

Abstract
The aim of the present study was to identify the relationship between progressive neurobehavioural decline and phospho-tau levels (p-tau181) in the cerebrospinal fluid (CSF) and the brain in transgenic rats expressing human truncated tau protein. Behavioural analyses, as quantified using the NeuroScale scoring method, revealed that the transgenic rats fell into two main groups based on the baseline behavioural functioning: (1) mild neurobehavioural impairment (MNI, score 3.3–26) and (2) severe neurobehavioural impairment (SNI, score 36–44). SNI transgenic rats showed a significant increase in brain sarkosyl insoluble p-tau181 when compared to their MNI counterparts. In order to determine whether CSF phosphotau reflects the behavioural decline and increase in sarkosyl insoluble tau in the brain, p-tau181 was measured in the CSF in a longitudinal study. The study showed a significant increase in CSF p-tau181 during the progression of the disease from MNI to SNI. Moreover, increased levels of p-tau181 in CSF correlated with an increase in the sarkosyl insoluble p-tau181 levels in the brain. The increase in the CSF level of p-tau181 during progressive behavioural decline suggests that it may represent a useful surrogate biomarker for preclinical drug development and a potential surrogate endpoint for clinical trials of disease-modifying therapy for Alzheimer's disease and related human tauopathies.
Keywords: Cerebrospinal fluid, Truncated tau, Rat model of tauopathy, CSF biomarker, Behavioural decline
Introduction
Alzheimer disease (AD) is the most common form of dementia, accounting for 50–70% of all cases of clinically diagnosed senile dementia. The number of AD cases is rapidly increasing as a consequence of increasing life span and a lack of effective treatment. Therefore, a lot of attention is currently being directed at the development of approaches that counteract the fundamental pathological processes of the disease. It is indisputable that therapeutic intervention is most effective and beneficial when administered early on in the progression of disease. Thus, there is an unmet need for diagnostic tools that accurately identify incipient AD [8, 10]. Tremendous efforts have been made in the past few years to identify biomarkers for preclinical AD with the focus mainly on cerebrospinal fluid (CSF). Several CSF biomarkers have been extensively studied, including amyloid β1–42 (Ab42) [7, 20], amyloid β1–40 (Ab40), amyloid β1·38 (Ab38) or Aβ-oligomers [10], total tau [19, 20], tau protein phosphorylated at several epitopes, such as threonine 181 [39], threonine 181 and 231 [6], threonine 231 [32], threonine 231 and 235, serine 199 [29], serine 396 and 404 [26] and isoprostane [36, 37]. From the list of candidate CSF biomarkers, three of them have been suggested to fulfil the criteria required by the working group on molecular and biochemical markers of AD: total tau (t-tau), amyloid β1–42 (Ab42) and phosphorylated tau protein (p-tau) [19]. Several independent studies have shown that the levels of CSF Aβ42, t-tau and p-tau are altered in AD and could predict the progression of mild cognitive impairment (MCI) to AD [9, 17, 18, 23, 24, 35, 40]. However, it has been shown that CSF levels of total tau, p-tau and Aβ42 considerably overlap between diseased and control cases [26, 27]. Disease heterogeneity is considered to be the major cause of the overlap in CSF biomarkers [28].
Besides their potential as tools in clinical diagnosis and dementia prognosis, CSF biomarkers are also useful in identifying and monitoring drug efficacy. The question whether CSF p-tau181 could be used as a surrogate marker for drug efficacy in clinical trials yielded contradictory results. Bouwman et al. [13] suggested that p-tau181 was not useful in a clinical setting because it was insensitive to disease progression, while Andersson et al. [1] reported its increase during cognitive decline and progression to dementia. Moreover, a significant reduction in CSF p-tau181 was found in AD patients after 1 year of treatment with memantine [14]. Furthermore, little is known about the potential of CSF p-tau181 in preclinical drug development. Thus, there is a need for studies investigating the CSF p-tau biomarker in transgenic rodent models of human tauopathy. Recently, we have presented the first rat model of human tauopathies expressing human truncated tau151–391 which derived from sporadic AD. This model reconstitutes the complete tau cascade of neurofibrillary degeneration, consisting of tau hyperphosphorylation, the formation of argyrophilic tangles and sarkosyl-insoluble tau [34, 43]. The larger size of transgenic rats compared to mice permits serial sampling of the CSF and monitoring of the longitudinal changes in CSF biomarkers. The aim of the present study was to investigate the relationship between progressive neurobehavioural decline and the CSF and brain p-tau181 levels in the transgenic rat model of tauopathy.
Materials and methods
Animals
The generation of a transgenic rat model of tauopathy expressing human truncated tau151–391 was described in details elsewhere [43]. The transgene expression levels, the load of neurofibrillary tangles and life span were previously determined. The median survival time for the transgenic rats (line SHR72) was 222.5 days (SD = 24.48) [34]. In the present study, heterozygous transgenic rats and non-transgenic SHR age-matched controls were used. All animals were housed under standard laboratory conditions with free access to water and food and were kept under diurnal lighting conditions (12 h light/dark cycles with light starting at 7:00 a.m.). All experiments on animals were carried out according to the institutional animal care guidelines conforming to international standards and were approved by the State Veterinary and Food Committee of Slovak Republic and by Ethics Committee of Institute of Neuroimmunology. Efforts were made to minimise the number of animals utilised and to limit discomfort, pain or any other suffering of the experimental animals used.
Neurobehavioural evaluation
Neurobehavioural response in the rats was assessed using the NeuroScale—a complex battery of behavioural tests especially designed for transgenic rats expressing human truncated tau protein. The NeuroScale is composed of sensorimotor (beam walking test), neuromuscular (prehensile traction test) and neurological tasks (placing, righting, postural, pinna, startle and hindlimb escape extension reflex) enriched with basic observational assessment [33].
The general observation involved assessment of posture and limb functions; the neurological examination included the basic reflex response, all graded on a 1-point scale (normal response 0; delayed or incomplete response 1 point). Special attention was paid to the assessment of the hindlimb escape extension reflex, which was graded on a 3-point scale (normal response 0–1; deficit 2–3 points).
For the beam walking test, three sorts of traversing segments were used (3 × 3 cm, 4 × 2 cm and a round beam of 3.5 cm diameter). The maximum number of points to be awarded was 10 (5 points for latency + 5 points for hindlimb slips on one beam type). The lower the score obtained from the beam walking test, the better was the sensorimotor–coordination ability of the tested animal. The highest achievable point total in this task was 30.
For the prehensile traction test, an animal was allowed to grasp with its forepaws a horizontal steel wire (3 mm in diameter) suspended 76 cm above a padded surface. Latency to fall from the wire was measured. The maximal number of points awardable was 5, reflecting serious impairment of neuromuscular functionality and muscular weakness. The longer latency to fall, the lower score obtained from the task, reflecting forelimb muscular strength and agility of the tested animal.
The NeuroScale score was calculated from the scores obtained in the individual tests. A maximum total score of 49 points was reached by summing up the contribution of the observational assessment, the neurological examination, the three series of beam walking tests and the prehensile traction test. The higher NeuroScale score, the more severe neurobehavioural impairment observed.
Collection of CSF from the cisterna magna
Cerebrospinal fluid was collected from the cisterna magna. Animals were anesthetized with ketamine–xylazine mixture (80, 24 mg/kg), fixed in a head holder and a midline incision in the skin was made up to the head area to permit easy access to the cisterna magna. Approximately 100 μl of CSF was collected from each animal. All CSF samples were kept on ice for a maximum of 5 min before centrifuged for 3 min at 5,000g at 4°C and stored at −70°C till used. All CSF samples were collected between 9:00 and 12:00 a.m.
Phospho-tau threonine 181 ELISA
A sandwich ELISA assay (Innotest Phospho-Tau 181P, Innogenetics, Belgium) was used to detect tau phosphorylated at threonine 181 (p-tau181) in CSF. 75 μl of the CSF sample was added to each well. The detection limit for this assay is 15.6 pg/ml. In this assay, tau is captured with human-specific backbone-directed antibody HT7 recognising human tau 159–163. The captured tau is then detected by AT270, which is specific for phospho-threonine 181 [39].
Tissue preparation
Under deep anaesthesia with xylazine and ketamine, the rats (n = 10) were perfused transcardially with phosphate buffered saline (PBS 0.01 M, pH 7.4) followed by 4% paraformaldehyde (PFA). After transcardial perfusion, brains were immediately removed and postfixed in 4% PFA, embedded in paraffin and serially cut into 8-μm-thick sections.
Immunohistochemistry
Immunohistochemical staining was performed on paraffin sections after deparaffinization. To analyse cortical changes in the motor cortex, sagittal sections of the brain were prepared. Coronal sections at the level of facial and gigantoreticular nucleus were used for description of pathological lesions in the brainstem. Brain sections were incubated for 20 min at room temperature in 0.01 M PBS, pH 7.4, containing 0.3% Triton X-100 and 1% H2O2, followed by a 30-min incubation in blocking solution (0.01 M PBS, containing 0.3% Triton X-100, 1% horse serum). This was followed by overnight incubation at 4°C in primary antibody AT8 (Innogenetics, Belgium) or CD68 (Serotec, UK) in blocking solution. After washing, the sections were incubated in biotinylated secondary antibody (Vectastain; Vector Laboratories, CA). The reaction product was visualised using avidin–biotin and Vector VIP as the chromogen (Vector Laboratories). Paraffin sections were counterstained with methyl green.
Luxol fast blue staining
Paraffin-embedded sections were stained with Luxol fast blue/cresyl violet as previously described [31]. Sections were stained in 0.1% Luxol fast blue solution for 2 h at 60°C and then washed in 95% ethanol and differentiated in a jar of 0.05% lithium carbonate followed by 70% ethanol. Afterwards, sections were counterstained with 0.1% cresyl violet in 1% acetic acid for 10 min, washed in distilled water, differentiated in 70% ethanol until only nuclei and Nissl substance were purple. Finally, these sections were examined using an Olympus BX51 light microscope.
Extraction of sarkosyl insoluble tau
Sarkosyl insoluble tau was isolated from brain tissues of transgenic rats as described previously [21]. Approximately, 0.6 g of brain tissue was homogenised in 10 volumes of 10 mM Tris (Applichem, Germany), pH 7.4, 0.8 M NaCl (Calbiochem, UK), 1 mM EGTA (Sigma, Germany) and 10% sucrose (Sigma, Germany) and centrifuged at 27,200g for 20 min. Then, 3 ml of cleared supernatant was adjusted to 1% (w/v) N-lauroylsarcosine (Sigma, Germany) and incubated 1 h at room temperature. After the incubation, the extract was spun at 12,3000g for 1 h at RT. The pellet was resuspended in 60 μl of 10 mM sodium phosphate, pH 7.4 (Roth, Germany), 150 mM NaCl (Calbiochem, UK) and analysed by Western blots.
Western blots
Sarkosyl soluble and insoluble tau proteins were separated by electrophoresis on 10% SDS-PAGE gel and then electrotransferred to nitrocellulose membrane (Schleicher and Schuell, Germany) in 10 mM N-cyclohexyl-3-aminopropanesulphonic acid (CAPS, pH 11, Roth, Germany). Membranes were stained with Ponceau S (Sigma, Germany) to verify the uniform transfer of the proteins, blocked in 5% non-fat dried milk (Roth, Germany) in 10 mM Tris–HCl, pH 7.4 (AppliChem, Germany), 0.15 M NaCl (Calbiochem, UK), 0.1% Tween 20 (Roth, Germany) and then incubated with monoclonal antibody AT270 recognising p-tau181 (Thermo Fisher Scientific, IL, USA), followed by horse-radish peroxidase-conjugated polyclonal goat anti-mouse IgG (Dako, Denmark). The blots were developed using Super-Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). The chemiluminescence signals from Western blots were digitized using LAS3000 CCD imaging system (Fujifilm, Japan), and relative quantification of the Western blots was performed by AIDA Biopackage (Advanced Image Data Analyser software; Raytest, Germany).
Statistical analysis
Statistical analysis was carried out using Prism statistical software package (GraphPad Software, CA, USA). Distribution of p-tau181 values differed from normal Gaussian distribution as revealed by the Shapiro–Wilk normality test. Therefore, non-parametric statistical methods were used to avoid the bias of non-normally distributed data and unequal variances. For the proteomic study, non-parametric Spearman's rank correlation was used. To compare two groups, the Mann–Whitney U test or unpaired t test was applied. The Wilcoxon's matched paired test was applied to compare the baseline and follow-up values of p-tau181 and scores obtained from neurobehavioural assessment. The results are presented as mean ± SEM unless otherwise specified. Differences were considered significant at the level of P<0.05.
Results
Neuropathological hallmarks of the transgenic rat model of tauopathy
The transgenic rats expressing human truncated tau protein displayed several features of human tauopathies, including tau hyperphosphorylation, development of neurofibrillary lesions and axonal degeneration. Immunohistochemical examination of transgenic rats revealed somatodendritic accumulation of abnormally hyperphosphorylated tau protein in cortical pyramidal neurons (Fig. 1a). Importantly, human truncated tau protein induced the formation of neurofibrillary tangles (NFTs) in the spinal cord and the brain stem of transgenic rats. Neurofibrillary tangles were morphologically similar to human NFTs and were immunoreactive with p-tau-dependent monoclonal antibody AT8 (Fig. 1c). Age-matched non-transgenic rat brains did not show either hyperphosphorylated tau in pyramidal neurons (Fig. 1b) or neurofibrillary tangles (Fig. 1d). Another pathological hallmark, axonal damage, appeared in the white matter of the spinal cord and brain stem. The progress and scale of axonal vacuolization were clearly shown by Luxol fast blue staining (Fig. 1e). Simultaneously, a slight decrease in myelin staining was observed in the same brain region of transgenic animals when compared to age-matched controls (Fig. 1f). The axonal vacuoles were not an artefact of tissue processing, because CD68-immunoreactive macrophages were closely associated with these lesions (Fig. 1g). Finally, extensive axonal damage was associated with the presence of central chromatolytic neurons that were characterised by displacement of the Nissl bodies toward the periphery of the nerve cell body (Fig. 1h). The changes consisted of central chromatolysis, seen predominantly in the brainstem, especially in the nuclei of the reticular formation.
Fig. 1.

Neuropathological features of transgenic rats expressing human misfolded truncated tau. a Immunohistochemical staining of the hyperphosphorylated tau protein in the brain of a 7-month-old transgenic rat showed massive AT8 immunoreactivity in cortical pyramidal neurons. b Hyperphosphorylated tau was absent in the brain of age-matched control rats. c Neurofibrillary lesions in the brainstem immunolabeled with phospho-dependent mAb AT8. d The brain of the age-matched control rat cortex lacking the intraneuronal fibrillar structures. e In transgenic rats, affected tissue shows a number of vacuoles (arrow), distributed in the reticular formation of the brainstem, which are absent in age-matched control rats (f), when stained with Luxol fast blue—cresyl violet. Higher magnification of the vacuole located in the bundle of myelinated nerve fibres is shown in the inset figure. g Axonal vacuoles and activated macrophages (brown coloured) in the tectospinal tract of transgenic rats. Numerous CD68-immunoreactive activated macrophages are distributed in the lesions. Some of the CD68 immunopositive macrophages are located in the vicinity of axonal vacuoles (arrow and the inset figure). h Higher magnification clearly shows central chromatolysis occurring in the brainstem neurons (arrow). Paraffin sections; scale bars 50 μm
Identification of two prominent stages of the neurobehavioural impairment
The complex neurobehavioural assessment—NeuroScale was performed to evaluate the neurobehavioural response of transgenic rats (n = 44). On the basis of the level of neurobehavioural decline we divided transgenic rats into two subgroups. The first subgroup of transgenic rats (n = 34) with lower NeuroScale score (range 3.3–26 points) represented animals with mild neurobehavioural impairment (MNI). The stage of MNI starts at 3 months of age and can last about 3 months. The second subgroup of transgenic rats (n = 10) displaying score above 30 points (range 36–44 points) involved animals suffering from severe neurobehavioural impairment (SNI). The stage of SNI lasts several days. Neurobehavioural scores of transgenic rats from two different subgroups are described in detail in Table 1.
Table 1.
Two clinical stages of neurobehavioural impairment of transgenic rats expressing human truncated tau
| Cross-sectional study | MNI, mean ± SD (range) | SNI, mean ± SD (range) | P level |
|---|---|---|---|
| n | 34 | 10 | |
| Age (months) | 183.3 ± 3.3 (180–187) | 183.3 ± 3.3 (180–187) | |
| NeuroScale (score) | 11.1 ± 4.94 (3.3–26) | 42 ± 2.75 (36–44) | <0.001 |
| Beam walking 3 × 3 cm (score) | 1 ± 1.15 (0–5) | 10 ± 0 (10–10) | <0.001 |
| Beam walking 4 × 2 cm (score) | 1 ± 1.4 (0–6) | 10 ± 0 (10–10) | <0.001 |
| Beam walking 3.5 cm (score) | 4.5 ± 2.75 (0–10) | 10 ± 0 (10–10) | <0.001 |
| Hind limb reflex (score) | 1.8 ± 0.47 (1.1–3) | 2.36 ± 0.59 (1.33–3) | <0.05 |
| Prehensile traction (score) | 2.5 ± 1.18 (0–4) | 4.4 ± 1.26 (1–5) | <0.001 |
| Neurological response (score) | 0 | 6 ± 0 (6–6) | <0.001 |
MNI mild neurobehavioural impairment, SNI severe neurobehavioural impairment
Sarkosyl soluble and insoluble rat brain p-tau181 correlate with neurobehavioural impairment
To analyse levels of p-tau181 in brain tissue, we carried out Western blot analysis of sarkosyl soluble and insoluble tau fractions, from transgenic rats suffering from MNI or SNI stage of clinical symptoms. Monoclonal antibody AT270 recognising tau phosphorylated at threonine 181 revealed similar levels of soluble p-tau181 in both groups (MNI and SNI) of transgenic rats (Fig. 2a, b). However, the staining pattern was significantly different between these two groups of transgenic rats. The soluble p-tau181 of SNI animals showed increased levels of highly phosphorylated tau species (upper band of approximately 35 kDa) and decreased levels of less phosphorylated tau species (lower bands). Furthermore, the analysis of sarkosyl-insoluble p-tau181 revealed significant increase in the insoluble tau in SNI transgenic rats when compared to their MNI counterparts (Fig. 2c, d).
Fig. 2.

Comparison of brain p-tau181 levels in transgenic rats suffering from mild neurobehavioural impairment (MNI n = 5) and severe neurobehavioural impairment (SNI n = 5). Sarkosyl soluble (a, b) and insoluble (c, d) phospho-tau isolated from the brainstem of transgenic rats was examined with tau antibody AT270 recognising p-tau181. Sarkosyl soluble brain p-tau181 levels do not show significant differences between MNI and SNI rats; however, the staining pattern differs between these two groups. In comparison with MNI, SNI transgenic rats showed increased levels of highly phosphorylated soluble tau species (the upper band marked with a dotted line). Interestingly, sarkosyl-insoluble brain p-tau181 levels were remarkably increased in the SNI stage (***P>0.001; unpaired t test)
Longitudinal CSF p-tau181 study in the transgenic rat model of tauopathy
Longitudinal study was designed to compare the CSF p-tau181 levels in transgenic rats (n = 8) at the baseline and follow-up. The study was initiated with transgenic rat males at the age of 186 days (n = 10). The animals underwent neurobehavioural examination to measure the intensity of complex neurobehavioural impairment quantitatively using the NeuroScale battery of behavioural tests. CSF samples were collected only from subjects suffering from mild neurobehavioural impairment (NeuroScale score range 7.8–21.1 points, n = 8). After 3–7 weeks, transgenic rats reached the SNI stage (NeuroScale score 45 points) characterised by pronounced impairment of several reflexes (hind limb escape extension reflex, placing and righting reflexes), bradykinesia and paraparesis. At this stage, the CSF samples were collected again and p-tau181 levels were determined. Statistical analysis showed a significant (P<0.01; Fig. 3a) increase in CSF p-tau181 at follow-up (SNI 140.8 ± 26.98) compared with the baseline (MNI 62.3 ± 12.38 pg/ml). The rate of increase over time in CSF p-tau181 levels was presented for each individual transgenic rat (Fig. 3b). The NeuroScale scores, as well as CSF p-tau181 levels at baseline and follow-up are summarised in Table 2.
Fig. 3.

Longitudinal study of the CSF p-tau181 levels in transgenic rats expressing human truncated tau protein. a Longitudinal study shows statistically significant increase in p-tau181 levels during the follow-up (**P>0.01; Wilcoxon matched paired test). Data are presented as follows: a line across the box indicates the median; box represents the interquartile range which contains 50% of the cases, whiskers indicate the range (minimal and maximal values) of data distribution. b The rate of change over time in CSF p-tau181 levels is presented for each individual transgenic rat. MNI mild neurobehavioural impairment (n = 8), SNI severe neurobehavioural impairment (n = 8)
Table 2.
Number of subjects per group, age, individual neurobehavioural score, complex NeuroScale score and p-tau181 levels in the longitudinal study
| Longitudinal study | MNI, mean ± SD (range) | SNI, mean ± SD (range) | P level |
|---|---|---|---|
| n | 8 | 8 | |
| Age (days) | 186 | 223.3 ± 11 (206–232) | |
| NeuroScale (score) | 12.8 ± 5.38 (7.8–21.1) | 45 ± 0 (45–45) | <0.05 |
| Beam walking 3 × 3 cm (score) | 2 ± 0.75 (1–3) | 10 ± 0 (10–10) | <0.05 |
| Beam walking 4 × 2 cm (score) | 2 ± 2 (0–5) | 10 ± 0 (10–10) | <0.05 |
| Beam walking 3.5 cm (score) | 3.7 ± 2.65 (0–7) | 10 ± 0 (10–10) | <0.05 |
| Hind limb reflex (score) | 2 ± 0.48 (1.33–3) | 3 ± 0 (3–3) | <0.05 |
| Prehensile traction (score) | 3.1 ± 0.99 (2–4) | 5 ± 0 (5–5) | <0.05 |
| Neurological response (score) | 0 | 7 ± 0 (7–7) | <0.05 |
| P-tau181 (pg/ml) | 62.3 ± 12.38 (50.95–83.41) | 140.8 ± 26.98 (119.7–203.5) | <0.01 |
MNI mild neurobehavioural impairment, SNI severe neurobehavioural impairment
The levels of CSF p-tau181 correlate with the brain sarkosyl-insoluble p-tau181
For the proteomic analyses of brain soluble and insoluble p- tau181 and CSF p- tau181 levels, five transgenic rats showing early symptoms (age 177,8 ± 1,09; NeuroScale <26 points) and five transgenic rats showing mature neurobehavioural impairment (age 209,8 ± 3,83; NeuroScale score >40 points) were used. Transgenic rats were behaviourally tested and afterwards the CSF from cisterna magna was collected. Finally, animals were sacrificed, and the brainstem was dissected and used for proteomic analyses. Statistical analysis showed a significant correlation between CSF p-tau181 levels and the sarkosyl-insoluble brain p-tau181 levels (P<0.05, r = 0.76) (Fig. 4).
Fig. 4.
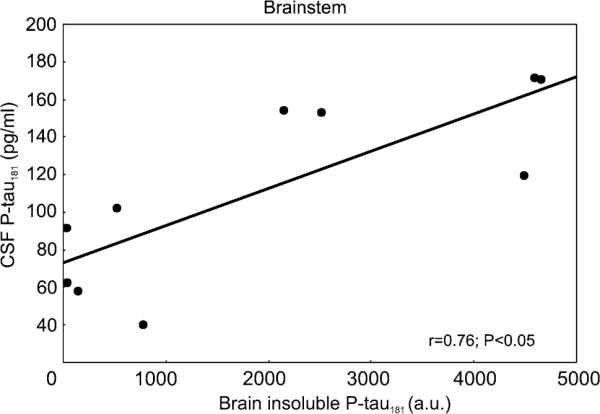
CSF p-tau181 levels (pg/ml) plotted against brain p-tau181 levels (a.u.) of transgenic rats (n = 10). Significant correlation between p-tau181 levels in the brain and in the CSF was observed. The Spearman's correlation coefficient (r) and corresponding P value are indicated
Discussion
The development of disease-modifying drugs emphasizes the need for improved diagnostic accuracy. Because biochemical changes in the brain are reflected in the composition of CSF, this fluid could be an appropriate source of biochemical markers for AD [19]. Indeed, diagnostic markers in CSF have become a fast growing research area [5]. During the last decade, several CSF biomarkers have been evaluated in numerous studies [6, 26, 28, 29, 32, 36, 37, 39, 41]. From the list of candidate CSF biomarkers, three have been suggested to fulfil the criteria required by the consensus group: t-tau, Aβ42 and p-tau [19]. Numerous studies have demonstrated elevated CSF concentrations of t-tau and reduced Aβ42 levels in AD [10, 19]. However, it has been suggested that elevated CSF p-tau is a more specific marker for AD than t-tau and Aβ42 [9]. The CSF p-tau levels seem to increase the specificity, because normal levels are found in other dementias and in cerebrovascular disease [4, 19, 26].
The issue whether CSF p-tau181 correlates with clinical symptoms in animal models for human tauopathies has not been addressed until now. To date most of the available information on the proteomic and genetic basis of AD and related human tauopathies has been generated in mice models. Although transgenic mouse models have been of great use, their potential is limited because of their small brain size and the difficulty in obtaining sufficient amounts of clean CSF. In comparison to mice, the size of a rat's brain is sufficient for intracerebroventricular injections and the relatively easy collection of CSF that allows the exploration of the CSF proteome. Recently, we have reported that transgenic rats expressing a non-mutated truncated tau species derived from sporadic AD faithfully replicate several crucial features of human tauopathies including tau hyperphosphorylation, the formation of NFTs composed of sarkosyl-insoluble tau complexes and axonal degeneration [34, 43]. These pathological changes led to the progressive decline of sensorimotor functions and the impairment of several reflexes [25, 33]. Moreover, the rat model demonstrates high reproducibility under defined conditions and limited variability caused by various genetic and medical backgrounds.
In this study, we report a correlation between the NeuroScale score reflecting progressive neurobehavioural decline and CSF p-tau181 levels. This is in line with findings from AD studies, where a significant correlation between the level of p-tau181 and the severity of dementia symptoms was identified [41]. These findings strongly suggest that p-tau181 should be a valuable marker for the severity and abundance of symptoms in the rat model.
Several studies described longitudinal changes of Aβ42, t-tau and p-tau levels in CSF in AD patients [22, 30], whereas others showed no significant changes of these CSF biomarkers over time [2, 3, 16, 38, 42]. Recently, it has been suggested that repeated assessment of CSF p-tau181 biomarker is not useful in a clinical setting because it is insensitive to disease progression over a 2-year period [12], while this feature was valued by others as an advantage for drug development [11]. The reason why CSF biomarkers do not seem to reflect disease progression over time is not known. One possibility is that brain-derived proteins are diluted in the CSF compartment [15]. Another explanation is that older non-demented individuals (over 65) tend to have higher p-tau levels than those younger, and so the elevation in this biomarker with the disease progression is masked by using older non-demented individuals as a control [13]. In contrast to the aforementioned findings, Andersson et al. [1] demonstrated that initially non-demented patients with severely impaired episodic memory (SIM) at baseline, who declined cognitively over time and progress to dementia, showed significantly increasing CSF p-tau181 levels during follow-up. The authors concluded that p-tau181 may be useful as a longitudinal marker of the neurodegenerative process. Despite these inconclusive findings, clinical trials using the CSF p-tau181 levels as outcome parameters have already been reported [14].
The present study has revealed that transgenic rats with mild neurobehavioural impairment at the baseline, which progress to severe neurobehavioural impairment, showed significantly elevated CSF p-tau181 levels during follow-up. Despite a limited sample size, our study is the first to describe the longitudinal changes of abnormally phosphorylated tau in the CSF in a transgenic animal model of human tauopathy. Moreover, increased levels of CSF p-tau181 correlated well with a significant elevation of the sarkosyl-insoluble p-tau181 levels in the brain. Our results indicate that the phospho-tau concentrations in the CSF could be used as a valid surrogate endpoint in therapeutic trials of AD drugs directly targeting tau protein. Thus, the CSF phospho-tau can be utilised as secondary outcomes indirectly measuring Alzheimer's disease pathology.
In conclusion, this study demonstrates for the first time (1) the presence of abnormally phosphorylated tau in CSF of a rodent model of human tauopathy (2) the correlation of CSF p-tau181 levels with sarkosyl-insoluble p-tau181 in the brain and (3) the correlation of CSF p-tau181 level with neurobehavioural impairment. These findings suggest that CSF p-tau181 has preclinical potential for drug development as a surrogate marker in preclinical trials with disease-modifying drug candidates.
Acknowledgments
This work was supported by Axon Neuroscience and research grants APVV-0631-07, APVV-0621-07, VEGA 2/0144/08, VEGA 2/0067/10, LPP-0354-06, LPP-0039-09, LPP-0363-06, LPP-0326-06 and NIH grant AG028538 (K.I.).
References
- 1.Andersson C, Blennow K, Almkvist O, et al. Increasing CSF phospho-tau levels during cognitive decline and progression to dementia. Neurobiol Aging. 2008;29:1466–1473. doi: 10.1016/j.neurobiolaging.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 2.Andreasen N, Hesse C, Davidsson P, et al. Cerebrospinal fluid beta-amyloid(1–42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999;56:673–680. doi: 10.1001/archneur.56.6.673. [DOI] [PubMed] [Google Scholar]
- 3.Andreasen N, Minthon L, Clarberg A, et al. Sensitivity, specificity, and stability of CSF-tau in AD in a community-based patient sample. Neurology. 1999;53:1488–1494. doi: 10.1212/wnl.53.7.1488. [DOI] [PubMed] [Google Scholar]
- 4.Andreasen N, Sjogren M, Blennow K. CSF markers for Alzheimer's disease: total tau, phospho-tau and Abeta42. World J Biol Psychiatry. 2003;4:147–155. doi: 10.1080/15622970310029912. [DOI] [PubMed] [Google Scholar]
- 5.Andreasen N, Blennow K. CSF biomarkers for mild cognitive impairment and early Alzheimer's disease. Clin Neurol Neurosurg. 2005;107:165–173. doi: 10.1016/j.clineuro.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 6.Blennow K, Wallin A, Agren H, et al. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol. 1995;26:231–245. doi: 10.1007/BF02815140. [DOI] [PubMed] [Google Scholar]
- 7.Blennow K, Vanmechelen E, Hampel H. CSF total tau, Abeta42 and phosphorylated tau protein as biomarkers for Alzheimer's disease. Mol Neurobiol. 2001;24:87–97. doi: 10.1385/MN:24:1-3:087. [DOI] [PubMed] [Google Scholar]
- 8.Blennow K, Hampel H. CSF markers for incipient Alzheimer's disease. Lancet Neurol. 2003;2:605–613. doi: 10.1016/s1474-4422(03)00530-1. [DOI] [PubMed] [Google Scholar]
- 9.Blennow K, Vanmechelen E. CSF markers for pathogenic processes in Alzheimer's disease: diagnostic implications and use in clinical neurochemistry. Brain Res Bull. 2003;61:235–242. doi: 10.1016/s0361-9230(03)00086-8. [DOI] [PubMed] [Google Scholar]
- 10.Blennow K. CSF biomarkers for Alzheimer's disease: use in early diagnosis and evaluation of drug treatment. Expert Rev Mol Diagn. 2005;5:661–672. doi: 10.1586/14737159.5.5.661. [DOI] [PubMed] [Google Scholar]
- 11.Blennow K, Zetterberg H, Minthon L, et al. Longitudinal stability of CSF biomarkers in Alzheimer's disease. Neurosci Lett. 2007;419:18–22. doi: 10.1016/j.neulet.2007.03.064. [DOI] [PubMed] [Google Scholar]
- 12.Bouwman FH, van der Flier WM, Schoonenboom NS, et al. Longitudinal changes of CSF biomarkers in memory clinic patients. Neurology. 2007;69:1006–1011. doi: 10.1212/01.wnl.0000271375.37131.04. [DOI] [PubMed] [Google Scholar]
- 13.Bouwman FH, Schoonenboom NS, Verwey NA, et al. CSF biomarker levels in early and late onset Alzheimer's disease. Neurobiol Aging. 2009;30:1895–1901. doi: 10.1016/j.neurobiolaging.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Degerman Gunnarsson M, Kilander L, Basun H, et al. Reduction of phosphorylated tau during memantine treatment of Alzheimer's disease. Dement Geriatr Cogn Disord. 2007;24:247–252. doi: 10.1159/000107099. [DOI] [PubMed] [Google Scholar]
- 15.de Leon MJ, Segal S, Tarshish CY, et al. Longitudinal cerebrospinal fluid tau load increases in mild cognitive impairment. Neurosci Lett. 2002;333:183–186. doi: 10.1016/s0304-3940(02)01038-8. [DOI] [PubMed] [Google Scholar]
- 16.de Leon MJ, DeSanti S, Zinkowski R, et al. Longitudinal CSF and MRI biomarkers improve the diagnosis of mild cognitive impairment. Neurobiol Aging. 2006;27:394–401. doi: 10.1016/j.neurobiolaging.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Diniz BS, Pinto JA, Jr, Forlenza OV. Do CSF total tau, phosphorylated tau, and beta-amyloid 42 help to predict progression of mild cognitive impairment to Alzheimer's disease? A systematic review and meta-analysis of the literature. World J Biol Psychiatry. 2007;9:1–11. doi: 10.1080/15622970701535502. [DOI] [PubMed] [Google Scholar]
- 18.Ewers M, Buerger K, Teipel SJ, et al. Multicenter assessment of CSF-phosphorylated tau for the prediction of conversion of MCI. Neurology. 2007;69:2205–2212. doi: 10.1212/01.wnl.0000286944.22262.ff. [DOI] [PubMed] [Google Scholar]
- 19.Formichi P, Battisti C, Radi E, et al. Cerebrospinal fluid tau, A beta, and phosphorylated tau protein for the diagnosis of Alzheimer's disease. J Cell Physiol. 2006;208:39–46. doi: 10.1002/jcp.20602. [DOI] [PubMed] [Google Scholar]
- 20.Frankfort SV, Tulner LR, van Campen JP, et al. Amyloid beta protein and tau in cerebrospinal fluid and plasma as bio-markers for dementia: a review of recent literature. Curr Clin Pharmacol. 2008;3:123–131. doi: 10.2174/157488408784293723. [DOI] [PubMed] [Google Scholar]
- 21.Greenberg SG, Davies PA. Preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hampel H, Buerger K, Kohnken R, et al. Tracking of Alzheimer's disease progression with cerebrospinal fluid tau protein phosphorylated at threonine 231. Ann Neurol. 2001;49:545–546. [PubMed] [Google Scholar]
- 23.Hansson O, Zetterberg H, Buchhave P, et al. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5:228–234. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- 24.Herukka SK, Hallikainen M, Soininen H, et al. CSF Abeta42 and tau or phosphorylated tau and prediction of progressive mild cognitive impairment. Neurology. 2005;64:1294–1297. doi: 10.1212/01.WNL.0000156914.16988.56. [DOI] [PubMed] [Google Scholar]
- 25.Hrnkova M, Zilka N, Minichova Z, et al. Neurodegeneration caused by expression of human truncated tau leads to progressive neurobehavioural impairment in transgenic rats. Brain Res. 2007;1130:206–213. doi: 10.1016/j.brainres.2006.10.085. [DOI] [PubMed] [Google Scholar]
- 26.Hu YY, He SS, Wang X, et al. Levels of nonphosphorylated and phosphorylated tau in cerebrospinal fluid Alzheimer's disease patients: an ultrasensitive bienzyme-substrate-recycle enzyme-linked immunosorbent assay. Am J Pathol. 2002;160:1269–1278. doi: 10.1016/S0002-9440(10)62554-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hulstaert F, Blennow K, Ivanoiu A, et al. Improved discrimination of AD patients using beta-amyloid(1–42) and tau levels in CSF. Neurology. 1999;52:1555–1562. doi: 10.1212/wnl.52.8.1555. [DOI] [PubMed] [Google Scholar]
- 28.Iqbal K, Flory M, Khatoon S, et al. Subgroups of Alzheimer's disease based on cerebrospinal fluid molecular markers. Ann Neurol. 2005;58:748–757. doi: 10.1002/ana.20639. [DOI] [PubMed] [Google Scholar]
- 29.Ishiguro K, Ohno H, Arai H, et al. Phosphorylated tau in human cerebrospinal fluid is a diagnostic marker for Alzheimer's disease. Neurosci Lett. 1999;270:91–94. doi: 10.1016/s0304-3940(99)00476-0. [DOI] [PubMed] [Google Scholar]
- 30.Kanai M, Matsubara E, Isoe K, et al. Longitudinal study of cerebrospinal fluid levels of tau, A beta1–40, and A beta1–42(43) in Alzheimer's disease: a study in Japan. Ann Neurol. 1998;44:17–26. doi: 10.1002/ana.410440108. [DOI] [PubMed] [Google Scholar]
- 31.Kluver H, Barrera E. A method for the combined staining of cells and fibers in the nervous system. J Neuropathol Exp Neurol. 1953;12:400–403. doi: 10.1097/00005072-195312040-00008. [DOI] [PubMed] [Google Scholar]
- 32.Kohnken R, Buerger K, Zinkowski R, et al. Detection of tau phosphorylated at threonine 231 in cerebrospinal fluid of Alzheimer's disease patients. Neurosci Lett. 2000;287:187–190. doi: 10.1016/s0304-3940(00)01178-2. [DOI] [PubMed] [Google Scholar]
- 33.Korenova M, Zilka N, Stozicka Z, et al. NeuroScale, the battery of behavioral tests with novel scoring system for phenotyping of transgenic rat model of tauopathy. J Neurosci Methods. 2009;177:108–114. doi: 10.1016/j.jneumeth.2008.09.027. [DOI] [PubMed] [Google Scholar]
- 34.Koson P, Zilka N, Kovac A, et al. Truncated tau expression levels determine life span of a rat model of tauopathy without causing neuronal loss or correlating with terminal neurofibrillary tangle load. Eur J Neurosci. 2008;28:239–246. doi: 10.1111/j.1460-9568.2008.06329.x. [DOI] [PubMed] [Google Scholar]
- 35.Mitchell AJ. CSF phosphorylated tau in the diagnosis and prognosis of mild cognitive impairment and Alzheimer's disease: a meta-analysis of 51 studies. J Neurol Neurosurg Psychiatry. 2009;80:966–975. doi: 10.1136/jnnp.2008.167791. [DOI] [PubMed] [Google Scholar]
- 36.Montine TJ, Beal MF, Cudkowicz ME, et al. Increased CSF F2-isoprostane concentration in probable AD. Neurology. 1999;52:562–565. doi: 10.1212/wnl.52.3.562. [DOI] [PubMed] [Google Scholar]
- 37.Pratico D, Clark CM, Lee VM, et al. Increased 8, 12-isoiPF2alpha-VI in Alzheimer's disease: correlation of a noninvasive index of lipid peroxidation with disease severity. Ann Neurol. 2000;48:809–812. [PubMed] [Google Scholar]
- 38.Sunderland T, Wolozin B, Galasko D, et al. Longitudinal stability of CSF tau levels in Alzheimer patients. Biol Psychiatry. 1999;46:750–755. doi: 10.1016/s0006-3223(99)00143-2. [DOI] [PubMed] [Google Scholar]
- 39.Vanmechelen E, Vanderstichele H, Davidsson P, et al. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett. 2000;285:49–52. doi: 10.1016/s0304-3940(00)01036-3. [DOI] [PubMed] [Google Scholar]
- 40.Vemuri P, Wiste HJ, Weigand SD, et al. MRI and CSF biomarkers in normal, MCI, and AD subjects: diagnostic discrimination and cognitive correlations. Neurology. 2009;73:287–293. doi: 10.1212/WNL.0b013e3181af79e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallin AK, Blennow K, Andreasen N, et al. CSF bio-markers for Alzheimer's disease: levels of beta-amyloid, tau, phosphorylated tau relate to clinical symptoms and survival. Dement Geriatr Cogn Disord. 2006;21:131–138. doi: 10.1159/000090631. [DOI] [PubMed] [Google Scholar]
- 42.Zetterberg H, Pedersen M, Lind K, et al. Intra-individual stability of CSF biomarkers for Alzheimer's disease over two years. J Alzheimers Dis. 2007;12:255–260. doi: 10.3233/jad-2007-12307. [DOI] [PubMed] [Google Scholar]
- 43.Zilka N, Filipcik P, Koson P, et al. Truncated tau from sporadic Alzheimer's disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006;580:3582–3588. doi: 10.1016/j.febslet.2006.05.029. [DOI] [PubMed] [Google Scholar]