

Abstract
A precise mixture of extracellular matrix (ECM) secreted by valvular cells forms a scaffold that lends the heart valve the exact mechanical and tensile strength needed for accurate hemodynamic performance. ECM proteins are a key component of valvular endothelial cell (VEC) - valvular interstitial cell (VIC) communication essential for maintenance of the valve structure. This study reports the healthy adult pulmonary and aortic valve proteomes characterized by LC/MS/MS, resulting in 2710 proteins expressed by 1513 genes, including over 300 abundant ECM proteins. Surprisingly, this study defines a distinct proteome for each semilunar valve. Protein-protein networking (PPN) was used as a tool to direct selection of proteomic candidates for biological investigation. Local PPN for nidogen 1 (Nid1), biglycan (Bgn), elastin microfibril interface-located protein 1 (Emilin-1) and milk fat globule-EGF factor 8 protein (Mfge8) were enriched with proteins essential to valve function and produced biological functions highly relevant to valve biology. Immunofluorescent investigations demonstrated that these proteins are functionally distributed within the pulmonary and aortic valve structure, indicative of important contribution to valve function. This study yields new insight into protein expression contributing to valvular maintenance and health and provides a platform for unbiased assessment of protein alterations during disease processes.
Keywords: Aortic valve, pulmonary valve, valve proteome, extracellular matrix, protein-protein-networks, immunofluorescence
Introduction
Congenital and acquired valve disease affect over ten million Americans, yet the only treatment is surgical valve replacement.1,2 Aortic valve dysfunction is the most commonly observed pathology. An extreme hemodynamic environment with a highly pressurized blood flow and continual alterations in shear stress during mechanical flexure are causative factors for the deterioration of aortic valve function.3 Numerous clinical observations have shown that mechanical forces play a significant role in influencing valvular health and structure.4–7 These investigations have illustrated that the interplay between mechanical force and valvular biology is an exquisitely tuned system that results in precise remodeling of the valvular extracellular matrix (ECM) and ensures robust function of the valve throughout the 3 billion heartbeats of an individual's lifetime. However, the cellular mechanisms that maintain the health and function of the valvular structure remain poorly defined.
Together, the two outflow tract valves, the pulmonary and the aortic valve, modulate blood outflow from the heart (Figure 1). The pulmonary valve sits at the junction of the right ventricle and pulmonary artery and facilitates blood flow to the pulmonary system to be oxygenated. Oxygenated blood is pumped from the left atria into the pressurized left ventricle and sent throughout the body via the aortic valve, situated between the root of the aorta and the left ventricle. While structurally similar, the pulmonary valve operates in a low oxygen tension, low pressure environment, while the aortic valve operates in a highly oxygenated, high pressure environment.
Figure 1.
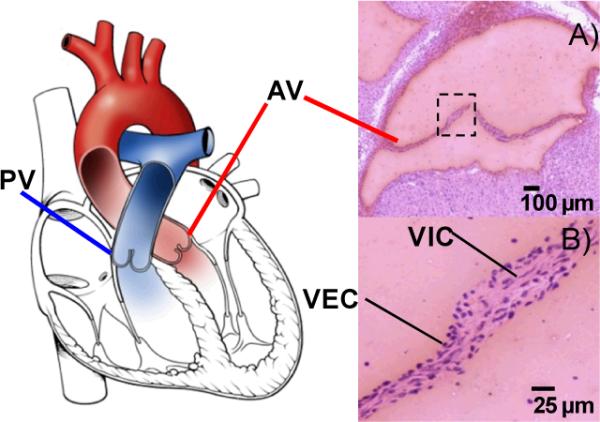
Illustration of the pulmonary (PV) and aortic valves (AV) within the heart. A) Hematoxylin and eosin stain of a coronal section from adult mouse aortic valve; B) Cellular anatomy of the valve. VEC-valvular endothelial cells, VIC- valvular interstitial cells.
Maintenance of the outflow heart valve structure for precise and robust distribution of blood load is a delicate interplay between mechanical force and cellular signaling. Constant bending, stretching, pulsatile, oscillatory and shear are all stimuli involved in regulation of valvular remodeling and valvular anatomy is arranged to respond to these forces (Figure 1).6,8 Valvular endothelial cells (VEC) lining the outer valve surface serve as a regulatory interface to the valvular interior, translating hemodynamic forces to cellular signaling;9 differential flow has been shown to produce distinct valvular endothelial phenotypes.9–11 Valvular interstitial cells (VICs) attached through focal adhesions and scattered throughout the internal valve structure communicate with valvular endothelial cells and each other through signaling mechanisms that are also sensitive to the mechanics of the environment.12,13 Cellular signaling occurs through a field of extracellular matrix, which forms the structural scaffold of the valve. Continual valvular interstitial cell remodeling of the ECM is essential for maintaining valvular sufficiency from birth to old age.
Collagens, glycosaminoglycans (GAGs) and elastin are species conserved primary components of the ECM and form a malleable valvular scaffold.14 The mature mammalian aortic valve has a dense collagenous region exposed to the arterial surface (fibrosa), an interior composed largely of GAGs (spongiosa), and a thin elastic layer lining the ventricular surface (ventricularis).6 These components arrange according to hemodynamic flow providing a precise combination of mechanical strength, compliance and elasticity, respectively, to the valve. Other major ECM components such as perlecan (growth factor regulation),15,16 fibronectin (valve repair),17 versican (adhesion, proliferation, ECM assembly)18,19 are essential to valve formation and remain part of the mature valve structure.20 Within the ECM meshwork of the scaffold, cytokines, signaling and growth factors, and proteases traffic between cellular components of the valve.21 Thus the ECM is a structural support system and an information highway within the valve that facilitates valvular competency.
While major components of the valvular structure have been defined utilizing candidate gene strategies, little work has been done to systematically define the valvular proteome. Proteomic analysis represents an unbiased approach for investigating the working biology of the valve as it is interactions at the protein level, e.g., focal adhesions, cell-cell signaling, that translate to regulation of signaling complexes and physical changes to the valve. Only a few reports have applied proteomic strategies to the investigation of valve biology. These reports include comparison of cultured aortic valve VIC during myofibroblast differentiation22 or comparison of mitral valve to atrium for identification of autoantigens.23 There are no accounts of the outflow tract valve proteome, despite interest in the application of proteomics to valvular disease and the obvious relevance to human pathology.24 Defining and comparing the protein mechanisms of the outflow tract valves has a high potential to provide new information on major protein components of valve structure and biology, providing vital information for the understanding of mechanisms of valve integrity.
For tissue that has not been well characterized, selecting good candidates for biological investigation can be particularly challenging as proteins may have context dependent regulatory activities, i.e different functions in different tissue environments. One approach is to mine the discovered proteome through the computation and examination of protein-protein networks. Protein-protein networks depict proteins as nodes connected through both predictive and published associations based on gene proximity, gene fusion, similarities in phylogenic profiles, predictive binding through conserved sequence domains, and experimentally proven evidence of interaction.25,26 The use of protein-protein networks to define and examine the functional relationship between novel proteins and proteins that are already well characterized within a tissue provides a mechanism for earmarking proteins for further exploration in a biological system.26,27 Protein-protein networks have been used to predict and confirm candidate disease genes,28 to predict phenotypic effects of gene mutation,29 and to infer protein function within specific tissue environments.27,30
In the present study, we combine proteomics and network connectivity to discover new ECM proteins participating in valvular function. We compare the proteomic profiles of aortic and pulmonary valves from the adult ICR mouse, characterizing proteomes by subcellular localization, biological and molecular function, and enriched pathways. From the proteomic analysis, ECM proteins shown to be in high abundance within the valvular proteomes were selected for protein-protein network analysis. Local networks of select extracellular matrix proteins were evaluated for connection to well characterized valvular proteins, and for biological function of locally interacting proteins in order to earmark new extracellular matrix proteins for biological investigation. Candidate proteins were validated by immunofluorescence and demonstrated expression pattern differences between the outflow tract valves. All proteins investigated proved highly relevant to valve biology and provided new insight into functional distribution of protein expression in regards to valve structure. These proteins were combined to calculate a new extracellular matrix protein-protein network that was found to be enriched with gene phenotypes involved in processes of degenerative valve disease or valve development. This study provides the first report of the outflow tract valvular proteomes and suggests that the aortic and pulmonary valves have distinct proteomes related to their unique hemodynamic environment.
Materials and Methods
Aortic and Pulmonary Valve Collection
All animal use and handling was in accordance with protocols approved by the Vanderbilt University Institutional Animal Care and Use Committee (IACUC). Female ICR mice between the ages of 8 and 12 weeks were euthanized by CO2 asphyxiation following IACUC guidelines. Nine animals were used for method development purposes. Three biological replicates of aortic valve and pulmonary valve were collected by microdissection. Each biological replicate consisted of pooled aortic or pulmonary valves from 15 female ICR mice. Microdissected valves were immediately stored at −80°C. Valves were analyzed within one week of microdissection.
Protein Extraction
Valve cusps were collected into 200 μL tubes at the time of microdissection. Each tube contained a pool of 15 cusps from either aortic or pulmonary valve and all proceeding sample handling was performed in that tube. Valve cusps were washed two times in 10 μL ice cold PBS, centrifuged 5 minutes at 10,000 g and supernatant removed. A total of 10 μL of cell lysis buffer consisting of 2% SDS, 75 mM DTT, 1 mM EDTA in 10 mM Tris, pH 7.2 was added to the valves. For detergent comparison, a secondary cell lysis buffer consisted of 1% Triton X-100, 150 nM NaCl, 1% deoxycholate, 5 mM EDTA, 1 mM vanadate in 10 mM Tris pH 7.2. Valves in cell lysis buffer were sonicated for 20 minutes, vortexed for 1 minute and centrifuged, repeating three times. Valves were incubated overnight in cell lysis buffer at 37.5°C and again sonicated for 20 minutes and centrifuged 5 minutes at 10,000g. Supernatant was collected, followed by rinsing valves in 5 μL cold PBS and collecting the rinse.
In-gel Digestion
For each set of 15 AV or PV, concentration was estimated using a Nanovue spectrometer (GE Healthcare, Piscataway, NJ) and comparing absorbance values at 280 nm against known concentrations of BSA bracketing the extracted protein solution. Results showed equal amounts of protein (~75 μg) collected from each valve type. Protein was dissolved in equal parts Laemmli buffer, boiled for 15 minutes, cooled on ice and loaded onto a 4–12% Bis-Tris minigel (Invitrogen, Carlsbad, CA). Electrophoresis was performed for 2 hours at 150 volts using MOPS-SDS running buffer. Gels were washed in ice cold water for 10 minutes and lanes cut into eight fractions. Each fraction was minced into 1 mm cubes and washed three times in 50 mM NH4HCO3, pH 8. Bands were dehydrated three times by 20 minute incubations in 50/50 (acetonitrile /50 mM NH4HCO3) followed by 100% acetonitrile and drying under vacuum for 25 minutes. Freshly prepared 20 mM DTT/ 50 mM NH4HCO3 pH 8 was added to each fraction and incubated for 1 hour at 55°C. Pieces were washed with 100% acetonitrile before adding a solution of 40 mM iodoacetamide in 50 mM NH4HCO3 pH 8 and incubating for 1 hour at room temperature. After carbamidomethylation, gel pieces were washed and dehydrated as described above. Proteins were digested by adding 200 μL of a 0.01 μg/μL trypsin solution to the pieces and incubating at 37°C overnight. Peptides were extracted once with 50/50 (5% formic acid)/ acetonitrile to halt digestion and twice with a 50/50 (50 mM NH4HCO3) /acetonitrile solution.
LC-MS/MS
Peptides from gel fractions were pressure loaded onto a 75 μm I.D. microcapillary column hand packed with 10 cm of 5 μm, 300 Å C18 (Rainin, Oakland, CA) and coupled to a 75 μm I.D. column with a pulled tip packed with 12 cm of 3 μm, 300 Å Jupiter C18 (Phenomenex, Torrence, CA). Peptide separation was performed via a 0.25% B/min gradient from 2% B to 35% B at ~250 nL/min using an Agilent 1100 series HPLC. Mobile phase A was 0.1% formic acid in water, mobile phase B was 0.1% formic acid in acetonitrile. Peptides eluted directly from the analytical column into an LTQ ion trap (Thermo Scientific, Inc., San Jose, CA). Source voltage was set to 2.2. kV. Each full MS scan (m/z range 350 –1800) was followed by nine MS/MS events on the nine most abundant peaks. Selected ions were dynamically excluded for 60 s.
Protein Identification
Raw tandem mass spectra were read as centroid peak lists and converted from raw files into the Sequest .dta format using “ScanSifter v 2.0”, a software tool under development at Vanderbilt University Medical Center. Spectra with less than 6 peak counts were removed from further analysis. DTAs were searched using Sequest version 27 against the IPI non-redundant mouse database version 3.39 (52,777 entries) downloaded 2/14/2008 paired with a contaminants database and concatenated with the reverse database. Search parameters were set to trypsin-strict as the enzyme specification, 2 missed cleavages, +/− 2.5 Da for the precursor and 0.6 Da for the fragment masses. Variable modifications used were carbamidomethylation of cysteines, and methoxidation. Peptides identified from the database search were clustered using IDPicker version 2.2.31 The initial protein list was produced using a peptide false discovery rate (FDR) ≤ 5.0 with a minimum of two peptides per protein, resulting in a protein result level FDR of 1.5%. The data associated with this manuscript may be downloaded from ProteomeCommons.org Tranche using the following hash: DqFFbDxlIGy/KyDSruNIXjG2ADQaOLlzqdBamTYPHqgRTM+5q7FGb7sAj/xI4N+nAiAtvEzuPay 8q+gSJIq7ukc5pTYAAAAAAAADqQ==. In depth details for each protein may be explored using IDPicker software available at www.mc.vanderbilt.edu/msrc.
Bioinformatics
Predictions of subcellular localizations were made using WoLF PSORT.32 Primary Gene Ontology (GO) processes and molecular functions and pathways were calculated using the Database for Annotation, Visualization and Integrated Discovery (DAVID) software using the mus musculus genome as a reference list.33,34 For calculation of biological processes associated with local networks where number of molecules was limited, biological processes are reported with Fisher's Exact test p-value <0.001. For all other molecules and proteome-wide analysis, biological processes and molecular functions are reported with Benjamini (FDR corrected) p-value <0.001; Pathways are reported with Benjamini (FDR corrected) p-value <0.005.35 Webgestalt was used to calculate and enrich for biological processes related to distinctive proteomes, comparing distinctive protein expression to the entire proteome assuming a hypergeometric distribution and adjusting the p-value by the method of Benjamini and Hochberg.35
Quantitation
Spectral counts were used to estimate quantitative aspects of the MS analysis. Spectral count data was normalized to make total spectral counts for replicate experiments comparable. A multiplication factor (F) was calculated for each replicate based on the replicate with the highest spectral counts using the equation F= (Maximum Total Spectral Count/Individual Replicate Spectral Count). Spectral count from each protein was multiplied by the multiplication factor as a normalization factor. To encompass situations where data dependent collection of data produced a zero spectral count for a replicate analysis of a protein, we increased all spectral counts by 1. The normalized data was then log transformed to achieve a better approximation of normal distribution. The normalized and log transformed data was analyzed using the Limma package36 in the open source software Bioconductor (www.bioconductor.org) to fit a linear model for each protein using an empirical bayes method to moderate standard errors. This method provides more stable inference and is particularly effective when the number of replicates is low. For each protein, the coefficient in the linear model estimates the log-ratio between the two groups, from which fold change was calculated from comparison of the average adjusted spectral count. A moderated t-statistic was used to eliminate large t-statistic values occurring due to small same-group variance. The false discovery rate of the quantitative p-value was calculated using the method of Benjamini and Hochberg,35 annotated as the adjusted p-value. The log odds of differential expression was calculated, represented by the B value. B-values less than zero imply that there is more evidence against differential expression; B-values larger than zero indicate that expression levels of a particular protein are more likely to be differentially expressed between groups.
To estimate and compare relative standings of proteins within each proteome, proteins were ranked within each proteome based on average spectral counts per protein per replicate using the Rank and Percentile function in Excel.
Network Analysis
Local protein interaction networks were calculated using Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) version 8.2.37 The full protein-protein network is presented as an interactive file in supplemental data. STRING nodes are proteins, edges represent predicted functional associations based on primary databases such as KEGG and GO, and primary literature. STRING complements these interactions with predictions based on genomic proximity, gene fusion products, evolutionary history, and similarity of co-expression patterning. Strength of association scores for each node are computed as a joint probability derived from curated databases of experimental information, text mining and computationally predicted by genetic proximity.37 STRING networks were calculated with default settings of medium confidence score 0.400, network depth of 0, and no more than 50 interactors.
The Ingenuity Pathways Analysis (IPA) software version 8.6 (www.ingenuity.com) was used to calculate literature supported networks for expression data. IPA software employs an expert curated knowledge base derived from biology literature. Network scores are the negative log of the probability calculated by Fisher's exact test and represent the probability that molecules in the network fit by random chance. Networks calculated by Ingenuity had scores>35.
Immunofluorescence
Immunofluorescence was performed on aortic and pulmonary valves cryosectioned from fresh frozen adult mouse heart (n= 3). Mfge8 (sc-33546), Emilin-1(sc-50430) and Biglycan (sc-27936) antibodies were purchased from Santa Cruz Biotechnology, Inc. Nidogen 1 (ab44944) antibody was purchased from Abcam. For Mfge8, Emilin-1, and NID-1 sections were fixed in 70% ethanol, and treated with 0.25% Triton X-100. Mfge8 was blocked for 4 hours in ice-cold 5% donkey serum/PBST. Emilin-1 and NID-1 were blocked in 5% goat serum/PBST. For biglycan, sections were fixed in 70% methanol and blocked for 4 hours with 5% rabbit serum/PBST. All sections were incubated overnight with primary antibodies (1:50 dilutions). Secondary antibodies were applied in ratios of 1:300 and sections were mounted with DAPI-containing medium (VectaShield) and visualized using a Nikon Eclipse E800 epifluorescence microscope.
Results
Isolation of Proteins from Decellularized Aortic and Pulmonary Valves
The abundance of structural proteins and small size of the microdissected valve cusps (600 µm × 250 µm × 25 µm) created challenges for sample handling and analysis. Collagens are a major constituents of the mouse heart valve,38 providing structural strength against tensile forces occurring during normal heart beating. We sought to identify extracellular matrix components embedded in the valvular collagen without over sampling of the collagens. One approach would be to decellularize the valves using detergents and collect the proteins in the supernatant. Decellularization of tissue has been utilized with success in the field of tissue engineering to remove cellular components from heart tissue without altering their structural integrity, allowing the construct to be reseeded with new cells.39,40 We investigated decellularization as a strategy for extracting cellular and extracellular contents of the valve employing either a Triton X-100/deoxycholate mixture or SDS cell lysis solution combined with sonication cycling and overnight incubation. Comparison by Coomassie staining showed that the SDS protocol produced the most intense staining, in agreement with previous reports on heart tissue decellularization where SDS produced a more complete removal of all cellular components without major disruption to structural proteins; this result correlated with previous reports on decellularization of cardiac tissue.39,40 Absence of nuclei from valve fragments assessed by hematoxylin staining of the stripped valve pellet and DAPI fluorescence staining of valve fragments after sonication demonstrated elimination of cellular components from the valve scaffold, indicating that proteins were isolated from valvular interstitial cells and valvular endothelial cells.
Characterization of the valvular proteomes
The sample preparation workflow, including microdissection and extraction, is summarized in Figure 2. A total of 2710 proteins represented by 1513 genes were identified, with 2202 mapping to the pulmonary valve and 2049 mapping to the aortic valve. Of these, 57% proteins were found to be common to both the aortic and pulmonary valve tissue, indicating significant differences between the two valvular proteomes (Figure 3A). Survey of subcellular localization based on full length sequence prediction annotated the majority of the identified proteins to the cytosol (Figure 3B). Approximately 12% of the proteins from each proteome were predicted to be localized to the extracellular matrix (256 PV proteins; 246 AV proteins), providing a sizable pool for new investigations into the ECM environment.
Figure 2.

Microdissection and decellularization of valve cusps. A) Dissected aortic sinus. Box encompasses aortic valve, zoomed in B and C; B) Close-up of in situ aortic valve; C) Dotted lines (black or white for differing cusps) outline the cusps of the in situ aortic valve; D) Dissected valve cusps collected for proteomic analysis. Bar - 250 μm E) Decellularization of cusps; F) Visualization of protein extract by SDS PAGE and Coomassie blue staining. MWL- molecular weight ladder, PV- pulmonary valve, AV- aortic valve.
Figure 3.

Protein identification and localization. A) Venn diagram of protein identification; B) Subcellular localization of proteins. Nucl-nuclear, Cyto-cytosol, Mito-mitochondrial, E. R.-endoplasmic reticulum, Pero- peroxisome, Cysk- cytoskeletal, Plas- plasma membrane, ECM-extracellular matrix.
Analysis of the protein datasets for Gene Ontology (GO) biological and molecular functions showed that molecular functions and biological processes were similar between aortic and pulmonary valves (Figure 4A). Major functions of `Protein binding' and `nucleotide binding' indicated active involvement of the proteome with genomic material. This interaction with the genomic material was also a striking feature within calculated biological processes. For example, a main biological process after `metabolism' was `cellular organization and biogenesis', a category with subsets involved in macromolecular complex assembly, chromatin organization, and organelle organization. Since the aortic and pulmonary valves are situated in different environments and thus may involve differential processes, we further examined biological processes significant to differential protein expression from each valve proteome (Figure 4B). The proteins identified only within the pulmonary valve clustered to oxidation/reduction (49 proteins, p-value<3.55E-13, GO:0055114), and establishment of localization (75 genes, p-value 5.00E-4, GO:0051234). Proteins identified within the aortic valve mapped to localization (64 proteins, p-value <5.00E-4, GO:0051179) and transport (57 proteins, p-value 3.00E-4, GO:0006810) as significant processes. Approximately 25% of all the proteins were not yet annotated by the GO consortium and are not shown in the figures.
Figure 4.

Characterization of proteomes by GO annotations and pathway analysis. A) GO annotation of molecular function and biological processes. Outer wheel-aortic valve, inner wheel -pulmonary valve. B) KEGG Pathways analysis of AV and PV proteomes. C) Pathways with Benjamini (FDR corrected) p-value<0.005 were considered significant fold enrichment, demarked by red line.
Figure 4C shows enrichment of pathways from the proteome of the two outflow tract valves. We observed that the pathway `Tight junction', important to valvular endothelial integrity, permeability and cell-cell communication was significantly enriched >2-fold for both AV and PV. The pathways of `Cell Communication', `Focal adhesion (FA)', `ECM-receptor interaction' and `Regulation of Actin Cytoskeleton' were enriched consistent to an involvement with mechanotransduction. We found that many of the ECM proteins related to focal adhesions were abundant by percentile rank of spectral count within each proteome. Examples of these proteins are vinculin (ranked 96.4% PV; 93.3% AV), talin-1 (ranked 71.5% PV; 95.7% AV), both regulated in response to cyclic stretch of the valve structure,41 and fibronectin (ranked 98.8% PV; 97.9% AV) involved in early FA formation41 and matrix remodeling.42
Quantitative Aspects of the Valvular Proteomes
A list of all identified proteins and quantitative aspects may be found in Supplemental Table 1. Using the log odds of differential (B value) as evidence of significant fold change by spectral counting, we observed that protein expression levels did not vary significantly between proteomes. A possible reason for this is that sample size was small (n=3, where each sample was a pool of 15 complete valve sets) and may have limited sensitivity in detection of regulatory changes. To approximate quantitative aspects within individual datasets, we ranked all proteins by average spectral count within the proteomes and examined ECM protein expression in relation to each of the total proteomes. Many of the extracellular matrix proteins were above the 90th percentile, indicating that these proteins were abundantly expressed within their respective proteomes. Comparison to known valvular ECM proteins revealed that many ECM proteins found highly abundant within each proteome have been reported as major constituents of valvular ECM.21 Consistent with previous biological reports, these include periostin, perlecan, and versican; all knockouts of these proteins display embryonic lethal phenotypes.20,21
Network connections earmark ECM proteins for further investigation
Our primary goal was to use an unbiased approach to identify new ECM components that participate in normal valve homeostasis and provide valuable information for the design of tissue engineered valve components. Network analysis was utilized to select ECM protein candidates for biological investigation from the proteomic results. In a protein-protein network, proteins are represented as nodes and edges connecting the proteins describe the relationship between proteins (e.g., direct binding, co-expression, gene fusion).37,43 Current network theory proposes that 1) proteins essential to a phenotype form densely connected hubs within a network44,45 and 2) closely associated proteins are involved in similar functions.26 Since protein dynamics and even protein function may differ per tissue, calculating the local networks and related functions from proteins identified within a tissue may yield information on the role of that protein specific to the tissue environment.27 For this study, we focused on earmarking new ECM protein candidates by protein-protein network analysis for further investigation because of the importance of the ECM to valvular structure and cell-cell messaging. ECM proteins were sorted by percentile rank within the proteome and filtered to the top 20th percentile of ECM proteins within the datasets. Select ECM proteins from this list are shown in Table 1.
Table 1.
Selected extracellular matrix proteins from pulmonary and aortic valves. Seq Cov- sequence coverage, No. Pep- Number of peptides per protein.
| Pulmonary Valve | Aortic Valve | ||||||
|---|---|---|---|---|---|---|---|
| Description | Accession | Molecular Weight (kDa) | pI | Seq Cov | No. Peps | Seq Cov | No. Peps |
| Perlecan | IPI00515360.8 | 469.7 | 6.0 | 22 | 76 | 23 | 71 |
| Collagen alpha3(VI) | IPI00877197.1 | 182.9 | 5.0 | 45 | 58 | 54 | 70 |
| Fibronectin 1 | IPI00352163.3 | 250.5 | 5.6 | 32 | 49 | 22 | 36 |
| Laminin subunit alpha-1 | IPI00113726.3 | 335.7 | 6.2 | 32 | 40 | 12 | 14 |
| Periostin | IPI00338018.1 | 93.1 | 7.3 | 53 | 29 | 60 | 36 |
| Collagen alpha-1(VI) chain | IPI00339885.2 | 204.8 | 5.8 | 31 | 27 | 34 | 30 |
| Laminin, beta 2 | IPI00109612.2 | 192.6 | 6.2 | 20 | 27 | 13 | 19 |
| Nidogen-1 | IPI00111793.1 | 133.6 | 5.1 | 27 | 26 | 12 | 11 |
| Collagen alpha-2 (VI) chain precursor | IPI00621027.2 | 107.8 | 5.8 | 24 | 22 | 23 | 22 |
| Versican | IPI00875672.1 | 364.6 | 4.3 | 10 | 22 | 10 | 26 |
| Hyaluronan and proteoglycan link protein 1 | IPI00131995.1 | 38.2 | 7.8 | 48 | 17 | 54 | 20 |
| Itih1 protein | IPI00322867.2 | 99.4 | 6.5 | 27 | 17 | 26 | 17 |
| Emilin-1 precursor | IPI00115516.1 | 105.2 | 5.0 | 23 | 16 | 28 | 19 |
| Serpin H1 precursor | IPI00114733.1 | 44.8 | 9.1 | 46 | 16 | 39 | 13 |
| Decorin precursor | IPI00123196.1 | 37.9 | 8.6 | 41 | 15 | 43 | 16 |
| Prostacyclin synthase | IPI00126498.1 | 54.3 | 5.9 | 32 | 14 | 39 | 16 |
| Collagen alpha-1 (XV) | IPI00409035.5 | 136.9 | 4.4 | 13 | 13 | 13 | 12 |
| Biglycan | IPI00123194.1 | 39.9 | 6.9 | 38 | 12 | 47 | 14 |
| Collagen alpha-1 (XVIII) | IPI00284816.4 | 179.3 | 5.2 | 9 | 12 | 9 | 10 |
| Latent-transforming growth factor beta-binding protein 4 | IPI00170216.1 | 176.1 | 5 0 | 10 | 12 | 9 | 11 |
| Lumican | IPI00313900.1 | 36.5 | 5.9 | 36 | 11 | 33 | 11 |
| Milk fat globule-EGF factor 8 | IPI00788387.1 | 49.0 | 5.9 | 30 | 11 | 43 | 20 |
| Angiotensin-converting enzyme | IPI00272690.2 | 150.9 | 6.1 | 10 | 11 | 9 | 9 |
| Fibulin-5 precursor | IPI00323035.3 | 47.6 | 4.1 | 29 | 10 | 28 | 10 |
| Mimecan | IPI00120848.1 | 31.8 | 5.4 | 35 | 10 | 35 | 9 |
| Pentraxin-related protein | IPI00114358.1 | 40.0 | 5.0 | 28 | 10 | 19 | 7 |
| Fibrinogen | IPI00115522.3 | 59.2 | 6.9 | 25 | 9 | 11 | 4 |
| Collagen alpha-2(I) chain | IPI00222188.4 | 129.5 | 9.5 | 7 | 9 | 6 | 7 |
| Dolichyl-diphosphooligosaccharide-protein glycosyltransferase | IPI00475154.1 | 66.9 | 5.4 | 22 | 9 | 21 | 9 |
| Tubulointerstitial nephritis antigen-like | IPI00115458.1 | 50.8 | 6.3 | 29 | 9 | 20 | 6 |
| Sarcalumenin | IPI00224456.2 | 99.2 | 4.1 | 10 | 9 | 13 | 11 |
| Fibrinogen gamma chain | IPI00122312.2 | 50.3 | 5.2 | 24 | 8 | 15 | 5 |
| Asporin | IPI00117957.1 | 40.2 | 8.6 | 27 | 8 | 35 | 14 |
| Integrin beta-1 precursor | IPI00132474.3 | 86.0 | 5.6 | 23 | 8 | 15 | 13 |
| Ubiquitin A-52 | IPI00138892.2 | 14.7 | 10.3 | 32 | 5 | 35 | 5 |
| Microfibril-associated glycoprotein 4 | IPI00133751.1 | 26.6 | 4.8 | 27 | 4 | 21 | 5 |
| Cartilage oligomeric matrix | IPI00882289.1 | 80.5 | 4.0 | 8 | 4 | 25 | 11 |
| Fibromodulin | IPI00120187.3 | 41.1 | 5.6 | 15 | 3 | 23 | 6 |
| Elastin | IPI00314186.1 | 69.2 | 10.8 | 10 | 3 | 3 | 2 |
We first selected for ECM proteins with edge connections to the major valvular proteins HSPG2, collagens, elastin, and fibulins, known to be essential to valvular phenotype by detailed biological studies.20 We then calculated related functions using proteins identified within the proteome, giving the local networks tissue expression specificity. Among the many potential candidate ECM proteins examined, four proteins, nidogen 1 (Ndg1), elastin microfibril interactor 1 (Emilin-1), biglycan (Bgn) and milk fat globule protein-EGF factor 8 protein (Mfge8), were selected based on numerous networked connections to major valvular proteins (Supplemental Figure 1). In each case, calculating the biological processes using proteins from the identified proteome narrowed the list of biological processes from those calculated using a nonspecific setting where all protein connections (reported or predicted across all tissue types) above the threshold setting were allowed. A result from this was that the local networks of Ndg1, Bgn and Emilin-1 produced a distinctive process of `ECM organization' with over 20% of proteins from each local network involved in this process, implying that the candidate proteins are also involved in this process (Figure 5).27,30 Other calculated functions from the local networks showed a high specificity for processes relevant to valvular biology. Major processes demonstrated unique potential for involvement in valve biology for each of the candidate proteins. Mfge8 showed significant functions related to stimulus response, including stress, relevant to the mechanical environment of the valve. The Bgn and Nid1 networks showed major processes of cell adhesion; adhesive properties of the ECM influence cell function and control valve quality.13 The least networked protein was Emilin-1, reflected in the low amount of associated genes and generalized functions. However, the genes making up the local network were considered highly relevant to valve biology. Emilin-1 directly interacts with elastin, a major component of mammalian valve structure, and the structural proteins fibrillin 1, 2, and 5.46
Figure 5.

Calculated biological functions for local networks surrounding the selected ECM proteins A) Ndg1; (45 genes), B) Bgn (21 genes), C) Mfge8 (31 genes); D) Emilin-1 (7 genes).
Immunoflourescence of ECM proteins earmarked by protein-protein networks
To begin to understand the role of the ECM proteins selected by protein-protein networking, we investigated the spatial distribution of Ndg1, Emilin-1, Bgn and Mfge8 within the pulmonary and aortic valve structure by immunofluorescence (Figure 6). Of note, none of these proteins have been previously explored for comparative localization between the pulmonary and aortic valve.
Figure 6.
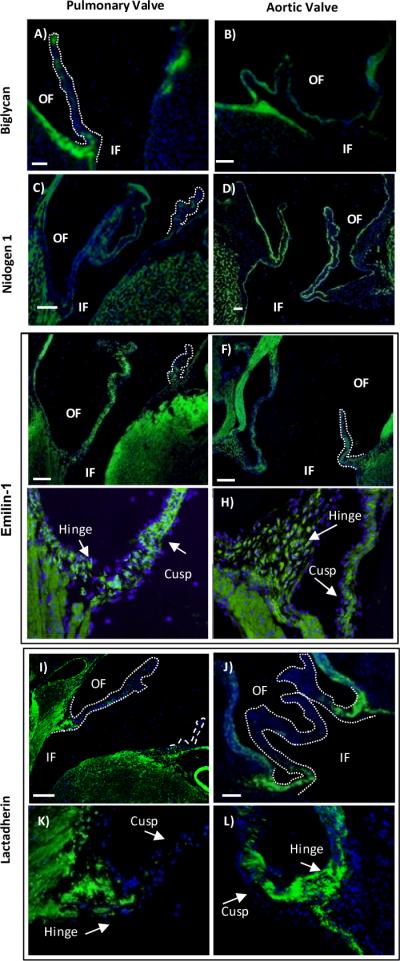
Immunofluorescence microscopy of select protein candidates earmarked by protein-protein networks, discussed in text. Blue-DAPI nuclear stain; green- fluorescein isothyocyanate (FITC) highlighting protein expression (Ndg1, Mfge8, Emilin-1) or Dylight 488-TFP (Bgn), false colored green ; OF- outflow surface; IF- Inflow surface. Bar- 100 μm. Dotted lines highlight the valve cusp.
Biglycan was expressed within the extracellular matrix of the valvular sinus. Expression within the walls of the pulmonary artery matched intensity of expression in the walls of the aorta, providing a control for comparison between the outflow tract valves. Within the cusps, Bgn expression varied (Figure 6A, B). Only patches of Bgn were detectable within the extracellular matrix of the pulmonary cusp. Within the aortic valve cusp, Bgn expression was uniform throughout the length of the aortic valve from hinge to cusp. Bgn appeared segregated to the central portion of the aortic cusp, a finding similar to that in human aortic valves.47
We observed that Ndg1 was expressed uniformly in a thin layer beneath the endothelial cells of the valve tissue (Figure 6C,D). This was an expected expression pattern, since Ndg1 is a reported basement membrane protein. Ndg1 was detected throughout the sinus walls and valve cusps with no obvious intensity differences between pulmonary and aortic heart valves. Ndg1 plays a key role within the basement membrane stabilizing tissues undergoing rapid growth and cell turnover. Since high cell turnover is associated with the arterial side of the valve, we measured expression depth of Ndg1 in the medial portion of the pulmonary and aortic valve. There was no significant difference in either valves between the ventricularis side, exposed to shear stress upon valve opening, versus the arterial side of the valve, exposed to turbulent shear stress upon valve closing. This result is comparable to reports from large mammals, where basement membrane thickness did not vary between the ventricular or arterial of the valve.48
We found that subcellular localization of Emilin-1 differed throughout the structure of the valve (Figure 6E–H). Within the cusps of both the pulmonary and aortic valves, Emilin-1 was mostly expressed throughout the extracellular matrix. However, towards the hinge portion Emilin-1 localized to the cytoplasm of the cell (Figure 6G,H). Both the pulmonary and aortic walls showed striated expression of Emilin-1 following the elastin laminae, correlating with previous reports of Emilin-1 within the arterial walls.46
Mfge8 expression localized to extracellular matrix within the valve hinges, but showed a significant variation between pulmonary and aortic valves (Figure 6I–L). In pulmonary valves, Mfge8 appeared in sparse patches of intense expression within the hinge and proximal regions of the cusp, but was absent in distal portions of the cusp. Aortic valves showed a more dense expression within the hinge region that appeared localized to the aortic side of the hinge (Figure 6K,L). Mfge8 expression in the aortic cusp extended to the medial section and in the distal region appeared in sparse patches of intense expression. Mfge8 appeared within the walls of the aorta, but did not appear within ventricular portions of the aortic valve sinus. We did not detect Mfge8 within the walls of the pulmonary valve sinus.
A new extracellular matrix network for the valvular structure
We used the IPA software to construct a network for extracellular matrix proteins identified by proteomics and investigated by immunofluorescence (Figure 7). The network contained 28 proteins, 20 of which were observed in the proteomic analysis. Primary functions of the combined network were `Tissue development' (16 proteins), `Cell-cell signaling and interaction' (13 proteins), and Cardiovascular system development and function' (10 proteins). Within both `Tissue development' and `Cell-cell signaling and interaction' functions was a cluster of proteins that have been shown to influence cell adhesive properties in other tissue systems, including vascular environments (Apoe, Bgn, Col18A1, Emilin-1, Fbln5, Hspg2, Nid1, Postn, Vcan). We further found enrichment in the total valvular proteomes of proteins affecting cellular adhesion (PV, 86 proteins p-value>2.58.0E-4, AV, 84 proteins, p-value 3.01E-5. Major complexes involved with signaling pathways linked to the calculated ECM network were the ERK, ERK(1/2), p38 MAPK, JNK, and NFKB pathways.
Figure 7.

Literature-supported network of proteins surrounding Ndg1, Bgn, Emilin-1, and Mfge8, calculated by Ingenuity Pathways Analysis. Gray nodes are proteins observed in combined outflow tract valve proteomes. Phenotypes of the network discussed in text. Apoe-apolipoprotein E; Bgn- biglycan; Col18A1- collagen 18A1; Col4A1- collagen 4A1; Dag1-dystroglycan associated protein 1; Eln- elastin; Emilin-1- elastin microfibril interacting protein 1; Emilin-2 - elastin microfibril interacting protein 1;Ephb2-ephyrin type-B receptor 2; Fbln2 – fibulin 2; Fbln5- fibulin 5; FBN1 – fibrillin 1; Hspg2- perlecan; Itgb1- integrin beta 1; Lama1- laminin alpha 1; Loxl1- Lysyl oxidase-like 1; Mfge8- milk fat globule-EGF factor 8 protein; Mtmr9 - myotubularin related protein 9; Nid1- nidogen 1; Nid2- nidogen 2; Nmi-N-myc-interactor; Postnperiostin; Prelp - proline, arginine-rich end leucine-rich repeat protein;Tgfb1-transforming growth factor beta 1; Tgfbr2- transforming growth factor beta receptor 2; Vcan- versican; Zp2- zona pellucid sperm-binding protein 2; Zp3 - zona pellucid sperm-binding protein 3.
Discussion
Extracellular matrix plays a critical role as a structural support for the valve and as a communication system between cellular components of the valve. Understanding functional localization of extracellular matrix components within the valve structure requires developing a baseline of protein expression patterns from healthy valve tissue. In this study, we isolated proteins from whole valve leaflets in order to identify and characterize new extracellular matrix proteins. Proteomic analysis revealed considerable insight into the global protein expression as well as identifying new ECM proteins involved in the biology of the adult mouse aortic and pulmonary valves.
An important observation was that only half of the proteins were in common between the PV and AV proteomes, indicating that these valves are different on a proteome-wide scale. Many of the differences in protein expression between each valve type could be explained in the context of the valvular environment. For instance, tenascin C expression within the aortic valve proteome corresponds with previous reports of a mechanosensitive expression of this protein within the valve structure.49, 50 In humans, tenascin C is expressed on the outflow tract side of the aortic leaflet49 and is upregulated in human calcific aortic valve stenosis.50 Another example of expression due to the valvular environment was the H2A histone family member X (H2AX) within the pulmonary valve proteome. H2AX is crucial as a DNA repair response and for maintaining proliferative capacity of vascular endothelial cells under hypoxic conditions via the ATR pathway.51 The thin heart valves obtain oxygen by diffusion through the tissue from the flowing blood.6 The PV valve population obviously experiences hypoxic conditions as oxygen depleted blood flows through the valve to become oxygenated in the pulmonary system, and H2AX expression could be a DNA repair response under these potentially damaging conditions. The biological processes from proteins clustered to individual proteomes also reflected differences in valvular environments. The pulmonary valve showed significant processes mapping to oxidation/reduction (Ox/Red), as well as protein transport. Under hypoxic conditions, Ox/Red activity slows, yet an adaptive increase of mitochondrial machinery has been reported for chronic hypoxic conditions in cardiac tissue;52 this may explain why this machinery is more dominant in the pulmonary valve proteome. Reactive oxygen species (ROS) are generated by oxidative/reductive machinery within the mitochondria and although the mechanism is controversial, ROS induced regulation of genetic factors is observed under hypoxic conditions.53 An intriguing idea is that the ROS due to the hypoxic environment plays a role in the differences between the valves. Future work on this topic will include comparison of the oxidative status within the valve proteomes. In both individual datasets, protein localization was a significant biological process. A major difference related to mechanical stress alterations during development and disease is the regulation of protein localization within the aortic valve structure.13,14,54 Differential localization of protein within the valve structures correlates to the need for functional protein expression in response to mechanical forces impacting the valve structure.13 The current immunofluorescent studies on proteomic data validate the expression of examined proteins within the outflow tract valves and show that it is the localization that varies significantly between the pulmonary and aortic valves. From these results, we believe that the striking difference between the proteomes represents a largely real difference and is not an artifact of dynamic acquisition during proteomic analysis.
The use of protein-protein interaction networks proved a valuable tool in selection of candidates for biological investigation. Relevant biological candidates were targeted for investigation using data from biological functions of local networks surrounding protein candidates and edge connections to developmentally important valve proteins. Subsequent immunofluorescence revealed functional localization of several ECM proteins previously unexplored in either pulmonary or aortic valve biology. Biglycan was intensely expressed within the ECM of the central portion of the aortic valve, but sparingly expressed within pulmonary valves, showing that in healthy valves, this protein is stimulated by the high pressure environment of the aortic valve. This correlates with previous investigations in human and porcine aortic valves, where upregulation of Bgn occurred in response to mechanical strain in culture and in situ.54 Biglycan has been implicated in adult aortic valve disease and appears to be a contributing factor to development of atherosclerotic lesions within the aorta;55 however, mechanistic details have yet to be elucidated.
We were especially intrigued by the differential subcellular localization of Emilin-1, localized to cuspal ECM and to the cytoplasm in the valve hinge. Extracellular matrix localization of Emilin-1 expression has been linked to attenuation of TGFβ1 processing within blood vessel walls.56 Upregulation of TGFβ1 promotes matrix deposition within the valve structure; overexpression of TGFβ1 leads to fibrosis, an event that is associated with degenerative valve disease.57 It is possible the distinct expression of Emilin-1 within the valve structure could be a novel mechanism of TGFβ-1 regulation within the healthy valve structure; detailed biological investigations using knockout animals are being performed to confirm this hypothesis.
Finally, immunoflourescence revealed an important functional localization of Mfge8. Mfge8 participates in apoptic cell engulfment by linking to cell surface exposed glycerophosphoserines and glycerophosphoethanolamines to form a bridge to phagocytic integrin αvβ3.58 In this study, Mfge8 expression was localized to the arterial side of the aortic hinge (Figure 6K,L) an area that experiences turbulent shear during each valve closure, a mechanical force that documented to induce cellular inflammation and apoptosis.9 The data indicated that this protein is abundant in both proteomes and the more extensive expression on the arterial surface of the aortic cusp appears to be significant to the mechanical environment of the aortic valve. From the combined data and literature, it is likely that Mfge8 plays a role in clearing apoptic cells due to turbulent stress impinging upon the aortic valve hinge.
A significant finding was that the calculated network of ECM proteins confirmed by immunoflourescence contained clusters of proteins associated with adhesive properties. Within the valvular environment, VIC adhesion receptor interactions with the ECM maintain the VIC phenotype and disruption of adhesive receptors activates processes of mineralization.59 Although poorly understood, calcification due to degenerative valve disease involves upregulation of adhesive molecules.60 This subset of proteins identified in the proteomics dataset of healthy semilunar valves could thus be useful towards providing a starting point for exploring the effects of altered adhesion within the degenerative valve.
In order to relate the calculated ECM protein network further to health of the valve, we examined genes from the calculated network for previously reported mouse phenotypes correlating to processes involved with developmental malformation of the heart, aberrant bone development and inflammation (both related to degenerative valve disease), and cardiac valve malformation (Shown in Figure 7). Since most phenotypes were not explored for valvular malformations, embryonic lethality was used as a potential indicator of valvular malformation; disruption of genes indispensible to early valve development is lethal to the continuing development of the embryo.20 Here, 8/29 proteins were embryonic lethal, including dystrophin-associated glycoprotein 1 (DAG1), uncharacterized in valve tissue, and involved in basement membrane structure through interactions with perlecan and laminin. Eleven of the genes were associated with developmental defects of the heart, including Emilin-1 and Nid1, indicating a potential for remaining networked genes to be associated with regulation of the valvular structure as well as global heart development. Finally, several genes produced phenotypes with increased inflammation and altered bone development. These included two proteins newly characterized here, Mfge8 (inflammation) and Bgn (abnormal bone development), both demonstrated within this work as having unique patterns in the aortic valve versus the pulmonary valve with potential disease relatedness. Inflammation of the valve tissue is a process associated with early valve degeneracy, and late stage valve disease involves calcification of the valve tissue, ultimately leading to the need for valve transplant. These proteins indicate that continued proteomic investigation of the degenerative heart valve will produce rich information on protein mechanisms influencing valvular disease.
Conclusions
This study represents the first detailed proteomic analysis of pulmonary and aortic heart valves. The detailing of in situ protein expression is crucial since valvular cells retain their phenotype for only a few passages in cell culture.61 Using mouse as a species to model valvular protein mechanisms allowed us to establish the groundwork for proteomic investigation of mouse models of specific valvular dysfunction as well as valvular tissue from large mammals, including humans. We utilized an SDS-based decellularization strategy to limit oversampling of abundant valvular collagen and to optimize number of new protein identifications. The produced pool of proteins supported the concept that there are critical differences between the two outflow tract valves, demonstrated by limited overlap between pulmonary and aortic proteins. These proteins appear to be related to differences in oxygen tension and mechanical force within the valvular environment.
Our main goal was to identify and characterize new primary ECM components of the valvular proteomes as these proteins play significant roles in regulating valve structure. Percentile ranking of proteins by spectral count proved effective at narrowing our search to the abundant ECM proteins within the proteome and showed that ECM proteins are the more abundant proteins within the valve proteome. The use of protein-protein networking as a tool to characterize protein expression was valuable in evaluating the potential importance of specific proteins within the valvular tissue. This was critical as protein function varies from tissue to tissue and valvular tissue is not well defined. This strategy of selecting protein candidates for biological evaluation based on abundance within the proteome, connection to well defined proteins of valvular biology, and calculated biological functions of local networks from the identified proteome allowed us to choose protein candidates with unique functional patterns between aortic and pulmonary valve. Importantly, although the ECM proteins investigated by immunoflourescence had similar expression levels in the aortic and pulmonary proteomes as reported by the proteomics study, these proteins were revealed to be functionally localized according to the environment of the valve. This characterization of the valvular proteomes provides the first insight into the mechanisms regulating distinct pulmonary and aortic valve functions and allows a new understanding of extracellular networks involved in communication and functional protein expression within the valve structure. These observations may be clinically relevant in explaining why valve function is frequently not preserved when pulmonary valves are utilized for correction of aortic valve disease.62
Supplementary Material
Acknowledgment
The authors wish to thank Erin Seeley for insightful review of the manuscript. P.M. A. was supported by an Interdisciplinary Postdoctoral Fellowship through the Systems-based Consortium for Organ Design and Engineering, 5RL1 HL092551-02. Funding for research provided by NIH grant 1-UL1-RR024920-01.
References
- 1.Birkmeyer N, O'Connor GT, Baldwin JC. Aortic valve replacement: current clinical practice and opportunities for quality improvement. Cur Opin Cardiol. 2001;16(2):152–157. doi: 10.1097/00001573-200103000-00013. [DOI] [PubMed] [Google Scholar]
- 2.Writing Group M, Rosamond W, Flegal K, Furie K, Go A, Greenlund K, Haase N, Hailpern SM, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O'Donnell C, Roger V, Sorlie P, Steinberger J, Thom T, Wilson M, Hong Y. American Heart Association Statistics Committee and Stroke Statistics, S., Heart Disease and Stroke Statistics--2008 Update: A Report From the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circ. 2008;117(4):e25–146. doi: 10.1161/CIRCULATIONAHA.107.187998. [DOI] [PubMed] [Google Scholar]
- 3.Sacks MS, Yoganathan AP. Heart valve function: a biomechanical perspective. Philos Trans R Soc B: Biol Sci. 2007;362(1484):1369–1391. doi: 10.1098/rstb.2007.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Novaro GM, Griffin BP. Calcific aortic stenosis: another face of atherosclerosis? Cleve Clin J Med. 2003;70(5):471–477. doi: 10.3949/ccjm.70.5.471. [DOI] [PubMed] [Google Scholar]
- 5.Butcher JT, S. C., Warnock JN. Mechanobiology of the aortic heart valve. J Heart Valve Dis. 2008;17(1):62–73. [PubMed] [Google Scholar]
- 6.Schoen FJ. Evolving concepts of cardiac valve dynamics: the continuum of development, functional structure, pathobiology, and tissue engineering. Circulation. 2008;118(18):1864. doi: 10.1161/CIRCULATIONAHA.108.805911. [DOI] [PubMed] [Google Scholar]
- 7.Robicsek F, Thubrikar MJ, Cook JW, Fowler B. The congenitally bicuspid aortic valve: how does it function? Why does it fail? Ann Thorac Cardiovasc Surgery. 2004;77(1):177–185. doi: 10.1016/s0003-4975(03)01249-9. [DOI] [PubMed] [Google Scholar]
- 8.Aikawa E, Whittaker P, Farber M, Mendelson K, Padera RF, Aikawa M, Schoen FJ. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113(10):1344–1352. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- 9.Butcher JT, Nerem RM. Valvular endothelial cells and the mechanoregulation of valvular pathology. Philos Trans R Soc B: Biol Sci. 2007;362(1484):1445–1457. doi: 10.1098/rstb.2007.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weinberg EJ, Mack PJ, Schoen FJ, García-Cardeña G, Kaazempur Mofrad MR. Hemodynamic environments from opposing sides of human aortic valve leaflets evoke distinct endothelial phenotypes in vitro. Cardiovasc Eng. 2010;10(1):5–11. doi: 10.1007/s10558-009-9089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simmons CA, Grant GR, Manduchi E, Davies PF. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circ Res. 2005;96(7):792–799. doi: 10.1161/01.RES.0000161998.92009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merryman WD, Liao J, Parekh A, Candiello JE, Lin H, Sacks MS. Differences in tissue-remodeling potential of aortic and pulmonary heart valve interstitial cells. Tissue Eng. 2007;13(9):2281–2289. doi: 10.1089/ten.2006.0324. [DOI] [PubMed] [Google Scholar]
- 13.David Merryman W. Mechano-potential etiologies of aortic valve disease. J Biomechanics. 2010;43(1):87–92. doi: 10.1016/j.jbiomech.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinton RB, Jr, Lincoln J, Deutsch GH, Osinska H, Manning PB, Benson DW, Yutzey KE. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98(11):1431–1438. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- 15.Handler M, Yurchenco PD, Iozzo RV. Developmental expression of perlecan during murine embryogenesis. Dev Dyn. 1998;210(2):130–145. doi: 10.1002/(SICI)1097-0177(199710)210:2<130::AID-AJA6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 16.Whitelock JM, Melrose J, Iozzo RV. Diverse Cell Signaling Events Modulated by Perlecan. Biochemistry. 2008;47(43):11174. doi: 10.1021/bi8013938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fayet C, Bendeck MP, Gotlieb AI. Cardiac valve interstitial cells secrete fibronectin and form fibrillar adhesions in response to injury. Cardiovasc Pathol. 2007;16(4):203–211. doi: 10.1016/j.carpath.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 18.Mjaatvedt CH, Yamamura H, Capehart AA, Turner D, Markwald RR, Splicing A, Myosins C, Mapping C, Cloning M, Mice MS. The Cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation. Dev Biol. 1998;202(1):56–66. doi: 10.1006/dbio.1998.9001. [DOI] [PubMed] [Google Scholar]
- 19.Wight TN. Versican: a versatile extracellular matrix proteoglycan in cell biology. Curr Opin Cell Biol. 2002;14(5):617–623. doi: 10.1016/s0955-0674(02)00375-7. [DOI] [PubMed] [Google Scholar]
- 20.Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. 2009;105(5):408–421. doi: 10.1161/CIRCRESAHA.109.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroeder JA, Jackson LF, Lee DC, Camenisch TD. Form and function of developing heart valves: coordination by extracellular matrix and growth factor signaling. J Mol Med. 2003;81(7):392–403. doi: 10.1007/s00109-003-0456-5. [DOI] [PubMed] [Google Scholar]
- 22.Pho M, Lee W, Watt DR, Laschinger C, Simmons CA, McCulloch CA. Cofilin is a marker of myofibroblast differentiation in cells from porcine aortic cardiac valves. Am J Physiol Heart Circ Physiol. 2008;294(4):H1767–H1778. doi: 10.1152/ajpheart.01305.2007. [DOI] [PubMed] [Google Scholar]
- 23.Faé KC, da Silva D. PDIA3, HSPA5 and vimentin, proteins identified by 2-DE in the valvular tissue, are the target antigens of peripheral and heart infiltrating T cells from chronic rheumatic heart disease patients. J Autoimmun. 2008;31(2):136–141. doi: 10.1016/j.jaut.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 24.Zerkowski HR, Grussenmeyer T, Matt P, Grapow M, Engelhardt S, Lefkovits I. Proteomics strategies in cardiovascular research. J Proteome Res. 2004;3(2):200–208. doi: 10.1021/pr034079t. [DOI] [PubMed] [Google Scholar]
- 25.Valencia A, Pazos F. Computational methods for the prediction of protein interactions. Curr Opin Struct Biol. 2002;12(3):368–373. doi: 10.1016/s0959-440x(02)00333-0. [DOI] [PubMed] [Google Scholar]
- 26.Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Mol Systems Biol. 2007;3:88–101. doi: 10.1038/msb4100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S, Chen H, Liu K, Sun Z. Inferring protein function by domain context similarities in protein-protein interaction networks. BMC bioinformatics. 2009;10(1):395–401. doi: 10.1186/1471-2105-10-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oti M, Snel B, Huynen MA, Brunner HG. Predicting disease genes using protein–protein interactions. J Med Genet. 2006;43(8):691–698. doi: 10.1136/jmg.2006.041376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fraser H, Plotkin J. Using protein complexes to predict phenotypic effects of gene mutation. Genome Biol. 2007;8(11):R252. doi: 10.1186/gb-2007-8-11-r252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li J, Zimmerman LJ, Park BH, Tabb DL, Liebler DC, Zhang B. Network-assisted protein identification and data interpretation in shotgun proteomics. Mol Syst Biol. 2009;5(1):303–314. doi: 10.1038/msb.2009.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang B, Chambers MC, Tabb DL. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J Proteome Res. 2007;6(9):3549–3557. doi: 10.1021/pr070230d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horton P, Park K-J, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucl Acids Res. 2007;35(suppl_2):W585–587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Da Wei Huang BTS, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 34.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):3. [PubMed] [Google Scholar]
- 35.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statistical Society. Series B (Methodological) 1995;57(1):289–300. [Google Scholar]
- 36.Ho MS, Bose K, Mokkapati S, Nischt R, Smyth N. Nidogens-Extracellular matrix linker molecules. Microsc Res Tech. 2008;71(5):387–395. doi: 10.1002/jemt.20567. [DOI] [PubMed] [Google Scholar]
- 37.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucl Acids Res. 2009;37:D412. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peacock JD, Lu Y, Koch M, Kadler KE, Lincoln J. Temporal and spatial expression of collagens during murine atrioventricular heart valve development and maintenance. Dev Dyn. 2008;237(10):3051–3058. doi: 10.1002/dvdy.21719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ott HC, Matthiesen TS, Goh SK, Black LD, Kren SM, Netoff TI, Taylor DA. Perfusion-decellularized matrix: using nature's platform to engineer a bioartificial heart. Nature Med. 2008;14(2):213–221. doi: 10.1038/nm1684. [DOI] [PubMed] [Google Scholar]
- 40.Gilbert TW, Sellaro TL, Badylak SF. Decellularization of tissues and organs. Biomaterials. 2006;27(19):3675–3683. doi: 10.1016/j.biomaterials.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 41.Romer LH, Birukov KG, Garcia JGN. Focal adhesions: paradigm for a signaling nexus. Circ Res. 2006;98(5):606–616. doi: 10.1161/01.RES.0000207408.31270.db. [DOI] [PubMed] [Google Scholar]
- 42.Larsen M, Artym VV, Green JA, Yamada KM. The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr Opin Cell Biol. 2006;18(5):463–471. doi: 10.1016/j.ceb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 43.Ma'ayan A. Insights into the organization of biochemical regulatory networks using graph theory analyses. J Biol Chem. 2009;284(9):5451–5455. doi: 10.1074/jbc.R800056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jeong H, Mason SP, Barabási AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411(6833):41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 45.Zotenko E, Mestre J, O'Leary DP, Przytycka TM. Why do hubs in the yeast protein interaction network tend to be essential: reexamining the connection between the network topology and essentiality. PLoS Comput Biol. 2008;4(8):e1000140. doi: 10.1371/journal.pcbi.1000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zanetti M, Braghetta P, Sabatelli P, Mura I, Doliana R, Colombatti A, Volpin D, Bonaldo P, Bressan GM. EMILIN-1 deficiency induces elastogenesis and vascular cell defects. Mol Cell Biol. 2004;24(2):638–650. doi: 10.1128/MCB.24.2.638-650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Latif N, Sarathchandra P, Taylor PM, Antoniw J, Yacoub MH. Localization and pattern of expression of extracellular matrix components in human heart valves. J Heart Valve Dis. 2005;14(2):218–227. [PubMed] [Google Scholar]
- 48.Brody S, Anilkumar T, Liliensiek S, Last JA, Murphy CJ, Pandit A. Characterizing nanoscale topography of the aortic heart valve basement membrane for tissue engineering heart valve scaffold design. Tissue Eng. 2006;12(2):413–421. doi: 10.1089/ten.2006.12.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schenke-Layland K, Stock UA, Nsair A, Xie J, Angelis E, Fonseca CG, Larbig R, Mahajan A, Shivkumar K, Fishbein MC. Cardiomyopathy is associated with structural remodelling of heart valve extracellular matrix. Eur Heart J. 2009;30(18):2254–2265. doi: 10.1093/eurheartj/ehp267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jian B, Jones PL, Li Q, Mohler Iii ER, Schoen FJ, Levy RJ. Matrix metalloproteinase-2 is associated with tenascin-C in calcific aortic stenosis. Am J Pathol. 2001;159(1):321–327. doi: 10.1016/S0002-9440(10)61698-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Economopoulou M, Langer HF, Celeste A, Orlova VV, Choi EY, Ma M, Vassilopoulos A, Callen E, Deng C, Bassing CH. Histone H2AX is integral to hypoxia-driven neovascularisation. Nature Med. 2009;15(5):553–558. doi: 10.1038/nm.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wust RCI, Jaspers RT, van Heijst AF, Hopman MTE, Hoofd LJC, van der Laarse WJ, Degens H. Region-specific adaptations in determinants of rat skeletal muscle oxygenation to chronic hypoxia. Am J Physiol Heart Circulatory Physiol. 2009;297(1):H364–H374. doi: 10.1152/ajpheart.00272.2009. [DOI] [PubMed] [Google Scholar]
- 53.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr Opin Cell Biol. 2009;21(6):894–899. doi: 10.1016/j.ceb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lehmann S, Walther T, Kempfert J, Rastan A, Garbade J, Dhein S, Mohr FW. Mechanical Strain and the Aortic Valve: Influence on Fibroblasts, Extracellular Matrix, and Potential Stenosis. Ann Thoracic Surg. 2009;88(5):1476–1486. doi: 10.1016/j.athoracsur.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 55.Olin KL, Potter-Perigo S, Barrett PHR, Wight TN, Chait A. Biglycan, a vascular proteoglycan, binds differently to HDL2 and HDL3: role of apoE. Arterioscler Thromb, Vasc Biol. 2001;21(1):129–135. doi: 10.1161/01.atv.21.1.129. [DOI] [PubMed] [Google Scholar]
- 56.Zacchigna L, Vecchione C, Notte A, Cordenonsi M, Dupont S, et al. Emilin1 Links TGF-[beta] Maturation to Blood Pressure Homeostasis. Cell. 2006;124(5):929–942. doi: 10.1016/j.cell.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 57.Hakuno D, Kimura N, Yoshioka M, Fukuda K. Molecular mechanisms underlying the onset of degenerative aortic valve disease. J Mol Med. 2009;87(1):17–24. doi: 10.1007/s00109-008-0400-9. [DOI] [PubMed] [Google Scholar]
- 58.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417(6885):182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 59.Gu X, Masters KS. Regulation of valvular interstitial cell calcification by adhesive peptide sequences. J Biomed Mater Res A. 2010;93A(4):1620–1630. doi: 10.1002/jbm.a.32660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yetkin E, Waltenberger J. Molecular and cellular mechanisms of aortic stenosis. Int J Cardiol. 2009;135(1):4–13. doi: 10.1016/j.ijcard.2009.03.108. [DOI] [PubMed] [Google Scholar]
- 61.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171(5):1407–1418. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pasquali SK, Shera D, Wernovsky G, Cohen MS, Tabbutt S, Nicolson S, Spray TL, Marino BS. Midterm outcomes and predictors of reintervention after the Ross procedure in infants, children, and young adults. J Thorac Cardiovasc Surg. 2007;133(4):893–899. doi: 10.1016/j.jtcvs.2006.12.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.