Y.Choi at The Rockefeller University. Mammalian expression plasmids encoding Flag epitope-tagged caspase 1-DN (C285A) and gD epitope-tagged caspase 8-DN (C360S) (Muzio et al., 1996) were gifts of Dr Dixit (Genentech). pRK5 was used as vector control in all experiments.
293 cell transfection experiments
A total of 8 × 104 293 cells were seeded per well into 24-well plates in Dulbecco’s modified Eagle’s medium (DMEM). After an overnight incubation, the cells were transfected using FuGENE 6 (Boehringer Mannheim) at a ratio of 3 µl of FuGENE 6/µg DNA, according to the manufacturer’s directions. The total amount of DNA was kept constant in all transfections by supplementing pRK5 vector DNA. At 24 h post-transfection, the medium was aspirated and sBLP was added in DMEM/5% fetal calf serum (FCS)/0.025% HSA. Luciferase reporter gene activity and apoptosis were assessed after 6 and 48 h, respectively.
Western blot analysis
Between 36 and 48 h post-transfection, cells were collected, washed and lysed [lysis buffer: 150 mM NaCl, 50 mM Tris pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.02% NaN3, 10 µg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride (PMSF)] for 5 min on ice. A Bradford protein assay (Bio-Rad) was performed on the post-nuclear supernatants (PNS). Equivalent amounts of protein were resolved by 10% SDS–PAGE, transferred to a PVDF membrane (Immobilon P, Millipore, Bedford, MA) and immunoblotted with one of the following antibodies: anti-gD (Yang et al., 1998), anti-AU1, anti-Myc and anti-HA (BaBCO, Richmond, CA), anti-FLag M2 (Sigma) or anti-caspase8 (kindly provided by D.W.Nicholson, Merck).
NF-κB luciferase reporter gene assays
Cells were transfected with the indicated plasmids plus 0.05 µg of pGL3-ELAM.tk and 0.005 µg of a Renilla luciferase reporter vector as an internal control. pGL3-ELAM.tk encodes firefly luciferase upstream of the NF-κB-dependent E-selectin promoter (Yang et al., 1998). Each luciferase activity was measured independently using the Dual Luciferase Reporter Assay System (Promega, Madison, WI). Results expressed as relative luciferase activity are determined by dividing the value of the NF-κB-inducible firefly luciferase by that of the constitutive Renilla luciferase reporter.
Apoptosis assays
Apoptosis was quantified by either TUNEL or annexin V staining. For TUNEL assays, culture supernatants and cells were collected and stained with the Apoptosis Detection System, Fluorescein Kit (Promega) and propidium iodide (PI) according to the manufacturer’s directions for flow cytometric analysis. Cells were considered apoptotic if they were TUNEL positive and/or had a DNA content of <2N, as determined by PI staining. A total of 5000 cells were counted per sample. For annexin V assays, culture supernatants and cells were collected and stained using annexin V–biotin (PharMingen, San Diego, CA) and streptavidin–TriColor (Caltag Laboratories, Burlingame, CA) according to the manufacturer’s directions and analyzed by flow cytometry. A total of 5000 GFP-positive cells (transfected) were counted per sample. Results presented as percentage specific apoptosis were determined using the following formula: [(% apoptotic cells in experimental – % apoptotic cells in control)/(100 – % apoptotic cells in control) × 100]. Unless indicated otherwise, results are the average ± SD of three independent samples.
THP-1 cytotoxicity assays
A total of 2.5 × 104 THP-1 cells were plated in serum-free RPMI-1640. Cells were pre-incubated with the compounds indicated in the figure legends for 1 h at 37°C. Test compounds were diluted into RPMI-1640/0.05% HSA and added to the cells. For mAb blocking experiments, both anti-TNF and anti-Fas where used in at least a 40-fold excess and the neutralizing activity was confirmed independently. Percentage cytotoxicity was determined between 6 and 8 h later by a colorimetric lactate dehydrogenase release (LDH) assay using the Cytotox 96 Assay kit (Promega). Results are the average ±÷SD of three independent samples.
IκB-α degradation western blot
A total of 1.5 × 106 THP-1 cells were plated in serum-free RPMI-1640. Test compounds were added as stated for THP-1 cytotoxicity assays. Cell lysates were prepared and a Bradford protein assay was performed on the PNS. Equivalent amounts of protein were resolved by 15% SDS–PAGE, transferred to PVDF and immunoblotted with a polyclonal rabbit anti-IκB-α antibody (Santa Cruz Biotechnology, Santa Cruz, CA) or with anti-PLCγ (a kind gift of E.Skolnik, NYU) as control.
Co-immunoprecipitation
This was performed essentially as described (Yang et al., 1999). Cells were transfected by CaPO4 precipitation. Immunoprecipitations were carried out with the indicated mAb (BaBCO, Richmond, CA). A polyclonal anti-FADD antibody was from StressGen Biotechnologies Corp. (Victoria, BC, Canada). Experiments were performed twice with similar results.
Interleukin-1β western blot
THP-1 cells (1 × 106) were pre-treated for 6 h with 32 nM of the phorbol ester, PMA, in serum-free RPMI-1640. Test compounds were added as stated for THP-1 cytotoxicity assays. After 3 h, the cells and supernatants were collected separately. Cell lysates were prepared as described above. Lysates and supernatants were resolved by 15% SDS–PAGE, transferred to nitrocellulose and immunoblotted with a polyclonal goat anti-hIL-1β antibody (R & D Systems, Minneapolis, MN).
Acknowledgments
Acknowledgements
We thank D.Littman, V.Dixit and Y.Weinrauch for critical reading of the manuscript. We especially thank V.Dixit and members of his laboratory who provided numerous reagents used in this study. A.O.A. was supported by a grant from the Life and Health Insurance Fund. This work was supported by a grant from the NIH to A.Z. (AI 37720-04).
References
- Adachi O., Kawai,T., Takeda,K., Matsumoto,M., Tsutsui,H., Sakagami,M., Nakanishi,K. and Akira,S. (1998) Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity, 9, 143–150. [DOI] [PubMed] [Google Scholar]
- Aliprantis A.O., Yang,R.B., Mark,M.R., Suggett,S., Devaux,B., Radolf,J.D., Klimpel,G.R., Godowski,P. and Zychlinsky,A. (1999) Cell activation and apoptosis by bacterial lipoproteins through Toll-like receptor-2. Science, 285, 736–739. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A. and Dixit,V.M. (1998) Death receptors: signaling and modulation. Science, 281, 1305–1308. [DOI] [PubMed] [Google Scholar]
- Baeuerle P.A. (1998) IκB–NF-κB structures: at the interface of inflammation control. Cell, 95, 729–731. [DOI] [PubMed] [Google Scholar]
- Baeuerle P.A. and Baichwal,V.R. (1997) NF-κ B as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv. Immunol., 65, 111–137. [PubMed] [Google Scholar]
- Baichwal V.R. and Baeuerle,P.A. (1997) Activate NF-κ B or die? Curr. Biol., 7, R94–R96. [DOI] [PubMed] [Google Scholar]
- Baker S.J. and Reddy,E.P. (1998) Modulation of life and death by the TNF receptor superfamily. Oncogene, 17, 3261–3270. [DOI] [PubMed] [Google Scholar]
- Bessler W.G. et al. (1997) Bacterial cell wall components as immunomodulators—I. Lipopeptides as adjuvants for parenteral and oral immunization. Int. J. Immunopharmacol., 19, 547–550. [DOI] [PubMed] [Google Scholar]
- Boldin M.P., Goncharov,T.M., Goltsev,Y.V. and Wallach,D. (1996) Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell, 85, 803–815. [DOI] [PubMed] [Google Scholar]
- Bortner C.D. and Cidlowski,J.A. (1999) Caspase independent/dependent regulation of K(+), cell shrinkage and mitochondrial membrane potential during lymphocyte apoptosis. J. Biol. Chem., 274, 21953–21962. [DOI] [PubMed] [Google Scholar]
- Brightbill H.D. et al. (1999) Host defense mechanisms triggered by microbial lipoproteins through Toll-like receptors. Science, 285, 732–736. [DOI] [PubMed] [Google Scholar]
- Cao Z., Xiong,J., Takeuchi,M., Kurama,T. and Goeddel,D.V. (1996) TRAF6 is a signal transducer for interleukin-1. Nature, 383, 443–446. [DOI] [PubMed] [Google Scholar]
- Chiao P.J., Miyamoto,S. and Verma,I.M. (1994) Autoregulation of Iκ B α activity. Proc. Natl Acad. Sci. USA, 91, 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan A.M., O’Rourke,K., Tewari,M. and Dixit,V.M. (1995) FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell, 81, 505–512. [DOI] [PubMed] [Google Scholar]
- Choi K.B., Wong,F., Harlan,J.M., Chaudhary,P.M., Hood,L. and Karsan,A. (1998) Lipopolysaccharide mediates endothelial apoptosis by a FADD-dependent pathway. J. Biol. Chem., 273, 20185–20188. [DOI] [PubMed] [Google Scholar]
- Delic J., Masdehors,P., Omura,S., Cosset,J.M., Dumont,J., Binet,J.L. and Magdelenat,H. (1998) The proteasome inhibitor lactacystin induces apoptosis and sensitizes chemo- and radioresistant human chronic lymphocytic leukaemia lymphocytes to TNF-α-initiated apoptosis. Br. J. Cancer, 77, 1103–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato J.A., Hayakawa,M., Rothwarf,D.M., Zandi,E. and Karin,M. (1997) A cytokine-responsive IκB kinase that activates the transcription factor NF-κB. Nature, 388, 548–554. [DOI] [PubMed] [Google Scholar]
- Dinarello C.A. (1997) Interleukin-1. Cytokine Growth Factor Rev., 8, 253–265. [DOI] [PubMed] [Google Scholar]
- Esser R.E., Watts,L.M., Angelo,R.A., Thornburg,L.P., Prior,J.J. and Palmer,J.T. (1993) The effects of fluoromethyl ketone inhibitors of cathepsin B on adjuvant induced arthritis. J. Rheumatol., 20, 1176–1183. [PubMed] [Google Scholar]
- Fantuzzi G. and Dinarello,C.A. (1999) Interleukin-18 and interleukin-1 β: two cytokine substrates for ICE (caspase-1). J. Clin. Immunol., 19, 1–11. [DOI] [PubMed] [Google Scholar]
- Fenteany G. and Schreiber,S.L. (1998) Lactacystin, proteasome function and cell fate. J. Biol. Chem., 273, 8545–8548. [DOI] [PubMed] [Google Scholar]
- Garcia-Calvo M., Peterson,E.P., Leiting,B., Ruel,R., Nicholson,D.W. and Thornberry,N.A. (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem., 273, 32608–32613. [DOI] [PubMed] [Google Scholar]
- Hardiman G., Rock,F.L., Balasubramanian,S., Kastelein,R.A. and Bazan,J.F. (1996) Molecular characterization and modular analysis of human MyD88. Oncogene, 13, 2467–2475. [PubMed] [Google Scholar]
- Henderson B., Poole,S. and Wilson,M. (1996) Bacterial modulins: a novel class of virulence factors which cause host tissue pathology by inducing cytokine synthesis. Microbiol. Rev., 60, 316–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman A.R. (1998) Global progress in infectious disease control. Vaccine, 16, 1116–1121. [DOI] [PubMed] [Google Scholar]
- Hirschfeld M., Kirschning,C.J., Schwandner,R., Wesche,H., Weis,J.H., Wooten,R.M. and Weis,J.J. (1999) Inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by toll-like receptor 2. J. Immunol., 163, 2382–2386. [PubMed] [Google Scholar]
- Hoshino K., Takeuchi,O., Kawai,T., Sanjo,H., Ogawa,T., Takeda,Y., Takeda,K. and Akira,S. (1999) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol., 162, 3749–3752. [PubMed] [Google Scholar]
- Hu S., Vincenz,C., Ni,J., Gentz,R. and Dixit,V.M. (1997) I-FLICE, a novel inhibitor of tumor necrosis factor receptor-1- and CD-95-induced apoptosis. J. Biol. Chem., 272, 17255–17257. [DOI] [PubMed] [Google Scholar]
- Huang D.C., Hahne,M., Schroeter,M., Frei,K., Fontana,A., Villunger,A., Newton,K., Tschopp,J. and Strasser,A. (1999) Activation of Fas by FasL induces apoptosis by a mechanism that cannot be blocked by Bcl-2 or Bcl-x(L). Proc. Natl Acad. Sci. USA, 96, 14871–14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaunin F., Burns,K., Tschopp,J., Martin,T.E. and Fakan,S. (1998) Ultrastructural distribution of the death-domain-containing MyD88 protein in HeLa cells. Exp. Cell Res., 243, 67–75. [DOI] [PubMed] [Google Scholar]
- Kawai T., Adachi,O., Ogawa,T., Takeda,K. and Akira,S. (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity, 11, 115–122. [DOI] [PubMed] [Google Scholar]
- Kirschning C.J., Wesche,H., Merrill Ayres,T. and Rothe,M. (1998) Human Toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J. Exp. Med., 188, 2091–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura M. (1999) NF-κB-mediated self defense of macrophages faced with bacteria. Eur. J. Immunol., 29, 1647–1655. [DOI] [PubMed] [Google Scholar]
- Kopp E.B. and Medzhitov,R. (1999) The Toll-receptor familly and control of innate immunity. Curr. Opin. Immunol., 11, 13–18. [DOI] [PubMed] [Google Scholar]
- Ling L., Cao,Z. and Goeddel,D.V. (1998) NF-κB-inducing kinase activates IKK-α by phosphorylation of Ser-176. Proc. Natl Acad. Sci. USA, 95, 3792–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinin N.L., Boldin,M.P., Kovalenko,A.V. and Wallach,D. (1997) MAP3K-related kinase involved in NF-κB induction by TNF, CD95 and IL-1. Nature, 385, 540–544. [DOI] [PubMed] [Google Scholar]
- Martin D.A., Siegel,R.M., Zheng,L. and Lenardo,M.J. (1998) Membrane oligomerization and cleavage activates the caspase-8 (FLICE/MACHα1) death signal. J. Biol. Chem., 273, 4345–4349. [DOI] [PubMed] [Google Scholar]
- Matzinger P. (1998) An innate sense of danger. Semin. Immunol., 10, 399–415. [DOI] [PubMed] [Google Scholar]
- Means T.K., Wang,S., Lien,E., Yoshimura,A., Golenbock,D.T. and Fenton,M.J. (1999) Human Toll-like receptors mediate cellular activation by Mycobacterium tuberculosis.J. Immunol., 163, 3920–3927. [PubMed] [Google Scholar]
- Medzhitov R. and Janeway,C.A.,Jr (1997a) Innate immunity: impact on the adaptive immune response. Curr. Opin. Immunol., 9, 4–9. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. and Janeway,C.A.,Jr (1997b) Innate immunity: the virtues of a nonclonal system of recognition. Cell, 91, 295–298. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. and Janeway,C.A.,Jr (1998) An ancient system of host defense. Curr. Opin. Immunol., 10, 12–15. [DOI] [PubMed] [Google Scholar]
- Medzhitov R., Preston-Hurlburt,P., Kopp,E., Stadlen,A., Chen,C., Ghosh,S. and Janeway,C.A.,Jr (1998) MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell, 2, 253–258. [DOI] [PubMed] [Google Scholar]
- Monack D.M., Mecsas,J., Ghori,N. and Falkow,S. (1997) Yersinia signals macrophages to undergo apoptosis and YopJ is necessary for this cell death. Proc. Natl Acad. Sci. USA, 94, 10385–10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss J.E., Aliprantis,A.O. and Zychlinsky,A. (1999) The regulation of apoptosis by microbial pathogens. Int. Rev. Cytol., 187, 203–259. [DOI] [PubMed] [Google Scholar]
- Muzio M. et al. (1996) FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell, 85, 817–827. [DOI] [PubMed] [Google Scholar]
- Muzio M., Ni,J., Feng,P. and Dixit,V.M. (1997) IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science, 278, 1612–1615. [DOI] [PubMed] [Google Scholar]
- Muzio M., Natoli,G., Saccani,S., Levrero,M. and Mantovani,A. (1998a) The human Toll signaling pathway: divergence of nuclear factor κB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6). J. Exp. Med., 187, 2097–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M., Stockwell,B.R., Stennicke,H.R., Salvesen,G.S. and Dixit,V.M. (1998b) An induced proximity model for caspase-8 activation. J. Biol. Chem., 273, 2926–2930. [DOI] [PubMed] [Google Scholar]
- Newton K., Harris,A.W., Bath,M.L., Smith,K.G.C. and Strasser,A. (1998) A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J., 17, 706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgard M.V., Arndt,L.L., Akins,D.R., Curetty,L.L., Harrich,D.A. and Radolf,J.D. (1996) Activation of human monocytic cells by Treponema pallidum and Borrelia burgdorferi lipoproteins and synthetic lipopeptides proceeds via a pathway distinct from that of lipopolysaccharide but involves the transcriptional activator NF-κ B. Infect. Immun., 64, 3845–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill L.A. and Greene,C. (1998) Signal transduction pathways activated by the IL-1 receptor family: ancient signaling machinery in mammals, insects and plants. J. Leukoc. Biol., 63, 650–657. [PubMed] [Google Scholar]
- Orth K., Palmer,L.E., Bao,Z.Q., Stewart,S., Rudolph,A.E., Bliska,J.B. and Dixon,J.E. (1999) Inhibition of the mitogen-activated protein kinase kinase superfamily by a Yersinia effector. Science, 285, 1920–1923. [DOI] [PubMed] [Google Scholar]
- Palombella V., Rando,O.J., Goldberg,A.L. and Maniatis,T. (1994) The ubiquitin–proteasome pathway is required for processing the NF-κB1 precursor protein and the activation of NF-κB. Cell, 78, 773–785. [DOI] [PubMed] [Google Scholar]
- Poltorak A. et al. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science, 282, 2085–2088. [DOI] [PubMed] [Google Scholar]
- Pozo O. and Lam,E. (1998) Caspases and programmed cell death in the hypersensitive response of plants to pathogens. Curr. Biol., 8, 1129–1132. [DOI] [PubMed] [Google Scholar]
- Relton J.K. and Rothwell,N.J. (1992) Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Res. Bull., 29, 243–246. [DOI] [PubMed] [Google Scholar]
- Richberg M.H., Aviv,D.H. and Dangl,J.L. (1998) Dead cells do tell tales. Curr. Opin. Plant Biol., 1, 480–485. [DOI] [PubMed] [Google Scholar]
- Rock F.L., Hardiman,G., Timans,J.C., Kastelein,R.A. and Bazan,J.F. (1998) A family of human receptors structurally related to Drosophila Toll. Proc. Natl Acad. Sci. USA, 95, 588–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell N., Allan,S. and Toulmond,S. (1997) The role of interleukin 1 in acute neurodegeneration and stroke: pathophysiological and therapeutic implications. J. Clin. Invest., 100, 2648–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckdeschel K., Harb,S., Roggenkamp,A., Hornef,M., Zumbihl,R., Kohler,S., Heesemann,J. and Rouot,B. (1998) Yersinia enterocolitica impairs activation of transcription factor NF-κB: involvement in the induction of programmed cell death and in the suppression of the macrophage tumor necrosis factor α production. J. Exp. Med., 187, 1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg J.O., Eizirik,D.L. and Sandler,S. (1997) IL-1 receptor antagonist inhibits recurrence of disease after syngeneic pancreatic islet transplantation to spontaneously diabetic non-obese diabetic (NOD) mice. Clin. Exp. Immunol., 108, 314–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savill J. (1997) Apoptosis in resolution of inflammation. J. Leukoc. Biol., 61, 375–380. [DOI] [PubMed] [Google Scholar]
- Schesser K., Spiik,A.K., Dukuzumuremyi,J.M., Neurath,M.F., Pettersson,S. and Wolf-Watz,H. (1998) The yopJ locus is required for Yersinia-mediated inhibition of NF-κB activation and cytokine expression: YopJ contains a eukaryotic SH2-like domain that is essential for its repressive activity. Mol. Microbiol., 28, 1067–1079. [DOI] [PubMed] [Google Scholar]
- Schwandner R., Dziarski,R., Wesche,H., Rothe,M. and Kirschning,C.J. (1999) Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. J. Biol. Chem., 274, 17406–17409. [DOI] [PubMed] [Google Scholar]
- Shimazu R., Akashi,S., Ogata,H., Nagai,Y., Fukudome,K., Miyake,K. and Kimoto,M. (1999) MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll- like receptor 4. J. Exp. Med., 189, 1777–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoholm A. (1998) Aspects of the involvement of interleukin-1 and nitric oxide in the pathogenesis of insulin-dependent diabetes mellitus. Cell Death Differ., 5, 461–468. [DOI] [PubMed] [Google Scholar]
- Staskawicz B.J., Ausubel,F.M., Baker,B.J., Ellis,J.G. and Jones,J.D. (1995) Molecular genetics of plant disease resistance. Science, 268, 661–667. [DOI] [PubMed] [Google Scholar]
- Strasser A., Harris,A.W., Huang,D.C., Krammer,P.H. and Cory,S. (1995) Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J., 14, 6136–6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O., Hoshino,K., Kawai,T., Sanjo,H., Takada,H., Ogawa,T., Takeda,K. and Akira,S. (1999) Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity, 11, 443–451. [DOI] [PubMed] [Google Scholar]
- Thornberry N.A. and Lazebnik,Y. (1998) Caspases: enemies within. Science, 281, 1312–1316. [DOI] [PubMed] [Google Scholar]
- Ting A.T., Pimentel-Muinos,F.X. and Seed,B. (1996) RIP mediates tumor necrosis factor receptor 1 activation of NF-κB but not Fas/APO-1-initiated apoptosis. EMBO J., 15, 6189–6196. [PMC free article] [PubMed] [Google Scholar]
- Wang C.Y., Mayo,M.W., Korneluk,R.G., Goeddel,D.V. and Baldwin,A.S.,Jr (1998) NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science, 281, 1680–1683. [DOI] [PubMed] [Google Scholar]
- Whitham S., Dinesh-Kumar,S.P., Choi,D., Hehl,R., Corr,C. and Baker,B. (1994) The product of the tobacco mosaic virus resistance gene N: similarity to Toll and the interleukin-1 receptor. Cell, 78, 1101–1115. [DOI] [PubMed] [Google Scholar]
- Yang R.B. et al. (1998) Toll-like receptor-2 mediates lipopoly saccharide-induced cellular signalling. Nature, 395, 284–288. [DOI] [PubMed] [Google Scholar]
- Yang R.B., Mark,M.R., Gurney,A.L. and Godowski,P.J. (1999) Signaling events induced by lipopolysaccharide-activated Toll-like receptor 2. J. Immunol., 163, 639–643. [PubMed] [Google Scholar]
- Yang Y., Shah,J. and Klessing,D.R. (1997) Signal perceptions and transduction in plant defense responses. Genes Dev., 11, 1621–1639. [DOI] [PubMed] [Google Scholar]
- Yeh W.C. et al. (1997) Early lethality, functional NF-κB activation and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity, 7, 715–725. [DOI] [PubMed] [Google Scholar]
- Yoshimura A., Lien,E., Ingalls,R.R., Tuomanen,E., Dziarski,R. and Golenbock,D. (1999) Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J. Immunol., 163, 1–5. [PubMed] [Google Scholar]
- Yuan J. (1997) Transducing signals of life and death. Curr. Opin. Cell Biol., 9, 247–251. [DOI] [PubMed] [Google Scholar]
- Zandi E., Rothwarf,D.M., Delhase,M., Hayakawa,M. and Karin,M. (1997) The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell, 91, 243–252. [DOI] [PubMed] [Google Scholar]
- Zhang F.X. et al. (1999) Bacterial lipopolysaccharide activates nuclear factor-κB through interleukin-1 signaling mediators in cultured human dermal endothelial cells and mononuclear phagocytes. J. Biol. Chem., 274, 7611–7614. [DOI] [PubMed] [Google Scholar]
- Zhang J., Cado,D., Chen,A., Kabra,N.H. and Winoto,A. (1998) Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature, 392, 296–300. [DOI] [PubMed] [Google Scholar]
- Zhang H., Peterson,J.W., Niesel,D.W. and Klimpel,G.R. (1997) Bacterial lipoprotein and lipopolysaccharide act synergistically to induce lethal shock and proinflammatory cytokine production. J. Immunol., 159, 4868–4878. [PubMed] [Google Scholar]