

Abstract
Translation of cellular mRNAs involves formation of a cap-binding translation initiation complex known as eIF4F, containing phosphorylated cap-binding protein eIF4E, eIF4E kinase Mnk1, eIF4A, poly(A)-binding protein and eIF4G. Adenovirus is shown to prevent cellular translation by displacing Mnk1 from eIF4F, thereby blocking phosphorylation of eIF4E. Over expression of an eIF4E mutant that cannot be phosphorylated by Mnk1 impairs translation of cellular but not viral late mRNAs. Adenovirus 100k protein is shown to bind the C-terminus of eIF4G in vivo and in vitro, the same region bound by Mnk1. In vivo, 100k protein displaces Mnk1 from eIF4G during adenovirus infection, or in transfected cells. Purified 100k protein also evicts Mnk1 from isolated eIF4F complexes in vitro. A mutant adenovirus with a temperature-sensitive 100k protein that cannot inhibit cellular protein synthesis at restrictive temperature no longer blocks Mnk1 binding to eIF4G, or phosphorylation of eIF4E. We describe a mechanism whereby adenovirus selectively inhibits the translation of cellular but not viral mRNAs by displacement of Mnk1 from eIF4G and inhibition of eIF4E phosphorylation.
Keywords: adenovirus/eIF4E/eIF4F/Mnk1/protein synthesis
Introduction
Most mRNAs possess a 5′ m7GpppN (cap) structure that is specifically bound by a complex of cap-binding translation initiation factors known as eIF4F. eIF4F functions as a cap-dependent RNA helicase that stimulates protein synthesis by unwinding the 5′ end of mRNAs, promoting 40S ribosome binding. Core eIF4F consists of three polypeptides: a 24 kDa cap-binding protein (eIF4E), a 45 kDa ATP-dependent RNA helicase (eIF4A) and a 220 kDa molecular adapter protein (eIF4GI) upon which a number of translation initiation factors assemble at the cap (Gingras et al., 1999). A closely related homolog of eIF4GI, referred to as eIF4GII, is thought to be present in cells at 10% of the level of eIF4GI, and is presently indistinguishable in function (Gradi et al., 1998). Both forms will be referred to as eIF4G. eIF4E is phosphorylated by a MAP kinase activating kinase (MAPKAP)/p38 kinase known as Mnk1 (Fukunaga and Hunter, 1997; Waskiewicz et al., 1997), which is also bound to eIF4G (Pyronnet et al., 1999; Waskiewicz et al., 1999). Mnk1 only efficiently phosphorylates eIF4E in vivo when both are bound to eIF4G (Pyronnet et al., 1999; Waskiewicz et al., 1999). Translation factor eIF3 binds to eIF4G and recruits the small 40S ribosome subunit. Poly(A)-binding protein (PABP) binds to the N-terminus of eIF4G (Tarun et al., 1997; Imataka et al., 1998), potentially circularizing capped and polyadenylated mRNAs (Wells et al., 1998). eIF4G is therefore a central adapter protein that couples cap recognition of the mRNA, 5′ mRNA unwinding activity, 40S ribosome subunit loading and surveillance of the poly(A) tail.
Phosphorylation of eIF4E at Ser209/Thr210 by Mnk1 is thought to stabilize the interaction between the capped mRNA and the initiation complex (Marcotrigiano et al., 1997; Pyronnet et al., 1999; Waskiewicz et al., 1999). Stimulation of cap-dependent mRNA translation by mitogenic growth factors, serum and nutrients correlates with increased phosphorylation of eIF4E (Gingras et al., 1999). There are exceptions, particularly increased phosphorylation of eIF4E in cells treated with arsenite, which impairs translation (Wang et al., 1998). Increased phosphorylation of eIF4E in this setting is likely to be a compensatory response to maintain some level of translation. Dephosphorylation of eIF4E strongly correlates with inhibition or impairment of cap-dependent mRNA translation during cell stress such as heat shock and nutrient deprivation (reviewed in Duncan, 1996), metaphase arrest of the cell cycle (Bonneau and Sonenberg, 1987; Huang and Schneider, 1991) and infection with certain viruses such as adenovirus (Ad) (Huang and Schneider, 1991) or influenza virus (Feigenblum and Schneider, 1993). Dephosphorylation of eIF4E probably prevents cap-dependent mRNA translation by reducing the interaction of eIF4F complexes with capped mRNA (Minich et al., 1994; Marcotrigiano et al., 1997).
Infection of cells with Ad results in inhibition of cellular protein synthesis several hours after the virus enters the late phase of infection (reviewed in Schneider, 1996). Ad inhibition of cellular protein synthesis involves a 90–95% block in the phosphorylation of eIF4E (Huang and Schneider, 1991; Zhang et al., 1994) and does not involve eIF4E sequestration by 4E-BPs (Feigenblum and Schneider, 1996; Gingras and Sonenberg, 1997). Expression of Ad late gene products is associated with inhibition of eIF4E phosphorylation and host protein synthesis (Zhang et al., 1994). Genetic evidence implicates the Ad L4-100 kDa non-structural polypeptide in preferential translation of viral late mRNAs (Hayes et al., 1990). The Ad 100k protein is an abundant viral late polypeptide encoded by the L4 late transcription unit. A large proportion of 100k protein is bound to cellular and viral mRNAs in the cytoplasm (Adam and Dreyfuss, 1987; Hayes et al., 1990; Riley and Flint, 1993). A temperature-sensitive (ts) Ad mutant in 100k protein (Ad5ts1) is defective in viral late protein synthesis (Hayes et al., 1990).
Ad late mRNAs translate despite inhibition of host cell protein synthesis, a property conferred by the viral 200 nucleotide 5′ noncoding region (5′NCR) known as the tripartite leader (Logan and Shenk, 1984). The tripartite leader is common to most viral late mRNAs and permits translation when eIF4E is largely dephosphorylated (Dolph et al., 1988, 1990; Jang et al., 1988), as occurs during Ad late infection. The tripartite leader directs a novel form of translation initiation known as ribosome shunting, whereby 40S ribosome subunits bind the cap and translocate to the downstream initiating AUG in a non-linear manner (Yueh and Schneider, 1996, 2000).
In this report we show that Ad inhibits the phosphorylation of eIF4E and blocks host cell protein synthesis during the late phase of infection by preventing the association of the eIF4E kinase, Mnk1, with translation initiation factor eIF4G. Loss of Mnk1 from the cap-initiation complex leads to dephosphorylation of eIF4E, thereby impairing eIF4F activity and inhibiting translation of cellular but not viral late mRNAs.
Results
Adenovirus does not block Mnk1 kinase activity during infection
Studies were performed to determine whether Ad prevents host cell protein synthesis by blocking Mnk1 kinase activity. 293 cells were transfected with plasmids expressing glutathione S-transferase (GST)–Mnk1, or the kinase-inactive Mnk1 mutant T2A2 (Waskiewicz et al., 1997). At 18 h post-transfection, cells were either infected with wild-type (wt) Ad (Ad5dl309) and mock infected for 36 h, or treated with EGF to stimulate eIF4E phosphorylation (Feigenblum et al., 1998). Equal amounts of GST–Mnk1 proteins were purified by glutathione–Sepharose chromatography, and tested for activity by the ability to phosphorylate equal amounts of recombinant eIF4E protein added to in vitro reactions with [γ-32P]ATP (Figure 1). Phosphorylation of eIF4E in vivo is strongly facilitated when Mnk1 and eIF4E are bound to eIF4G (Pyronnet et al., 1999; Waskiewicz et al., 1999). In vitro, Mnk1 can phosphorylate eIF4E in the absence of eIF4G. Mnk1 isolated from uninfected cells was capable of phosphorylating eIF4E, whereas the T2A2 catalytic mutant of Mnk1 could not. Mnk1 isolated from Ad late infected cells slightly stimulated kinase activity. Since Ad does not impair Mnk1 activity, studies were carried out to determine whether the composition of the cap-initiation complex/eIF4F is altered in Ad late infected cells.
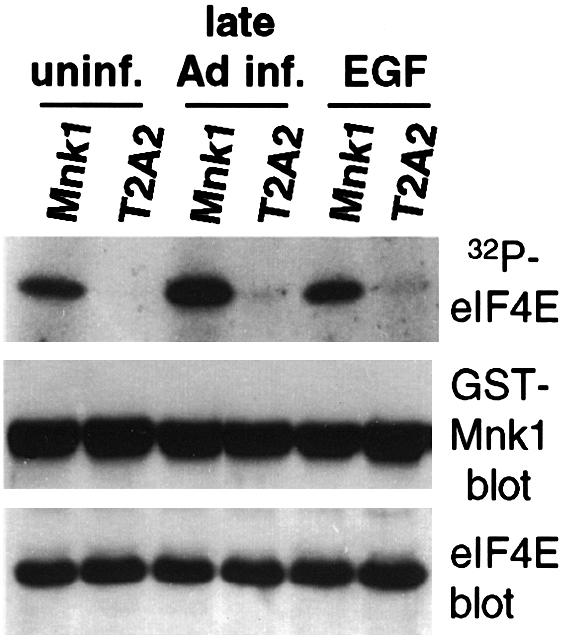
Fig. 1. Ad does not impair Mnk1 kinase activity. 293 cells were transfected with plasmids expressing GST–Mnk1 or GST–T2A2, a kinase-deficient mutant of Mnk1 (Waskiewicz et al., 1997). Cells were either uninfected (uninf.), treated with 30 ng/ml EGF for 15 min (Feigenblum et al., 1998), or infected with wtAd for 36 h (late Ad inf.). GST–Mnk1 or GST–T2A2 was recovered from equal amounts of cell lysates by glutathione–Sepharose chromatography and incubated with 1 µg of purified recombinant eIF4E and [γ-32P]ATP, resolved by SDS–PAGE and autoradiographed (top panel), or subjected to immunoblot analysis with anti-GST antibodies (middle panel). eIF4E levels are shown by immunoblot (bottom panel). Quantitation was performed by digital densitometry.
Ad disrupts the interaction of Mnk1 with cap-initiation complexes
We investigated whether Ad degrades or reduces the abundance of protein factors that comprise the cap-initiation complex. Cells were transfected with a GST–Mnk1 expression vector, infected by wtAd and cytoplasmic extracts prepared 36 h later, after inhibition of cellular protein synthesis. Equal amounts of proteins were resolved by SDS–PAGE and levels of eIF4E, eIF4G, eIF4A, Mnk1 and PABP were examined by immunoblot analysis (Figure 2). Ad late infection did not significantly alter the steady-state cytoplasmic abundance of any of the proteins that comprise the eIF4F complex.

Fig. 2. Immunoblot analysis of eIF4F complexes during Ad late infection. 293 cells were uninfected (uninf.) or infected for 36 h with wtAd (late Ad inf.). Equal amounts of protein lysates (10 µg) were resolved by SDS–10%PAGE and immunoblotted with specific antisera as described in Materials and methods. Antisera were: polyclonal anti-eIF4GI, polyclonal anti-GST (for Mnk1), monoclonal anti-eIF4A, polyclonal anti-eIF4E and monoclonal anti-PABP.
Studies were therefore performed to determine whether the association of Mnk1 with the eIF4F complex is disrupted by Ad infection. 293 cells were transfected with a GST–Mnk1 expression plasmid for 18 h, then infected with Ad or mock infected. Ad inhibition of cellular protein synthesis was examined by labeling cells with [35S]methionine followed by SDS–PAGE (Figure 3A). Cellular protein synthesis was partially inhibited by 24 h, and fully blocked by 36 h of infection. Interaction of Mnk1 with eIF4G was examined at these same time points (Figure 3B). Equal amounts of Mnk1 were recovered by glutathione–Sepharose chromatography from uninfected and Ad late infected cells, and associated proteins were examined by immunoblotting. Uninfected and early (12 h) Ad infected cells contained equal levels of eIF4G–eIF4E–Mnk1 complexes, which were decreased 5-fold at 24 h of infection, and ∼15-fold by 36 h. The progressive decrease in Mnk1 interaction with eIF4G is in accord with the kinetics for inhibition of cellular mRNA translation. To determine whether dissociation of Mnk1 from eIF4F complexes destabilizes the interaction of other members of the eIF4F complex, eIF4G was immunoprecipitated and interacting proteins identified by immunoblot analysis (Figure 3C). There was no significant decrease in the level of eIF4E, eIF4A and PABP bound to eIF4G compared with uninfected cells. Results shown later (Figure 6B) demonstrate the singular absence of Mnk1 in these eIF4F complexes. To determine whether dephosphorylation of eIF4E results from displacement of Mnk1 from eIF4G, uninfected and Ad infected cells were labeled with 32PO4 during early (12 h) and late (36 h) infection. eIF4E was purified by cap-affinity chromatography, resolved by SDS–PAGE and detected by autoradiography (Figure 3D). Ad late infection was associated with a 15-fold decrease in the phosphorylation of eIF4E, but no change in its abundance. Thus, loss of Mnk1 from the eIF4F complex during Ad infection parallels the kinetics for inhibition of cellular mRNA translation and dephosphorylation of eIF4E.

Fig. 3. Composition of eIF4F–Mnk1 complexes in Ad late infected cells. 293 cells were transfected with plasmids expressing GST or GST–Mnk1 proteins. At 18 h post-transfection, cells were uninfected or infected with wtAd and harvested at 12, 24 or 36 h post-infection (p.i.). (A) Cells were labeled with [35S]methionine and equal amounts of protein resolved by SDS–15%PAGE and fluorographed. (B) GST fusion proteins were recovered from equal amounts of cell lysates by glutathione–Sepharose chromatography, and immunoblot analysis of associated proteins performed using antisera to eIF4GI, GST (for Mnk1) and eIF4E. (C) Endogenous eIF4GI was immunoprecipitated (eIF4G immunop.) from equal amounts of cell lysates and associated proteins resolved by SDS–10%PAGE. Proteins were detected by immunoblot analysis using specific antisera as above. (D) Equal numbers of uninfected cells and cells infected for 12 h (early Ad infection) or 36 h (late Ad infection) were labeled with 200 µCi/ml of 32PO4 in phosphate-free medium for 2 h. eIF4E was recovered from equal amounts of cell extracts by m7GTP–Sepharose affinity chromatography, equal fractions resolved by SDS–15%PAGE and autoradiographed (upper panel), or transferred to Immobilon-P (Millipore) and detected by immunoblot analysis with specific eIF4E antisera (bottom panel). Quantitation was performed by digital densitometry.

Fig. 6. Characterization of 100k protein–eIF4F complexes in the absence of virus infection. (A) 293 cells were transfected with plasmids expressing GST, GST–Mnk1, GST–T2A2 protein or Flag-100k protein. At 36 h post-transfection, lysates were prepared from equal numbers of cells, GST fusion proteins were recovered by glutathione–Sepharose chromatography, resolved by SDS–10%PAGE, and associated proteins detected by immunoblot with antisera to endogenous eIF4GI, GST (for GST, GST–Mnk1, GST–T2A2) or endogenous eIF4E. (B) 293 cells were transfected with plasmids expressing Flag or Flag-100k protein, endogenous eIF4GI was immunoprecipitated with specific antisera (IP eIF4G), or preimmune sera (preimm). Immunoblotting was performed using antisera described in the legend to Figure 2. (C and D) Cells were transfected with plasmids expressing Flag-100k protein and full-length HA-eIF4GI (F4G, lacking the PABP binding site amino acids 157–1560); N-terminal fragment (N4G, 157–613), middle fragment (M4G, 565–1045) or C-terminal fragment (C4G, 1045–1560). (C) Flag-100k protein was immunoprecipitated (IP Flag-100k) with anti-Flag antibodies, proteins were resolved and immunoblotted for interaction with HA-eIF4G using anti-HA antibodies. Flag-100k proteins were detected using anti-Flag antibodies (bottom panel). (D) HA-eIF4G proteins were immunoprecipitated (IP HA-eIF4G) using anti-HA antibodies, proteins were resolved and immunoblotted as above.
We next determined whether dissociation of Mnk1 from the eIF4F complex, and dephosphorylation of eIF4E, are sufficient to block cellular but not viral late mRNA translation. The effect of eIF4E dephosphorylation on a representative cellular and viral late mRNA was examined. Two sets of model reporter mRNAs were expressed in transfected cells, one expressing β-galactosidase from an Ad late tripartite leader 5′UTR (3LDR), and the other from a non-viral eIF4F-dependent 5′UTR (CR3) (Feigenblum and Schneider, 1996). Two mutants of eIF4E that cannot be phosphorylated were developed. The single mutant contains a Ser209→Ala substitution, eliminating the major site of eIF4E phosphorylation. A double mutant additionally contains a Thr210→Ala mutation, eliminating spillover phosphorylation to this adjacent site. These mutations are identical to those used previously to demonstrate that Ser209/Thr210 are the in vivo sites of eIF4E phosphorylation (Flynn and Proud, 1995; Joshi et al., 1995). 293T cells were cotransfected with vectors expressing wt or mutant eIF4E, and tripartite leader or eIF4F-dependent β-galactosidase reporters. Translation was measured by immunoblot analysis of β-galactosidase reporter protein. Translation of the eIF4F-dependent mRNA was reduced 4-fold by expression of the single eIF4E mutant and ∼8-fold by expression of the double mutant (Figure 4A). In contrast, translation of the tripartite leader mRNA was unaffected by expression of either eIF4E mutant. These mutant forms of eIF4E were previously shown to bind eIF4G and enter into eIF4F complexes (Pyronnet et al., 1999; R.Cuesta and R.J.Schneider, unpublished results). These data support the conclusion that dephosphorylation of eIF4E is largely responsible for inhibition of cellular but not viral late mRNA translation.
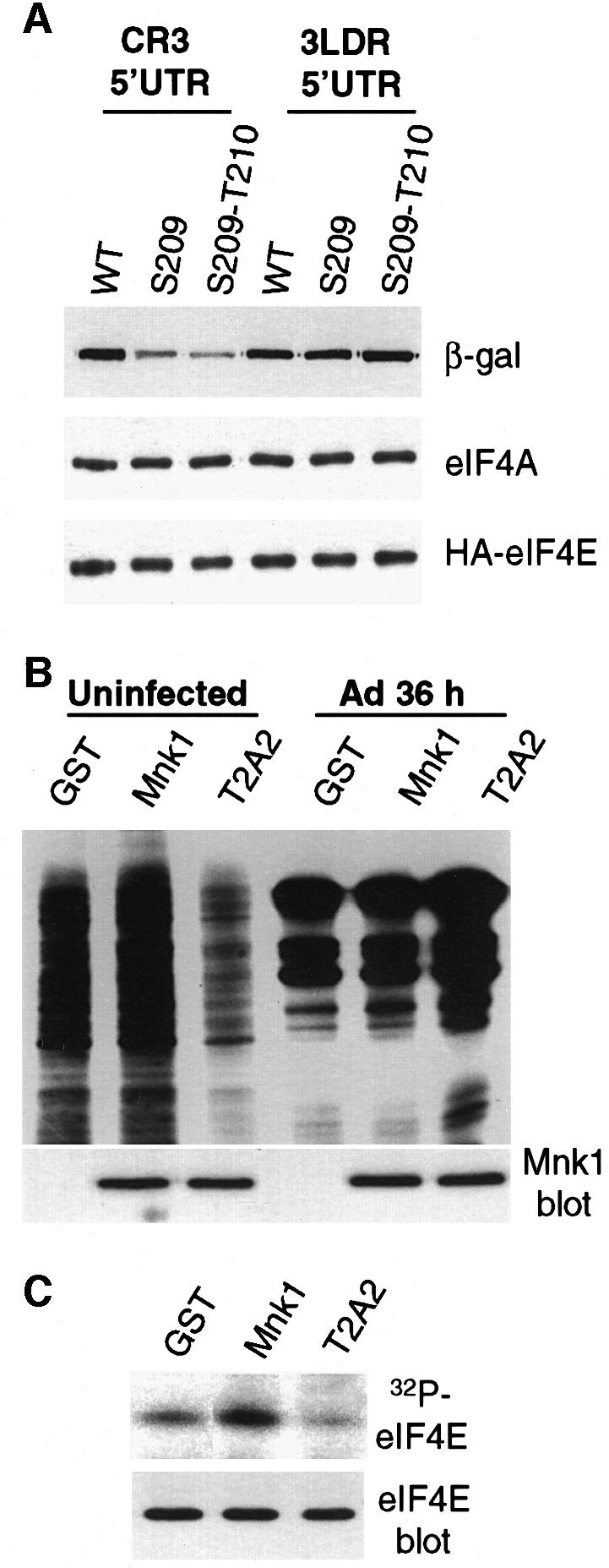
Fig. 4. Effect of eIF4E phosphorylation on translation of model Ad late and cellular mRNAs. (A) 293T cells were cotransfected with plasmids expressing β-galactosidase mRNAs containing either the Ad late tripartite leader 5′UTR (3LDR), or an eIF4F-dependent 5′UTR (CR3), and plasmids expressing HA-tagged wt eIF4E, Ser209→Ala eIF4E or Ser209→Ala/Thr210→Ala eIF4E. At 36 h post-transfection, equal amounts of cell lysates were resolved by SDS–12%PAGE and immunoblotted using specific antisera for β-galactosidase protein (β-gal), HA-eIF4E and eIF4A (for protein levels). (B) 293 cells were transfected with plasmids expressing GST, GST–Mnk1 or the GST–T2A2 Mnk1 mutant. At 18 h post-transfection, cells were infected with wtAd for 36 h, or were uninfected, then labeled with [35S]methionine, or (C) labeled with 32PO4. Samples were analyzed as in the legend to Figure 3. Mnk proteins were detected by immunoblot analysis with antisera to GST. eIF4E phosphorylation and abundance were examined as in the legend to Figure 3.
Overexpression of a kinase-deficient mutant of Mnk1 that cannot phosphorylate eIF4E also impaired translation of cellular but not Ad late mRNAs. 293 cells were transfected with plasmids expressing wt GST–Mnk1, the T2A2 Mnk1 mutant or GST alone at ∼40% efficiency (based on cotransfection with a green fluorescent protein expression vector; data not shown). Cells were infected with wt Ad 18 h later or mock infected, metabolically labeled with [35S]methionine 36 h after infection, and equal amounts of protein resolved by SDS–PAGE (Figure 4B). Overexpression of wt Mnk1 stimulated total translation by uninfected cells (Figure 4B) and phosphorylation of eIF4E 2-fold (Figure 4C), but had no effect on Ad translation. Overexpression of the T2A2 Mnk1 mutant reduced overall protein synthesis by 8- to 10-fold (Figure 4B) and endogenous eIF4E phosphorylation by ∼3-fold (Figure 4C), consistent with inhibition of (eIF4F)-dependent mRNA translation. By normalizing to a 40% transfection efficiency, these data suggest that in transfected cells overexpression of the T2A2 Mnk1 mutant impairs translation by ∼20-fold and eIF4E phosphorylation by ∼8-fold, consistent with previously published findings (Waskiewicz et al., 1999). In contrast, translation of Ad late mRNAs actually increased ∼2-fold with overexpression of the T2A2 Mnk1 mutant. The effect of mutant Mnk1 expression on eIF4E phosphorylation during Ad infection cannot be determined because eIF4E phosphorylation is inhibited by the virus regardless of the expression of Mnk proteins (data not shown). Overexpressed Mnk proteins formed complexes with eIF4G (Figures 3B and 6A). Collectively, these data demonstrate that displacement of Mnk1 from eIF4G, and dephosphorylation of eIF4E, are sufficient to prevent cellular protein synthesis but do not impair translation of Ad late mRNAs.
Ad late L4-100k protein associates with eIF4F complexes and promotes Mnk1 dissociation in vivo
Since one or more unknown Ad late gene products was previously implicated in inhibition of cellular protein synthesis (Zhang et al., 1994; Feigenblum et al., 1998), we investigated whether an Ad late polypeptide directly interacts with components of the eIF4F complex to promote dissociation of Mnk1. Uninfected and Ad late infected cells were metabolically labeled with [35S]methionine during inhibition of cellular translation, specifically to identify newly synthesized viral late proteins. Cells were lysed, extracts were solubilized in mild non-ionic detergent, then digested with RNaseA in low salt conditions to degrade RNA and poly(A) (Tarun et al., 1997), thereby preserving authentic protein–protein interactions. eIF4E was immunoprecipitated, resolved by SDS–PAGE and fluorographed. A 100 kDa protein that was labeled with [35S]methionine only in Ad late infected cells specifically co-immunoprecipitated with eIF4E/eIF4F complexes (Figure 5A, left panel). Immunopre cipitation of eIF4G yielded a 100 kDa protein as well (data not shown). The Ad late L4-100k protein was previously implicated in preferential translation of viral late mRNAs (Hayes et al., 1990). Immunoprecipitation using specific antisera for 100k protein demonstrated its presence in these samples (Figure 5A, right panel). The higher molecular weight band is the viral hexon protein (mol. wt 120 kDa), which is often recognized by preimmune serum.
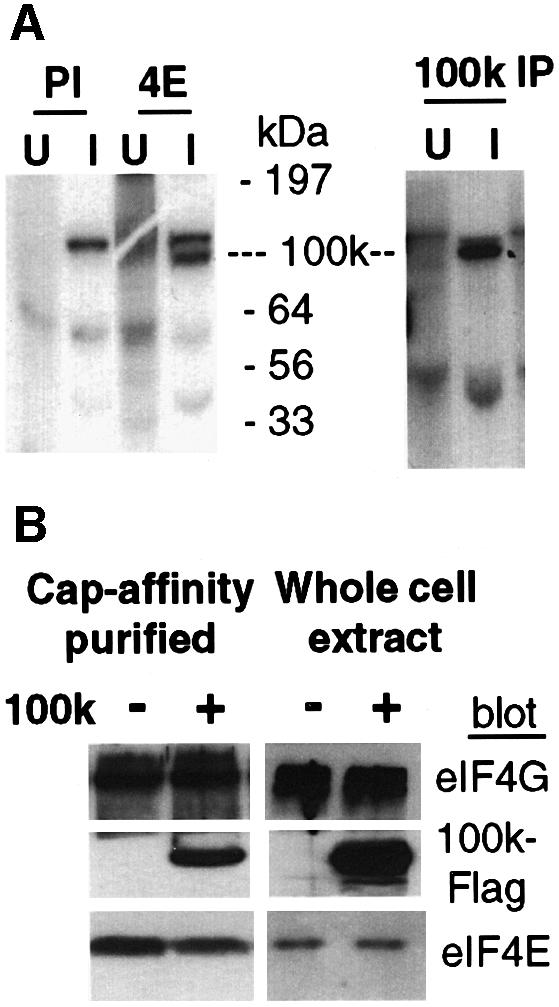
Fig. 5. Association of Ad L4-100k protein with eIF4F complexes. 293 cells were uninfected (U) or infected (I) with wtAd. (A) Cells were labeled with 50 µCi/ml of [35S]methionine for 4 h at 24 h post-infection, lysates were prepared and equal amounts of protein were immunoprecipitated with preimmune sera (PI) or with antibodies specific for eIF4E (4E) or Ad5-100k protein (100k IP), followed by SDS–15%PAGE and autoradiography. (B) 293 cells were either mock transfected or transfected with a cDNA clone expressing Flag epitope tagged 100k protein. Equal amounts of lysates were prepared and eIF4F complexes were purified by cap-affinity chromatography (left panel), or directly resolved by SDS–10%PAGE (right panel). Proteins were detected by immunoblot analysis using antisera as in the legend to Figure 2.
A full-length cDNA clone of the Ad5 100k coding region was prepared by synthetic oligonucleotide priming and reverse transcription of Ad5 late mRNA, then constructed with an N-terminal foreign Flag epitope. 293 cells were transfected with vectors expressing Flag-100k; eIF4F complexes were isolated by m7GTP(cap)-affinity chromatography, eluted, resolved by SDS–PAGE and examined by immunoblot analysis with antisera specific to endogenous eIF4GI, eIF4E and Flag-100k proteins. Whole cell extracts contained equal levels of eIF4F proteins prior to cap-affinity chromatography (Figure 5B, right panel). Cap-binding complexes purified from control cells (without 100k expression) contained eIF4GI and eIF4E (Figure 5B, left panel). Cap-binding complexes purified from cells expressing Flag-100k protein contained eIF4GI, eIF4E and 100k protein. 100k protein therefore interacts with cap-binding eIF4E/4G complexes. Sepharose beads lacking m7GTP residues did not retain eIF4G, eIF4E or 100k proteins (data not shown). As shown later, 100k protein was found to interact by direct binding to the C-terminus of eIF4G (Figure 6C and D).
Studies examined whether interaction of 100k protein with eIF4F complex in vivo is associated with loss of Mnk1. 293 cells were cotransfected with vectors expressing Flag-100k protein and either GST, GST–Mnk1 or the GST–T2A2 Mnk1 mutant. GST fusion proteins were purified by glutathione–Sepharose chromatography, and the association of eIF4G and eIF4E with Mnk1 was determined, with and without expression of 100k protein (Figure 6A). In the absence of 100k protein, equal amounts of eIF4G and eIF4E were associated with wt and mutant Mnk1 proteins. Expression of 100k protein was associated with a 10-fold reduction in eIF4G and eIF4E interaction with Mnk1. 100k protein did not copurify with Mnk1 protein (data not shown). The reciprocal experiment was carried out by immunoprecipitation of endogenous eIF4GI from cells transfected with a plasmid expressing Flag or Flag-100k protein. Strong binding was evident between endogenous eIF4GI and Flag-100k protein that was obtained from Flag-100k transfected cells, which was not detected in Flag transfected cells or by preimmune serum (Figure 6B). eIF4G immunoprecipitates from Flag-100k expressing cells contained all of the normal constituents of eIF4F (eIF4E, eIF4A, PABP) but not Mnk1 protein. These data demonstrate the specific dissociation of Mnk1 from eIF4F/eIF4G by expression of 100k protein.
The interaction partner for 100k protein, and the site of interaction, were then identified in vivo. In unpublished studies, we found a strong RNA-independent interaction, which was resistant to high salt wash, between 100k protein and eIF4G. We therefore characterized the site of 100k protein binding on eIF4G in vivo. 293 cells were transfected with plasmids expressing Flag-100k protein and HA-tagged regions of eIF4GI, corresponding to the C-terminal third (eIF4A, Mnk1 binding sites), middle third (eIF4A, eIF3 binding sites) or N-terminal third (eIF4E binding site, lacking the PABP binding element). HA-eIF4GI fragments accumulated at similar levels in transfected cells (Figure 6D). Equal amounts of Flag-100k protein were immunoprecipitated with anti-Flag antibody and examined by immunoblot analysis for interaction with HA-eIF4G (Figure 6C). 100k protein bound to the full-length (F4G) and C-terminus (C4G) of eIF4G, but not to the N-terminus (N4G), and possibly weakly to the middle fragment (M4G) (Figure 6C). HA-eIF4G proteins were immunoprecipitated from these same samples using anti-HA monoclonal antibodies, followed by immunoblotting for HA (eIF4G) or Flag (100k) proteins (Figure 6D). There was strong binding of 100k protein to the C4G fragment and to F4G, but not to N4G or M4G. Collectively, the results of Figure 6 demonstrate that 100k protein binds specifically to the C-terminus of eIF4G, the same region bound by Mnk1, and that binding by 100k protein is associated with selective loss of Mnk1 protein from eIF4G.
100k protein directly binds eIF4G and evicts Mnk1 in vitro
Studies were performed to determine whether 100k protein directly interacts with eIF4G in vitro and if so, whether this is mechanistically linked to loss of Mnk1 from eIF4F complexes. Regions corresponding to the N-terminus (N4G), middle (M4G) and C-terminus (C4G) of eIF4G, full-length 100k protein and Mnk1 were expressed in bacteria as GST fusion proteins, purified, and the GST moiety removed from eIF4G proteins by digestion with thrombin. Equal amounts of recombinant eIF4G proteins were incubated in vitro with purified 100k or Mnk1 proteins. 100k or Mnk1 proteins were recovered by glutathione–Sepharose chromatography, and the bound proteins were identified by immunoblot analysis with specific antisera (Figure 7A). The integrity and abundance of purified proteins are shown in the first lane of each panel. The smaller forms of 100k protein result from degradation during purification and are unavoidable. Purified 100k protein bound directly to the C4G fragment of eIF4G as observed in vivo, but not to the M4G or N4G fragments (compare middle lanes of each panel). Control studies showed that the level of interaction between C4G and 100k protein was similar to that of Mnk1 and C4G (Figure 7A, compare top panels). There was no interaction between 100k protein and GST alone (data not shown). Thus, purified 100k protein binds in vitro to the C-terminal region of purified eIF4G.

Fig. 7. 100k protein binds the C-terminus of eIF4G and evicts Mnk1 protein from eIF4F complexes in vitro. (A) Soluble recombinant proteins were purified from bacteria corresponding to full-length GST–100k, GST–Mnk1 and fragments of eIF4G for the N-terminus (N4G), middle (M4G) and C-terminus (C4G). It was not possible to isolate sufficient amounts of full-length eIF4G due to degradation. GST was removed from eIF4G by thrombin digestion. Equal amounts of purified eIF4G proteins (2 µg) and GST–100k or GST–Mnk1 proteins (0.3 µg) were incubated in vitro, GST fusion proteins were recovered by glutathione–Sepharose chromatography and bound proteins identified by SDS–10%PAGE and immunoblot analysis with specific antibodies to the eIF4G fragments, or to GST (for Mnk1 or 100k). (B) 293 cells were transfected with plasmids expressing GST or GST–Mnk1, extracts were prepared and equal amounts of eIF4F isolated by immuno precipitation of eIF4G (eIF4G IP). Equal amounts (2 µg) of purified recombinant GST–100k or GST fusion proteins were incubated in vitro with eIF4F complexes, and associated proteins detected by immunoblot analysis as shown. Equal amounts of whole cell lysates (‘lysate’) were resolved in lanes 4 and 5 to establish protein levels.
Studies were performed in vitro to determine whether binding of 100k protein to eIF4G directly evicts Mnk1 from the eIF4F complex. eIF4F complexes were isolated in a manner similar to that used to show eviction of PABP from eIF4F by the reovirus NSP3 protein (Piron et al., 1998). eIF4F was isolated from extracts of 293 cells transfected with plasmids expressing GST alone as a control, or GST–Mnk1, by immunoprecipitation of eIF4G. Isolated eIF4F complexes were then incubated with purified GST or GST–100k protein, and the effect on eIF4F composition was determined by immunoblot analysis (Figure 7B). Addition of GST alone had no effect on eIF4F, which retained PABP, Mnk1, eIF4A and eIF4E proteins (Figure 7B, lane 3). Addition of GST–100k protein did not alter the interaction of eIF4G with PABP or eIF4E, but was associated with a significant loss dissociation of GST–Mnk1 from eIF4G (∼10-fold; Figure 7B, lane 2) and a 2- to 4-fold decrease in eIF4A binding to eIF4G. GST–Mnk1 was not expressed in vivo in the eIF4F samples shown in lane 1, as a control. Interestingly, under these conditions there was less eIF4A retained and a greater amount of 100k protein bound to eIF4G. The potential difference between in vivo and in vitro results will be discussed later. Control studies (Figure 7B, lanes 4 and 5) showed equal levels of proteins in cell lysates prior to inhibition of eIF4F complexes. These data therefore demonstrate in vitro that 100k protein binds to the C-terminal region of eIF4G, which is coupled to the dissociation of Mnk1 protein from eIF4F complexes.
Role of L4-100k protein in viral late translational control
Previous studies showed that an Ad variant that contains a temperature-sensitive mutation in 100k protein (Ad5ts1) is impaired in translation of viral late mRNAs at restrictive temperature (Hayes et al., 1990). In those studies, shutoff of cellular protein synthesis by wt Ad was weak, leaving unresolved the effect of 100k protein on inhibition of cellular translation. We investigated whether the ts1-100k protein fails to facilitate displacement of Mnk1 from eIF4GI at restrictive temperature. 293 cells were transfected with a GST–Mnk1 expression vector, followed 18 h later by infection with wtAd or Ad5ts1. Cells were maintained at 33°C (non-restrictive) or 39.5°C (restrictive) for the ts1-100k mutation. Labeling of cells with [35S]methionine showed that at 33°C, wtAd and Ad5ts1 were indistinguishable in synthesis of viral late polypeptides and inhibition of cellular protein synthesis (Figure 8A). At restrictive temperature (39.5°C), Ad5ts1 did not inhibit cellular protein synthesis, and was reduced 3- to 5-fold in viral late mRNA translation. The association of Mnk1 with eIF4G was examined in wtAd and Ad5ts1 infected cells (Figure 8B). GST–Mnk1 was recovered from cells infected with wtAd or Ad5ts1 at 33 or 39.5°C, and association with endogenous eIF4G and eIF4E was determined by immunoblot analysis. At non-restrictive temperature, both wt and ts1-100k protein promoted an ∼10-fold loss of Mnk1 from eIF4G/eIF4E, whereas at restrictive temperature (39.5°C), ts1-100k protein did not. Immunoprecipitation of eIF4G from these same samples, followed by immunoblot analysis, demonstrated that eIF4G–eIF4E (Figure 8C) and eIF4G–eIF4A–PABP (not shown) remained associated regardless of temperature. eIF4E phosphorylation was examined in cells labeled in vivo with 32PO4 at 33 or 39.5°C. eIF4E was recovered and analyzed by SDS–PAGE and autoradiography, or by immunoblot analysis (Figure 8D). At restrictive temperature, Ad5ts1 did not block eIF4E phosphorylation, consistent with the inability to promote loss of Mnk1 from eIF4G and to inhibit cellular protein synthesis. At 33°C, Ad5ts1 largely, but not completely, blocked eIF4E phosphorylation, as observed previously (Zhang et al., 1994). The ts1-100k protein is probably slightly defective at non-restrictive temperature, accounting for the failure to block eIF4E phosphorylation fully. These results confirm the critical requirement for the Ad 100k protein in inhibition of cellular protein synthesis by targeted dissociation of Mnk1 from eIF4G, leading to dephosphorylation of eIF4E and inhibition of eIF4F-dependent cellular mRNA translation.

Fig. 8. The ts-1 100k protein mutant is translationally inactive. 293 cells were transfected with a GST–Mnkl expression plasmid followed 18 h later by infection with wtAd or Ad5ts1. Cells were maintained at non-restrictive temperature (33°C) for 49 h, or restrictive temperature (39.5°C) for 29 h of infection. (A) Cells were labeled with [35S]methionine for 60 min, lysates prepared and equal amounts of protein resolved by SDS–10%PAGE and fluorographed. (B) Mnk1 was recovered from equal amounts of cell lysates by glutathione–Sepharose chromatography, or (C) eIF4G was immunoprecipitated, proteins were resolved by SDS–10%PAGE and detected by immunoblotting with specific antisera as shown. (D) Cells were labeled in vivo with [32P]orthophosphate at 33 or 39.5°C, eIF4E was recovered by m7GTP chromatography from equal amounts of lysate, resolved by SDS–15%PAGE and autoradiographed (upper panel), or subjected to immunoblot analysis with specific antisera as described in the legend to Figure 3 (lower panel). The protein migrating immediately above eIF4G is non-specific and also reacts with this preimmune serum.
Discussion
We have described a novel mechanism by which Ad inhibits translation of cellular mRNAs during the late phase of virus infection. Previous studies demonstrated that Ad blocks the phosphorylation of eIF4E, which is associated with impaired or altered function of eIF4F and ablation of cellular mRNA translation (Huang and Schneider, 1991; Zhang et al., 1994). Here it is shown that Ad blocks binding of the eIF4E kinase Mnk1 to eIF4G. Association of Mnk1 with eIF4G facilitates phosphorylation of eIF4E in vivo (Pyronnet et al., 1999; Waskiewicz et al., 1999), and phosphorylation of eIF4E has been strongly associated with translation of most cellular mRNAs (reviewed in Gingras et al., 1999). Importantly, we provided direct evidence that translation of an eIF4F-dependent mRNA is impaired by dephosphorylation of eIF4E, whereas an Ad late tripartite leader mRNA is not (Figure 4A). Ad does not impair the kinase activity of Mnk1, which actually increases slightly during Ad late infection (Figure 1). Ad also does not alter the cytoplasmic abundance of translation initiation factors, or Mnk1 and PABP, during the late phase of infection (Figure 2 and data not shown). However, isolation and comparison of eIF4F complexes revealed that Mnk1 interaction with eIF4G was progressively disrupted during Ad late infection, consistent with inhibition of cellular mRNA translation (Figure 3A–C) and eIF4E phosphorylation (Figure 3D). Studies also showed that overexpression of either a phosphorylation-deficient eIF4E or the Mnk1 T2A2 kinase mutant is associated with impaired cellular mRNA translation, but not that of Ad late tripartite leader mRNAs (Figure 4A and B). A previous report (Waskiewicz et al., 1999) and Figure 4C provided evidence that the kinase-deficient T2A2 mutant impairs eIF4E phosphorylation in vivo. These experiments directly demonstrate that phosphorylation of eIF4E is important for translation of eIF4F-dependent (cellular) mRNAs but not Ad late mRNAs. Nevertheless, Ad late mRNAs do require some form of eIF4F (Thomas et al., 1992), which, as shown here, is likely to contain dephosphorylated eIF4E.
Dephosphorylated eIF4E is thought to weaken the interaction of the capped mRNA with eIF4F (Minich et al., 1994), due to a failure to generate a clamping salt bridge about the capped mRNA (Marcotrigiano et al., 1997). Thus, capped cellular mRNAs will not translate efficiently in the absence of strong binding to eIF4F. The dephosphorylation of eIF4E in the eIF4F complex has important consequences for translation of Ad late mRNAs. Ad late mRNAs are capped, as are cellular mRNAs, and they initiate ribosome binding through a cap-dependent process (reviewed in Schneider, 1996). However, Ad late mRNAs can translate by a shunting mechanism, in which 40S ribosome subunits are loaded in a cap-dependent manner, but are then translocated without scanning (or with limited scanning) to the downstream initiation codon (Yueh and Schneider, 1996, 2000). Given the low abundance of phosphorylated eIF4E during Ad late infection, it is likely that eIF4F that contains dephosphorylated eIF4E is the form used for translation of Ad late mRNAs. Thus, although Ad late mRNAs require eIF4F for translation, and involve cap recognition, a weak interaction of eIF4F with the capped end of Ad late mRNAs probably favors translation by ribosome shunting.
We found that the Ad late 100k protein is directly involved in inhibition of host cell protein synthesis. 100k protein interacts with eIF4G during Ad late infection (Figure 5) and when expressed independently in transfected cells (Figures 5B and 6B). In vitro, 100k protein bound the C-terminal fragment of eIF4G, the same domain bound by Mnk1 (Figure 7A). The interaction of 100k protein with eIF4G in vivo was coupled to loss of Mnk1 binding, and did not detectably impair eIF4G binding to eIF4E, eIF4A and PABP (Figures 3, 6 and 8). These data suggest that 100k binding to eIF4G is mechanistically linked to loss of Mnk1 from eIF4G. Addition of purified recombinant 100k protein to isolated eIF4F complexes resulted in the displacement of Mnk1 (Figure 7B), and Mnk1 and 100k proteins are likely to occupy a related binding site on the C-terminus of eIF4G (Figure 7A). Consequently, eviction of Mnk1 from eIF4G possibly occurs by competitive displacement. It is also possible that in vivo, 100k protein might bind more strongly to eIF4G that lacks Mnk1, thereby blocking rebinding of Mnk1 to eIF4G. This could explain the inhibition of cellular mRNA translation despite overexpression of wt Mnk1, as shown in Figure 4B. It is particularly interesting that in vitro, but not in vivo, 100k binding to eIF4G detectably reduced the association of eIF4A with eIF4G (Figure 7B). eIF4A binds eIF4G at middle and C-terminal eIF4G sites (Imataka and Sonenberg, 1997). 100k protein may therefore partially prevent the binding of eIF4A to the C-terminal site, which is more readily detected in vitro due to the lack of eIF4A recycling into eIF4G. The loss of eIF4A from eIF4F may also contribute to inhibition of cellular mRNA translation upon 100k protein interaction with eIF4G. A reduced level of eIF4A protein in eIF4F is consistent with the reduced requirement for RNA unwinding displayed by the Ad tripartite leader (Yueh and Schneider, 1996, 2000).
Our results can account for the observation that 100k protein binds in vivo to both viral and cellular mRNAs (Adam and Dreyfuss, 1987). It is likely that the eIF4F–100k complex can bind to both Ad and cellular mRNAs, although non-viral mRNAs would be deprived of functional eIF4F complexes and their translation would be blocked. However, Ad late mRNAs can continue to translate by ribosome shunting, although it is possible that both productively translated and non-translated mRNAs would be expected to be bound to 100k protein through eIF4G.
Materials and methods
Plasmids, antisera and cells
Plasmids pEBG-Mnkl, pGEX-Mnk1, pEBG-Mnkl (T2A2) (Waskiewicz et al., 1997) (kindly provided by J.A.Cooper), pcDNA-HA-eIF4GI (Imataka and Sonenberg, 1997) (a gift of N.Sonenberg), pCMV-Ad βgal and pCR3-βgal (Feigenblum and Schneider, 1996) were described previously. pGEX-3X and pGEX-4T-1 were from Amersham. Plasmids expressing N-terminal, central and C-terminal segments of eIF4G were constructed as follows. Plasmid pcDNA-HA-eIF4GI [157–613] was generated by digestion of plasmid pcDNA-HA-eIF4GI with BamHI, the fragment was repaired with Klenow enzyme, digested with EcoRI and subcloned into the pcDNA3-HA vector (Imataka and Sonenberg, 1997) at EcoRI–XhoI after repair with Klenow enzyme. pcDNA-HA-eIF4GI (565–1045) was constructed by cloning the EcoRI–XhoI fragment prepared by PCR amplification of the eIF4G sequence, using the primers 5′-CAACGAATTCGGAAGCCTCCAAACC-3′ and 5′-AAAACTCGAGTCAGAGCTGGTTGTTAG-3′, into an EcoRI–XhoI-digested pcDNA-HA. Plasmid pcDNA-HA-eIF4GI (1045–1560) was generated as follows. The C-terminal third of eIF4G was amplified by PCR using the primers 5′-CAACGAATTCTCTTTGCACCTGGAGG-3′ and 5′-CAACCTCGAGTCAGACTCCTCCTCTG-3′. The resultant PCR product was digested with EcoRI and XhoI and subcloned into EcoRI–XhoI-digested pcDNA3-HA. Plasmids expressing GST fusion N-terminal (157–626), middle (627–1045) and C-terminal (1045–1560) segments of eIF4GI were constructed as follows. An EcoRI–XhoI fragment was prepared by PCR amplification of the corresponding eIF4GI sequence using specific primers, and cloned into an EcoRI–XhoI-digested pGEX-4T-I vector (Amersham Pharmacia Biotech). The primers used to amplify each segment were: 5′-CCCCGAATTCATGTCTGGGGCCCGC-3′ and 5′-CAACCTCGAGTCAGAAGTCTGGGCC-3′ for the N-terminal; 5′-CAACGAATTCACTCCATCCTTTGCC-3′ and 5′-AAAACTCGAGTCAGAGCTGGTTGTTAG-3′ for the middle fragment; and 5′-CAACGAATTCCTCTTTGCACCTGGAG-3′ and 5′-CAACCTCGAGTC AGACTCCTCCTCTG-3′ for the C-terminal segment of eIF4GI.
Plasmid pFlag-100kDa was constructed in several steps by cloning different overlapping PCR amplification products of the Ad5-L4 transcription unit, using specific oligonucleotide primers, into vector pFLAG-CMV2 (Sigma). Specific details are available upon request. To construct pGEX-100kDa, the PCR-amplified 100 kDa ClaI–SalI fragment used to generate pFlag-100k was digested with ClaI–SalI, repaired with Klenow fragment and subcloned into pGEX-3X at a repaired EcoRI site. pcDNA-HA-4E, pcDNA-HA-4E(S209A) and pcDNA-HA-4E(S209A/T210A) were constructed by cloning an EcoRI–XhoI fragment prepared by PCR amplification of the human eIF4E cDNA, eIF4E(S209) and eIF4E(S209A/T210A) using primers 5′-CAACGAATTCAGATGGCGACTGTCG-3′ and 5′-CCGGCTCGAGTTAAACAACAAACC-3′ into EcoRI–XhoI-digested pcDNA-HA. Rabbit polyclonal antisera were raised against full-length recombinant human eIF4E protein, and against the N-terminal, central and C-terminal protein fragments of human eIF4GI. Mouse monoclonal anti-human PABP was provided by G.Dreyfuss (University of Pennsylvania), mouse monoclonal anti-rabbit eIF4A was provided by W.Merrick (Case Western Reserve), and rabbit anti-100k protein was provided by J.Flint (Princeton University). Other antisera were from commercial sources and include affinity-purified rabbit polyclonal antibody to GST (#Z-5; Santa Cruz Biotech.), horseradish peroxidase-conjugated donkey anti-rabbit or sheep anti-mouse secondary antibodies (Amersham), mouse anti-HA monoclonal antibody (12CA5; Boehringer Mannheim) and the enhanced chemiluminescence system (Amersham). 293 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% calf serum (Hyclone).
GST fusion protein purification
To obtain GST fusion proteins from mammalian cells, 293 cells were transiently transfected with plasmids, lysed in Triton lysis buffer (1% Triton X-100, 50 mM NaF, 10 mM HEPES pH 7.4, 2 mM EDTA, 2 mM sodium orthovanadate, 0.1% β-mercaptoethanol, 1 µg/ml aprotinin, 1 µg/ml leupeptin, l mM phenylmethylsulfonyl fluoride), and lysates clarified by centrifugation for 15 min at 14 000 g at 4°C. GST fusion proteins were purified with glutathione–Sepharose 4B (Pharmacia) for 1 h at 4°C, beads were collected by centrifugation and washed three times with Triton lysis buffer (for copurification experiments), or once with lysis buffer containing 0.5 M LiCl and three times with lysis buffer without LiCl (for in vitro kinase assays). GST fusion proteins were purified from Escherichia coli BL21 as described by Smith and Johnson (1988). Removal of the GST affinity tail was carried out by thrombin cleavage.
Immunoprecipitation and western immunoblot analysis
293 cells were lysed in Nonidet P-40 lysis buffer (0.5% NP-40, 50 mM HEPES pH 7.0, 250 mM NaCl, 2 mM EDTA, 2 mM sodium orthovanadate, 25 mM glycerophosphate, l µg/ml aprotinin, l µg/ml leupeptin, l mM phenylmethylsulfonyl fluoride). Lysates were clarified by centrifugation for 15 min at 14 000 g. Lysates were incubated for 2 h at 4°C with 2 µg/ml RNaseA and antiserum. Affinity chromatography on m7GTP–Sepharose was carried out as previously described (Huang and Schneider, 1991).
Metabolic labeling of cells
Cells were labeled with 50 µCi of [35S]methionine per milliliter (Easytag Express Protein Labeling Mix, Dupont/NEN) in DMEM without methionine for the times indicated. Cells were lysed in 0.5% NP-40 lysis buffer at 4°C, sonicated for 2× 30 s, and equal amounts of protein analyzed by SDS–PAGE and fluorography. Specific activity was determined by trichloroacetic acid precipitation and liquid scintillation. For 32PO4 labeling, cells were washed twice with phosphate-free DMEM (Gibco), incubated at 37°C for 30 min in this medium supplemented with 1% dialyzed fetal bovine serum, then labeled for 2 h with 200 µCi of [32P]orthophosphate (0.l mCi/ml; DuPont/NEN) in the same medium. Cells were lysed in 0.5% NP-40 buffer and eIF4E was purified by m7GTP–Sepharose chromatography.
Kinase assays
Wt Mnkl and catalytically inactive Mnkl (T2A2) were assayed for kinase activity in vitro by addition of 1 µg of purified eIF4E to a purified GST–bead mixture. Kinase–substrate reactions were carried out at 30°C for 20 min in kinase buffer [20 mM HEPES pH 7.4, 10 mM MgCl2, 10 mM β-glycerophosphate, 2 mM sodium orthovanadate, l mM dithiothreitol (DTT), 25 µM ATP, 10 µCi [γ-32P]ATP (3000 Ci/mmol)]. Products were resolved by SDS–PAGE and visualized by autoradiography.
In vitro protein studies
Purified recombinant proteins were mixed in buffer A (50 mM Tris–HCl pH 7.4, 100 mM NaCl, 1 mM DTT, 0.1% NP-40) for 1 h at 4°C, then 20 µl of 50% glutathione–Sepharose slurry were added and incubated for 1 h at 4°C. The mixtures were washed five times with 1 ml of buffer A. Bound proteins were analyzed by SDS–PAGE followed by immunoblotting with specific antibodies.
Acknowledgments
Acknowledgements
The authors thank N.Sonenberg for the HA-eIF4GI full-length clone, J.Cooper for GST–Mnk1 clones, and G.Dreyfuss, J.Flint and W.Merrick for antisera. This work was supported by NIH grant CA 42357 (to R.J.S.) and by a fellowship from Fundacion Ramon Areces, Spain (to R.C.).
References
- Adam S.A. and Dreyfuss,G. (1987) Adenovirus proteins associated with mRNA and hnRNA in infected Hela cells. J. Virol., 61, 3276–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau A.M. and Sonenberg,N. (1987) Involvement of the 24kd cap-binding protein in regulation of protein synthesis in mitosis. J. Biol. Chem., 262, 11134–11139. [PubMed] [Google Scholar]
- Dolph P.J., Racaniello,V., Villamarin,A., Palladino,F. and Schneider,R.J. (1988) The adenovirus tripartite leader eliminates the requirement for cap binding protein during translation initiation. J. Virol., 62, 2059–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolph P.J., Huang,J. and Schneider,R.J. (1990) Translation by the adenovirus tripartite leader: elements which determine independence from cap-binding protein complex. J. Virol., 64, 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R.F. (1996) Translational control during heat shock. In Hershey,J.W.B., Mathews,M.B. and Sonenberg,N. (eds), Translational Control During Heat Shock. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 271–294. [Google Scholar]
- Feigenblum D. and Schneider,R.J. (1993) Modification of eukaryotic initiation factor 4F during infection by influenza virus. J. Virol., 67, 3027–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenblum D. and Schneider,R.J. (1996) Cap-binding protein (eukaryotic initiation factor 4E) and 4E-inactivating protein BP-1 independently regulate cap-dependent translation. Mol. Cell. Biol., 16, 5450–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenblum D., Walker,R. and Schneider,R.J. (1998) Adenovirus induction of an interferon-regulatory factor during entry into the late phase of infection. J. Virol., 72, 9257–9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn A. and Proud,C.G. (1995) Serine 209, not serine 53, is the major site of phosphorylation in initiation factor eIF-4E in serum-treated chinese hamster ovary cells J. Biol. Chem., 270, 21684–21688. [DOI] [PubMed] [Google Scholar]
- Fukunaga R. and Hunter,T. (1997) MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J., 16, 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A.-C. and Sonenberg,N. (1997) Adenovirus infection inactivates the translational inhibitors 4E-BP1 and 4E-BP2. Virology, 237, 182–186. [DOI] [PubMed] [Google Scholar]
- Gingras A.-C., Raught,B. and Sonenberg,N. (1999) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem., 68, 913–963. [DOI] [PubMed] [Google Scholar]
- Gradi A., Imataka,H., Svitkin,Y.V., Rom,E., Raught,B., Morino,S. and Sonenberg,N. (1998) A novel functional human eukaryotic translation initiation factor 4G. Mol. Cell. Biol., 18, 334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes B.W., Telling,G.C., Myat,M.M., Williams,J.F. and Flint,S.J. (1990) The adenovirus L4 100 kilodalton protein is necessary for efficient translation of viral late mRNA species. J. Virol., 64, 2732–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. and Schneider,R.J. (1991) Adenovirus inhibition of cellular protein synthesis involves inactivation of cap binding protein. Cell, 65, 271–280. [DOI] [PubMed] [Google Scholar]
- Imataka H. and Sonenberg,N. (1997) Human eukaryotic translation initiation factor 4G (eIF4G) possesses two separate and independent binding sites for eIF4A. Mol. Cell. Biol., 17, 6940–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imataka H., Gradi,A. and Sonenberg,N. (1998) A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J., 17, 7480–7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S.K., Krausslich,H.G., Nicklin,M.J.H., Duke,G.M., Palmenberg,A.C. and Wimmer,E. (1988) A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol., 62, 2636–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi B. et al. (1995) Phosphorylation of eukaryotic protein synthesis initiation factor 4E at Ser-209. J. Biol. Chem., 270, 14597–14603. [DOI] [PubMed] [Google Scholar]
- Logan J. and Shenk,T. (1984) Adenovirus tripartite leader sequence enhances translation of mRNAs late after infection. Proc. Natl Acad. Sci. USA, 81, 3655–3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotrigiano J., Gingras,A.-C., Sonenberg,N. and Burley,S.K. (1997) Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell, 89, 951–961. [DOI] [PubMed] [Google Scholar]
- Minich W.B., Balasta,M.L., Goss,D.J. and Rhoads,R.E. (1994) Chromatographic resolution of in vivo phosphorylated and nonphosphorylated eukaryotic translation initiation factor eIF-4E: increased cap affinity of the phosphorylated form. Proc. Natl Acad. Sci. USA, 91, 7668–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piron M., Vende,P., Cohen,J. and Poncet,D. (1998) Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J., 17, 5811–5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyronnet S., Imataka,H., Gingras,A.-C., Fukunaga,R., Hunter,T. and Sonenberg,N. (1999) Human eukaryotic translation initiation factor 4G (eIF4G) recruits Mnk1 to phosphorylate eIF4E. EMBO J., 18, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley D. and Flint,S.J. (1993) RNA-binding properties of a translational activator, the adenovirus L4 100-kilodalton protein. J. Virol., 67, 3586–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R.J. (1996) Adenovirus and vaccinia virus translational control. In Hershey,J.W.B., Mathews,M.B. and Sonenberg,N. (eds), Adenovirus and Vaccinia Virus Translational Control. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 575–605. [Google Scholar]
- Smith D.B. and Johnson,K.S. (1988) Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene, 67, 31–40. [DOI] [PubMed] [Google Scholar]
- Tarun S.Z., Wells,S.E. and Sachs,A.B. (1997) Translation initiation factor eIF-4G mediates in vitro polyA tail dependent translation. Proc. Natl Acad. Sci. USA, 94, 9046–9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A.M., Scheper,G.C., Kleijn,M., DeBoer,M. and Voorma,H.O. (1992) Dependence of the adenovirus tripartite leader on the p220 subunit of eukaryotic initiation factor 4F during in vitro translation. Eur. J. Biochem., 207, 471–477. [DOI] [PubMed] [Google Scholar]
- Wang X., Flynn,A., Waskiewicz,A.J., Webb,B.L.J., Vries,R.G., Baines,I.A., Cooper,J.A. and Proud,C.G. (1998) The phosphorylation of eukaryotic initiation factor eIF-4E in response to phorbol esters, cell stresses and cytokines is mediated by distinct MAP kinase pathways. J. Biol. Chem., 273, 9373–9377. [DOI] [PubMed] [Google Scholar]
- Waskiewicz A.J., Flynn,A., Proud,C.G. and Cooper,J.A. (1997) Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J., 16, 1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waskiewicz A.J., Johnson,J.C., Penn,B., Mahalingham,M., Kimball,S.R. and Cooper,J.A. (1999) Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol. Cell. Biol., 19, 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells S.E., Hillner,P.E., Vale,R.D. and Sachs,A.B. (1998) Circulariz ation of mRNA by eukaryotic translation initiation factors. Mol. Cell, 2, 135–140. [DOI] [PubMed] [Google Scholar]
- Yueh A. and Schneider,R.J. (1996) Selective translation by ribosome jumping in adenovirus infected and heat shocked cells. Genes Dev., 10, 1557–1567. [DOI] [PubMed] [Google Scholar]
- Yueh A. and Schneider,R.J. (2000) Translation by ribosome shunting on adenovirus and Hsp70 mRNas facilitated by complementarity to 18S rRNA. Genes Dev., 14, 414–421. [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Feigenblum,D. and Schneider,R.J. (1994) A late adenovirus factor induces eIF-4E dephosphorylation and inhibition of cell protein synthesis. J. Virol., 68, 7040–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]