

Abstract
Traits that have been stringently selected to conform to specific criteria in a closed population are phenotypic stereotypes. In dogs, Canis familiaris, such stereotypes have been produced by breeding for conformation, performance (behaviors), etc. We measured phenotypes on a representative sample to establish breed stereotypes. DNA samples from 147 dog breeds were used to characterize single nucleotide polymorphism allele frequencies for association mapping of breed stereotypes. We identified significant size loci (quantitative trait loci [QTLs]), implicating candidate genes appropriate to regulation of size (e.g., IGF1, IGF2BP2 SMAD2, etc.). Analysis of other morphological stereotypes, also under extreme selection, identified many additional significant loci. Behavioral loci for herding, pointing, and boldness implicated candidate genes appropriate to behavior (e.g., MC2R, DRD1, and PCDH9). Significant loci for longevity, a breed characteristic inversely correlated with breed size, were identified. The power of this approach to identify loci regulating the incidence of specific polygenic diseases is demonstrated by the association of a specific IGF1 haplotype with hip dysplasia, patella luxation, and pacreatitis.
Keywords: association, canine, disease, longevity, morphology, QTL
Dog breeds are closed breeding populations (genetic isolates) that have been under stringent selection for breed standards (morphological or behavioral traits; Parker et al. 2004; Ostrander and Wayne 2005; Calboli et al. 2008). Many breeds have been through a significant bottleneck (Sutter and Ostrander 2004; Lindblad-Toh et al. 2005), and all have been subject to drift. Allele frequencies in some regions of the genome have been driven to extremes in many breeds. Where these regions represent ancient haplotypes common to all dog breeds, they should inform the same phenotype. The breed selection process has had unintended consequences on the health and longevity of purebred dogs, with high rates of specific diseases in certain breeds due to increased frequency of risk alleles. Disease frequency is, therefore, a breed characteristic phenotype that can be used for genetic analysis.
Because fixed portions of a breed's genome will remain fixed as long as the breeding population remains closed (barring mutation), the phenotypes that they regulate will continue to produce consistent phenotypes, and therefore, the phenotype and genotype need not be measured on the same animal. Both the allele frequency of a single nucleotide polymorphism (SNP) in fixed regions of the genome and the phenotype are enduring characteristics of a breed. As a result, associating breed-specific genotypes with breed-specific phenotypes in multiple breeds (across-breed mapping) presents a powerful tool for identifying quantitative trait loci (QTLs) that may form the genetic basis for the phenotypic diversity observed in dog breeds.
Across-breed association analysis requires a large number of breeds for which the genome of each breed has been characterized by a common set of well-distributed, highly informative SNPs. In addition, a quantitative evaluation of the fixed phenotypes associated with each breed must be available. Phenotypes that have been under stringent selection, such as morphology and behavior, are ideal for this purpose. Using across-breed mapping, we have analyzed the genetic basis for various morphological features (including size), some behaviors, and the relationship between size and longevity. Here, we discuss those results and then present preliminary evidence that the technique can be used to identify fixed genotypes that influence the frequency with which polygenic disease is expressed in a breed.
Methods
Phenotypes
A total of 147 domestic dog breeds were characterized for a variety of sex-averaged phenotypes: height, weight, other morphology characters, longevity, and behavior. Phenotypic values used for the different breeds are given in Supplementary Table 1 of Jones et al. (2008). They can be obtained at http://www.genetics.org/cgi/content/full/genetics.108.087866/.
Disease frequencies for hip dysplasia, patella luxation, and pancreatitis were obtained from the Veterinary Medicine Data Base (VMDB) (http://www.vmdb.org/vmdb.html). This database consists of approximately 1.5 million canine disease reports from 22 veterinary medical schools. Cases are coded according to various symptoms, breed, sex, and age. We have calculated breed frequency of any disease as the fraction of the total visits for a particular breed that resulted in a diagnosis of the particular disease of interest.
Genotypes
Details of genotyping are described in Jones et al. (2008). Further details of the marker set and allele frequencies are found at http://www.genetics.org/cgi/content/full/genetics.108.087866/.
Breeds were characterized using a common set of SNP markers. For the experiments described, 2801 dogs representing 147 breeds were used. One hundred and twenty-nine of these breeds were represented by 10 or more dogs. DNA from each dog was genotyped using 1536 markers, of which 674 were spaced across the 38 canine autosomes. A total of 862 additional markers were concentrated in regions of interest that showed maximal variation in allele frequency between breeds. The median distance between markers was 409 kb although only approximately 26% of the genome was within 250 kb of a marker.
SNP Association
Correlations between breed allele frequency (xi) and breed-characterized phenotypes (yi) were tested using a weighted Pearson product correlation.
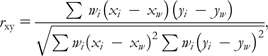 |
where, 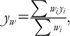
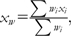 and
ni = number of animals for breed i. Two measures of significance were important: single SNP P value and genome-wide P value (e.g., the probability of a particular rxy value in a single test and the multitest correction when testing all SNPs across the genome).
and
ni = number of animals for breed i. Two measures of significance were important: single SNP P value and genome-wide P value (e.g., the probability of a particular rxy value in a single test and the multitest correction when testing all SNPs across the genome).
Permutation tests were used to establish the null distribution of the rxy statistic for each SNP and for each phenotype. A generalized extreme value distribution was fit to the empirical “null” data using the gevFit function of the fExtremes package (Wuertz 2006) for R (R Development Core Team 2006). The Kolmogorof–Smirnoff test (Conover 1971) of the R package (ks.test) was used to test the goodness of fit. Distributions with a ks.test P value of 0.01 or less were considered poorly estimated and dropped. The significance of rxy values was estimated using the cumulative probability function (pgev) and −log10 transformed for convenience (log P). For each permutation, the phenotype was randomized with respect to the genotypes. The maximum score across all SNPs in the data set was recorded as the single genome-scan maximum. Genome-scan maximum values from 1000 permutations were used to estimate the null distribution of a genome-wide scan. The 90%, 95%, and 99% percentiles of this distribution were used as the thresholds from genome-wide significance of 0.1, 0.05, and 0.01, respectively. Where necessary, 10 000 permutations were used to estimate genome-wide significance thresholds of 0.001.
Results and Discussion
Morphological QTLs were identified by association across breeds using breed stereotypes (fixed phenotypes) based on data described in our previous publication (Jones et al. 2008). Unlike within-breed mapping of segregant phenotypes, across-breed mapping identifies loci within a much smaller linkage disequilibrium (LD) distance 400–500 kb (Jones et al. 2008). Thus, relevant candidate genes are identified from a much smaller genome region. In all, 30 morphological QTLs were identified. A subset of these, listed in Table 1, contained relevant candidate genes. These represent immediate targets for fine mapping and/or validation in specific breeds (e.g., hair length and FGF5 in Dachshunds and Corgis [Housley and Venta 2006]).
Table 1.
Details of QTLs for size-related traits and other aspects of morphology
| Trait | Chrom | Pos | Log P | GWT | No. of genes | Candidate genes |
| Weight | CFA 7 | 46696633 | 7.20 | 0.001 | 7 | SMAD2, NPR2 |
| CFA 10 | 11465975 | 3.63 | 0.05 | 5 | HMGA2 | |
| CFA 15 | 37006865 | 3.86 | 0.05 | 2 | SOCS2 | |
| CFA 15 | 44228026 | 4.59 | 0.01 | 5 | IGF1 | |
| CFA 34 | 21414695 | 3.36 | 0.1 | 9 | IGF2BP2 | |
| Short coat | CFA 25 | 17862111 | 3.88 | 0.01 | 5 | TNFRSF19 |
| CFA 32 | 7806734 | 5.43 | 0.001 | 1 | Fgf5 | |
| Tail curve | CFA 25 | 51048799 | 4.36 | 0.01 | 4 | COL6A3 |
| Heada | CFA 9 | 25422459 | 4.01 | 0.01 | 16 | IGFBP4 |
| Lega | CFA 3 | 64678450 | 4.27 | 0.01 | 8 | RNF4, MXD |
| Necka | CFA 9 | 24032840 | 5.00 | 0.001 | 17 | STAT3 |
Traits are listed on the left (for details see Jones et al. 2008). Chromosomes (Chrom, CFA) and CanFam2 base pair position (Pos) of the SNP at which significance was estimated are shown in columns 2 and 3. Log P is the negative logarithm of the P value for the single SNP test. The genome-wide significance threshold (GWT) exceeded is shown in the GWT column. GWTs for log P varied between 3.26 and 3.29 for P < 0.1, 3.45–3.50 for P < 0.05, 3.8–4.1 for P < 0.01, and 4.35–4.7 for P < 0.001. Number of genes shows a count of the number of genes within 200 kb of the SNP. The names of candidate genes within this LD interval are listed in the last column. More significant loci are in bold. Italics denote genes.
The ratio of the head, leg, or neck to overall body size.
To test the potential of the technique, we have analyzed several behavioral phenotypes. Using an ethological approach, we asked a dog trainer with more than 30 years experience training and judging various dog breeds to score all the breeds for boldness, herding, pointing, and trainability. In all, 10 loci were identified 5 of which contained the suggestive candidate genes presented in Table 2. Four of these candidate genes relate to various loci affecting the nervous system and/or have been implicated in behavior: MC2R on CFA 1 (27381939 bp) is a melanocortin receptor, and C18orf1 (27572327 bp) has been implicated in schizophrenia. DRD1, on CFA 4 (40743436 bp), encodes a dopamine subtype receptor. CNIH, on CFA 8 (33396000), has been implicated in cranial nerve development. Finally, PCDH9, on CFA 22 (24273482 bp), encodes a protein localized to synaptic junctions and believed to be involved in specific neural connections and signal transduction. Although the behaviors involved are poorly defined, the presence of major candidate genes appropriate to behavior is encouraging. The apparent exception is IGF1 that might be expected to affect boldness on the basis of size (large dogs bold, small timid). However, the locus on CFA 15 does not appear to be related to size, as approximately equal numbers of large and small breeds were found to be bold (Jones et al. 2008), and boldness and size were not correlated (r = 0.18; P = 0.3).
Table 2.
QTLs associated with behavior
| Trait | Chromosome | Position | Log P | GWT | No. of genes | Candidate genes |
| Herding | CFA 1 | 27630805 | 7.20 | 0.001 | 4 | MC2R, C18orf1 |
| Boldness | CFA 4 | 40782966 | 4.15 | 0.05 | 7 | DRD1 |
| Pointing | CFA 8 | 33344686 | 5.33 | 0.05 | 6 | CNIH |
| Boldness | CFA 15 | 44137464 | 5.05 | 0.001 | 5 | IGF1 |
| Boldness | CFA 22 | 25446003 | 6.09 | 0.001 | 1 | PCDH9 |
As in Table 1, a genome-wide SNP scan was used to associate SNP markers with several behavioral phenotypes: pointing, herding, and boldness. Phenotypic data is available in Jones et al. (2008). From left to right, columns list the trait, chromosome, CanFam2 nucleotide position on the chromosome, the log P value of the significance, the genome-wide threshold (GWT) of significance, number of known genes in the LD interval (400 kb), and possible candidate genes. The GWTs for the 3 traits were herding: 0.01 < P < 0.05 = 4.38; P < 0.01 = 5.04; pointing: 0.01 < P < 0.05 = 4.69; P < 0.01 = 5.69; Boldness: 0.01 < P < 0.05 = 4.09; P < 0.01 = 4.81. More significant loci are in bold. Italics denote genes.
Tables 1 and 2 present previous across-breed mapping results (Jones et al. 2008) for morphology and behavior. Across-breed mapping successfully overlapped previously identified loci for the traits analyzed and suggested a number of new candidate loci appropriate to the stereotypic phenotype in question. In all cases, we were able to rule out spurious effects due to a simple measure of between-breed relatedness (i.e., average breed genome similarity [Jones et al. 2008]). However, the genotyping database that we used only interrogated about 25% of the genome. Thus, many potential loci will not have been identified. More importantly, false positives due to nonsystenic LD and more complex breed relationships cannot be ruled out without complete genome coverage.
Many of the limitations of the current data set will be addressed by an effort, termed CanMap, (http://www.sciencemag.org/cgi/content/full/sci;317/5845/1668) currently being completed. The goal of the CanMap project is to produce dense SNP profiles of a dozen dogs from each of nearly a hundred breeds. Unrelated dogs from each breed (deemed unrelated if they shared no common grandparents) will have been genotyped to provide much more complete genome coverage.
Despite the power of across-breed mapping to identify genomic regions of interest, the potential for false positives, whereby causative regions of the genome cannot be distinguished from noncausative, will always necessitate validation using within-breed segregation analysis. Most often, across-breed mapping identifies markers that tend to be near or at fixation (homozygous) in breeds with the associated phenotype. Breeds in which the phenotype is still segregating will not contribute to the power of QTL identification. However, they will provide a resource in which the association can be validated using within-breed segregation analysis. Such breeds are readily identified from the across-breed SNP genotyping database (e.g., alelle frequencies near 0.5). It should be possible now to validate the most significant (P ≤0.001) loci in Tables 1 and 2 using breeds in which the implicated SNPs are segregating (e.g., the locus on CFA 32 for short coat (Table 2) was identified by segregation analysis using Dachshunds or Corgis (Housley and Venta 2006).
Another facet of across-breed mapping is the ability to investigate phenotypes that vary between breeds but do not appear to segregate within any breed. One such trait is longevity. It has long been known that, on average, dogs from small breeds live longer than those from large breeds (Egenvall et al. 2005). However, a detailed study of longevity and size within breeds has failed to provide any evidence for a similar relationship of size and longevity. Indeed, in that study, there was a trend suggesting that within-breed larger dogs may live longer (Galis et al. 2007).
Using mean breed longevity (Jones et al. 2008), across-breed mapping identified the 2 major size loci on CFA 7 and CFA 15 as highly significant loci regulating longevity (Table 3). Given the inverse correlation of size and longevity, this may seem like a trivial result. However, the difference in significance between size and age of death in the IGF1 region of CFA 15 suggests that more than just an effect on size is involved. The fact that within breeds increasing size fails to decrease longevity also suggests that size per se is not a causal factor in altering longevity. Moreover, the association of IGF1 with breed longevity is not unexpected considering that it has been implicated in regulating longevity in a number of organisms (Kenyon 2001; Bartke 2005).
Table 3.
QTLs associated with age of death (AOD) and the probability that size is also associated with that SNP. Trait, chromosome (CFA), CanFam2 nucleotide position on the chromosome, log P, and genome-wide significance threshold (GWT) are as in Table 1
| Trait | Chrom | Position | Log P | GWT |
| AOD | CFA 7 | 46696633 | 7.06 | 0.001 |
| Size | CFA 7 | 46696633 | 7.73 | 0.001 |
| AOD | CFA 15 | 44228026 | 8.94 | 0.001 |
| Size | CFA 15 | 44228026 | 4.59 | 0.010 |
Log P GWT for AOD was 3.95 for P < 0.01 and for size was 4.00 for P < 0.01.
One explanation may be that introducing loci that drastically change growth rate may detrimentally perturb physiological balance, exposing the system to various senescent pathologies without the compensating mutations that normally would arise in the course of a prolonged evolutionary process. More generally, strong selection for morphological or behavioral traits has generated a variety of genome configurations. Some of these, though functional through reproductive maturity, may increase risk of complex disease (Egenvall 2005). Across-breed mapping can be used to determine the impact of the breed-fixed genome configurations on disease risk, provided that accurate estimates of breed disease frequencies are available.
As a first test of this hypothesis, we have analyzed the association of IGF1 with variation in the frequency of certain diseases. It is known that breed size is correlated with the frequency of certain orthopedic diseases found in many dog breeds. Recently, pancreatitis has been correlated with the concentration of IGF1 in rodent serum, and it has been possible to decrease the incidence of pancreatitis by increasing serum levels of IGF1 (Warzecha et al. 2003; Dembinski et al. 2006). We therefore expected that across-breed mapping might associate IGF1 haplotypes with such diseases. We obtained disease frequencies (Figure 1) for hip dysplasia, patella luxation, and pancreatitis from the VMDB (see Methods). Figure 2 presents the frequency of these diseases in small and large dog breeds. Here, small and large breeds are defined by the frequency of an SNP allele on CFA 15 (44228468 bp) that is part of the nucleotide sequence common to all small dogs (Sutter et al. 2007). For all 3 diseases, the association with IGF1 is very significant (genome-wide significance P < 0.01). These associations were not the result of genotypic relatedness between breeds as they became more significant after correcting for breed relationships. This implies that selection for size has influenced the frequency of these diseases.
Figure 1.
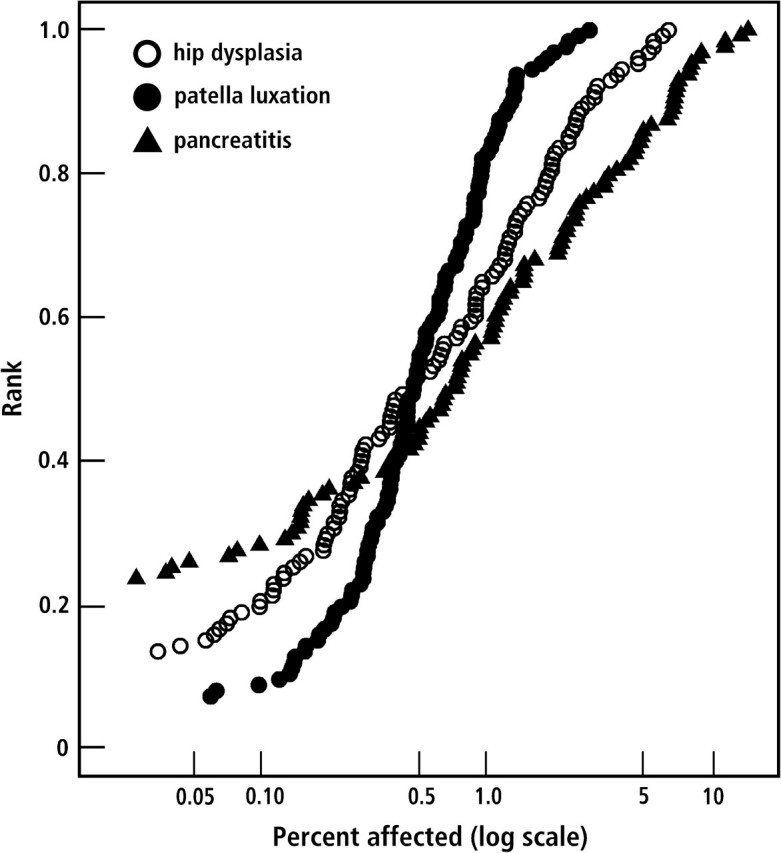
Disease frequencies are graphed as cumulative distributions (x axis, log scale) for hip dysplasia (open circles), patella luxation (closed circles), and pancreatitis (closed triangles). Records from 22 veterinary hospitals were obtained from the VMDB (see Methods). In all, 129 breeds were included that had records for more than 100 individuals. Cumulative rankings start at different places on the y axis corresponding to the number of breeds for which there were no diagnoses recorded for the disease—for example, 30 breeds without hip dysplasia, 17 breeds without patella luxation, and 9 breeds without pancreatitis.
Figure 2.

Frequencies of hip dysplasia, patella luxation, and pancreatitis in large and small breeds of dogs. Frequencies of these diseases (Figure 1) were divided between the 2 groups of breeds. Cumulative distributions are shown for each group. We defined small breeds as those with a frequency of more than 0.85 for the small IGF1 haplotype (Sutter et al. 2007). Conversely, large breeds were defined as those with a frequency of less than 0.15 for this same haplotype.
Across-breed mapping combined with within-breed mapping is a powerful approach for exploration of the wide range of behavior, morphology, and disease frequencies observed across the spectrum of dog breeds. The CanMap project promises the characterization of the allele frequencies for many breeds, thus creating an invaluable enduring fixed resource. As we obtain a more detailed picture of the complex relationships between breeds, across-breed mapping will become more sensitive and less prone to false positives. Characterization of breed phenotypes can be applied to the CanMap resource as an easy starting point from which to carry out genetic analysis of many complex phenotypes. Results from across-breed mapping will provide a narrower range of focus for validation in specific breeds. Different breeds may be fixed for different haplotypes affecting a complex phenotype. These provide snapshots of genotypes, each of which display a simplified aspect of the interactive network. Integration of these should become a powerful tool for understanding the network as a whole.
References
- Bartke A. Minireview: role of the growth hormone/insulin-like growth factor system in mammalian aging. Endocrinology. 2005;146:3718–3723. doi: 10.1210/en.2005-0411. [DOI] [PubMed] [Google Scholar]
- Calboli FC, Sampson J, Fretwell N, Balding DJ. Population structure and inbreeding from pedigree analysis of purebred dogs. Genetics. 2008;179:593–601. doi: 10.1534/genetics.107.084954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover WJ. Two-sample “Smirnov” test. Practical nonparametric statistics. New York: John Wiley And Sons; 1971. 309–314. [Google Scholar]
- Dembinski A, Warzecha Z, Ceranowicz P, Cieszkowski J, Pawlik WW, Tomaszewska R, Kusnierz-Cabala B, Naskalski JW, Kuwahara A, Kato I. Role of growth hormone and insulin-like growth factor-1 in the protective effect of ghrelin in ischemia/reperfusion-induced acute pancreatitis. Growth Horm IGF Res. 2006;16:348–356. doi: 10.1016/j.ghir.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Egenvall A, Bonnett BN, Hedhammar A, Olson P. Mortality in over 350,000 insured Swedish dogs from 1995-2000: II. Breed-specific age and survival patterns and relative risk for causes of death. Acta Vet Scand. 2005;46:121–136. doi: 10.1186/1751-0147-46-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galis F, Van der Sluijs I, Van Dooren TJ, Metz JA, Nussbaumer M. Do large dogs die young? J Exp Zoolog B Mol Dev Evol. 2007;308:119–126. doi: 10.1002/jez.b.21116. [DOI] [PubMed] [Google Scholar]
- Housley DJ, Venta PJ. The long and the short of it: evidence that FGF5 is a major determinant of canine ‘hair'-itability. Anim Genet. 2006;37:309–315. doi: 10.1111/j.1365-2052.2006.01448.x. [DOI] [PubMed] [Google Scholar]
- Jones P, Chase K, Martin A, Davern P, Ostrander EA, Lark KG. Single-nucleotide-polymorphism-based association mapping of dog stereotypes. Genetics. 2008;179:1033–1044. doi: 10.1534/genetics.108.087866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C. A conserved regulatory system for aging. Cell. 2001;105:165–168. doi: 10.1016/s0092-8674(01)00306-3. [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, 3rd, Zody MC, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- Ostrander EA, Wayne RK. The canine genome. Genome Res. 2005;15:1706–1716. doi: 10.1101/gr.3736605. [DOI] [PubMed] [Google Scholar]
- Parker HG, Kim LV, Sutter NB, Carlson S, Lorentzen TD, Malek TB, Johnson GS, DeFrance HB, Ostrander EA, Kruglyak L. Genetic structure of the purebred domestic dog. Science. 2004;304:1160–1164. doi: 10.1126/science.1097406. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. ISBN 3-900051-07-0 [Internet] Vienna (Austria): R Foundation for Statistical Computing; 2006. Available from: URL http://www.R-project.org. [Google Scholar]
- Sutter NB, Bustamante CD, Chase K, Gray MM, Zhao K, Zhu L, et al. A single IGF1 allele is a major determinant of small size in dogs. Science. 2007;316:112–115. doi: 10.1126/science.1137045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter NB, Ostrander EA. Dog star rising: the canine genetic system. Nat Rev Genet. 2004;5:900–910. doi: 10.1038/nrg1492. [DOI] [PubMed] [Google Scholar]
- Warzecha Z, Dembinski A, Ceranowicz P, Konturek SJ, Tomaszewska R, Stachura J, Konturek PC. IGF-1 stimulates production of interleukin-10 and inhibits development of caerulein-induced pancreatitis. J Physiol Pharmacol. 2003;54:575–590. [PubMed] [Google Scholar]
- Wuertz D. fExtremes: Rmetrics—extreme financial market data. R package version 240.10068. 2006. Available from: URL http://www.rmetrics.org. [Google Scholar]