

Abstract
Objective
Aberrant posttranscriptional regulation of matrix metalloproteinases (MMPs) by microRNA has emerged as an important factor in human diseases. The aim of this study was to determine whether the expression of MMP-13 in human osteoarthritis (OA) chondrocytes is regulated by microRNA.
Methods
Chondrocytes were stimulated with interleukin-1β (IL-1β) in vitro. Total RNA was prepared using TRIzol reagent. Polymerase chain reaction (PCR)–based arrays were used to determine the expression profile of 352 human microRNA. Gene expression was quantified using TaqMan assays, and microRNA targets were identified using bioinformatics. Transfection with reporter construct and microRNA mimic was used to verify suppression of target messenger RNA (mRNA). Gene expression of argonaute and Dicer was determined by reverse transcription–PCR, and expression of protein was determined by immunoblotting. The role of activated MAP kinases (MAPKs) and NF-κB was evaluated using specific inhibitors.
Results
In IL-1β–stimulated OA chondrocytes, 42 microRNA were down-regulated, 2 microRNA were up-regulated, and the expression of 308 microRNA remained unchanged. In silico analysis identified a sequence in the 3′-untranslated region (3′-UTR) of MMP-13 mRNA complementary to the seed sequence of microRNA-27b (miR-27b). Increased expression of MMP-13 correlated with down-regulation of miR-27b. Overexpression of miR-27b suppressed the activity of a reporter construct containing the 3′-UTR of human MMP-13 mRNA and inhibited the IL-1β–induced expression of MMP-13 protein in chondrocytes. NF-κB and MAPK activation down-regulated the expression of miR-27b.
Conclusion
Our data demonstrated the expression of miR-27b in both normal and OA chondrocytes. Furthermore, IL-1β–induced activation of signal transduction pathways associated with the expression of MMP-13 down-regulated the expression of miR-27b. Thus, miR-27b may play a role in regulating the expression of MMP-13 in human chondrocytes.
Osteoarthritis (OA) is the most prevalent disease of the articulating joints and is characterized by pain, tenderness, limitation of movement, crepitus, and a variable degree of inflammation without systemic effects (1,2). The pathogenesis of OA is complex and involves the interaction of multiple factors ranging from genetic predisposition to altered mechanical loading and changes in the gene expression repertoire of the articular chondrocytes (3,4). In addition, symptoms of local inflammation and synovitis are present in many patients with OA and are also seen in animal models of OA (for review, see refs. 3–5). Proteolytic degradation of cartilage is a hallmark of OA, and activated chondrocytes are now known to produce matrix-degrading enzymes such as collagenase 3 (matrix metalloproteinase 13 [MMP-13]) in OA joints (5,6). MMP-13 has a broad substrate specificity and can cleave types I, II, III, IV, X, and XIV collagen, aggrecan, and fibronectin, with the highest activity toward type II collagen (7–9). The role of MMP-13 in OA was shown using a transgenic mouse model in which induced expression of MMP-13 resulted in pathologic changes in the joints, similar to human OA (10). In addition, the proinflammatory cytokine interleukin-1β (IL-1β) and MMP-13 localize to the site of cartilage degradation in OA joints, providing evidence of their key roles in the pathogenesis of OA (11; for review, see ref. 12). Furthermore, inhibition of IL-1 ameliorated OA-like pathology in animal models, and the role of IL-1 in OA pathogenesis was further substantiated by studies in IL-1–deficient mice (13,14).
Several recent studies demonstrated microRNA-mediated RNA interference as a novel evolutionarily conserved mechanism for the regulation of gene expression at the posttranscription level. The physiologic function of the majority of microRNA is unknown, but their importance in maintaining cartilage homeostasis has become evident from studies showing a progressive loss of proliferating chondrocytes leading to severe skeletal growth defects and premature death in Dicer-null mice (15). In rheumatoid arthritis (RA), microRNA-146 (miR-146) and miR-155 were found to be overexpressed, and this correlated with the reduced expression of MMP-3 in synovial fibroblasts, suggesting involvement of microRNA in RA (16,17). Altered expression of miR-203 has also been shown in psoriasis, where it is thought to exert a proinflammatory effect by targeting suppressor of cytokine signaling 3 (18). Peripheral blood leukocytes from patients with systemic lupus erythematosus express high levels of miR-21 and miR-198, while the expression of miR-184 and miR-17-5p is low (19). Other studies have shown that miR-146a contributes to the abnormal expression of type I interferon in lupus by targeting the signaling proteins (20). Studies have also shown that microRNA are involved in the regulation of MMP-2 expression in heart and the invasion in glioma by targeting MMP regulators (21,22).
Recent studies showed the microRNA expression profile in human OA chondrocytes and correlated the expression of MMP-13 with specific microRNA (23–25). No consensus on a specific microRNA being the regulator of MMP-13 expression was reached, because one group of investigators reported that miR-9 negatively regulates the expression of MMP-13 (24), while another group observed that miR-22 blocks cartilage degradation by inhibiting MMP-13 expression indirectly by regulating the production of bone morphogenetic protein 7 (BMP-7) (23). Studies have shown that BMP-7 accelerates cartilage degradation via induction of MMP-13 expression in chondrocytes; therefore, inhibition of BMP-7 may be chondroprotective (26,27). Although we could not find the miR-9 seed sequence in the 3′-untranslated region (3′-UTR) of human MMP-13 messenger RNA (mRNA) (results not shown), we cannot rule out a role of miR-9 in regulating MMP-13 expression via a different pathway. Intense expression of miR-146a in low-grade OA cartilage and its induction by IL-1β in human chondrocytes has also been shown (25). In this study, we determined the effect of IL-1β on the microRNA expression profile and determined whether the expression of MMP-13 is regulated by specific microRNA in human OA chondrocytes.
Materials and Methods
Patients
Permission to use discarded human tissue was obtained from the institutional review board of the University of South Carolina, prior to initiation of the studies. OA cartilage samples were obtained from 20 patients (mean ± SD age 61.12 ± 4.75 years) who underwent total joint arthroplasty at Richland Hospital (Columbia, SC). OA was diagnosed according to the American College of Rheumatology criteria (28,29). Normal cartilage samples were obtained from trauma patients with no known history of OA or RA (n = 3).
Preparation of chondrocytes
Specimens that included full-thickness cartilage and subchondral bone were washed with sterile phosphate buffered saline, and macroscopic cartilage degeneration was determined by staining with India ink (30). Portions of cartilage with a smooth articular surface were used to prepare chondrocytes by enzymatic digestion and were cultured as previously described (31,32). Primary OA chondrocytes at 80% confluence were used for all of the experiments.
Chondrocyte treatment and preparation of microRNA
OA chondrocytes were serum starved overnight and then stimulated with IL-1β (5 ng/ml or 10 ng/ml; R&D Systems) for the indicated periods of time, and total RNA was prepared using TRIzol reagent (Invitrogen). MicroRNA were purified using the mirVana Kit, essentially according to the manufacturer's instructions (Applied Biosystems). For some studies, microRNA were prepared directly from normal and OA cartilage samples. Briefly, cartilage was ground to a fine powder in liquid nitrogen and then processed to purify the microRNA, as described above.
MicroRNA expression analysis
MicroRNA isolated from OA chondrocytes, stimulated or not stimulated with IL-1β, were poly(A) tailed and complementary DNA (cDNA) synthesized using the RT2 miRNA First Strand Kit (SABiosciences). Poly(A)-tailed cDNA was mixed with RT2 SYBR Green qPCR Master Mix (SABiosciences) in 96-well plates containing predispensed primers specific for 352 known human microRNA, 4 housekeeping genes (HKGs; hsa-SNORD48, hsa-SNORD47, hsa-SNORD44, and hsa-U6), 2 RNA-quality, and 2 polymerase chain reaction (PCR)–positive controls. Real-time PCR amplification and data capture were performed using the StepOne Real-Time PCR System (Applied Biosystems).
Microarray data analyses
PCR array data were analyzed using the SA Biosciences Web site (http://www.sabiosciences.com/pcrarraydataanalysis.php). The ΔΔCt method (33) was used to calculate the relative expression of microRNA in control and IL-1β–stimulated chondrocytes. Values are expressed as the fold change in gene expression and were derived using the following formula: [(2−ΔCt(GOI) exp/2−ΔCt(HKG) exp)/(2−ΔCt(GOI) cont/2−ΔCt(HKG) cont) = 2−ΔCt(GOI) − Ct(HKG) exp/2−ΔCt(GOI) − Ct(HKG) cont = 2−ΔΔCt exp/2−ΔΔCt cont], where GOI = gene of interest, exp = experimental sample, and cont = control sample. MicroRNA with fold changes of >2 were considered to be differentially expressed and were chosen for further analysis.
Reverse transcription—PCR (RT-PCR) analysis of argonaute (AGO) and DICER1 gene expression
Total RNA (1 μg) was reverse transcribed using random primers and the reagents provided with the SuperScript cDNA Synthesis Kit (Invitrogen). The cDNA mixture was then diluted 50-fold, and 5 μl was used in the PCR containing 2 μl 10× AmpliTaq buffer (Applied Biosystems), 0.2 mM each dNTP, 1.5 mM MgCl2, 0.2 μM each primer, and 1 unit AmpliTaq DNA polymerase. Reaction mixtures were heated at 95°C for 5 minutes, followed by 35 cycles of 95°C for 1 minute, 53°C for 30 seconds, and 72°C for 30 seconds and then stored at 4°C until analyzed. Expression of the DICER1 and AGO1–4 genes was determined by RT-PCR using the primers shown in Table 1. Amplified DNA fragments were resolved by 1.2% agarose gel electrophoresis, cloned using the TOPO-TA Cloning System (Invitrogen), and sequenced; the sequences were compared with the catalogued sequences in the GenBank/European Molecular Biology Laboratory DNA databases, using the BLASTN program.
Table 1. Sequences of primers used for RT-PCR analysis of gene expression*.
| Gene name | Primer sequences | Accession number | Location on transcript, bp | Product size, bp |
|---|---|---|---|---|
| AGO1 | F: 5′-ACCCCAAAAACAGTGTCGAG-3′ | NM_012199 | 1,578–1,597 | 325 |
| R: 5′-GAGGCAGAGGTTGGAAGAG-3′ | 1,902–1,883 | |||
| AGO2 | F: 5′-ACAGTGCTGAAGGAAGCCATACCT-3′ | NM_012154 | 2,508–2,531 | 152 |
| R: 5′-AATCCCACTCGGTACACAATCGCT-3′ | 2,659–2,636 | |||
| AGO3 | F: 5′-AGCTCCATGAATGGAAATCG-3′ | NM_177422 | 247–266 | 212 |
| R: 5-CAACCACCTCCCTGTTCACT-3′ | 458–439 | |||
| AGO4 | F: 5′-GGATTCCACTCACAGGGAGA-3′ | NM_017629 | 4,124–4,143 | 243 |
| R: 5′-TTTATCCCCTTTTCCATCCC-3′ | 4,366–4,347 | |||
| DICER1 | F: 5′-TATGAACGCTTTTGTGCTGC-3′ | NM_030621 | 8,485–8,504 | 254 |
| R: 5′-GCTTTTTCAAGACACGCCTC-3′ | 8,738–8,719 | |||
| GAPDH | F: 5′-TCGACAGTCAGCCGCATCTTCTTT-3′ | NM_002046 | 45–68 | 94 |
| R: 5′-ACCAAATCCGTTGACTCCGACCTT-3′ | 138–155 | |||
| MMP-13 | F: 5′-CGCGTCATGCCAGCAAATTCCATT-3′ | NM_002427 | 1,409–1,432 | 322 |
| R: 5′-TCCATGTGTCCCATTTGTGGTGTG-3′ | 1,730–1,708 |
RT-PCR = reverse transcription–polymerase chain reaction; F = forward; R = reverse.
Quantitative RT-PCR analysis of MMP-13 and miR-27b expression
Expression of MMP-13 mRNA was quantified using the TaqMan Gene Expression Assay according to the manufacturer's instructions (Applied Biosystems). Expression of mature microRNA was quantified using TaqMan MicroRNA Assays (Applied Biosystems). Purified microRNA from control and experimental samples were reverse transcribed using 50 nM miR-27b stem loop RT primer, 10× RT buffer, 10 mM each dNTP, 50 units MultiScribe reverse transcriptase, and 20 units of RNase inhibitor. Quantitative PCR was performed in 10-μl reactions containing 2 μl of RT product, 5 μl of 2× TaqMan Universal Master Mix, 0.2 μM TaqMan probe, and 0.9 μM forward and reverse primers. Reaction mixtures were incubated at 95°C for 10 minutes, followed by 40 cycles of 95°C for 30 seconds and 60°C for 1 minute. Expression of RNU6B and GAPDH was used as endogenous control. A threshold cycle was observed in the exponential phase of amplification, and quantification of relative expression levels was determined by the ΔΔCt method (33). The value of each control sample was set at 1 and was used to calculate the fold change in miR-27b expression.
Western blotting
Culture supernatants were concentrated using Microcon YM-10 centrifugal filters (10-kd molecular cutoff; Amicon Bioseparations), and equal volumes of the concentrated samples were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (10% resolving gel with 4% stacking) and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked with nonfat dry milk powder in Tris buffered saline containing 0.1% Tween 20 and probed with diluted (1:1,000) polyclonal antibodies specific for MMP-13 (SC-30073; Santa Cruz Biotechnology). For the analysis of Dicer-1 protein expression, chondrocytes were lysed in radioimmunoprecipitation assay lysis buffer with complete protease inhibitor cocktail (Roche), and the blot was probed using a diluted (1:100) anti–Dicer-1 monoclonal antibody (ab14601; Abcam).
Immunoreactive proteins were visualized using horseradish peroxidase–linked secondary antibodies and enhanced chemiluminescence (GE Healthcare). Images were captured using the Mini-Medical series (AFP Imaging) and analyzed using Un-Scan-It software (Silk Scientific). Each band was scanned 5 times with background correction, and the values (pixels/band) were averaged and expressed as the mean ± SD.
MMP-13 enzyme-linked immunosorbent assay (ELISA)
OA chondrocytes were stimulated with IL-1β (5 ng/ml or 10 ng/ml) for 6 hours or 24 hours, and the MMP-13 protein in the culture supernatants was quantified using an ELISA kit according to the manufacturer's instruction (AnaSpec). Plates were read at 450 nm using a Synergy HT Microplate Reader (BioTek Instruments), and the MMP-13 concentration in the samples was calculated using a standard curve.
Luciferase reporter assay
The pMIR-REPORT miRNA Expression Reporter Vector System (Applied Biosystems) was used to construct the reporter plasmid containing 322 bp of the 3′-UTR of human MMP-13 mRNA encompassing the nucleotide sequence complementary to the miR-27b seed sequence. Total RNA (1 μg) was reverse transcribed into cDNA as described above, and the 3′-UTR was amplified using the primers shown in Table 1 and purified using the QIAquick PCR Purification Kit (Qiagen). Purified DNA was digested with Hind III and Spe I restriction endonucleases (New England Biolabs) in buffer 4 overnight, gel purified, and ligated 3′ to the luciferase reporter gene in the Hind III– and Spe I–digested vector to generate pMIR-REPORT-Luc-MMP-13 plasmid (reporter plasmid). The pMIR-REPORT-Luc-MMP-13 mutant plasmid contained the miR-27b seed sequence in reverse orientation. Competent Escherichia coli cells were transformed, single colonies were picked from ampicillin agar plates, and plasmid DNA was prepared using a Plasmid Miniprep kit (Qiagen).
Transfection of HeLa cells with the reporter plasmid and microRNA mimic
HeLa cells were transfected with 150 ng of reporter plasmid, 10 nM and 100 nM pre–miR-27b (Qiagen) or negative control microRNA (Applied Biosystems), and 3 ng of Renilla luciferase control vector (Promega), using Lipofectamine 2000 (Invitrogen) in a 24-well plate. The luciferase activity assay was performed 24 hours after transfection, using the Dual-Luciferase Reporter Assay System (Promega) and an LB 7505 luminometer (Berthold) equipped with double injectors. Firefly luciferase activity was normalized to Renilla luciferase activity. Each experiment was performed 3 times in triplicate.
Modulation of miR-27b expression by microRNA mimics and inhibitors in human OA chondrocytes
Human primary OA chondrocytes were transfected with mimic and inhibitor of miR-27b (Qiagen) at a 100 nM concentration, using the calcium phosphate precipitation method (34). Following transfection, cells were stimulated with IL-1β for 24 hours or were not stimulated, cell supernatants were harvested, and MMP-13 production was quantified by ELISA. Total RNA prepared from chondrocytes was used to check the expression of miR-27b.
Statistical analysis
Comparisons were performed using the Origin 6.1 software package (1 paired 2-tailed t-test with one-way analysis of variance and Tukey's post hoc analysis). P values less than 0.05 were considered significant, and P values less than 0.001 were considered highly significant.
Results
Expression of DICER1 and AGO genes in human OA chondrocytes
Dicer-1 is an RNase III endonuclease that converts pre-microRNA to mature microRNA after transport from the nucleus to the cytosol. Our results showed that human OA chondrocytes express transcripts for the DICER1 and AGO2–4 genes, although the expression of AGO4 was very weak (Figure 1A). Expression of AGO1 was not detected in the samples analyzed but was detected easily by the same primers in the brain RNA used as positive control (Figure 1B). Expression of Dicer-1 protein was detected in all of the samples analyzed (Figure 1C, and results not shown). These results confirmed that the proteins associated with the biogenesis of microRNA were expressed in human OA chondrocytes.
Figure 1.
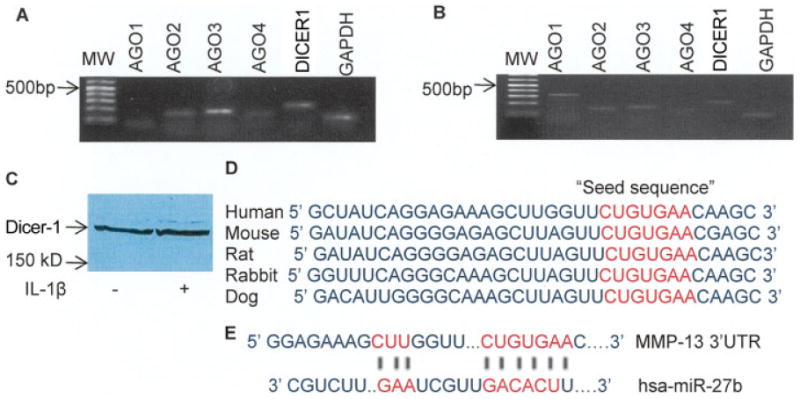
A and B, Expression of the argonaute (AGO) and DICER1 genes by human osteoarthritis (OA) chondrocytes, as determined by reverse transcription–polymerase chain reaction. Expression of AGO1 was not detected in human OA chondrocytes (A) but was readily detected in human brain mRNA that was used as positive control (B). C, Expression of Dicer-1 protein (217 kd) in human OA chondrocytes. The expression of Dicer-1 was not significantly affected by stimulation with interleukin-1β (IL-1β). D, Cross-species conservation of the microRNA-27b (miR-27b) seed sequence in the 3′-untranslated region (3′-UTR) of matrix metalloproteinase 13 (MMP-13) mRNA. E, TargetScanS-predicted duplex of hsa-miR-27b with the seed sequence in the 3′-UTR of human MMP-13 mRNA.
IL-1β–induced differential expression of microRNA in human OA chondrocytes
To identify the microRNA that are regulated by IL-1β, we determined the expression profile of 352 human microRNA in IL-1β–stimulated OA chondrocytes and compared it with the microRNA expression profile in unstimulated OA chondrocytes (n = 3), with each sample analyzed independently. After 6 hours of stimulation of OA chondrocytes with IL-1β, we identified 44 differentially expressed microRNA (Table 2). Our results also showed that expression of 2 microRNA, miR-491_3p and miR-146a, was up-regulated and that of 42 microRNA was significantly down-regulated (P < 0.05), including miR-423_3p (∼13-fold), miR-610 (∼6-fold), miR-637 (∼3-fold), miR-32 (∼3-fold), miR142_5p (∼3-fold), and miR-27b (∼3-fold).
Table 2. Differentially expressed microRNA after 6 hours of stimulation of OA chondrocytes with IL-1β*.
| Expression profile, microRNA | Fold change |
|---|---|
| Up-regulated | |
| hsa-miR-491_3p | 2.02 |
| hsa-miR-146a | 2.00 |
| Down-regulated | |
| hsa-miR-187 | −2.37 |
| hsa-miR-219_5p | −2.60 |
| hsa-miR-127_5p | −2.33 |
| hsa-miR-518_3p | −2.60 |
| hsa-miR-520d_3p | −2.13 |
| hsa-miR-550 | −2.60 |
| hsa-miR-544 | −3.97 |
| hsa-miR-502_5p | −3.94 |
| hsa-miR-638 | −2.28 |
| hsa-miR-643 | −2.55 |
| hsa-miR-603 | −2.69 |
| hsa-miR-602 | −2.17 |
| hsa-miR-563 | −2.46 |
| hsa-miR-558 | −2.11 |
| hsa-miR-575 | −2.88 |
| hsa-miR-181c | −2.15 |
| hsa-miR-637 | −3.44 |
| hsa-miR-564 | −2.56 |
| hsa-miR-337_5p | −2.00 |
| hsa-miR-298 | −2.10 |
| hsa-miR-622 | −2.81 |
| hsa-miR-659 | −2.60 |
| hsa-miR-208 | −2.09 |
| hsa-miR-142_5p | −3.22 |
| hsa-miR-29b | −2.39 |
| hsa-miR-144 | −2.86 |
| hsa-miR-19b | −2.06 |
| hsa-miR-32 | −3.41 |
| hsa-miR-141 | −2.17 |
| hsa-miR-27b | −3.21 |
| hsa-miR-302a | −2.20 |
| hsa-miR-423_3p | −12.99 |
| hsa-miR-503 | −2.84 |
| hsa-miR-130b | −2.15 |
| hsa-miR-34a | −2.92 |
| hsa-miR-33a | −2.03 |
| hsa-miR-301b | −2.26 |
| hsa-miR-138 | −2.02 |
| hsa-miR-497 | −2.33 |
| hsa-miR-545 | −2.84 |
| hsa-miR-372 | −2.53 |
| hsa-miR-610 | −6.19 |
OA = osteoarthritis; IL-1β = interleukin-1β.
Presence of the miR-27b seed sequence in the 3′-UTR of human MMP-13 mRNA
We used the miRanda, TargetScanS, and PicTar algorithms to identify the mRNA targeted by differentially expressed microRNA. The criteria for selecting targets were to choose a gene that is expressed in chondrocytes, that is known to be present in OA, and that the designated microRNA target mRNA pair must be common in 2 of 3 algorithms. TargetScanS and PicTar both identified sequences conserved in the 3′-UTR of MMP-13 mRNA that were complementary to the miR-27b seed sequence (Figure 1D). The seed sequence present in the 3′-UTR of human MMP-13 mRNA and the predicted duplex of miR-27b are shown in Figure 1E. The free energy scores for miR-27b–MMP-13 mRNA hybrids were −53.1 Kcal/mole (RNAhybrid; http://bibiserv.techfak.uni-bielefeld.de) and −24.4 Kcal/mole (PicTar), suggesting that miR-27b was most likely to interact with the 3′-UTR of MMP-13 mRNA and consequently down-regulate its expression posttranscriptionally.
Correlation between IL-1β–stimulated MMP-13 production and miR-27b expression in normal and OA chondrocytes
We treated OA chondrocytes with IL-1β and determined MMP-13 protein expression and the fold change in miR-27b gene expression, by Western blotting or MMP-13 ELISA and TaqMan assay, respectively. IL-1β stimulation of chondrocytes resulted in the secretion of MMP-13 protein in the culture supernatant at 6 hours and 24 hours (Figures 2A and B, respectively). Importantly, IL-1β stimulation resulted in a significant down-regulation of miR-27b expression at 6 hours (P < 0.001) and 24 hours (P < 0.05) in primary OA chondrocytes maintained in a G0 state of the cell cycle (n = 20). Expression of miR-27b was found to be 67% lower in OA cartilage samples when compared with its expression level in normal cartilage samples (n = 3 each; P < 0.05) (Figure 2C). An inverse correlation between miR-27b expression and MMP-13 production was also observed when normal chondrocytes were stimulated with IL-1β (P < 0.05) (Figures 2D and E, respectively). Kinetic analysis showed a time-dependent increase in MMP-13 protein expression in IL-1β–stimulated OA chondrocytes (P < 0.001 versus unstimulated control for all) (Figure 2F). A strong correlation between the level of miR-27b expression and MMP-13 protein secretion was observed at later time points (P < 0.001) (Figure 2G). However, no significant change in miR-27b expression at 2 hours and 4 hours after stimulation with IL-1β was noted, possibly due to slow degradation of the presynthesized microRNA pool.
Figure 2.
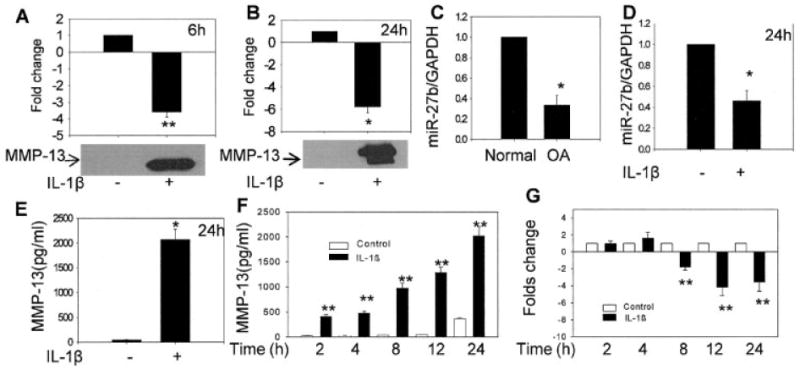
Correlation between IL-1β–stimulated MMP-13 production and miR-27b expression in normal and OA chondrocytes. A and B, IL-1β–induced production of MMP-13 (bottom) and down-regulation of miR-27b expression (top) in human OA chondrocytes. Primary OA chondrocytes were stimulated with IL-1β for 6 hours (n = 3) or 24 hours (n = 8). Expression of miR-27b was determined using TaqMan assay and was compared with the expression levels in controls. The immunoblot shown in B may be attributable to autocatalysis of MMP-13. C and D, Relative expression of miR-27b in normal and OA cartilage (n = 3 each) and in normal chondrocytes stimulated with IL-1β for 24 hours, respectively. E, MMP-13 production in normal chondrocytes stimulated with IL-1β for 24 hours. F, Kinetics of IL-1β–induced MMP-13 production in OA chondrocytes. G, Time-dependent down-regulation of miR-27b expression in OA chondrocytes stimulated with IL-1β for 2, 4, 8, 12, or 24 hours. Bars show the mean and SEM results of 3 independent experiments, each of which was run in duplicate. * = P < 0.05; ** = P < 0.001 versus control. See Figure 1 for definitions.
Inhibition of pMIR-REPORT-Luc-MMP-13 reporter activity by overexpression of miR-27b
No high-throughput method is available to experimentally validate the function of microRNA, and the major approach to validate microRNA–mRNA interactions uses in vitro gain-of-function and loss-of-function analysis (35). We also used this strategy to determine whether miR-27b interacts with its putative target sequence in the 3′-UTR of human MMP-13 mRNA. Because HeLa cells do not express miR-27b (Haqqi TM, et al: unpublished observations), they were transiently cotransfected with the pMIR-REPORT-Luc-MMP-13 plasmid, pMIR-REPORT-Luc-MMP-13 mutant, and miR-27b mimic or a negative control microRNA (10 nM and 100 nM) and the internal control plasmid pRL-SV-40. A marked reduction in luciferase activity levels in cells overexpressing miR-27b was observed (P < 0.05 versus nontransfected control) (Figure 3A). In contrast, cotransfection of the mutant reporter plasmid with miR-27b mimic or the reporter plasmid with the negative control microRNA had no effect on luciferase activity in the transfected cells (Figure 3A). These results demonstrated that miR-27b binds the seed sequence present in the 3′-UTR of human MMP-13 mRNA.
Figure 3.
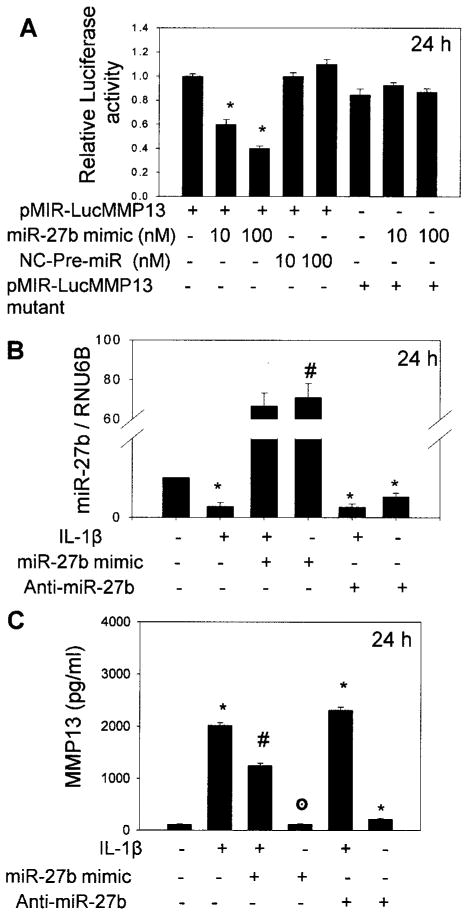
Validation of miR-27b binding with the putative binding site in the 3′-UTR of MMP-13 mRNA. A, Luciferase activity in HeLa cells transfected with pMIR-REPORT-Luc-MMP13, pMIR-REPORT-Luc-MMP13 mutant, a negative control (NC) miRNA, and miR-27b mimic. B, Relative expression level of miR-27b in human OA chondrocytes (n = 3) transfected with miR-27b mimic or miR-27b inhibitor and then stimulated or not stimulated with IL-1β for 24 hours. C, Expression of MMP-13 protein in culture supernatants after 24 hours of IL-1β stimulation, as estimated by enzyme-linked immunosorbent assay. Chondrocytes were transfected with pre–miR-27b or miR-2b inhibitor. Bars show the mean and SEM results of 3 independent experiments, each of which was run in triplicate. * = P < 0.05 versus control; # = P < 0.05 versus stimulation with IL-1β only; ⊙ = P > 0.05 versus control. See Figure 1 for other definitions.
Regulation of the expression of MMP-13 protein in human chondrocytes by miR-27b
For these studies, human OA chondrocytes were transfected with pre–miR-27b or anti–miR-27b and were then either stimulated or not stimulated with IL-1β, and the amount of secreted MMP-13 protein in culture supernatant was determined by ELISA. Significant overexpression of miR-27b was observed in pre–miR-27b–transfected chondrocytes (P < 0.05 versus stimulation with IL-1β only) but not in chondrocytes transfected with miR-27b inhibitor (Figure 3B). OA chondrocytes with overexpression of miR-27b produced ∼38% less MMP-13 protein upon stimulation with IL-1β compared with nontransfected chondrocytes (P < 0.05) (Figure 3C). Inhibition of miR-27b in IL-1β–stimulated chondrocytes resulted in an increase of ∼13% in MMP-13 protein expression compared with control OA chondrocytes (P < 0.05) (Figure 3C). OA chondrocytes transfected with miR-27b inhibitor alone produced ∼51% more MMP-13 protein without stimulation with IL-1β compared with nontransfected controls (Figure 3C).
Negative regulation of miR-27b expression by activation of NF-κB
In view of the published studies showing that expression of some of the microRNA are regulated by NF-κB (36), we determined whether NF-κB regulates miR-27b expression in OA chondrocytes. OA chondrocytes were pretreated for 2 hours with the NF-κB inhibitors MG132 (37) or Bay 11-7082 (38) and then stimulated with IL-1β for 24 hours. The expression levels of MMP-13 mRNA and miR-27b were determined using TaqMan assays. Compared with the expression in unstimulated controls, the expression of miR-27b in IL-1β–stimulated OA chondrocytes was down-regulated by ∼80% (P < 0.05) (Figure 4A), but MMP-13 gene expression was enhanced ∼64-fold (P < 0.05) (Figure 4C), and high levels of MMP-13 protein were detected in the culture supernatant (P < 0.05) (Figure 4E). These results are interesting, because they indicated that activated NF-κB up-regulated the expression of MMP-13 but acted as a negative regulator of miR-27b expression in OA chondrocytes.
Figure 4.
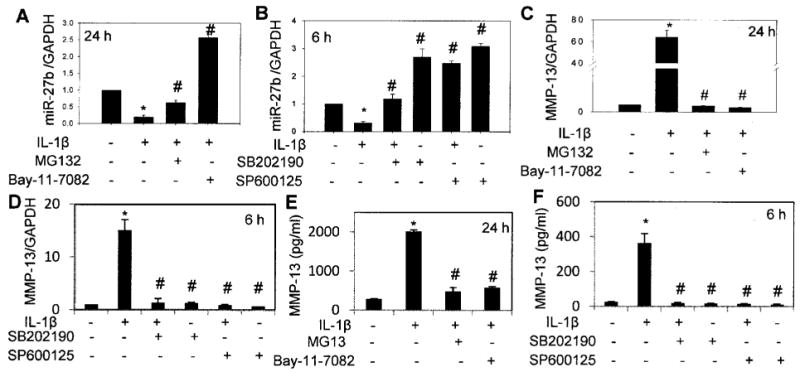
Role of NF-κB and MAPK pathways in the regulation of miR-27b expression. Primary OA chondrocytes were pretreated for 2 hours with small molecule inhibitors of NF-κB MG132 or Bay 11-7082 and MAPK inhibitors SB202190 and SP600125 and then stimulated with IL-1β as described in Materials and Methods. A and B, Expression of miR-27b in OA chondrocytes treated with NF-κB inhibitors and in OA chondrocytes treated with MAPK inhibitors, respectively. Bars show the mean and SEM results of 3 independent experiments (n = 3), each of which was run in duplicate. C and D, MMP-13 mRNA expression in IL-1β–stimulated OA chondrocytes treated with the inhibitors described in A and B (n = 3). Bars show the mean and SEM. E and F, MMP-13 protein concentration in culture supernatants of the chondrocytes used in C and D. Bars in E and F show the mean and SD. * = P < 0.05 versus unstimulated control; # = P < 0.05 versus chondrocytes stimulated with IL-1β only. See Figure 1 for definitions.
Regulation of miR-27b expression by JNK and p38 MAPKs
Previous studies have shown that the transcriptional response of the MMP-13 gene to IL-1β is controlled by the p38 MAPK and JNK pathways but not by ERK (39). However, the role of MAPKs in regulating the expression of microRNA in human chondrocytes has not yet been reported. To determine the impact of IL-1β–induced activation of p38 MAPK and JNK on the expression of miR-27b in OA chondrocytes, we pretreated chondrocytes for 2 hours with the p38 MAPK inhibitor SB202190 or the JNK inhibitor SP600125 and then stimulated the OA chondrocytes with IL-1β for 6 hours. In the chondrocytes pretreated with SP600125, stimulation with IL-1β showed an ∼88% increase in miR-27b expression (P < 0.05) (Figure 4B) and an ∼14-fold increase in MMP-13 mRNA expression compared with controls (P < 0.05) (Figure 4D). OA chondrocytes pretreated with SB202190 and then stimulated with IL-1β showed an ∼28% increase in miR-27b expression compared with chondrocytes not treated with SB202190 but stimulated with IL-1β (P < 0.05) (Figure 4B). Pretreatment of OA chondrocytes with SB202190 suppressed IL-1β–induced MMP-13 mRNA expression by ∼90% (P < 0.05) (Figure 4D) compared with IL-1β–stimulated OA chondrocytes. These data indicate that negative regulation of miR-27b expression upon activation of the signal transduction pathways essential for optimum expression of MMP-13 apparently is necessary for the unobstructed translation and release of MMP-13 protein from human OA chondrocytes.
Discussion
MMPs are important regulators of tissue remodeling and repair, but their overexpression or expression for too long in a joint can cause pathologic manifestations in chronic joint diseases such as OA. Thus, cells that produce MMPs must employ a multilayered mechanism to keep the production of MMPs under control. Here, we report the identification of microRNA-27b as a potential regulator of MMP-13 expression in human chondrocytes and provide the data linking it to OA, by demonstrating its presence and modulation by IL-1β in human chondrocytes derived from normal and OA cartilage. Using PCR-based expression profiling, we also identified several microRNA genes that were modulated by IL-1β in OA chondrocytes. Dysregulation of microRNA has been linked to many disease conditions, including cancer (40), cardiovascular disease (41), obesity (42), OA (23–25), rheumatoid arthritis (16), and inflammation (43). MicroRNA have also been shown to regulate the expression of proteins involved in pathways activated by the Toll-like receptors and IL-1β (44).
The human genome contains 2 miR-27 genes [miR-27a and miR-27b] on chromosomes 19 and 9, respectively, and their mature products differ by only 1 nucleotide in the 3′ region. Interestingly, no change in miR-27a expression was noted in this study (results not shown), and we conclude that miR-27a is not an IL-1β–responsive gene. Genetic silencing of DICER and DROSHA led to significantly reduced miR-27b expression in endothelial cells (45). The gene for miR-27b (located at q22.32) is clustered with miR-23 and miR-24-1 on human chromosome 9, indicating that they may share a similar regulatory mechanism. We did not observe a significant change in the expression of miR-23b and miR-24-1 in OA chondrocytes after IL-1β stimulation (data not shown), demonstrating that miR-27b expression was independently regulated. Similar results have been reported for the expression of murine miR-23b, miR-27b, and miR-24-1, which have the same organization in the mouse genome (46).
Our results indicate that the expression of miR-27b was constitutively high in unstimulated normal or OA chondrocytes that either did not produce MMP-13 or produced barely detectable levels of MMP-13. However, when these cells were stimulated with IL-1β, the expression of miR-27b was severely down-regulated, and enhanced production of MMP-13 protein was noted. We also compared the expression level of miR-27b using microRNA prepared directly from normal and OA cartilage samples and observed that the expression of miR-27b was low in OA cartilage samples (Figure 2C). In addition, IL-1β stimulation at different time points also resulted in a time-dependent inverse correlation between the expression of MMP-13 and miR-27b. In order to explore the mechanisms involved, we first performed computer-based sequence analysis using miRanda, TargetScanS, and PicTar algorithms.
We observed that the miR-27b seed sequence (the first 7 nucleotides) was complementary to the 1547–1553 nucleotide sequence of human MMP-13 mRNA and is conserved, operationally defined as present in orthologous locations in the 3′-UTR of human, mouse, rat, rabbit, and dog MMP-13 mRNA (Figure 1D). In addition, the context scores of miR-27b were low among most of the microRNA predicted to target the conserved sites in the 3′-UTR of human MMP-13 mRNA. This indicated that miR-27b was most likely to interact (although not with perfect complementarity) with the 3′-UTR of MMP-13 mRNA and down-regulate its expression at the posttranscriptional level. Most animal microRNA bind with mismatches and bulges, but a key feature of target recognition involves Watson–Crick base-pairing of microRNA nucleotides 2–8. This type of complementarity excludes AGO-mediated cleavage of mRNA while promoting the repression of mRNA translation and is the predominant mechanism for regulation by animal microRNA (47).
To determine the potential interaction between miR-27b and human MMP-13 mRNA, the region of the MMP-13 3′-UTR mRNA containing the sequence complementary to the miR-27b seed sequence was cloned downstream of the Renilla luciferase coding sequence and cotransfected with miR-27b mimic into HeLa cells, which do not express miR-27b (Haqqi TM, et al: unpublished observations). In HeLa cells cotransfected with pre–miR-27b and the reporter vector, luciferase enzyme activity was decreased by 60% when compared with cells cotransfected with the reporter vector and negative control microRNA (Figure 3A). In contrast, in HeLa cells cotransfected with mutant reporter vector and miR-27b mimic, luciferase enzyme activity was not affected. These results indicate that miR-27b can interact with the 3′-UTR of human MMP-13 mRNA and efficiently inhibit translation from the chimeric transcript. This hypothesis is further supported by the results of transfection of primary OA chondrocytes, in which overexpression of miR-27b led to a significant reduction in IL-1β–induced MMP-13 production, while inhibition of miR-27b in IL-1β–stimulated chondrocytes increased the MMP-13 protein expression in the culture supernatants (Figure 3C). This is interesting, because such a direct inhibitory effect of a microRNA on human MMP-13 expression has not been previously reported.
Because both MAPKs and NF-κB have been implicated in MMP-13 expression in IL-1β–stimulated chondrocytes (39), we investigated the possible role of the NF-κB and MAPK pathways in the regulation of miR-27b expression in OA chondrocytes. Previously, a role of JNK in the regulation of the miR-155–mediated inflammatory response in macrophages was reported (48). Furthermore, the role of MAPKs in regulating microRNA expression in cardiac disease has recently been proposed (49). We observed that activation of NF-κB acts as a negative regulator of miR-27b expression in OA chondrocytes. In OA chondrocytes treated with 2 different NF-κB inhibitors, the expression of miR-27b was enhanced but that of MMP-13 protein was reduced. This suggests that NF-κB activation may be necessary for the suppressive effect of IL-1β on miR-27b expression. It is possible that inhibition of MMP-13 expression in IL-1β–stimulated OA chondrocytes treated with NF-κB inhibitors could be attributable to an earlier inhibitory effect on NF-κB–mediated transcriptional induction of MMP-13 mRNA. However, this possibility could not be distinguished in the present study, due to the limitations of the system employed. Similar results were obtained with p38 MAPK and JNK inhibitors. Because these pathways are essential for the optimum expression of MMP-13, negative regulation of miR-27b expression by activated MAPKs and NF-κB may be necessary for the efficient production of MMP-13 in IL-1β–stimulated OA chondrocytes.
In conclusion, these results identify a novel microRNA, miR-27b, as a posttranscriptional regulator of MMP-13 expression in OA chondrocytes. Up-regulating the expression of miR-27b or preventing its down-regulation in vivo may present a novel therapeutic and/or preventive approach in OA.
Acknowledgments
Supported in part by the NIH (grant R01-AT-003267 from the National Center for Complementary and Alternative Medicine) and the University of South Carolina.
Footnotes
Author Contributions: All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Haqqi had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Akhtar, Haqqi.
Acquisition of data. Akhtar, Haqqi.
Analysis and interpretation of data. Akhtar, Rasheed, Ramamurthy, Anbazhagan, Voss, Haqqi.
References
- 1.Pun YL, Moskowitz RW, Lei S, Sundstrom WR, Block SR, McEwen C, et al. Clinical correlations of osteoarthritis associated with a single-base mutation (arginine519 to cysteine) in type II procollagen gene: a newly defined pathogenesis. Arthritis Rheum. 1994;37:264–9. doi: 10.1002/art.1780370216. [DOI] [PubMed] [Google Scholar]
- 2.Kuettner KE, Goldberg VM. Introduction. In: Kuettner KE, Goldberg VM, editors. Osteoarthritic disorders. Rosemont (IL): American Academy of Orthopaedic Surgeons; 1995. pp. xxi–xxv. [Google Scholar]
- 3.Malemud CJ, Islam N, Haqqi TM. Pathophysiological mechanisms in osteoarthritis lead to novel therapeutic strategies. Cells Tissues Organs. 2003;174:34–48. doi: 10.1159/000070573. [DOI] [PubMed] [Google Scholar]
- 4.Westacott CI, Sharif M. Cytokines in osteoarthritis: mediators or markers of joint destruction. Sem Arthritis Rheum. 1996;25:254–72. doi: 10.1016/s0049-0172(96)80036-9. [DOI] [PubMed] [Google Scholar]
- 5.Goldring MB. The role of cytokines as inflammatory mediators in osteoarthritis: lessons from animal models. Connect Tissue Res. 1999;40:1–11. doi: 10.3109/03008209909005273. [DOI] [PubMed] [Google Scholar]
- 6.Pelletier JP, Martel-Pelletier J, Abramson SB. Osteoarthritis, an inflammatory disease: potential implications for the selection of new therapeutic targets. Arthritis Rheum. 2001;44:1237–47. doi: 10.1002/1529-0131(200106)44:6<1237::AID-ART214>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 7.Knauper V, Lopez-Otin C, Smith B, Knight G, Murphy G. Biochemical characterization of human collagenase-3. J Biol Chem. 1996;271:1544–50. doi: 10.1074/jbc.271.3.1544. [DOI] [PubMed] [Google Scholar]
- 8.Knauper V, Cowell S, Smith B, Lopez-Otin C, O'Shea M, Morris H, et al. The role of the C terminal domain of human collagenase-3 (MMP-13) in the activation of procollagenase-3, substrate specificity, and tissue inhibitor of metalloproteinase interaction. J Biol Chem. 1997;272:7608–16. doi: 10.1074/jbc.272.12.7608. [DOI] [PubMed] [Google Scholar]
- 9.Fosang AJ, Last K, Knauper V, Murphy G, Neame PJ. Degradation of cartilage aggrecan by collagenase-3 (MMP-13) FEBS Lett. 1996;380:17–20. doi: 10.1016/0014-5793(95)01539-6. [DOI] [PubMed] [Google Scholar]
- 10.Neuhold LA, Killar L, Zhao W, Sung ML, Warner L, Kulik J, et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest. 2001;107:35–44. doi: 10.1172/JCI10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tetlow LC, Adlam DJ, Woolley DE. Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 2001;44:585–94. doi: 10.1002/1529-0131(200103)44:3<585::AID-ANR107>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 12.Pujol JP, Chadjichristos C, Legendre F, Bauge C, Beauchef G, Andriamanalijaona R, et al. Interleukin-1 and transforming growth factor-β1 as crucial factors in osteoarthritic cartilage metabolism. Connect Tissue Res. 2008;49:293–7. doi: 10.1080/03008200802148355. [DOI] [PubMed] [Google Scholar]
- 13.Blom AB, van der Kraan PM, van den Berg WB. Cytokine targeting in osteoarthritis. Curr Drug Targets. 2007;8:283–92. doi: 10.2174/138945007779940179. [DOI] [PubMed] [Google Scholar]
- 14.Lotz M. Cytokines in cartilage injury and repair. Clin Orthopedics Res. 2001;391:108–15. doi: 10.1097/00003086-200110001-00011. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi T, Lu J, Cobb BS, Rodda SJ, McMahon AP, Schipani E, et al. Dicer-dependent pathways regulate chondrocyte proliferation and differentiation. Proc Natl Acad Sci U S A. 2008;105:1949–54. doi: 10.1073/pnas.0707900105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakasa T, Miyaki S, Okubo A, Hashimoto M, Nishida K, Ochi M, et al. Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheum. 2008;58:1284–92. doi: 10.1002/art.23429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, et al. Altered expression of microRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008;58:1001–9. doi: 10.1002/art.23386. [DOI] [PubMed] [Google Scholar]
- 18.Sonkoly E, Wei T, Janson PC, Saaf A, Lundeberg L, Tengvall-Linder M, et al. MicroRNAs: novel regulators involved in the pathogenesis of psoriasis? PLoS One. 2007;2:e610. doi: 10.1371/journal.pone.0000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan YH, et al. Microarray analysis of microRNAs expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus. 2007;16:939–46. doi: 10.1177/0961203307084158. [DOI] [PubMed] [Google Scholar]
- 20.Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, et al. MicroRNA-146a contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009;60:1065–75. doi: 10.1002/art.24436. [DOI] [PubMed] [Google Scholar]
- 21.Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, et al. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82:21–9. doi: 10.1093/cvr/cvp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, Linsley PS, et al. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol. 2008;28:5369–80. doi: 10.1128/MCB.00479-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iliopoulos D, Malizos KN, Oikonomou P, Tsezou A. Integrative microRNAs and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS One. 2008;3:e3740. doi: 10.1371/journal.pone.0003740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones SW, Watkins G, Le Good N, Roberts S, Murphy CL, Brockbank SM, et al. The identification of differentially expressed microRNAs in osteoarthritic tissue that modulate the production of TNF-α/MMP-13. Ostearthritis Cartilage. 2009;17:464–72. doi: 10.1016/j.joca.2008.09.012. [DOI] [PubMed] [Google Scholar]
- 25.Yamasaki K, Nakasa T, Miyaki S, Ishikawa M, Deje M, Adachi N, et al. Expression of microRNA-146a in osteoarthritis cartilage. Arthritis Rheum. 2009;60:35–41. doi: 10.1002/art.24404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muddasani P, Norman JC, Ellman M, van Wijnen AJ, Im HJ. Basic fibroblast growth factor activates the MAPK and NFκB pathways that converge on Elk-1 to control production of matrix metalloproteinase-13 by human adult articular chondrocytes. J Biol Chem. 2007;282:31409–21. doi: 10.1074/jbc.M706508200. [DOI] [PubMed] [Google Scholar]
- 27.Im HJ, Muddasani P, Natarajan V, Schmid TM, Block JA, Davis F, et al. Basic fibroblast growth factor stimulates matrix metalloproteinase-13 via the molecular cross-talk between the mitogen-activated protein kinases and protein kinase Cδ pathways in human adult articular chondrocytes. J Biol Chem. 2007;282:11110–21. doi: 10.1074/jbc.M609040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altman R, Alarcon G, Appelrouth D, Bloch D, Borenstein D, Brandt K, et al. The American College of Rheumatology criteria for the classification and reporting of osteoarthritis of the hip. Arthritis Rheum. 1991;34:505–14. doi: 10.1002/art.1780340502. [DOI] [PubMed] [Google Scholar]
- 29.Altman R, Asch E, Bloch D, Bole G, Borenstein D, Brandt K, et al. Development of criteria for the classification and reporting of osteoarthritis: classification of osteoarthritis of the knee. Arthritis Rheum. 1986;29:1039–49. doi: 10.1002/art.1780290816. [DOI] [PubMed] [Google Scholar]
- 30.Armstrong CG, Mow VC. Variations in the intrinsic mechanical properties of human articular cartilage with age, degeneration and water content. J Bone Joint Surg. 1982;64:88–94. [PubMed] [Google Scholar]
- 31.Singh R, Ahmed S, Islam N, Goldberg VM, Haqqi TM. Epigallocatechin-3-gallate inhibits interleukin-1β–induced expression of nitric oxide synthase and production of nitric oxide in human chondrocytes: suppression of nuclear factor κB activation by degradation of the inhibitor of nuclear factor κB. Arthritis Rheum. 2002;46:2079–86. doi: 10.1002/art.10443. [DOI] [PubMed] [Google Scholar]
- 32.Shukla M, Gupta K, Rasheed Z, Khan KA, Haqqi TM. Bioavailable constituents/metabolites of pomegranate (Punica granatum L) preferentially inhibit COX2 activity ex vivo and IL-1β-induced PGE2 production in human chondrocytes in vitro. J Inflamm (Lond) 2008;5:9–18. doi: 10.1186/1476-9255-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qureshi HY, Ahmad R, Zafarullah M. High-efficiency transfection of nucleic acids by the modified calcium phosphate precipitation method in chondrocytes. Anal Biochem. 2008;382:138–40. doi: 10.1016/j.ab.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 35.Krutzfeldt J, Poy MN, Stoffel M. Strategies to determine the biological function of microRNAs. Nat Genet. 2006;38(Suppl):S14–9. doi: 10.1038/ng1799. [DOI] [PubMed] [Google Scholar]
- 36.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-κB-dependent induction of microRNAs miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12481–6. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ortiz-Lazareno PC, Hernandez-Flores G, Dominguez-Rodriguez JR, Lerma-Diaz JM, Jave-Suarez LF, Aguilar-Lemarroy A, et al. MG132 proteasome inhibitor modulates proinflammatory cytokines production and expression of their receptors in U937 cells: involvement of nuclear factor-κB and activator protein-1. Immunology. 2008;124:534–41. doi: 10.1111/j.1365-2567.2008.02806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, et al. Novel inhibitors of cytokine-induced IκBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- 39.Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor κB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum. 2000;43:801–11. doi: 10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 40.Bartels CL, Tsongalis GJ. MicroRNAs: novel biomarkers for human cancer. Clin Chem. 2009;55:623–31. doi: 10.1373/clinchem.2008.112805. [DOI] [PubMed] [Google Scholar]
- 41.Mishra PK, Tyagi N, Kumar M, Tyagi SC. MicroRNAs as a therapeutic target for cardiovascular diseases. J Cell Mol Med. 2009;13:778–89. doi: 10.1111/j.1582-4934.2009.00744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin Q, Gao Z, Alarcon RM, Ye J, Yun Z. A role of miR-27 in the regulation of adipogenesis. FEBS J. 2009;276:2348–58. doi: 10.1111/j.1742-4658.2009.06967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheedy FJ, O'Neill LA. Adding fuel to fire: microRNAs as a new class of mediators of inflammation. Ann Rheum Dis. 2008;67:iii50–5. doi: 10.1136/ard.2008.100289. [DOI] [PubMed] [Google Scholar]
- 44.Marcucci G, Radmacher MD, Maharry K, Mrozek K, Ruppert AS, Paschka P, et al. MicroRNA expression in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1919–28. doi: 10.1056/NEJMoa074256. [DOI] [PubMed] [Google Scholar]
- 45.Kuehbacher A, Urbich C, Zeiher A, Dimmeler S. Role of dicer and drosha for endothelial microRNA expression and angiogenesis. Circulation Res. 2007;101:59–68. doi: 10.1161/CIRCRESAHA.107.153916. [DOI] [PubMed] [Google Scholar]
- 46.Sun F, Wang J, Pan Q, Yu Y, Zhang Y, Wan Y, et al. Characterization of function and regulation of miR-24-1 and miR-31. Biochem Biophy Res Commun. 2009;380:660–5. doi: 10.1016/j.bbrc.2009.01.161. [DOI] [PubMed] [Google Scholar]
- 47.Bushati N, Cohen SM. MicroRNA function. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 48.O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 2007;104:1604–9. doi: 10.1073/pnas.0610731104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]