

Abstract
The matrix (MA) domain of the HIV-1 precursor Gag (PrGag) protein plays multiple roles in the viral replication cycle. One essential role is to target PrGag proteins to their lipid raft-associated phosphatidylinositol-(4,5)-bisphosphate (PI[4,5]P2) assembly sites at the plasma membranes (PMs) of infected cells. In addition to this role, several reports have implicated nucleic acid binding properties to retroviral MAs. Evidence indicates that RNA binding enhances the binding specificity of MA to PI(4,5)P2-containing membranes, and supports a hypothesis in which RNA binding to MA acts as a chaperone that protects MA from associating with inappropriate cellular membranes prior to PrGag delivery to PM assembly sites. To gain a better understanding of HIV-1 MA-RNA interactions, we have analyzed the interaction of HIV MA with RNA ligands that previously were selected for their high affinities to MA. Binding interactions were characterized via bead binding, fluorescence anisotropy, gel shift, and analytical ultracentrifugation methods. Moreover, MA residues that are involved in RNA binding were identified from NMR chemical shift data. Our results indicate that the MA RNA and PI(4,5)P2 binding sites overlap, and suggest models for Gag-membrane and Gag-RNA interactions, and for the HIV assembly pathway.
Keywords: Human immunodeficiency virus type-1 (HIV-1); Gag; Matrix (MA); phosphatidylinositol-(4,5)-bisphosphate (PI[4,5]P2); nuclear magnetic resonance (NMR)
INTRODUCTION
The human immunodeficiency virus type 1 (HIV-1) matrix domain (MA) is the N-terminal cleavage product of the HIV-1 precursor Gag (PrGag) protein. Initially, PrGag is synthesized on cytosolic ribosomes and becomes cotranslationally modified by the N-terminal attachment of a myristoyl group by N-myristoyl-transferase 1–3. Myristoylated Gag precursors associate with the inner layer of the plasma membrane (PM), where they oligomerize, assemble, and bud off from cells as immature virions. During the process, cleavage of PrGag by the viral protease (PR) generates the mature MA protein as well as capsid (CA), nucleocapsid (NC), p6 and two spacer peptides 4,5. The MA domain plays multiple roles in the viral replication cycle. One essential role is to target PrGag proteins to their lipid raft-associated phosphatidylinositol-(4,5)-bisphosphate (PI[4,5]P2) assembly sites at the PMs of infected cells 6–15. A second role is the incorporation of the viral surface/transmembrane (SU/TM) envelope (Env) protein complex into virions 16–21. In addition to these activities, several reports have implicated nucleic acid binding properties to retroviral MAs 22–27, and it is possible that such binding might facilitate PrGag delivery to the PM, virus assembly, or nuclear import of viral preintegration complexes (PICs) 28–34.
A number of structural studies have been conducted on HIV-1 MA 25, 35–39. In addition to its N terminal myristate, which fosters membrane binding 2, 40–41, MA is composed of six helices and three β sheet strands 35–39. Sedimentation equilibrium data have shown that while myristoylated MA exists in a monomeric-trimeric state at equilibrium, unmyristoylated MA occurs as a monomer even at high concentrations 37. NMR studies suggest that upon Gag multimerization the myristoyl group is exposed, and fosters Gag binding to membranes 37–39. The membrane binding face of HIV-1 MA is basic, promoting interactions with negatively charged phospholipid headgroups at the inner leaflets of PMs 35–39. The significance of such interactions has been highlighted through molecular genetic experiments which demonstrated that depletion of PM PI(4,5)P2 by overexpression of polyphosphoinositide 5-phosphatase IV, reduced virus assembly efficiency, resulting in the delivery of viral proteins to intracellular compartments 42–43. NMR investigations have indicated that HIV-1 MA preferentially binds to soluble PI(4,5)P2 mimics through contacts with the lipid headgroup and its 2′ acyl chain, and that binding promotes both exposure of the MA myristate group and protein oligomerization 38–39. Consistent with the above observations, we have shown that MA proteins tend to organize as hexamers of trimers on lipid membranes containing PI(4,5)P2 44, and that the binding specificity of MA is enhanced by cholesterol 25.
In addition to the MA membrane-binding capabilities described above, several lines of experimentation have shown that HIV MA has a nucleic acid binding capacity that may include the highly basic region of MA 22–26, 33, 34, 45–47. Interestingly, high affinity RNA ligands to HIV-1 MA have been selected by screening of random 76mer and 31mer RNA libraries 23, 24. Moreover, we have shown that nucleic acid binding enhances the binding specificity of MA to PI(4,5)P2-containing membranes. This was evident by the fact that PI(4,5)P2-containing liposomes successfully competed with nucleic acids for MA binding, whereas other liposomes did not 25. Recent experiments also showed that RNAse treatment of Gag in vitro translation lysates reduced the selectivity of Gag binding to PI(4,5)P2 liposomes 26. Taken together, these results suggest that the ability of MA to bind to RNA increases the selectivity of MA for appropriate PM assembly sites. These results support a hypothesis in which RNA binding to MA acts as a chaperone that protects MA from associating with inappropriate cellular membranes prior to PrGag delivery to the PM.
To gain a better understanding of HIV-1 MA-RNA interactions, we have applied a multidisciplinary approach to analyze HIV MA interactions with RNA ligands that previously were selected for their high affinities to MA 23. We characterized binding interactions via bead binding, fluorescence anisotropy, gel shift, and analytical ultracentrifugation methods. Moreover, MA residues that are involved in RNA binding were identified from NMR chemical shift data. Our results indicate that the MA RNA and PI(4,5)P2 binding sites overlap, and suggest models for Gag membrane and RNA interactions, and for the HIV assembly pathway.
RESULTS
Analysis of MA binding to RNA
A number of studies have shown that HIV MA has a nucleic acid binding capacity that may include the highly basic region of MA 22–26, 47. We demonstrated previously that MA binds to nucleic acids, and that PI(4,5)P2-containing liposomes successfully competed with nucleic acids for MA binding, whereas alternate liposomes did not. Other results 26 have shown that RNAse treatment of in vitro translated Gag protein preparations decreased the binding specificity to membranes containing PI(4,5)P2, suggesting that RNA influences the membrane binding specificity of MA. To further investigate the role of MA binding to RNA, we used a fluorescently tagged 25mer RNA, Sel25, that was selected previously for high affinity binding to MA 23. Using this ligand, we performed binding studies with the myristoylated matrix protein that we will refer to here as MyrMA, and to the unmyristoylated protein that we will refer to as MA. Interactions between matrix proteins and Sel25 RNA were studied via bead binding assays and binding was quantitated by fluorescent bead brightness which is indicative of RNA binding 25. Our experiments indicated that fluorescently tagged Sel25 bound well to either HIV-1 MA (Figure 1A) or MyrMA (Figure 1B) coated beads, but not to CA-coated beads (Figure 1C).
Figure 1. Fluorescent RNA bead binding assay.

In panels (A–C), binding assays were performed with 0.16 μM FITC-Sel25 RNA and beads coated with (A) MyrMA, (B) MA or (C) CA proteins. After binding and washing steps, beads with bound RNA were imaged by fluorescence microscopy under identical gain and exposure settings, and photographed: note that bead brightness is indicative of RNA binding. In panels (D–F) levels of binding were quantitated for beads coated with the indicated proteins and FITC-Sel25 RNA (black bars) or FITC-Ran25 RNA (white bars) at the indicated 0.5, 0.16, and 0.05 μM RNA concentrations. For quantitation, background-subtracted bead brightness values were calculated, averaged from multiple beads for each incubation, and normalized to MyrMA plus FITC-Sel25 RNA (0.5 μM) signals. For capsid beads, averages derive from four readings each; for matrix beads, averages derive from 12 separate beads collected from experiments on three separate dates.
To confirm these results and to check Sel25 specificity for binding, we examined MA, MyrMA and CA binding to a randomized sequence of Sel25 (Ran25), as well as Sel25 at multiple concentrations. As expected, both RNA ligands bound poorly to CA (Figure 1F). In contrast, both MyrMA and MA bound well to these RNAs and showed higher binding affinities for Sel25 than Ran25. Interestingly, Sel25 versus Ran25 binding to MA appeared to be more selective at higher concentrations than binding to MyrMA. These results suggest that the presence or the absence of the MA myristoyl group may influence binding selectivity of RNA to matrix proteins.
To determine whether RNA binding influences membrane binding to the matrix proteins, we carried out membrane competition binding experiments. Interestingly, when MyrMA beads plus fluorescent Sel25 RNAs were incubated with PI(4,5)P2-containing liposomes, bound RNA levels were reduced significantly (Figure 2A). We also tested whether untagged RNAs or phospholipid headgroups could compete successfully with tagged Sel25 for MA binding (Figure 2B). Control experiments showed that 20 and 80 μM Sel25 RNA inhibited binding of the fluorescent ligand whereas the Ran25 RNA did not (Figure 2B). We also observed that 80 μM concentrations of a 15mer RNA that contained the Sel25 binding consensus sequence (Sel15) were able to reduce tagged Sel25 binding levels. To extend our investigations, we tested whether soluble phosphatidylserine (PS) with two six carbon acyl chains (PSC6) or PI(4,5)P2 analogues with saturated four (PIPC4) or eight (PIPC8) carbon chains could inhibit RNA-MA interactions. As shown in Figure 2B, PSC6 and PIPC4 failed to compete with tagged Sel25 for MA binding, whereas 80 μM PIPC8 reduced binding by approximately 50%. These results are consistent with the notion that longer acyl chains of the PIPC8 analogue contribute to PI(4,5)P2-MA binding25, 38.
Figure 2. Competition binding assays.

(A) MyrMA beads were incubated with 0.16 μM of FITC-Sel25 RNA either without liposomes (control) or with rhodamine-DOPE-tagged 20% cholesterol-70% PC-10% PI(4,5)P2 liposomes (liposomes). (B) MA beads were incubated with 0.16 μM FITC-Sel25 RNA in the absence (control) or presence of the indicated concentrations of untagged Sel25 RNA (Sel), Ran25 RNA (Ran), Sel15 RNA (Sel15), Ran15 RNA (Ran15), di-C6 phosphatidyl serine (PSC6), di-C4 PI(4,5)P2 (PIP2C4), or di-C8 PI(4,5)P2 (PIP2C8). After incubations and washes, beads were imaged and RNA binding (brightness) was quantitated as described for Figure 1. Values each derive from 6–12 beads, collected from experiments on three separate dates.
Characterization of MA-RNA binding
To further characterize the manner by which RNA binds to MA, we probed binding using fluorescence anisotropy (FA). To do so, we titrated increasing concentrations of MA or MyrMA to 5 nM samples of fluorescent Sel25 in either pH 7.8 or pH 5.5 buffers. Binding curves (Figures 3A–D) showed tagged Sel25 affinities for MyrMA and MA were in the 0.5–1 μM range, consistent with previous results 23. We also performed FA competition binding assays. For these experiments, we employed 5 nM FITC-Sel25 and 1 μM of either MyrMA or MA with increasing concentrations of unlabeled Sel15 or its randomized counterpart, Ran15. As expected from bead binding results (Figure 2B), Sel15 inhibited FITC-Sel25 binding to MA or MyrMA at both pH 7.8 and pH 5.5 (Figures 3E–H). The Ran15 ligand only slightly reduced the levels of tagged Sel25 binding to the matrix proteins, indicating that Sel15 is a specific binding competitor. However, of note was the fact that differences between Sel15 and Ran15 effects were more pronounced with MA than MyrMA at both pH 5.5 and 7.8 (Figures 3E–H). These differences correlate with MA versus MyrMA bead binding results (Figure 1), and again suggest that the myristate group decreases the selectivity of RNA binding
Figure 3. Fluorescence Anisotropy RNA binding to MA.

(A–D) Fluorescence Anisotropy (FA) measurements were conducted using 5 nM FITC-Sel25 RNA in buffers at the indicated pHs for either MyrMA (A,C) or MA (B,D). Affinities determined were 813 ± 74 nM for MyrMA pH 7.8; 1032 ± 80 nM for MA pH 7.8; 410 ± 72 nM for MyrMA pH 5.5; and ~1000 nM for MA pH 5.5. Changes in the fluorescence polarization were fitted assuming 1:1 binding curves. (E–H) FA competition binding assays used 5 nM FITC-Sel25 RNA ligand, 1 μM MyrMA or MA, and increasing concentrations of untagged Sel15 or Ran15 RNAs. Measurements were obtained in triplicate, and dissociation curves were fitted to exponential decay best fit curves.
In addition to FA RNA competition studies, we measured competition between the tagged Sel25 RNA and membrane phospholipid mimics. To do so, MyrMA plus fluorescent Sel25 incubations were supplemented with increasing concentrations of soluble PI(4,5)P2 and PS derivatives. As a control, we used unlabeled Sel25 as a competitor. As expected, Sel25 RNA readily reduced FITC-Sel25-MyrMA binding levels (Figure 4). We also observed that the PI(4,5)P2 headgroup mimic reduced MyrMA-RNA binding levels to a greater extent than the PS mimic (Figure 4). These results are in agreement with our bead competition binding results (Figure 2), and support the hypothesis that the matrix protein RNA-binding and PI(4,5)P2-binding sites overlap.
Figure 4. Fluorescence Anisotropy competition binding assays.
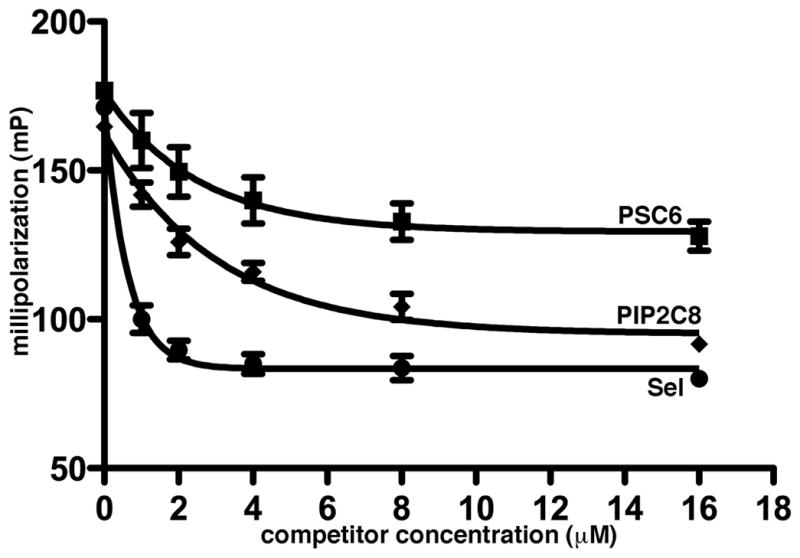
FA competition binding assays were performed with 5 nM FITC-Sel25 and 1 μM MyrMA plus increasing concentrations of untagged Sel25, di-C6 phosphatidyl serine (PSC6) or di-C8 PI(4,5)P2 (PIP2C8). Measurements were obtained in triplicate and dissociation curves were fitted to exponential decay best fit curves.
As a complementary approach to the characterization of matrix protein binding to RNA, we employed native electrophoretic mobility shift assays (EMSA), using a native polyacrylamide gel electrophoresis (PAGE) system. As illustrated in Figure 5, MyrMA or MA binding to Sel25 RNA was much more apparent than binding to Ran25 RNA, supporting bead binding (Figure 1), and FA results (Figure 3). In particular, well-defined Sel25 shift bands were observed with MyrMA (Figure 5A) and MA (Figure 5C), along with lower mobility smears. In contrast, Ran25 shift bands (Figures 5B, D) were much fainter than their Sel25 counterparts, and appeared only as diffuse smears. Additional experiments were conducted with the shorter RNAs, the Sel15 and Ran15 ligands. With these partners, little evidence of Ran15 binding was seen (Figure 5F), while the Sel15 shifts appeared as distinct bands (Figure 5E), consistent with the interpretation that matrix proteins can bind Sel15 with a defined, possibly 1:1 stoichiometry.
Figure 5. Electrophoretic mobility shift assay.

Electrophoretic mobility shift assays (EMSA) used 12% native polyacrylamide gels and were stained for detection of free and bound RNAs after electrophoresis. (A–F) Native PAGE data were obtained with 15 μM Sel25 (A, C), Ran25 (B,D), Sel15 (E), and Ran15 (F) RNAs with increasing concentrations (0, 7.5, 15, 22.5, 30, 37.5 μM) of MyrMA (A–B) or MA (C–F) proteins. Bound (B) and free (F) RNAs are as indicated.
To confirm our hypotheses as to the stoichiometry of MA-RNA binding, we examined MA interactions with FITC-tagged Sel15 RNA via analytical equilibrium centrifugation. As a control, we examined sedimentation of the fluorescent RNA by itself (Figure 6A). Assuming partial specific volume of 0.53 cm3/g for the RNA, its mass corresponded to 5098 ± 900 daltons, close to that of the calculated tagged Sel15 mass (5362.4 daltons). We obtained the oligomerization state MA-RNA complex indirectly by focusing on the apparent size of fluorescently tagged RNA-bound complex in the presence of an excess of MA (Figure 6B). For the RNA-MA complex, the measured mass corresponded to 18013 ± 700 daltons, assuming a partial specific volume of 0.678 cm3/g. This value is slightly lower than the calculated 20963.9 dalton value for a 1:1 complex, but this may be attributed to the fact that incomplete binding yielded a lower weight average than would a completely bound FITC-Sel15 complex. Taken together, our gel shifts (Figure 6) and centrifugation (Figure 7) data indicate that MA and Sel15 RNA form a 1:1 complex.
Figure 6. Equilibrium sedimentation.

Equilibrium sedimentation profiles for (A) 10 μM FITC-Sel15 and (B) 10 μM FITC-Sel15 plus 40 μM MA were obtained by analytical ultracentrifugation. Absorbance was measured at 490 nm to detect the FITC signal of the tagged RNA. Sedimentation corresponded to a mass of 5098 ± 900 daltons for FITC-Sel15 alone, assuming a partial specific volume of 0.53 cm3/g. For the MA-RNA complex, assuming a partial specific volume of 0.678 cm3/g, the measured mass corresponded to 18013 ± 700 daltons, indicative of 1:1 RNA: protein binding profile.
Figure 7. NMR of MA-PI(4,5)P2 binding.

(A) 1H-15N heteronuclear single quantum coherence spectrum (HSQC) of 15N-labeled MA at 60 μM was obtained at pH 5.5 and 35°C. Indicated in circles are some of the unambiguously residue assignments. (B–E) 15N NMR chemical shift titration data of 60 μM MA and 0, 60, 120 and 240 μM di-C4-PI(4,5)P2, di-C4-PI(4,5)P2: MA= 0:1, 1:1, 2:1, and 4:1 were obtained at pH 5.5 and 35°C. Binding curves are plotted for representative residues and are in agreement with previously obtained results.
Identification of binding site residues
What MA surfaces are sensitive to RNA binding? In an attempt to analyze this, NMR binding studies were performed. Our initial efforts with MyrMA and with Sel25 RNA caused protein aggregation, NMR signal line broadening and loss of signals. So, to avoid these complications, MA and Sel15 were used. For our purposes, the MA NMR structure, and databases for 1H, and 15N assignments were available 35, 37, and the availability of chemical shift assignments provided us a baseline for our comparisons. We first performed NMR HSQC experiments on MA only. The 2D 1H-15N HSQC spectra were collected for uniformly labeled MA, and the assignments (Figure 7A, Supplementary Table 1) were cross-checked with the available assignments of MA. Most of the residue assignments were accounted and matched the reported assignments for MA 35, 37–39. We were unable to locate a few residues, such as residues 2–5, 9, 26, 30 and 36, which may be attributed to the strength and resolution of the instrument used in these experiments. Before we analyzed MA-RNA interactions, we duplicated previous work on MA-di-C4 PI(4,5)P2 binding, so as to validate our approach. In this controlled study, we observed similar chemical shifts as reported previously 38. For example, the addition of di-C4 PI(4,5)P2 to MA led to significant chemical shift changes for residues such as R22, N73, S77 (Figures 7B, D, E), and not for residue E42 ( Figure 7C) that is far from the PI(4,5)P2 binding site of MA. These results confirmed past investigations, and validated our methodologies.
Next, the interactions between MA and RNA were monitored by tracking chemical shifts in 2D HSQC spectra during titrations with RNA. This approach allowed us the identification of MA residues involved in RNA binding. As illustrated in Figure 8A, high quality 2D HSQC spectra were obtained for MA upon titrations with Sel15. While the majority of signals were not sensitive to Sel15 addition, titrations of Sel15 led to significant changes in the backbone 1H and 15N chemical shifts of a subset of residues located at several domains of MA (Figure 8A, Table 1; Supplementary Table 1). In particular, we observed significant shifts (≥ 0.15 ppm) for residues Ser-6, Leu-13, Trp-16 (Table 1) located at the N-terminus helix I of MA: these residues were shown previously to interact with the myristoyl group, supporting the idea (Figures 1, 3) that the myristoyl group may influence binding selectivity of RNA to MA. Significant 1H and 15N chemical shifts also were observed (Table 1) for residues Gln-28, His-33, Glu-40, Glu-42, Ile-60, Leu-68, Thr-70, Glu-73, Arg-76, Ser-77, Tyr-79 and Asn-80. Examples of data for Gly-10, which was unaffected by RNA binding (Supplementary Table 1), and for affected residues Gln-28, Glu-42 and Ser-77 are shown in Figures 8B–E. Interestingly, several shifted residues locate to the matrix protein β-II-V cleft, and some of them (residues 33, 73, 76 and 79) were shown previously to contribute to the PI(4,5)P2 binding site. Non-linear least square fits of the titration data gave dissociation constant (Kd) values of 30 ± 9 μM (Figures 8F–I), which is higher than our FA data would suggest (Figure 3; discussed below). Nevertheless, our results are in agreement with recent work that implicates residues 28–33 and to a lesser extent residues 70–79 in MA-DNA interactions 34. We also observed 1H and 15N chemical shifts corresponding to residues Val-94, Thr-97, and Lys-103 and Ile-104. These residues are located on helix VI, away from the β-II-V cleft, and may be due to overall conformational changes of MA upon RNA binding. These observations are discussed in more detail below.
Figure 8. MA binding to Sel15 RNA.

(A) shown is an overlay of 2D 1H-15N HSQC spectra upon titrations with Sel15 RNA at the following RNA: MA ratios: 0:1 (black), 0.25:1 (blue), 0.5:1(green), 1:1 (yellow), 2:1 (orange), and 4:1 (red). Indicated in circles are some of the unambiguously assigned residues. (B–E) representatives of overlay expanded portions of the 2D 1H-15N HSQC are shown, with same titration ratios as in (A). (F–I) Representative binding curves of 1H-15N NMR chemical shift titration data of MA are depicted. The Δδ values are defined as ΔδHN = ((Δδ 1H)2 + (Δδ 15N)2 )½, and binding isotherms were fitted assuming a 1:1 binding stoichiometry. Note that G10 is provided as an example of a residue that is not affected by RNA binding, whereas Q28, E42 and S77 are examples of residues that were affected by binding.
Table 1. MA residues showing significant chemical shifts on RNA binding.
Shown are MA residues that displayed significant 1H -15N NMR chemical shifts on Sel15 binding. The association of Sel15 RNA and MA was monitored by observing changes in 1H–15N cross-peaks of HSQC spectra of 15N labeled MA upon titration with Sel15 RNA. Residues that exhibited chemical shifts of ≥ 0.15 ppm upon titrations with Sel15 RNA are listed. The extent of the chemical-shift changes of the combined 1H and 15N (ΔδHN) was calculated according to the formula ΔδHN = ((Δδ 1H)2 + (Δδ 15N)2 )1/2, as determined from data at a final RNA: MA ratio of 4:1.
| Residue | Δδ1H-15N (≥ 0.15 ppm) |
|---|---|
| 6 Ser | 0.28 |
| 13 Leu | 0.15 |
| 16 Trp | 0.26 |
| 28 Gln | 0.45 |
| 33 His | 0.21 |
| 40 Glu | 0.47 |
| 42 Glu | 0.32 |
| 60 Ile | 0.16 |
| 68 Leu | 0.24 |
| 70 Thr | 0.15 |
| 73 Glu | 0.31 |
| 76 Arg | 0.32 |
| 77 Ser | 0.15 |
| 79 Tyr | 0.44 |
| 80 Asn | 0.18 |
| 94 Val | 0.22 |
| 97 Thr | 0.22 |
| 103 Lys | 0.18 |
| 104 Ile | 0.33 |
DISCUSSION
MA serves multiple roles in the HIV replication cycle. Among these is its role in regulating PrGag-membrane interactions 6–10, 12–15, 26, 38–39, 42. Another function is the incorporation of the viral Env protein complex into virus particles 16–21. A number of reports also have implicated nucleic acid binding properties to retroviral MAs 22–27. While the NC domain is the most prominent RNA-binding element of HIV Gag, experiments have shown that either MA or NC must be present on Gag for efficient RNA binding and assembly 22. These results indicate that MA can substitute at least in part for the NC virus assembly function, probably by virtue of its ability to concentrate PrGag proteins on an RNA scaffold 22. We and others have demonstrated that HIV MA has a nucleic acid binding capacity that may include the highly basic region of MA 22–26, 47. Previously, we reported that HIV MA has an RNA binding capacity. Furthermore, we demonstrated that MA binds to nucleic acids, and that PI(4,5)P2-containing liposomes successfully compete with nucleic acids for MA binding, whereas other liposomes do not 25. These and other results 26 suggested that RNA may provide a chaperone function in preventing Gag proteins from binding to membranes until they reach PI(4,5)P2-rich plasma membranes.
Our current results clearly indicate that MA binds to an RNA that previously has been shown to have high affinity binding to MA (Figure 1). These investigations identify RNA as a competitor for membrane binding, and assays indicate that PI(4,5)P2-containing liposomes significantly reduce RNA binding to MA (Figure 2A). Bead competition binding experiments showed that soluble PIPC8 reduced Sel25 binding to MA, whereas a PSC6 mimic did not (Figure 2); while FA competition data indicated that PIPC8 reduced MyrMA-RNA binding levels to a greater extent than did the PS mimic (Figure 4). These results are consistent with the notion that RNA-MA interactions increase the ability of MA to distinguish between phospholipid headgroups.
Our NMR studies are consistent with the above results and identify residues on MA that are sensitive for RNA binding. In particular, we observed significant NMR shifts for residues located to the matrix protein β-II-V cleft corresponding to residues Gln-28, His-33, Glu-40, Glu-42, Ile-60, Leu-68, Thr-70, Glu-73, Arg-76, Ser-77, Tyr-79 and Asn-80 (see Figures 9A, C). Some of these residues (residues 33, 73, 76 and 79) previously were shown to contribute to the PI(4,5)P2 binding site (Figures 9B, D; 38). This observed overlap of PIP2-MA and RNA-MA binding sites reinforces a chaperone function hypothesis. These results also are in agreement with recent studies which implied that MA residues 28–33 and (to a lesser extent) residues 70–79 contribute to MA-DNA interactions of the preintegration complexes at the entry place of virus replication 34.
Figure 9. MA binding sites of RNA and PI(4,5)P2.

Shown is a surface representation of MA structure (pdb ID: 1UPH) from the side (A, B) and slightly tilted from the side (C, D). In panels A and C, residues that exhibited significant chemical shift changes (ΔδHN ≥ 0.15 ppm) on Sel15 RNA binding are colored in red. In panels B and D, di-C4-PI(4,5)P2 binding residues are highlighted in green. For convenience a subset of the binding residues is labeled.
In terms of the nature of MA-RNA binding, bead binding, fluorescence anisotropy, and gel shift results indicate that MA-RNA binding can be selective, and that a randomized version of Sel RNA (Ran RNA) bound poorly to MA (Figures 1, 3, 5). Our results also showed a slight selectivity of Sel RNA binding to the unmyristoylated MA protein, suggesting a myristate effect on MA-RNA binding specificity (Figure 1, 3). These results are consistent with the identification of chemical shifts on Sel15-MA binding corresponding to MA residues Ser-6, Leu-13 and Trp-16. However, the significance of the contributions of myristoyl binding residues to Sel RNA binding is unclear, since the Sel RNA sequence might not be the preferred MA-binding RNA in vivo. Another point worth noting is that while the gel shift and sedimentation equilibrium results suggest that MA and Sel15 bind with 1:1 stoichiometry, we can not exclude the possibility of multimeric binding to Sel25 substrates (Figures 5A–B). Moreover, some aggregation at high MA to RNA ratios was observed during NMR titrations, and that may have contributed to the higher calculated NMR dissociation constants than those inferred from FA data. We envision that multimerization could contribute to specificity and selectivity of RNA binding in vivo. It is pertinent to note also that NMR titrations indicated residues 94, 97, 103 and 104 were affected by RNA titrations. These residues are located on MA helix VI and may involve a conformational change of MA upon RNA binding that also could affect binding specificity.
Our results support a hypothesis in which RNA binding to MA acts as a chaperone that protects MA from associating with inappropriate cellular membranes prior to PrGag delivery to the PM. However, the exact identity of the RNAs that bind to MA in vivo remains to be determined. Our results demonstrate that MA binds to an RNA that has previously been shown to have high affinity binding to MA, and a version of this sequence is found in HIV RNA 23. However, while mutations of consensus nucleotides involved in MA-RNA binding reduced binding in vitro, they were less severe in reducing viral infectivity in vivo 23. It is possible that MA may bind to other sequences on viral or cellular RNAs. Moreover, it is likely that viral genomic RNA simultaneously can bind both NC and MA, which is consistent with previous observations and modeling of a proposed compact conformation of PrGag 47–49. Whatever the role of MA-nucleic acid binding in the HIV-1 replication cycle is, it can not be essential under all circumstances, since viruses that have large deletions in the MA coding region may be infectious in certain cell culture models 31, 50. Nevertheless, the MA domains of natural HIV-1 isolates and the RNA-binding residues we have identified are highly conserved. Thus, it seems likely that further examination of HIV-1 MA interactions with nucleic acids will be of interest.
MATERIALS AND METHODS
Protein preparation
Myristoylated HIV-1 MA protein (MyrMA), as well as the unmyristoylated MA protein (MA), were expressed in Escherichia coli strain BL21(DE3)/pLysS (Novagen) along with Saccharomyces cerevisiae N-methyltransferase from pET-11a-based vectors kindly provided by Michael F. Summers (University of Maryland Baltimore County) as described previously 37–39, 44. The MA protein was prepared similarly to MyrMA, except that myristic acid was excluded from the bacterial growth media during the induction phase. The proteins were desalted by three rounds of buffer exchange using dialysis buffer of 10 mM sodium phosphate (pH 7.8) and 50 mM NaCl, supplemented with β-mercaptoethanol (BME; 1 mM final concentration), aliquoted, and stored at −80°C under nitrogen gas. 15N isotopically labeled proteins were prepared by growing cells on 15NH4Cl (Cambridge Isotopes; NLM-467-5) as the sole nitrogen source as described previously 35, 37–39. For NMR studies, the proteins were dialyzed against 50 mM sodium phosphate (pH 5.5), 100 mM NaCl and 5 mM dithiothreitol (DTT), and then concentrated by Pierce ultrafiltration concentrators against the same buffer. Protein purities were evaluated after fractionation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)25, 51–55 and by Coomassie blue staining 25, 51–53.
RNA Samples
The RNAs used for our studies were selected for high affinity binding to MA 23. The consensus sequence for the high affinity RNA ligand to HIV-1 MA derived from screening of random 76 mer and 31 mer libraries 23, 24, yielding a consensus, 5′GGAAU UAAUA GUAGC3′ (Sel15). As a control, we obtained a randomized (Ran15) version of the 15mer, with a sequence of 5′GAAGG AGAUU UAUAC3′. We also prepared a 25mer stem-loop (Sel25) version of Sel15 that retained its high MA binding affinity: its sequence is 5′ GGACA GGAAU UAAUA GUAGC UGUCC3′. As another control we prepared a randomized version (Ran25) of Sel25, with the sequence of 5′GGACA GAAGG AGAUU UAUAC UGUCC3′. In addition to the untagged RNAs, we procured fluorescently tagged versions of Sel15, Ran15, Sel25, and Ran25. All RNAs were obtained from Invitrogen.
Liposome preparation
Liposomes were prepared as previously reported 25 from stocks of cholesterol (Sigma); 1,2-dioleoyl-sn-glycerol-3-phosphocholine (PC; Avanti); brain PI(4,5)P2 (Avanti); 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Rhodamine-DOPE; Avanti). For liposome preparation, lipids were mixed and dried in glass vials with a stream of nitrogen gas. Dried lipids were supplemented with liposome buffer (10 mM HEPES [pH 7.4], 50 mM NaCl, 0.002% sodium azide), and suspended by twenty 30 sec rounds of sonication in a Branson 1210 bath sonicator, with incubations on ice between each sonication round. Final liposome lipid concentrations were 2 mg/ml, and liposomes were stored for up to 1 week under nitrogen at 4°C. By weight, PI(4,5)P2 liposomes were composed of 10% (wt/wt) PI(4,5)P2, 20% cholesterol, 69.8% PC, and 0.2% Rhodamine-DOPE.
Bead binding assays
For bead binding assays, 0.2 ml of packed nickel-nitrilotriacetic acid (Qiagen) beads were washed with 0.5 ml of wash buffer (25 mM sodium phosphate [pH 7.8], 50 mM NaCl, 0.1 mg/ml bovine serum albumin [BSA; Sigma A4503]), suspended in 0.5 ml of wash buffer supplemented with 20 μl of 10 mg/ml BSA, incubated for 5 min at 4°C, and washed three times with 0.3 ml wash buffer. After the washes, packed beads were supplemented with 0.2 ml wash buffer minus or plus 2 – 3 μg of His-tagged MA or CA. Proteins and beads were incubated for 2 h at 4°C, after which beads were pelleted to remove unbound protein, washed twice with 0.3 ml wash buffer, and suspended in a total volume of 360 μl wash buffer on ice: the estimated bead-bound MA or CA concentration used in each assay was 300 nM.
For RNA binding assays, beads in 60 μl wash buffer were supplemented with fluorescently tagged RNAs to final concentrations of 0 – 0.5 μM, and incubated for 1 h at 4°C, after which beads were pelleted (1 min; 13,700 × g), quickly washed twice with 300 μl wash buffer, and resuspended in 60 μl of wash buffer on ice. For RNA-liposome competition assays, beads were incubated with 0.16 μM of fluorescent RNA either without liposomes or with 725 nM PI(4,5)P2 in Rhodamine-DOPE-tagged 20% cholesterol-70% PC-10% PI(4,5)P2 liposomes (prepared as described above). Beads were incubated for 16 – 18 h at 4°C, after which beads were pelleted (1 min; 13,700 × g), quickly washed twice with 300 μl wash buffer, and resuspended in 60 μl of wash buffer on ice. For RNA and lipid competition assays, MA beads were incubated with 0.16 μM fluorescent RNAs in the absence (control) or presence of the indicated concentrations of untagged RNAs or 1,2-dihexanoyl-sn-glycero-3-phospho-L-serine (PSC6; Avanti), di-C4 PI(4,5)P2 (PIP2C4; Echelon) or di-C8 PI(4,5)P2 (PIP2C8; Avanti). Competitors were added to incubations from stocks in water to achieve final concentrations of 20 – 80 μM, respectively.
For viewing of fluorescently tagged beads, samples were mixed by pipetting up and down, 10 μl samples were applied to microscope slides, and covered with 22 × 22 mm coverslips. Fluorescent green beads were imaged on a Zeiss AxioPlan fluorescence microscope using a 20x (LDA-Plan) objective and Zeiss filter set 10 (excitation band-pass, 450 to 490; beam splitter Fourier transform, 510; emission band-pass, 515 to 565). After collection of multiple bead images per sample as grayscale tagged-image-file-format (TIFF) files using Improvision OpenLab software, images were ported to NIH Image J 25, 53 for analysis for each bead, normalized bead brightness values were determined as described previously 25. Briefly, areas and average brightness values were collected from circled beads. Average background brightness values were calculated by dividing total image average brightness values (with bead area brightness values zeroed out) by total image areas minus bead areas. Normalized bead brightness values (average bead brightness minus background brightness) were averaged for all beads of a given incubation in an experiment, and results of experiments were normalized to results with control beads of MyrMA or MA bound to Sel25 RNA.
Fluorescence Polarization
Fluorescence polarization was used to measure binding of MA to RNA. Measurements were conducted using 5 nM FITC-labeled RNAs in either pH 7.8 buffer (25 mM sodium phosphate [pH 7.8], 50 mM NaCl) or pH 5.5 buffer (50 mM sodium phosphate [pH 5.5], 100 mM NaCl, 5 mM DTT). Measurements were obtained using a Pan Vera Beacon 2000 fluorescence polarizer (Invitrogen) with a 490 nm excitation wavelength. All readings were obtained in triplicate at room temperature. To determine binding affinities of MA to RNA variants, one ml of 5 nM FITC-Sel25, FITC-Ran25, FITC-Sel15, or FITC-Ran15 RNAs were placed in 12 × 75 mm disposable borosilicate glass tubes. Binding reactions were titrated by successive additions of 30 μM MA or MyrMA to achieve final concentrations of 0 –5120 nM. Polarization values correspond to emitted light intensities as defined by the ratio (parallel − perpendicular)/(parallel + perpendicular) 56. Binding isotherms were fitted assuming 1:1 binding curves using Prism (www.graphpad.com). Competition assays employed 5 nM FITC-Sel25, 1 μM MyrMA and increasing concentrations of Sel25, PSC6, or PIPC8.
Analytical ultracentrifugation
Sedimentation equilibrium measurements were performed with a Beckman model XLA analytical ultracentrifuge. Samples of 10 μM FITC-Sel15 RNA only or 10 μM FITC-Sel15 RNA plus 4 fold molar excess of MA were prepared in 25 mM sodium phosphate [pH 7.8], 50 mM NaCl. Centrifugation runs were carried out at 16°C in an An60-Ti rotor using 12 mm cells and Epon, 2 channels, centerpieces at 20,000 rpm; monitoring the absorbance of the free and bound FITC- labeled RNA at 490 nm. Partial specific volumes were calculated from the protein sequence 57, 58 and estimated for RNA. The radial distribution was analyzed with a program written for Scientist (Micromath), and molecular mass estimates derived from the slopes of ln (concentration) versus (radius) plots after baseline adjustment to optimize linearity.
Electrophoretic mobility shift assays (EMSA)
RNA and MA samples were prepared in stocks containing 50 mM sodium phosphate (pH 5.5), 100 mM NaCl. For binding reactions, 3 μl of 100 μM RNA (15 μM final concentration) was mixed with increasing amounts of 150 μM MA to yield final concentrations of 0–37.5 μM MA or MA:RNA ratios ranging from 0:1 to 2.5: 1. Samples in final volumes of 20 μl were incubated at 35 °C for 30 min. Following incubations, samples were supplemented with 6 μl of 50% glycerol, and then loaded onto 12% native polyacrylamide gels in 0.5 × Tris-borate buffer (44.5 mM Tris base, 44.5 mM boric acid, pH 8.0)59. Gels were electrophoresed at 4 °C at 30–35 mA. For visualization of bound and free RNAs, gels were stained with Stains-all (Sigma), destained by light exposure, and scanned immediately on an Epson Perfection 1240U scanner. Band intensities were obtained with Image J software25, 53.
NMR Spectroscopy
NMR data were collected at 35 °C on a Bruker Avance III (600 MHz 1H) spectrometer, processed with Bruker-Topspin 2.1 software (BRUKER, http://www.bruker-biospin.com), and analyzed with NMRVIEW 60. All of the NMR samples were prepared in a buffer containing 50 mM sodium phosphate at pH 5.5, 100 mM NaCl and 5 mM DTT using 15N uniformaly labeled protein samples of 60–80 μM (10% 2H2O). Amide [1HN–15N] backbone assignments for MA were obtained using standard two dimensional heteronuclear sequential quantum correlation 2D [1H–15N] HSQC with pulse program hsqceftf3gp as described previously 61. The 2D HSQC spectrum was recorded as 2048 t2 × 256 t1 data points with 90° pulses for 1H and 15N of 10 μs and 36 μs respectively. The 2D HSQC spectrum was processed by Fourier transformation after zero-filling to 2048 × 1024 real data points, followed by phase and base line correction. The 2D HSQC spectrum was recorded first with MA only. Signal assignments and verification of MA were matched with the chemical shift assignments that have been deposited at the BioMagResBank 35, 37. The interactions of di-C4-PI(4,5)P2 and MA were monitored by the chemical shift changes of 1HN–15N cross-peaks in the HSQC spectra of 15N labeled MA upon titration with di-C4-PI(4,5)P2 as described previously 38. Similarly, the association of Sel15 RNA and MA was monitored by observing the changes in the 1HN–15N cross-peaks in the HSQC spectra of 15N labeled MA upon titration with of unlabeled Sel15 stock of 10 mM in 50 mM sodium phosphate at pH 5.5, 100 mM NaCl and 5 mM DTT. Residues that exhibited significant chemical-shift changes upon titrations with either di-C4-PI(4,5)P2 or Sel15 RNA were analyzed for the extent of the chemical-shift changes of the combined 1H and 15N (ΔδHN) by calculating ΔδHN = ((Δδ 1H)2 + (Δδ 15N)2 )1/2 37,38. Binding isotherms from 1H–15N NMR HSQC titration experiments were calculated using Prism software (www.graphpad.com). Surface representations of the MA structure (pdb ID: 1UPH) were generated with PYMOL (http://pymol.sourceforge.net).
Supplementary Material
Acknowledgments
We are grateful to Michael F. Summers for bacterial expression vectors. We also appreciate the help and support of Claudia López, Jacob Eccles, Colleen Noviello, Rachel Sloan, Daniel Pauw, Robin Barklis and Mike Webb. This research was supported by National Institutes of Health grants GM060170 and AI071798 to E.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mervis R, Ahmad N, Lillehij E, Raum M, Salazar F, Chan H, Venkatesan S. The gag gene products of human immunodeficiency virus type 1: alignment within the gag open reading frame, identification of posttranslational modifications, and evidence for alternative gag proteins. J Virol. 1988;62:3993–4002. doi: 10.1128/jvi.62.11.3993-4002.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryant M, Ratner L. Myristylation-dependent replication and assembly of human immunodeficiency virus. Proc Natl Acad Sci USA. 1990;87:523–527. doi: 10.1073/pnas.87.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tritel M, Resh MD. Kinetic analysis of human immunodeficiency virus type 1 assembly reveals the presence of sequential intermediates. J Virol. 2000;74:5845–5855. doi: 10.1128/jvi.74.13.5845-5855.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swanstrom R, Wills J. Synthesis, assembly and processing of viral proteins. In: Coffin J, Hughes S, Varmus H, editors. Retroviruses. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1997. pp. 263–334. [PubMed] [Google Scholar]
- 5.Freed E. HIV-1 gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- 6.Bhatia A, Campbell N, Panganiban A, Ratner L. Characterization of replication defects induced by mutations in the basic domain and C-terminus of HIV-1 matrix. Virology. 2007;369:47–54. doi: 10.1016/j.virol.2007.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouamr F, Scarlata S, Carter C. Role of myristoylation in HIV-1 Gag assembly. Biochemistry. 2003;42:6408–6417. doi: 10.1021/bi020692z. [DOI] [PubMed] [Google Scholar]
- 8.Dalton A, Ako-Adjei D, Murray P, Murray D, Vogt V. Electrostatic interactions drive membrane association of the human immunodeficiency virus type 1 Gag MA domain. J Virol. 2007;81:6434–6445. doi: 10.1128/JVI.02757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ehrlich L, Fong S, Scarlata S, Zybarth G, Carter C. Partitioning of HIV-1 Gag and Gag-related proteins to membranes. Biochemistry. 1996;35:3933–3943. doi: 10.1021/bi952337x. [DOI] [PubMed] [Google Scholar]
- 10.Jouvenet N, Neil S, Bess C, Johnson M, Virgen C, Simon S, Bieniasz P. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006;4:2296–2310. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morikawa Y, Hockley D, Nermut M, Jones I. Roles of the matrix, p2, and N-terminal myristoylation in human immunodeficiency virus type 1 Gag assembly. J Virol. 2000;74:16–23. doi: 10.1128/jvi.74.1.16-23.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray P, Li L, Wang J, Tang C, Honig B, Murray D. Retroviral matrix domains share electrostatic homology: models for membrane binding function throughout the viral life cycle. Structure. 2005;13:1521–1531. doi: 10.1016/j.str.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Scarlata S, Ehrlich L, Carter C. Membrane-induced alterations in HIV-1 Gag and matrix protein-protein interactions. J Mol Biol. 1998;277:161–167. doi: 10.1006/jmbi.1997.1615. [DOI] [PubMed] [Google Scholar]
- 14.Scholz I, Still A, Dhenub T, Coday K, Webb M, Barklis E. Analysis of human immunodeficiency virus matrix domain replacements. Virology. 2008;371:322–335. doi: 10.1016/j.virol.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spearman P, Wang J, Vander Heyden N, Ratner L. Identification of human immunodeficiency virus type 1 Gag protein domains essential to membrane binding and particle assembly. J Virol. 1994;68:3232–3242. doi: 10.1128/jvi.68.5.3232-3242.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu X, Yuan X, Matsuda Z, Lee TH, Essex M. The matrix protein of human immunodeficiency virus type 1 is required for incorporation of viral envelope protein into mature virions. J Virol. 1992;66:4966–4971. doi: 10.1128/jvi.66.8.4966-4971.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis M, Jiang J, Zhou J, Freed E, Aiken C. A mutation in the human immunodeficiency virus type 1 Gag protein destabilizes the interaction of envelope protein subunits gp120 and gp41. J Virol. 2006;80:2405–2417. doi: 10.1128/JVI.80.5.2405-2417.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dorfman T, Mammano F, Haseltine W, Gottlinger H. Role of the matrix protein in the virion association of the human immunodeficiency virus type 1 envelope glycoprotein. J Virol. 1994;68:1689–1696. doi: 10.1128/jvi.68.3.1689-1696.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freed E, Martin M. Virion incorporation of envelope glycoproteins with long but not short cytoplasmic tails is blocked by specific, single amino acid substitutions in the human immunodeficiency virus type 1 matrix. J Virol. 1995;69:1984–1989. doi: 10.1128/jvi.69.3.1984-1989.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freed E, Martin M. Domains of the human immunodeficiency virus type 1 matrix and gp41 cytoplasmic tail required for envelope and incorporation into virions. J Virol. 1996;70:341–351. doi: 10.1128/jvi.70.1.341-351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wyma D, Kotov A, Aiken C. Evidence for a stable interaction of gp41 with Pr55Gag in immature human immunodeficiency virus type 1 particles. J Virol. 2000;74:9381–9387. doi: 10.1128/jvi.74.20.9381-9387.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ott D, Coren L, Gagliardi T. Redundant roles for nucleocapsid and matrix RNA-binding sequences in human immunodeficiency virus type 1 assembly. J Virol. 2005;79:13839–13847. doi: 10.1128/JVI.79.22.13839-13847.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purohit P, Dupont S, Stevenson M, Green M. Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA. 2001;4:576–84. doi: 10.1017/s1355838201002023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lochrie M, Waugh S, Pratt D, Clever J, Parslow T, Polisky B. In vitro selection of RNAs that bind to the human immunodeficiency virus type-1 gag polyprotein. Nucleic Acids Res. 1997;25:2902–2910. doi: 10.1093/nar/25.14.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alfadhli A, Still A, Barklis E. Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J Virol. 2009;83:12196–12203. doi: 10.1128/JVI.01197-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chukkapalli V, Oh SJ, Ono A. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci USA. 2010;107:1600–1605. doi: 10.1073/pnas.0908661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monde K, Chukkapalli V, Ono A. Assembly and Replication of HIV-1 in T Cells with Low Levels of Phosphatidylinositol-(4,5)-Bisphosphate. J Virol. 2011;85:3584–3595. doi: 10.1128/JVI.02266-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bukrinsky MI, Haggerty S, Dempsey MP, Sharova N, Adzhubel A, Spitz L, Lewis P, Goldfarb D, Emerman M, Stevenson M. A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature. 1993;365:666–669. doi: 10.1038/365666a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.VonSchwedler U, Kornbluth R, Trono D. The nuclear localization signal of the matrix protein of human immunodeficiency virus type 1 allows the establishment of infection in macrophages and quiescent T lymphocytes. Proc Natl Acad Sci USA. 1994;91:6992–6996. doi: 10.1073/pnas.91.15.6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller MD, Farnet CM, Bushman F. Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J Virol. 1997;71:5382–5390. doi: 10.1128/jvi.71.7.5382-5390.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reil H, Bukovsky A, Gelderblom HR, Göttlinger HG. Efficient HIV-1 replication can occur in the absence of the viral matrix protein. EMBO J. 1998;17:2699–2708. doi: 10.1093/emboj/17.9.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haffar OK, Popov S, Dubrovsky L, Agostini I, Tang H, Pushkarsky T, Nadler SG, Bukrinsky M. Two nuclear localization signals in the HIV–1 matrix protein regulate nuclear import of the HIV-1 pre-integration complex. J Mol Biol. 2000;299:359–368. doi: 10.1006/jmbi.2000.3768. [DOI] [PubMed] [Google Scholar]
- 33.Hearps AC, Wagstaff KM, Piller SC, Jans DA. The N-terminal basic domain of the HIV-1 matrix protein does not contain a conventional nuclear localization sequence but is required for DNA binding and protein self-association. Biochemistry. 2008;47:2199–2210. doi: 10.1021/bi701360j. [DOI] [PubMed] [Google Scholar]
- 34.Cai M, Huang Y, Craigie R, Clore GM. Structural basis of the association of HIV-1 matrix protein with DNA. PLoS One. 2010;5:e15675. doi: 10.1371/journal.pone.0015675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Massiah MA, Starich MR, Paschall C, Summers MF. Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J Mol Biol. 1994;244:198–223. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- 36.Hill C, Worthylake D, Bancroft D, Christensen A, Sundquist W. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc Natl Acad Sci USA. 1996;93:3099–3104. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers M. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci USA. 2004;101:517–522. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saad J, Miller J, Tai J, Kim A, Ghanum R, Summers M. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci USA. 2006;103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saad J, Loeliger E, Luncsford P, Liriano M, Tai J, Kim A, Miller J, Joshi A, Freed E, Summers M. Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J Mol Biol. 2007;366:574–585. doi: 10.1016/j.jmb.2006.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Göttlinger HG, Sodroski JG, Haseltine WA. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1989;86:5781–5785. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spearman P, Wang JJ, Vander Heyden N, Ratner L. Identification of human immunodeficiency virus type 1 Gag protein domains essential to membrane binding and particle assembly. J Virol. 1994;68:3232–42. doi: 10.1128/jvi.68.5.3232-3242.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chukkapalli V, Hogue I, Boyko V, Hu W, Ono A. Interaction between the human immunodeficiency virus type 1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient Gag membrane binding. J Virol. 2008;82:2405–2417. doi: 10.1128/JVI.01614-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ono A, Ablan S, Lockett S, Nagashima K, Freed E. Phosphatidyl (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci USA. 2004;101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alfadhli A, Barklis R, Barklis E. HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology. 2009;387:466–472. doi: 10.1016/j.virol.2009.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cimarelli A, Luban J. Translation elongation factor 1-alpha interacts specifically with the human immunodeficiency virus type 1 Gag polyprotein. J Virol. 1999;73:5388–5401. doi: 10.1128/jvi.73.7.5388-5401.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang CY, Chang YF, Wang SM, Tseng YT, Wang CT. HIV-1 matrix protein repositioning in nucleocapsid region fails to confer virus-like particle assembly. Virology. 2008;378:97–104. doi: 10.1016/j.virol.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones CP, Datta SA, Rein A, Rouzina I, Musier-Forsyth K. HIV-1 Gag extension: conformational changes require simultaneous interaction with membrane and nucleic acid. J Mol Biol. 2011;406:205–214. doi: 10.1016/j.jmb.2010.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Datta S, Zhao P, Clark S, Tarasov J, Alexandratos S, Campbell M, Kvaratskhelia J, Lebowitz, Rein A. Interactions between HIV-1 Gag molecules in solution: an inositol phosphate-mediated switch. J Mol Biol. 2007;365:799–811. doi: 10.1016/j.jmb.2006.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Datta S, Curtis W, Ratcliff P, Clark R, Crist J, Lebowitz, Krueger S, Rein A. Conformation of the HIV-1 Gag protein in solution. J Mol Biol. 2007;365:812–824. doi: 10.1016/j.jmb.2006.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang CT, Barklis E. Assembly, processing, and infectivity of human immunodeficiency virus type 1 Gag mutants. J Virol. 1993;67:4264–4273. doi: 10.1128/jvi.67.7.4264-4273.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alfadhli A, Dhenub TC, Still A, Barklis E. Analysis of human immunodeficiency virus type 1 Gag dimerization-induced assembly. J Virol. 2005;79:14498–14506. doi: 10.1128/JVI.79.23.14498-14506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alfadhli A, Huseby D, Kapit E, Colman D, Barklis E. Human immunodeficiency virus type 1 matrix protein assembles on membranes as a hexamer. J Virol. 2007;81:1472–1478. doi: 10.1128/JVI.02122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barklis E, Alfadhli A, McQuaw C, Yalamuri S, Still A, Barklis R, Kukull B, Lopez C. Characterization of the in vitro HIV-1 capsid assembly pathway. J Mol Biol. 2009;387:376–389. doi: 10.1016/j.jmb.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang CT, Zhang Y, McDermott J, Barklis E. Conditional infectivity of a human immunodeficiency virus matrix domain deletion mutant. J Virol. 1993;67:7067–7076. doi: 10.1128/jvi.67.12.7067-7076.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zuber G, McDermott J, Karanjia S, Zhao W, Schmid MF, Barklis E. Assembly of retrovirus capsid-nucleocapsid proteins in the presence of membranes or RNA. J Virol. 2000;74:7431–7441. doi: 10.1128/jvi.74.16.7431-7441.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lundblad JR, Laurance M, Goodman RH. Fluorescence polarization analysis of protein-DNA and protein-protein interactions. Mol Endocrinol. 1996;10:607–612. doi: 10.1210/mend.10.6.8776720. [DOI] [PubMed] [Google Scholar]
- 57.Perkins SJ. Protein volumes hydration effects. The calculations of partial specific volumes, neutron scattering matchpoints and 280-nm absorption coefficients for proteins and glycoproteins from amino acid sequences. Eur J Biochem. 1986;157:169–180. doi: 10.1111/j.1432-1033.1986.tb09653.x. [DOI] [PubMed] [Google Scholar]
- 58.Alfadhli A, Steel E, Finlay L, Bächinger HP, Barklis E. Hantavirus nucleocapsid protein coiled-coil domains. J Biol Chem. 2002;277:27103–27108. doi: 10.1074/jbc.M203395200. [DOI] [PubMed] [Google Scholar]
- 59.Zhou J, McAllen J, Tailor Y, Summers MF. High affinity nucleocapsid protein binding to the μ RNA packaging signal of Rous sarcoma virus. J Mol Biol. 2005;349:976–988. doi: 10.1016/j.jmb.2005.04.046. [DOI] [PubMed] [Google Scholar]
- 60.Johnson BA, Blevins RA. NMRview: a computer program for the visualization and analysis for NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 61.Davis AL, Keeler J, Laue ED, Moskau D. Experiments for recording pure-absorption heteronuclear correlation spectra using pulsed field gradients. J Magn Reson. 1992;98:207–216. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.