

Abstract
Alzheimer disease (AD) is multi-factorial and heterogeneous. Independent of the aetiology, this disease is characterized clinically by chronic and progressive dementia and histopathologically by neurofibrillary degeneration of abnormally hyperphosphorylated tau seen as intraneuronal neurofibrillary tangles, neuropil threads and dystrophic neurites, and by neuritic (senile) plaques of β-amyloid. The neurofibrillary degeneration is apparently required for the clinical expression of AD, and in related tauopathies it leads to dementia in the absence of amyloid plaques. While normal tau promotes assembly and stabilizes microtubules, the abnormally hyperphosphorylated tau sequesters normal tau, MAP1 and MAP2, and disrupts microtubules. The abnormal hyperphosphorylation of tau also promotes its self-assembly into tangles of paired helical and or straight filaments. Tau is phosphorylated by a number of protein kinases. Glycogen synthase kinase-3 (GSK-3) and cyclin dependent protein kinase 5 (cdk5) are among the kinases most implicated in the abnormal hyperphosphorylation of tau. Among the phosphatases which regulate the phosphorylation of tau, protein phosphatase-2A (PP-2A), the activity of which is down-regulated in AD brain, is by far the major enzyme. The inhibition of abnormal hyperphosphorylation of tau is one of the most promising therapeutic targets for the development of disease modifying drugs. A great advantage of inhibiting neurofibrillary degeneration is that it can be monitored by evaluating the levels of total tau and tau phosphorylated at various known abnormally hyperphosphorylated sites in the cerebrospinal fluid of patients, obtained by lumbar puncture. There are at least five subgroups of AD, each is probably caused by a different etiopathogenic mechanism. The AD subgroup identification of patients can help increase the success of clinical trials and the development of specific and potent disease modifying drugs.
Keywords: Alzheimer disease, tauopathies, microtubule assembly, microtubule associated protein tau, abnormally hyperphosphorylated tau, protein phosphatase-2A, memantine, neurofibrillary pathology
Introduction
Alzheimer disease (AD), the single major cause of dementia in the middle- to old-aged individuals, is histopathologically characterized by intraneuronal neurofibrillary degeneration of the abnormally hyperphosphorylated tau and extracellular β-amyloidosis. Neurofibrillary degeneration is seen as neurofibrillary tangles, neuropil threads and as dystrophic neurites surrounding the amyloid core in the neuritic (senile) plaques. β-amyloidosis is seen as plaque core amyloid in the brain parenchyma and as congophilic angiopathy in the cerebral vessels. Although the two hallmark lesions of AD, the β-amyloidosis and neurofibrillary degeneration were described by Alois Alzheimer in 1907, it was only in the 1980s that their molecular composition was discovered. Glenner and Wong, in 1984, discovered by amino acid sequencing Aβ peptide as the major component of cerebrovascular amyloid in AD brain. Subsequent studies confirmed Aβ peptide as the major component of neuritic (senile) plaques [1, 2]. The major protein subunit of neurofibrillary tangles/paired helical filaments was isolated in 1974 from bulk-separated tangles from AD brain and identified by Western blots as microtubule associated protein tau in 1986 [3, 4]. The same year, we demonstrated (1) that tau in AD brain was abnormally hyperphosphorylated and, in this state, was polymerized into paired helical filaments (PHF)/neurofibrillary tangles [5], and (2) that, unlike normal tau, cytosolic abnormally hyperphosphorylated tau in AD brain was unable to promote microtubule assembly [6]. These findings laid down the foundation of very exciting studies on the molecular mechanisms of AD and molecular biomarkers associated with plaques and tangles, and development of disease modifying drugs.
Key findings on tau include (i) the cloning of tau gene from mouse brain [7] and alternate splicing of its pre-mRNA, generating different isoforms in bovine brain [8] discovered by Kirschner's lab; (ii) the presence of six molecular isoforms of tau by alternate splicing in the human brain by Goedert et al.[9]; (iii) the identification of the cyclin-dependent protein kinase 5 (cdk5) and glycogen synthase kinase-3 (GSK-3) as the major tau kinase activities that can abnormally hyperphosphorylate tau by Imahori's group [10–12]; (iv) the mapping of the phosphorylation sites of normal [13] and PHF tau [73], and ubiquitination of PHF tau [14] by Ihara's lab; (v) the identification of Ser262 as one of the major functional phosphorylation sites of tau and its phosphorylation by microtubule-affinity regulation kinase (MARK) by Mandelkow's lab [15, 16]; (vi) the higher-than-normal adult level of phosphorylation in foetal tau by the group of Lee and Trojanowski [17]; (vii), the selective decrease in the activities of protein phosphatase (PP)-2A and PP-1 in AD brain, and PP2A as the major tau phosphatase by Gong from our group [18, 19]; (viii) the in vitro assembly of tau into filaments and the promotion of this assembly by phosphorylation of this protein by Avila's laboratory [20]; (ix) the discovery of the sequestration of normal mitogen activated proteins (MAPs) by the abnormally hyperphosphorylated tau by Alonso from our group [21–23] and (x) the use of the CSF level of tau as a biomarker for AD by the Innogenetics group [24].
AD is multi-factorial and heterogeneous. Identification of various etiopathogenic mechanisms and of various subgroups of the disease is critical for the development of potent disease-modifying drugs. In less than 1% of the AD cases the disease co-segregates with certain mutations in β-amyloid precursor protein, presenilin-1 and presenlin-2 [25]. Over 99% of the AD cases are not associated with any known mutations, and the nature of the aetiological agent is not yet understood, but might involve metabolic and signal transduction abnormalities [26]. These different etiological factors, nevertheless, may lead to some common downstream pathogenic events that ultimately produce the disease clinically. Independent of the aetiology, AD is histopathologically characterized by the presence of numerous neurofibrillary tangles and neuritic (senile) plaques with neurofibrillary changes in the dystrophic neurites [27]. In a large number of the mature tangles, tau is ubiquitinated [14, 28, 29]. Several detailed reviews on each of these aspects of AD have recently been published [26, 30–32]. Here, we update some of the major findings on neurofibrillary degeneration of the abnormally hyperphosphorylated tau concerning the pivotal role of this lesion in the disease and as a molecular biomarker, and as a drug target.
Significance
Studies on the clinical-to-pathological correlation have consistently demonstrated that the number of neurofibrillary tangles, and not the plaques, correlates best with the presence and or the degree of dementia in AD [33–35]. Whereas neurofibrillary degeneration appears to be required for the clinical expression of the disease, the dementia, β-amyloidosis alone in the absence of neurofibrillary degeneration does not produce the disease clinically. In fact some of the normal aged individuals have as much β-amyloid plaque burden in the brain as typical cases of AD, except that in the former case plaques lack dystrophic neurites with neurofibrillary changes surrounding the betaamyloid cores [33, 34, 36–38]. On the other hand, neurofibrillary degeneration of the AD type, but in the absence of β-amyloidosis, is seen in several conditions such as Guam Parkinsonism-dementia complex, dementia pugilistica, frontotem-poral dementia with Parkinsonism linked to chromosome-17 (FTDP-17), corticobasal degeneration, Pick disease, and progressive supranuclear palsy. All of these neurodegenerative disorders, collectively called tauopathies, are clinically characterized by dementia. Furthermore, in inherited cases of FTDP-17, certain missense mutations in the tau gene, including those that affect the alternate splicing of its mRNA, favouring the 4-repeat tau isoforms, co-segregate with the disease [39–41]. These mutated taus and the 4-repeat tau become more favourable substrates for abnormal hyperphosphorylation [42].
Etiopathogenesis
Currently, the most popular hypothesis on the etiopathogenesis of AD is the Amyloid Cascade Hypothesis, according to which the generation of Aβ is the primary pathological event which leads to neurofibrillary degeneration and dementia [43, 44]. Consistent with this hypothesis, both intracerebral infusion of Aβ in FTDP-17 tau mutation P301L-expressing transgenic mice, as well as crossing these animals with APPTg2576 (APP Swedish plus London mutations), were found to exacerbate neurofibrillary pathology [45, 46] and, in the triple transgenic mice 3XTgAD (APPSWE-PS1M146V-tau P301L), β-amyloid deposition was found to precede the neurofibrillary pathology and these animals showed more neurofibrillary pathology than the double transgenic Tg2X mice [47, 48]. However, to date, the data from human conditions apparently do not support the amyloid cascade hypothesis – (1) some of the normal aged individuals show similar level and topography of compact Aβ plaques as typical cases of AD, except that the plaques in the former lack dystrophic neurites with neurofibrillary pathology;(2) the plaques and neurofibrillary tangles are seen in disproportionate numbers in AD, especially in the plaque-dominant and tangle-dominant AD subgroups; (3) typically, a considerably higher brain Aβ burden is seen in hereditary cerebral haemorrhage with amyloidosis, Dutch type (HCHWA-D) but without any accompanying neurofibrillary degeneration [49], and (4) the tauopathies, such as FTDP-17, Pick disease, corticobasal degeneration, supranuclear palsy, dementia pugilistica and Guam Parkinsonism dementia complex, are characterized by dementia associated with neurofibrillary degeneration of abnormally hyperphosphorylated tau in the absence of β-amyloid plaques. Furthermore, recent studies have shown that PS-1 not only promotes or acts as a γ-secretase activity (the cleavage of APP which produces Aβ), but also activates the phosphatidylinositol 3-kinase (PI3K), which downstream through protein kinase B (Akt) inhibits the GSK-3, a major tau kinase. Some of the AD-causing mutations in PS-1 result in loss of its ability to activate PI3K pathway, resulting in a sustained activity of GSK-3 and, consequently, abnormal hyperphosphorylation of tau [50]. Finally, several of the AD-causing PS-1 mutations have been reported to produce no change to a decrease in Aβ generation in cultured cells [51]. In our view, AD may be caused by a number of different factors and the amyloid cascade hypothesis is too simplistic and narrow to explain this multifactorial disease. We have proposed [26] that different signal transduction and metabolic factors, through different disease mechanisms, apparently lead to the same two disease characteristic lesions – neurofibrillary degeneration of abnormally hyperphosphorylated tau and β-amyloidosis (Fig. 1).
1.
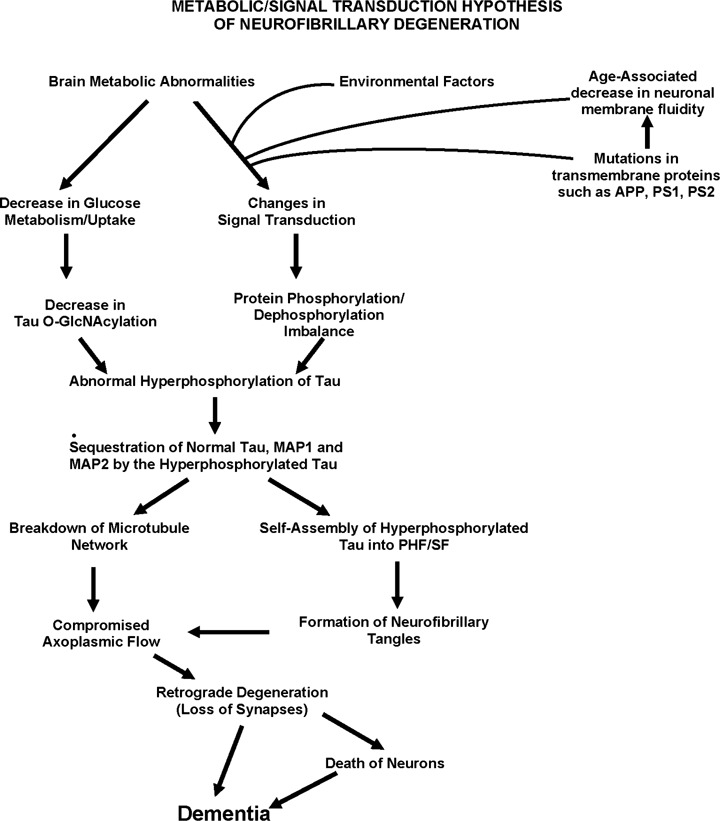
A schematic showing different major steps of the ‘Metabolic/Signal Transduction Hypothesis’. Alzheimer disease (AD) and other tauopathies require a genetic predisposition and are triggered by a variety of environmental factors, affecting one or more specific signal transduction pathways which result in a protein phosphorylation/dephosphorylation imbalance and the abnormal hyperphosphorylation of tau that leads to neurofibrillary degeneration and dementia. In AD, the protein phosphorylation/dephosphorylation imbalance in the affected neurons is generated at least in part by a decrease in the activities of tau phosphatases, that is PP-2A and PP-1;the activities of tau kinases, such as cdk5, GSK-3, CaM kinase II and PKA might also be increased in the affected neurons. This protein phosphorylation/dephosphorylation imbalance probably involves an alteration of a specific signal transduction pathway(s) produced by an increase in the levels of an extracellular signal, for example, fibroblast growth factor 2 (FGF2) or an alteration in the molecular topology of the neuronal cell membrane or both. With age, the molecular topology of the cell membranes is altered due to a decrease in membrane fluidity. The mutations in transmembrane proteins, such as β-APP, PS1 and PS2, increase the vulnerability of the cell membrane to alteration in pathological signal transduction. The increased risk for AD in the carriers of APOE4 allele as opposed to APOE2 or APOE3 alleles might also involve alteration of signal transduction through the interaction of APOE4 with the neuronal cell membrane. Any mutation or posttranslational modification of tau that will make it a better substrate for abnormal hyperphosphorylation will also increase the risk for the disease. High cholesterol might be involved in decreasing membrane fluidity. Decreased glucose metabolism/uptake might lead to the abnormal hyperphosphorylation of tau through a decrease in its O-GlcNAcylation.(Reproduced with permission from Iqbal and Grundke-Iqbal, Acta Neuropathologica, 2005, 109:25–31.)
Mechanism by which abnormal hyperphosphorylation of tau leads to neurofibrillary degeneration
Microtubule associated protein tau is highly hydrophilic and is, thus, highly soluble and heat stable. To date, not only in AD but also in every known human tauopathy, the tau pathology is made up of the abnormally hyperphosphorylated protein. While conformational changes [52–54] and truncation of tau [55–57] have been reported in AD, the most established and the most compelling cause of dysfunctional tau in AD and related tauopathies is the abnormal hyperphosphorylation of this protein [5, 6, 23]. Tau, a phosphoprotein which normally contains 2–3 mol. of phosphate/mol. of the protein [58], is abnormally hyperphosphorylated in AD brain [6] and, in this state, is the major protein subunit of the paired helical filaments/neurofibrillary tangles [4, 5, 59, 60]. Two major known functions of tau are its ability to promote assembly and to maintain structure of microtubules [61]. These functions of tau are regulated by its degree of phosphorylation [23, 62–64]. In AD brain there is as much normal tau as in age-matched control human brain, but, in addition, the diseased brain contains 4–8-fold of abnormally hyperphosphorylated tau [65, 66]. As much as 40% of the abnormally hyperphosphorylated tau is present in the cytosol and not polymerized into paired helical filaments/neurofibrillary tangles [58].
The tau polymerized into neurofibrillary tangles is apparently inert and neither binds to tubulin nor promotes its assembly into microtubules [63, 64, 67]. In contrast, the AD cytosolic abnormally hyperphosphorylated tau (AD P-tau) not only is unable to bind to tubulin and promote microtubule assembly, but also inhibits assembly and disrupts microtubules [23, 68]. This toxic property of the pathological tau involves the sequestration of normal tau by the diseased protein [22, 23]. The AD P-tau also sequesters the other two major neuronal microtubule associated proteins MAP1 A/B and MAP2 [23]. This toxic behavior of the AD P-tau appears to be solely due to its abnormal hyperphosphorylation because dephosphorylation of diseased tau converts it into a normal-like protein [23, 68–71]. Furthermore, in vitro dephosphorylation of neurofibrillary tangles disaggregates filaments and, as a result, the tau released behaves like normal protein in promoting microtubule assembly [70]. Thus, two characteristics of AD abnormally hyperphosphorylated tau are (1) that it sequesters normal MAPs and disrupts microtubules and (2) that it self-assembles into paired helical and or straight filaments.
Tau mutations, which cause FTDP-17, result either in increase in 4-repeat:3-repeat tau ratio or in missense mutations in the protein. Both 4-repeat tau and the mutated protein are more easily abnormally hyperphosphorylated than the normal wild-type protein [42, 72]. Thus, inhibition of the abnormal hyperphosphorylation of tau is likely to inhibit neurofibrillary degeneration and consequently the diseases characterized by this lesion.
Signal transduction pathways involved
Tau kinases
The state of phosphorylation of a phosphoprotein is a function of the balance between the activities of the protein kinases and the PPs that regulate its phosphorylation. Tau, which is phosphorylated at over 38 serine/threonine residues in AD [73, 74], is a substrate for several protein kinases [75, 76]. Among these kinases, GSK-3, cyclin dependent protein kinase-5 (cdk5), casein kinase-1 (CK-1), protein kinase A (PKA), calcium and calmodulin-dependent protein kinase-II (CaMKII), casein kinase-1 (CK-1), MAP kinase ERK 1/2 and stress-activated protein kinases have been most implicated in the abnormal hyperphosphorylation of tau [77, 78]. A large number of the abnormally hyperphosphorylated sites in tau are proline-directed, that is serine/threonine followed by proline which are canonical sites of proline-directed protein kinases (PDPKs).
GSK-3β and cdk5 phosphorylate tau at a large number of sites, most of which are common to the two enzymes [79, 80]. The expressions of GSK-3β and cdk5 are high in the brain [81–83] and both enzymes have been shown to be associated with all stages of neurofibrillary pathology in AD [84, 85]. Overexpression of GSK-3β in cultured cells and in transgenic mice results in hyperphosphorylation of tau at several of the same sites seen in AD and inhibition of this enzyme by lithium chloride attenuates phosphorylation in these models [86–93].
Cdk5 requires for its activity interaction with p39 or p35 or, better, their proteolytic products p29 or p25, respectively, which are generated in post mitotic neurons by digestion with calpains [94, 95]. Overexpression of p25 in transgenic mice, which results in an increase in the activity of this enzyme, also produces hyperphosphorylation of tau [96, 97].
The MAP kinase family, which includes ERK1, ERK2, p70S6 kinase and the stress-activated kinases JNK and p38 kinase, have been shown to phosphorylate tau at several of the same sites as the abnormally hyperphosphorylated tau and so has been the association of these enzymes with the progression of neurofibrillary degeneration in AD [78, 98–103].
Unlike the PDPKs, the non-PDPKs have been shown to phosphorylate tau at only a few of the sites. CaM Kinase II phosphorylates tau at Ser-262/356 and at Ser-416 [104–107]. Both PKA and MARK kinase have also been shown to phosphorylate tau at Ser-262 [16, 108, 109]. However, phosphorylation of tau by these non-PDPKs markedly increases the phosphorylation of tau by PDPKs, GSK-3β and cdk5 [79, 110–112]. The priming of tau by PKA appears to be sufficient to promote the abnormal hyperphosphorylation of tau by the basal level of GSK-3β activity in normal adult rat brain and leads to an impairment of spatial memory in these animals [113]. Although, to date, the activities of these protein kinases, except GSK-3β, have not been reproducibly shown to be up-regulated in AD brain, transient stimulation of these enzymes, especially the priming kinases such as PKA or CaMKII, might be sufficient to result in the abnormal hyperphosphorylation of tau.
Tau phosphatases
The activities of PP-2A and PP-1 are compromised by ∼ 20% in AD brain [19, 114], and the phosphorylation of tau that suppresses its microtubule binding and assembly activities in adult mammalian brain is regulated by PP-2A and not by PP-2B [107, 115] and PP-2A accounts for over 70% of all phosphoseryl/-phosphothreonyl activity in human brain [116]. PP-2A also regulates the activities of several tau kinases in brain. Inhibition of PP-2A activity by okadaic acid in cultured cells and in metabolically active rat brain slices results in abnormal hyperphosphorylation of tau at several of the same sites as in AD, not only directly by a decrease in dephosphorylation but also indirectly by promoting the activities of CaM Kinase II [107], PKA [117, 118], MAP kinase kinase (MEK1/2), extracellular regulated kinase (ERK 1/2) and P70S6 kinase [78, 102]. Thus, barring the fact that tau is not the only neuronal substrate of these protein kinases and phosphatases, it should be possible to inhibit the abnormal hyperphosphorylation of tau by inhibiting the activity of one or more tau kinases and or restoring or up-regulating the activity of PP-2A.
Although the brain has several tau phosphatase activities [119, 120], PP-2A and PP-1 make more than 90% of the serine/threonine PP activity in mammalian cells [121]. The intracellular activities of these enzymes are regulated by endogenous inhibitors. PP-1 activity is regulated mainly by a 18.7 kD heat stable protein called inhibitor-1 (I-1) [122, 123]. In addition, a structurally related protein, DARPP-32 (dopamine and cAMP-regulated phosphoprotein of apparent molecular weight 32,000) is expressed predominantly in the brain [124]. I-1 and DARPP-32 are activated on phosphorylation by protein kinase A and inactivated by calcineurin, and at basal calcium level by PP-2A [125]. Thus, inhibition of PP-2A activity would keep I-1, DARPP-32 in active form and thereby result in a decrease in PP-1 activity. In AD brain a reduction in PP-2A activity might have decreased the PP-1 activity by allowing the up-regulation of the I-1/DARPP-32 activity. In the subgroup of AD cases and or at moderate to severe stages of the disease, when there is a persistent excitotoxicity and increase in the intraneuronal calcium, DARPP-32 is probably mainly dephosphorylated and thereby inactivated as PP-1 inhibitor by calcineurin.
PP-2A inhibitors
PP-2A is inhibited in the mammalian tissue by two heat-stable proteins: (i) the I1PP2A, a 30 kD cytosolic protein [126] that inhibits PP-2A with a Ki of 30 nM and (ii) the I2PP2A, a 39 kD nuclear protein that inhibits PP-2A with a Ki of 23 nM [126]. Both I1PP2A and I2PP2A have been cloned from human kidney [127, 128] and brain [129]. I1PP2A has been found to be the same protein as the putative histocompatibility leukocyte antigen class II-associated protein-1 (PHAP-1). This protein, which has also been described as mapmodulin, pp32 and LANP [130] is 249 amino acids long and has apparent molecular weight of 30 kD on SDS-PAGE. I2PP2A, which is the same as template-activating factor (TAF)-1β or PHAPII, is a nuclear protein that is a homologue of the human SETα protein [131]. In AD brain there is a shift from nuclear to cytoplasmic localization of I2PP2A[132]. Both I1PP2A and I2PP2A interact with the catalytic subunit of PP2A [133]. The level of I1PP2A is ∼ 20% increased in AD brains as compared with age-matched control brains which probably is a cause of the decrease in PP-2A activity in AD brain.
Involvement of more than one kinase and phosphorylation site in abnormal hyperphosphorylation of tau
Hyperphosphorylation promotes the assembly of tau into PHF/SF [134]. In vitro studies have demonstrated that phosphorylation of tau to ∼4-6 moles/mole of the protein converts it into an ADP-tau-like state, that is, where instead of promoting it inhibits microtubule assembly by sequestering normal tau and other MAPs. On further hyperphosphorylation to ∼9-12 moles phosphate/mole of the protein, tau self-assembles into PHF/straight filaments (SF). The FTDP-17 mutated taus are more readily hyperphosphorylated than the normal/wild-type human brain tau, become inhibitory and self-assemble into PHF/SF at a lower stoichiometry of phosphorylation than the corresponding wild-type protein [42].
Abnormally hyperphosphorylated tau from AD brain cytosol, the AD P-tau, self-assembles into bundles of PHF/SF [134, 135]. On treatment with PP-2A, which dephosphorylates most of the known abnormally hyperphosphorylated sites, including Thr231 and Ser262, the AD P-tau loses its ability to both inhibit microtubule assembly and to self-assemble into PHF/SF [135]. Re-phosphorylation of the PP-2A de-phosphorylated AD P-tau, the PP2A-AD-P-tau, by PKA followed by CaMKinase-II and GSK-3β, or cdk5, or cdk5 followed by GSK-3β, results in phosphorylation of Thr231 and Ser262 among several other sites, and restores its ability to inhibit microtubule assembly and self-assemble into PHF/SF. The bundles of filaments formed under these conditions are congophilic and very reminiscent of neurofibrillary tangles seen in AD brain. Re-phosphorylation of PP-2A-AD P-tau by none of the above kinases individually, however, phosphorylates at both Thr231 and Ser262 and restores its self-assembly into PHF/SF. Thus, these studies [135] revealed that more than one specific combination of kinases are involved in converting normal tau into an AD P-tau-like state, and that PP-2A can alone convert the pathological state of the protein to a normal-like state.
Role of decreased brain glucose metabolism in neurofibrillary degeneration
In addition to abnormal hyperphosphorylation, tau is also abnormally glycosylated and the latter appears to precede the former in AD brain [136, 137]. In vitro studies indicate that the abnormal glycosylation promotes tau phosphorylation with PKA, GSK-3β and ckd5, and inhibits dephosphorylation of tau with PP2A and PP5 [138, 139]. In addition, like some other neuronal phosphoproteins, tau is also O-GlcNAcylated [140, 141]. In contrast to classical N- or O-glycosylation, O-GlcNAcylation which involves the addition of a single sugar at serine/threonine residues of a protein, dynamically post-translationally modifies cytoplasmic and nuclear proteins in a manner analogous to protein phosphorylation [142]. O-GlcNAcylation and phosphorylation reciprocally regulate each other. In AD, probably due to impaired glucose uptake/metabolism, there is a global decrease in O-GlcNAcylation, including that of tau. Decreased glucose metabolism in cultured cells and in mice, which decreases the O-GlcNAcylation of tau, produces abnormal hyperphosphorylation of this protein [143]. Thus, inhibition of O-GlcNAcylase, the enzyme which hydrolyses the removal of this sugar moiety from proteins, is a promising therapeutic target for AD and related tauopathies. Inhibition of O-GlcNAcylase with PUGNac or NAG-AE inhibits hyperphosphorylation of tau by increasing its O-GlcNAcylation.
Subgroups of AD
Given the multi-factorial nature of AD, identification of different disease subgroups which might represent different etiopathogenic mechanisms will not only improve the accuracy of the diagnosis but also help develop and measure the efficacy of different therapeutic drugs towards these disease subgroups. Because of clinical heterogeneity, the diagnosis of AD remains probable till postmortem histopathological examination, and is made primarily by exclusion of other causes of dementia [144]. AD histopathology shows considerable qualitative and quantitative heterogeneity. AD can be neocortical type, limbic type and plaque-dominant type, and it may present with numerous neurofibrillary tangles exclusively confined to the hippocampus and entorhinal cortex [145]. The two most common confounding diagnoses are cerebral vascular disease (multi-infarct dementia) and dementia with Lewy bodies.
Increased rates of ventricular volume and whole brain atrophy have been demonstrated in AD [146]. The whole brain atrophy in AD brain results in a loss of brain mass of as much as ∼2–3% per year compared with ∼0.4–0.5% in age-matched control subjects [147]. A number of animal and human studies have suggested that Aβ1-42 levels in cerebrospinal fluid (CSF) reflect the amyloid βpathology in the brain. Reduction of Aβ1-40 and Aβ1-42 in the brain of adult rats treated orally with γ-secretase inhibitors have been found to result in decreased levels of Aβ in both brain and CSF [148, 149]. An inverse relation between in vivo amyloid load and CSF levels of Aβ1-42 has been found in humans [150]. Antemortem CSF levels of Aβ1-42, total tau and phosphotau-Thr231 have been reported to reflect the histopathological changes observed postmortem in the brains of AD cases [151, 152]. The CSF levels of tau have been shown to be markedly increased in patients with diffuse axonal injury in head trauma which revert on clinical improvement [153]. Thus, bulk of the evidence supports that CSF reflects the state of the brain protein metabolism.
Development of therapeutic drugs requires our ability to accurately diagnose the disease and its specific subtypes, and the availability of specific outcome measures. We postulate that more than one disease mechanism and signalling pathway are involved in producing the AD pathology, especially the neurofibrillary degeneration of abnormally hyperphosphorylated tau, and that various subgroups of AD can be identified based on the CSF levels of proteins associated with senile (neuritic) plaques and neurofibrillary tangles. Towards testing this hypothesis, we immunoassayed the levels of tau, ubiquitin and A_1-42 in retrospectively collected lumbar CSFs of 468 patients clinically diagnosed as AD (353 CSFs) and as non-AD neurological and non-neurological cases (115 CSFs) and, based on the level of these molecular markers, all subjects were subjected to the latent profile analysis to determine the assignment of each subject to a particular cluster. We found that AD subdivides into at least five subgroups based on the CSF levels of A1-42, tau and ubiquitin, and that each subgroup presented a different clinical profile [154];these five subgroups are:
Subgroups AELO, ATEO, HARO and ATURO accounted for approximately 50%, 22%, 5% and 1%, respectively, of the AD cases studied (Fig 2). Subgroup LEBALO, which contained a majority of AD cases with Lewy bodies, accounted for ∼19% of the AD cases.
2.
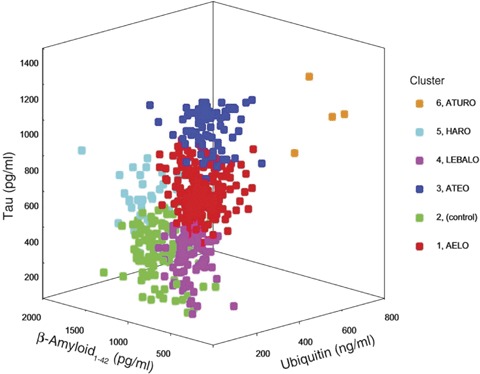
Three-dimensional representation of the five Alzheimer's disease subgroups (Clusters 1, 3, 4, 5 and 6) and the control subjects (Cluster 2). AELO = AD with low Aβ1-42, high incidence of apolipoprotein E4 and late onset; ATEO = AD with low Aβ1-42, high tau, and early onset; ATURO = AD with low Aβ1-42, high tau, high ubiquitin, and recent onset; HARO = AD with high Aβ1-42 and recent onset;LEBALO = AD with high incidence of Lewy bodies, low Aβ1-42, and late onset. (Reproduced with permission from Iqbal et al., Annals of Neurology, 2005, 58:748–757.)
To classify diagnosed AD cases into the proposed subgroups, we sought a simple set of rules using the level of only one indicator protein at any stage in the classification process. Ideally, it would classify cases with a sensitivity and a specificity of no less than 90% of each category and a comparable overall level of correct classification. The algorithm must unambiguously categorize all cases. A decision tree based on the algorithm was derived based on examination of cluster characteristics and experimental runs that came closest to fulfilling those criteria (see Fig. 2). The respective sensitivities and specificities were: AELO—90%, 92%; ATEO—90%, 95%; LEBALO—88%, 99%; HARO—100%, 99% and ATURO—100%, 100%. This study demonstrated that CSF levels of Aβ1-42, tau and ubiquitin could diagnose AD in five different subgroups at sensitivities and specificities of greater than 88% and, overall, 86% of cases were classified correctly. This rate of diagnostic accuracy not only is superior to using one of these markers individually or in combination of twos, but also exceeds the biomarker criteria of the Consensus Report [155].
Our recent studies have revealed that more than one signalling pathway is involved in neurofibrillary degeneration. We have found that tau can be abnormally hyperphosphorylated to self-assemble into bundles of paired helical filaments with more than one combination of protein kinases and that this phosphorylation of tau can be regulated by PPs, especially PP-2A [135]. Thus, it is likely that, in future, additional subgroups of AD may be identified from phosphorylation patterns of CSF tau of AD patients.
The CSF analysis not only helps identify a specific AD subgroup of a patient but also can serve as the outcome measure of a drug treatment. We discovered that memantine inhibited abnormal hyperphosphorylation of tau in rat hippocampal slices in culture [117], and that this effect of the drug was through disinhibition of PP-2A activity [156] which we previously showed to be down regulated in AD brain [19]. Based on our finding on the restoration of the PP-2A activity by memantine, Gunnarrsson et al.[157] investigated and found a significant decrease in phosphotau level in the CSF of patients one year after treatment with memantine.
Because of the involvement of different etiopathogenic mechanisms in AD, the identification of different subgroups of this single major cause of age-associated dementia is critical for development of potent and specific drugs that can prevent and cure this disease. Currently, several hundred drugs for AD are under development by the pharmaceutical industry. Stratification of the test subjects in clinical trials by disease subgroups may increase the chance of success up to several fold. The future of therapeutic drugs for AD may depend on recognition of different subgroups of the disease.
Therapeutic approaches to inhibit neurofibrillary degeneration
Neurofibrillary degeneration of abnormally hyperphosphorylated tau is downstream to alterations in more than one signalling pathway, but pivotally involved in the pathogenesis of AD and related tauopathies. Inhibition of this lesion is likely to arrest these diseases, which are products of multiple etiopathogenic mechanisms. The most promising therapeutic approaches to inhibit neurofibrillary degeneration and consequently AD and related tauopathies are (1) to inhibit the abnormal hyperphosphorylation of tau, (2) to inhibit sequestration of normal MAPs by the AD P-tau, (3) to inhibit misfolding of tau, and (4) to directly stabilize microtubules. The inhibition of abnormal hyperphosphorylation of tau can be carried out best apparently by inhibiting activities of both GSK-3 and cdk5, by activating the PP-2A activity or by increasing brain glucose uptake/metabolism which could enhance O-GlcNAcylation and consequently the inhibition of the abnormal hyperphosphorylation of tau.
Most inhibitors of GSK-3 also inhibit cdk5 and vice versa. To overcome this problem, several highly selective inhibitors of each of these kinases have been recently developed by the pharmaceutical industry, and some of these compounds are at different stages of human clinical trials for the treatment of AD. Our in vitro studies on the generation of abnormally hyperphosphorylated tau [135] suggest that compounds which inhibit both GSK-3 and cdk5 might even be more effective than the highly selective inhibitors of one of these enzymes in inhibiting neurofibrillary degeneration.
Memantine, a low to moderate affinity NMDA receptor antagonist, which improves mental function and the quality of daily living of patients with moderate to severe AD [158, 159], restores the PP-2A activity, and reduces the abnormal hyperphosphorylation of tau at Ser-262 and the associated neurodegeneration in hippocampal slice cultures from adult rats, and PC-12 cells in culture [117, 156]. Furthermore, the restoration of the PP-2A activity to normal levels by memantine also results in the restoration of the expression of MAP2 in the neuropil and a reversal of the hyperphosphorylation and the accumulation of neurofilament H and M subunits. Memantine, however, is a positively charged molecule and probably enters a neuron only during excitotoxicity when the N-methyl-D-aspartate (NMDA) receptor channels are open. Therefore, its therapeutic benefit might be limited to only those patients and/or the advanced states of the disease when there is a persistent excitotoxicity. Generation of cell permeable memantine-like compounds can help develop potent therapeutic drugs for AD and related tauopathies. The restoration of the PP-2A activity appears to be due to the binding of memantine to I2PP2A and disinhibition of its activity towards PP-2A [156]. The CSF level of phosphotau is significantly reduced in AD patients after one year treatment with memantine [157]. All these findings taken together suggest that PP-2A is a promising therapeutic target for AD and related tauopathies.
Recent studies suggest inhibition of calpains as another approach for the inhibition of neurofibrillary degeneration [160]. Calpain, the activity of which is up-regulated in AD brain, activates cdk5 through cleavage of its activators p39 and p35 to p29 and p25, respectively [94, 95]. Calpain also cleaves and activates calcineurin, which regulates the phosphorylation of CREB [160]. Thus, inhibition of calpain can be neuroprotective, both by inhibition of cdk5 activity and by increase in CREB activity.
The abnormally hyperphosphorylated tau causes neurofibrillary degeneration by sequestration of normal MAPs. A competitive inhibition of this sequestration by small molecules can arrest this pathology. Though inhibition of protein–protein interaction is generally a somewhat challenging task, there is a great advantage in developing drugs against such specific targets.
Since O-GlcNAcylation and phosphorylation reciprocally regulate each other, an approach, independent from modulation of tau kinases and phosphatases, is to restore the O-GlcNAcylation of tau, which is compromised in AD [143] to normal level. This could probably be achieved by increasing brain glucose levels through up-regulating the activity of neuronal glucose transporters such as Glut3 and or by mediation of the activity of the O-GlcNAcylase with PUGNec- and NAG-AE-like compounds.
The accumulation of abnormally hyperphosphorylated tau in AD and related tauopathies indicates that either the ubiquitin-proteasome system and or the chaperones in the affected neurons are overwhelmed. Isopeptidase activity might be increased, keeping the hyperphosphorylated tau from polyubiq-uitination for degradation by the ubiquitin protea-some pathway. Thus, isopeptidase inhibitors and drugs that promote heat-shock protein mediated clearance of tau may also inhibit neurofibrillary degeneration. Recently, HSP90 inhibitors have been shown to result in clearance of hyperphosphorylated tau through increase in the expression of chaperones HSP70-interacting protein (CHIP), a tau ubiquitin ligase, that re-folds the misfolded proteins [161, 162]. Most recently, immunization of P301L transgenic mice with a small tau phosphopeptide has been reported to clear the hyperphosphorylated tau [163]. Another approach to overcome the inhibitory activity of the hyperphosphorylated tau is the use of microtubule stabilizing drugs like taxol [118, 162, 164].
A large majority of therapeutic drugs currently under development for AD are focused on inhibiting β-amyloid. However, there is increasing interest in developing drugs that can inhibit tau pathology. After all, neurofibrillary degeneration of abnormally hyperphosphorylated tau is apparently required for the clinical expression of AD and tau pathology alone in the absence of any β-amyloid causes frontotemporal dementia and other tauopathies. Independent of whether the amyloid cascade hypothesis proves to be true or untrue, inhibition of neurofibrillary degeneration is likely to inhibit the clinical phonotype of AD and related tauopathies. A polytherapy targeting both β-amyloid and neurofibrillary degeneration might be synergistically beneficial in AD patients.
At present, a large majority of the anti-neurofibrillary degeneration drugs under development are GSK-3 inhibitors; a few target ERK-2, tau phosphatases, and aggregation of the hyperphosphorylated tau into filaments. In the authors' opinion, inhibition of the abnormal hyperphosphorylation of tau appears to be the most promising therapeutic target for AD and related tauopathies. Inhibition of GSK-3 activity and modulation of PP-2A, which is the major tau phosphatase and the activity of which is compromised in AD brain, are among the most attractive approaches to inhibit the abnormal hyperphosphorylation of tau. In the case of PP-2A, the restoration of its activity to normal level should have low risk of any deleterious side effects. GSK-3 is involved in several important signalling pathways and inhibiting its activity carries risks, but then, apparently there is a safe therapeutic window for this enzyme activity;LiCl, a GSK-3 inhibitor, has been successfully prescribed for bipolar disorders for many years.
An advantage of targeting neurofibrillary degeneration for the development of therapeutic drugs is that the efficacy of such neuroprotective drugs can be directly monitored by assaying the CSF level of total tau as a marker of neurodegeneration and of various phosphotaus as markers of inhibition of the abnormal hyperphosphorylation of tau.
A retrospective study of 2,661 autopsied brains has revealed that neurofibrillary degeneration precedes by several years the clinical expression, that is, dementia, in AD [165]. It will be very important to be able to detect neurofibrillary degeneration, probably by determining the CSF levels of total tau and phosphotaus at the presymptomatic stage of the disease. Inhibition of neurofibrillary degeneration in pre-symptomatic individuals can most probably prevent AD and related tauopathies. APOE4 carriers who are at ∼3.5-fold (oneAPOE4 allele) to ∼10-fold (APOE4/4) higher risk than the non-carriers of this allele for developing late onset AD [166], individuals with Down syndrome who invariably develop AD histopathology in the fourth decade of life, and individuals with a strong family history of AD or related tauopathies are among others who can benefit from employing therapeutic inhibitors of neurofibrillary degeneration as a prevention measure.
In short, neurofibrillary degeneration may result from more than one etiopathogenic mechanism, and a large number of therapeutic approaches are available to inhibit this pivotal lesion of AD and related tauopathies. Inhibition of neurofibrillary degeneration is very promising both as a treatment as well as a prevention measure for AD and related tauopathies.
Acknowledgments
We are grateful to Janet Murphy for secretarial assistance. Studies in our laboratories were supported in part by the New York State Office of Mental Retardation and Developmental Disabilities and NIH grants AG019158 and AG028538, and Alzheimer's Association (Chicago, IL) grant IIRG-00-2002.
Glossary
- AELO
AD with low Aβ1-42, high incidence of APOE4, and Late Onset
- ATEO
AD with low Aβ1-42, High Tau and Early Onset
- LEBALO
AD with high incidence of Lewy Bodies, low Aβ1-42, and Late Onset
- HARO
AD with High Aβ1-42 and Recent Onset
- ATURO
AD with low Aβ1-42, high Tau, high Ubiquitin and Recent Onset
References
- 1.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–9. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, Iqbal K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer's disease. Arch Biochem Biophys. 1993;301:41–52. doi: 10.1006/abbi.1993.1112. [DOI] [PubMed] [Google Scholar]
- 3.Iqbal K, Wisniewski HM, Shelanski ML, Brostoff S, Liwnicz BL, Terry RD. Protein changes in senile dementia. Brain Res. 1974;77:337–43. doi: 10.1016/0006-8993(74)90798-7. [DOI] [PubMed] [Google Scholar]
- 4.Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084–9. [PubMed] [Google Scholar]
- 5.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–7. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, Wisniewski HM, Alafuzoff I, Winblad B. Defective brain microtubule assembly in Alzheimer's disease. Lancet. 1986;2:421–6. doi: 10.1016/s0140-6736(86)92134-3. [DOI] [PubMed] [Google Scholar]
- 7.Lee G, Cowan N, Kirschner M. The primary structure and heterogeneity of tau protein from mouse brain. Science. 1988;239:285–8. doi: 10.1126/science.3122323. [DOI] [PubMed] [Google Scholar]
- 8.Himmler A, Drechsel D, Kirschner MW, Martin DW., Jr Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol. 1989;9:1381–8. doi: 10.1128/mcb.9.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–26. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 10.Ishiguro K, Ihara Y, Uchida T, Imahori K. A novel tubulin-dependent protein kinase forming a paired helical filament epitope on tau. J Biochem. 1988;104:319–21. doi: 10.1093/oxfordjournals.jbchem.a122465. [DOI] [PubMed] [Google Scholar]
- 11.Ishiguro K, Omori A, Sato K, Tomizawa K, Imahori K, Uchida T. A serine/threonine proline kinase activity is included in the tau protein kinase fraction forming a paired helical filament epitope. Neurosci Lett. 1991;128:195–8. doi: 10.1016/0304-3940(91)90259-v. [DOI] [PubMed] [Google Scholar]
- 12.Ishiguro K, Shiratsuchi A, Sato S, Omori A, Arioka M, Kobayashi S, Uchida T, Imahori K. Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 1993;325:167–72. doi: 10.1016/0014-5793(93)81066-9. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe A, Hasegawa M, Suzuki M, Takio K, Morishima-Kawashima M, Titani K, Arai T, Kosik KS, Ihara Y. In vivo phosphorylation sites in fetal and adult rat tau. J Biol Chem. 1993;268:25712–7. [PubMed] [Google Scholar]
- 14.Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science. 1987;235:1641–4. doi: 10.1126/science.3029875. [DOI] [PubMed] [Google Scholar]
- 15.Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–63. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 16.Drewes G, Trinczek B, Illenberger S, Biernat J, Schmitt-Ulms G, Meyer HE, Mandelkow EM, Mandelkow E. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem. 1995;270:7679–88. doi: 10.1074/jbc.270.13.7679. [DOI] [PubMed] [Google Scholar]
- 17.Matsuo ES, Shin RW, Billingsley ML, Van deVoorde A, O'Connor M, Trojanowski JQ, Lee VM. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer's disease paired helical filament tau. Neuron. 1994;13:989–1002. doi: 10.1016/0896-6273(94)90264-x. [DOI] [PubMed] [Google Scholar]
- 18.Gong CX, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer's disease abnormally phosphorylated tau by protein phosphatase-2A. Neuroscience. 1994;61:765–72. doi: 10.1016/0306-4522(94)90400-6. [DOI] [PubMed] [Google Scholar]
- 19.Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61:921–7. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- 20.Montejo de Garcini E, Serrano L, Avila J. Self assembly of microtubule associated protein tau into filaments resembling those found in Alzheimer disease. Biochem Biophys Res Commun. 1986;141:790–6. doi: 10.1016/s0006-291x(86)80242-x. [DOI] [PubMed] [Google Scholar]
- 21.Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration:sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci USA. 1997;94:298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alonso AD, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–7. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 23.Alonso AD, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:5562–6. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vandermeeren M, Mercken M, Vanmechelen E, Six J, Van De Voorde A, Martin JJ, Cras P. Detection of tau proteins in normal and Alzheimer's disease cere-brospinal fluid with a sensitive sandwich enzyme-linked immunosorbent assay. J Neurochem. 1993;61:1828–34. doi: 10.1111/j.1471-4159.1993.tb09823.x. [DOI] [PubMed] [Google Scholar]
- 25.Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65:664–70. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iqbal K, Grundke-Iqbal I. Metabolic/signal transduction hypothesis of Alzheimer's disease and other tauopathies. Acta Neuropathol. 2005;109:25–31. doi: 10.1007/s00401-004-0951-y. [DOI] [PubMed] [Google Scholar]
- 27.Braak H, Braak E, Grundke-Iqbal I, Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer's disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett. 1986;65:351–5. doi: 10.1016/0304-3940(86)90288-0. [DOI] [PubMed] [Google Scholar]
- 28.Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci USA. 1987;84:3033–6. doi: 10.1073/pnas.84.9.3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang GP, Khatoon S, Iqbal K, Grundke-Iqbal I. Brain ubiquitin is markedly elevated in Alzheimer disease. Brain Res. 1991;566:146–51. doi: 10.1016/0006-8993(91)91692-t. [DOI] [PubMed] [Google Scholar]
- 30.Selkoe DJ. Defining molecular targets to prevent Alzheimer disease. Arch Neurol. 2005;62:192–5. doi: 10.1001/archneur.62.2.192. [DOI] [PubMed] [Google Scholar]
- 31.Iqbal K, Grundke-Iqbal I. Developing pharmacological therapies for Alzheimer disease. Cell Mol Life Sci. 2007;64:2234–44. doi: 10.1007/s00018-007-7221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hardy J. A hundred years of Alzheimer's disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 33.Alafuzoff I, Iqbal K, Friden H, Adolfsson R, Winblad B. Histopathological criteria for progressive dementia disorders: clinical-pathological correlation and classification by multivariate data analysis. Acta Neuropathol. 1987;74:209–25. doi: 10.1007/BF00688184. [DOI] [PubMed] [Google Scholar]
- 34.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–9. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 35.Tomlinson BE, Blessed G, Roth M. Observations on the brains of demented old people. J Neurol Sci. 1970;11:205–42. doi: 10.1016/0022-510x(70)90063-8. [DOI] [PubMed] [Google Scholar]
- 36.Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen SH. Alzheimer's disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol. 1988;132:86–101. [PMC free article] [PubMed] [Google Scholar]
- 37.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–89. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 38.Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P, Renbing X, Peck A. Clinical, pathological, and neurochemical changes in dementia: a subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol. 1988;23:138–44. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- 39.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, De Graaff E, Wauters E, Van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, Van Swieten J, Mann D, Lynch T, Heutink P. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 40.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–25. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 41.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–41. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alonso AD, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004;279:34873–81. doi: 10.1074/jbc.M405131200. [DOI] [PubMed] [Google Scholar]
- 43.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 44.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 45.Gotz J, Chen F, Van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–5. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 46.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–5. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 47.Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003;24:1063–70. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 48.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 49.Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, Van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–6. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 50.Baki L, Shioi J, Wen P, Shao Z, Schwarzman A, Gama-Sosa M, Neve R, Robakis NK. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. EMBO J. 2004;23:2586–96. doi: 10.1038/sj.emboj.7600251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shioi J, Georgakopoulos A, Mehta P, Kouchi Z, Litterst CM, Baki L, Robakis NK. FAD mutants unable to increase neurotoxic Abeta 42 suggest that mutation effects on neurodegeneration may be independent of effects on Abeta. J Neurochem. 2007;101:674–81. doi: 10.1111/j.1471-4159.2006.04391.x. [DOI] [PubMed] [Google Scholar]
- 52.Jicha GA, Lane E, Vincent I, Otvos L, Jr, Hoffmann R, Davies P. A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J Neurochem. 1997;69:2087–95. doi: 10.1046/j.1471-4159.1997.69052087.x. [DOI] [PubMed] [Google Scholar]
- 53.Jicha GA, Berenfeld B, Davies P. Sequence requirements for formation of conformational variants of tau similar to those found in Alzheimer's disease. J Neurosci Res. 1999;55:713–23. doi: 10.1002/(SICI)1097-4547(19990315)55:6<713::AID-JNR6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 54.Jicha GA, Rockwood JM, Berenfeld B, Hutton M, Davies P. Altered conformation of recombinant fron-totemporal dementia-17 mutant tau proteins. Neurosci Lett. 1999;260:153–6. doi: 10.1016/s0304-3940(98)00980-x. [DOI] [PubMed] [Google Scholar]
- 55.Novak M, Jakes R, Edwards PC, Milstein C, Wischik CM. Difference between the tau protein of Alzheimer paired helical filament core and normal tau revealed by epitope analysis of monoclonal antibodies 423 and 7.51. Proc Natl Acad Sci USA. 1991;88:5837–41. doi: 10.1073/pnas.88.13.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA. 2003;100:10032–7. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cotman CW, Poon WW, Rissman RA, Blurton-Jones M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J Neuropathol Exp Neurol. 2005;64:104–12. doi: 10.1093/jnen/64.2.104. [DOI] [PubMed] [Google Scholar]
- 58.Kopke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993;268:24374–84. [PubMed] [Google Scholar]
- 59.Iqbal K, Grundke-Iqbal I, Smith AJ, George L, Tung YC, Zaidi T. Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc Natl Acad Sci USA. 1989;86:5646–50. doi: 10.1073/pnas.86.14.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–8. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 61.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975;72:1858–62. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. 1984;259:5301–5. [PubMed] [Google Scholar]
- 63.Iqbal K, Zaidi T, Bancher C, Grundke-Iqbal I. Alzheimer paired helical filaments. Restoration of the biological activity by dephosphorylation. FEBS. Lett. 1994;349:104–8. doi: 10.1016/0014-5793(94)00650-4. [DOI] [PubMed] [Google Scholar]
- 64.Khatoon S, Grundke-Iqbal I, Iqbal K. Guanosine triphosphate binding to betasubunit of tubulin in Alzheimer's disease brain: role of microtubule-associated protein tau. J Neurochem. 1995;64:777–87. doi: 10.1046/j.1471-4159.1995.64020777.x. [DOI] [PubMed] [Google Scholar]
- 65.Khatoon S, Grundke-Iqbal I, Iqbal K. Brain levels of microtubule-associated protein tau are elevated in Alzheimer's disease: a radioimmuno-slot-blot assay for nanograms of the protein. J Neurochem. 1992;59:750–3. doi: 10.1111/j.1471-4159.1992.tb09432.x. [DOI] [PubMed] [Google Scholar]
- 66.Khatoon S, Grundke-Iqbal I, Iqbal K. Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett. 1994;351:80–4. doi: 10.1016/0014-5793(94)00829-9. [DOI] [PubMed] [Google Scholar]
- 67.Alonso AD, Li B, Grundke-Iqbal I, Iqbal K. Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci USA. 2006;23:8864–9. doi: 10.1073/pnas.0603214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li B, Chohan MO, Grundke-Iqbal I, Iqbal K. Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol. 2007;113:501–11. doi: 10.1007/s00401-007-0207-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pei JJ, Gong CX, Iqbal K, Grundke-Iqbal I, Wu QL, Wirblad B, Cowburn RF. Subcellular distribution of protein phosphatases and abnormally phosphorylated tau in the temporal cortex from Alzheimer's disease and control brains. J Neural Transm. 1998;105:69–83. doi: 10.1007/s007020050039. [DOI] [PubMed] [Google Scholar]
- 70.Wang JZ, Gong CX, Zaidi T, Grundke-Iqbal I, Iqbal K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J. Biol. Chem. 1995;270:4854–60. doi: 10.1074/jbc.270.9.4854. [DOI] [PubMed] [Google Scholar]
- 71.Wang JZ, Grundke-Iqbal I, Iqbal K. Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res. Mol. Brain Res. 1996;38:200–8. doi: 10.1016/0169-328x(95)00316-k. [DOI] [PubMed] [Google Scholar]
- 72.Bhaskar K, Yen SH, Lee G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem. 2005;280:35119–25. doi: 10.1074/jbc.M505895200. [DOI] [PubMed] [Google Scholar]
- 73.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, Ihara Y. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J Biol Chem. 1995;270:823–9. doi: 10.1074/jbc.270.2.823. [DOI] [PubMed] [Google Scholar]
- 74.Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer's disease brain using nanoelectrospray mass spectrometry. J Neurochem. 1998;71:2465–76. doi: 10.1046/j.1471-4159.1998.71062465.x. [DOI] [PubMed] [Google Scholar]
- 75.Singh TJ, Grundke-Iqbal I, McDonald B, Iqbal K. Comparison of the phosphorylation of microtubule-associated protein tau by non-proline dependent protein kinases. Mol Cell Biochem. 1994;131:181–9. doi: 10.1007/BF00925955. [DOI] [PubMed] [Google Scholar]
- 76.Johnson GV, Hartigan JA. Tau protein in normal and Alzheimer's disease brain: an update. J Alzheimers Dis. 1999;1:329–51. doi: 10.3233/jad-1999-14-512. [DOI] [PubMed] [Google Scholar]
- 77.Iqbal K, Alonso Adel C, Chen S, Chohan MO, El-Akkad E, Gong CX, Khatoon S, Li B, Liu F, Rahman A, Tanimukai H, Grundke-Iqbal I. Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta. 2005;1739:198–210. doi: 10.1016/j.bbadis.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 78.Pei JJ, Gong CX, An WL, Winblad B, Cowburn RF, Grundke-Iqbal I, Iqbal K. Okadaic-acid-induced inhibition of protein phosphatase 2A produces activation of mitogen-activated protein kinases ERK1/2, MEK1/2, and p70 S6, similar to that in Alzheimer's disease. Am J Pathol. 2003;163:845–58. doi: 10.1016/S0002-9440(10)63445-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang JZ, Wu Q, Smith A, Grundke-Iqbal I, Iqbal K. Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett. 1998;436:28–34. doi: 10.1016/s0014-5793(98)01090-4. [DOI] [PubMed] [Google Scholar]
- 80.Anderton BH, Betts J, Blackstock WP, Brion JP, Chapman S, Connell J, Dayanandan R, Gallo JM, Gibb G, Hanger DP, Hutton M, Kardalinou E, Leroy K, Lovestone S, Mack T, Reynolds CH, Van Slegtenhorst M. Sites of phosphorylation in tau and factors affecting their regulation. Biochem Soc Symp. 2001:73–80. doi: 10.1042/bss0670073. [DOI] [PubMed] [Google Scholar]
- 81.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. Embo J. 1990;9:2431–8. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsai LH, Takahashi T, Caviness VS, Jr, Harlow E. Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development. 1993;119:1029–40. doi: 10.1242/dev.119.4.1029. [DOI] [PubMed] [Google Scholar]
- 83.Lew J, Huang QQ, Qi Z, Winkfein RJ, Aebersold R, Hunt T, Wang JH. A brain-specific activator of cyclin-dependent kinase 5. Nature. 1994;371:423–6. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- 84.Pei JJ, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF. Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer's disease neurofibrillary degeneration. Brain Res. 1998;797:267–77. doi: 10.1016/s0006-8993(98)00296-0. [DOI] [PubMed] [Google Scholar]
- 85.Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–9. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- 86.Lovestone S, Hartley CL, Pearce J, Anderton BH. Phosphorylation of tau by glycogen synthase kinase-3 beta in intact mammalian cells: the effects on the organization and stability of microtubules. Neuroscience. 1996;73:1145–57. doi: 10.1016/0306-4522(96)00126-1. [DOI] [PubMed] [Google Scholar]
- 87.Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–8. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- 88.Wagner U, Utton M, Gallo JM, Miller CC. Cellular phosphorylation of tau by GSK-3 beta influences tau binding to microtubules and microtubule organisation. J Cell Sci. 1996;109:1537–43. doi: 10.1242/jcs.109.6.1537. [DOI] [PubMed] [Google Scholar]
- 89.Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem. 1997;272:25326–32. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- 90.Spittaels K, Van den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K, Lasrado R, Vandezande K, Laenen I, Boon T, Van Lint J, Vandenheede J, Moechars D, Loos R, Van Leuven F. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. 2000;275:41340–9. doi: 10.1074/jbc.M006219200. [DOI] [PubMed] [Google Scholar]
- 91.Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear betacatenin, tau hyperphosphorylation and neurodegen-eration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Perez M, Hernandez F, Lim F, Diaz-Nido J, Avila J. Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J Alzheimers Dis. 2003;5:301–8. doi: 10.3233/jad-2003-5405. [DOI] [PubMed] [Google Scholar]
- 93.Tatebayashi Y, Haque N, Tung YC, Iqbal K, Grundke-Iqbal I. Role of tau phosphorylation by glycogen synthase kinase-3beta in the regulation of organelle transport. J Cell Sci. 2004;117:1653–63. doi: 10.1242/jcs.01018. [DOI] [PubMed] [Google Scholar]
- 94.Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent prote-olytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–72. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- 95.Patzke H, Tsai LH. Calpain-mediated cleavage of the cyclin-dependent kinase-5 activator p39 to p29. J Biol Chem. 2002;277:8054–60. doi: 10.1074/jbc.M109645200. [DOI] [PubMed] [Google Scholar]
- 96.Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–83. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- 97.Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, Davies P, Burns M, Veeranna, Nixon R, Dickson D, Matsuoka Y, Ahlijanian M, Lau LF, Duff K. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–65. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- 98.Drewes G, Lichtenberg-Kraag B, Doring F, Mandelkow EM, Biernat J, Goris J, Doree M, Mandelkow E. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 1992;11:2131–8. doi: 10.1002/j.1460-2075.1992.tb05272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ledesma MD, Correas I, Avila J, Diaz-Nido J. Implication of brain cdc2 and MAP2 kinases in the phosphorylation of tau protein in Alzheimer's disease. FEBS Lett. 1992;308:218–24. doi: 10.1016/0014-5793(92)81278-t. [DOI] [PubMed] [Google Scholar]
- 100.Roder HM, Eden PA, Ingram VM. Brain protein kinase PK40erk converts TAU into a PHF-like form as found in Alzheimer's disease. Biochem Biophys Res Commun. 1993;193:639–47. doi: 10.1006/bbrc.1993.1672. [DOI] [PubMed] [Google Scholar]
- 101.Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer's disease brains at different stages of neurofibrillary degeneration. J Alzheimers Dis. 2001;3:41–8. doi: 10.3233/jad-2001-3107. [DOI] [PubMed] [Google Scholar]
- 102.An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal IG, Winblad B, Pei JJ. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol. 2003;163:591–607. doi: 10.1016/S0002-9440(10)63687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kins S, Kurosinski P, Nitsch RM, Gotz J. Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am J Pathol. 2003;163:833–43. doi: 10.1016/S0002-9440(10)63444-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Steiner B, Mandelkow EM, Biernat J, Gustke N, Meyer HE, Schmidt B, Mieskes G, Soling HD, Drechsel D, Kirschner MW, et al. Phosphorylation of microtubule-associated protein tau: identification of the site for Ca2(+)-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tangles. EMBO J. 1990;9:3539–44. doi: 10.1002/j.1460-2075.1990.tb07563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Singh TJ, Wang JZ, Novak M, Kontzekova E, Grundke-Iqbal I, Iqbal K. Calcium/calmodulin-dependent protein kinase II phosphorylates tau at Ser-262 but only partially inhibits its binding to microtubules. FEBS Lett. 1996;387:145–8. doi: 10.1016/0014-5793(96)00485-1. [DOI] [PubMed] [Google Scholar]
- 106.Sironi JJ, Yen SH, Gondal JA, Wu Q, Grundke-Iqbal I, Iqbal K. Ser-262 in human recombinant tau protein is a markedly more favorable site for phosphorylation by CaMKII than PKA or PhK. FEBS Lett. 1998;436:471–5. doi: 10.1016/s0014-5793(98)01185-5. [DOI] [PubMed] [Google Scholar]
- 107.Bennecib M, Gong CX, Grundke-Iqbal I, Iqbal K. Inhibition of PP-2A upregulates CaMKII in rat forebrain and induces hyperphosphorylation of tau at Ser 262/356. FEBS Lett. 2001;490:15–22. doi: 10.1016/s0014-5793(01)02127-5. [DOI] [PubMed] [Google Scholar]
- 108.Scott CW, Spreen RC, Herman JL, Chow FP, Davison MD, Young J, Caputo CB. Phosphorylation of recombinant tau by cAMP-dependent protein kinase. Identification of phosphorylation sites and effect on microtubule assembly. J Biol Chem. 1993;268:1166–73. [PubMed] [Google Scholar]
- 109.Drewes G, Ebneth A, Preuss U, Mandelkow EM, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- 110.Cho JH, Johnson GV. Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J Biol Chem. 2003;278:187–93. doi: 10.1074/jbc.M206236200. [DOI] [PubMed] [Google Scholar]
- 111.Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphorylation of tau protein by casein kinase-1 converts it to an abnormal Alzheimer-like state. J Neurochem. 1995;64:1420–3. doi: 10.1046/j.1471-4159.1995.64031420.x. [DOI] [PubMed] [Google Scholar]
- 112.Sengupta A, Wu Q, Grundke-Iqbal I, Iqbal K, Singh TJ. Potentiation of GSK-3-catalyzed Alzheimer-like phosphorylation of human tau by cdk5. Mol. Cell. Biochem. 1997;167:99–105. doi: 10.1023/a:1006883924775. [DOI] [PubMed] [Google Scholar]
- 113.Liu SJ, Zhang JY, Li HL, Fang ZY, Wang Q, Deng HM, Gong CX, Grundke-Iqbal I, Iqbal K, Wang JZ. Tau becomes a more favorable substrate for GSK-3 when it is prephosphorylated by PKA in rat brain. J. Biol. Chem. 2004;279:50078–88. doi: 10.1074/jbc.M406109200. [DOI] [PubMed] [Google Scholar]
- 114.Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau:decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–8. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- 115.Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer's disease. J Biol Chem. 2000;275:5535–44. doi: 10.1074/jbc.275.8.5535. [DOI] [PubMed] [Google Scholar]
- 116.Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22:1942–50. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 117.Li L, Sengupta A, Haque N, Grundke-Iqbal I, Iqbal K. Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett. 2004;566:261–9. doi: 10.1016/j.febslet.2004.04.047. [DOI] [PubMed] [Google Scholar]
- 118.Tanaka T, Zhong J, Iqbal K, Trenkner E, Grundke-Iqbal I. The regulation of phosphorylation of tau in SY5Y neuroblastoma cells: the role of protein phosphatases. FEBS. Lett. 1998;426:248–54. doi: 10.1016/s0014-5793(98)00346-9. [DOI] [PubMed] [Google Scholar]
- 119.Cheng LY, Wang JZ, Gong CX, Pei JJ, Zaidi T, Grundke-Iqbal I, Iqbal K. Multiple forms of phosphatase from human brain: isolation and partial characterization of affi-gel blue binding phosphatases. Neurochem Res. 2000;25:107–20. doi: 10.1023/a:1007547701518. [DOI] [PubMed] [Google Scholar]
- 120.Cheng LY, Wang JZ, Gong CX, Pei JJ, Zaidi T, Grundke-Iqbal I, Iqbal K. Multiple forms of phosphatase from human brain: isolation and partial characterization of affigel blue nonbinding phosphatase activities. Neurochem Res. 2001;26:425–38. doi: 10.1023/a:1010963401453. [DOI] [PubMed] [Google Scholar]
- 121.Oliver CJ, Shenolikar S. Physiologic importance of protein phosphatase inhibitors. Front Biosci. 1998;3:D961–72. doi: 10.2741/a336. [DOI] [PubMed] [Google Scholar]
- 122.Cohen P. The structure and regulation of protein phosphatases. Annu Rev Biochem. 1989;58:453–508. doi: 10.1146/annurev.bi.58.070189.002321. [DOI] [PubMed] [Google Scholar]
- 123.Cohen P, Alemany S, Hemmings BA, Resink TJ, Stralfors P, Tung HY. Protein phosphatase-1 and protein phosphatase-2A from rabbit skeletal muscle. Methods Enzymol. 1988;159:390–408. doi: 10.1016/0076-6879(88)59039-0. [DOI] [PubMed] [Google Scholar]
- 124.Walaas SI, Greengard P. Protein phosphorylation and neuronal function. Pharmacol Rev. 1991;43:299–349. [PubMed] [Google Scholar]
- 125.Nishi A, Snyder GL, Nairn AC, Greengard P. Role of calcineurin and protein phosphatase-2A in the regulation of DARPP-32 dephosphorylation in neostriatal neurons. J Neurochem. 1999;72:2015–21. doi: 10.1046/j.1471-4159.1999.0722015.x. [DOI] [PubMed] [Google Scholar]
- 126.Li M, Guo H, Damuni Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry. 1995;34:1988–96. doi: 10.1021/bi00006a020. [DOI] [PubMed] [Google Scholar]
- 127.Li M, Makkinje A, Damuni Z. Molecular identification of I1PP2A, a novel potent heat-stable inhibitor protein of protein phosphatase 2A. Biochemistry. 1996;35:6998–7002. doi: 10.1021/bi960581y. [DOI] [PubMed] [Google Scholar]
- 128.Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem. 1996;271:11059–62. doi: 10.1074/jbc.271.19.11059. [DOI] [PubMed] [Google Scholar]
- 129.Tsujio I, Zaidi T, Xu J, Kotula L, Grundke-Iqbal I, Iqbal K. Inhibitors of protein phosphatase-2A from human brain structures, immunocytological localization and activities towards dephosphorylation of the Alzheimer type hyperphosphorylated tau. FEBS Lett. 2005;579:363–72. doi: 10.1016/j.febslet.2004.11.097. [DOI] [PubMed] [Google Scholar]
- 130.Ulitzur N, Rancano C, Pfeffer SR. Biochemical characterization of mapmodulin, a protein that binds microtubule-associated proteins. J Biol Chem. 1997;272:30577–82. doi: 10.1074/jbc.272.48.30577. [DOI] [PubMed] [Google Scholar]
- 131.Von Lindern M, Van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G. Can, a putative onco-gene associated with myeloid leukemogenesis, may be activated by fusion of its 3′half to different genes: characterization of the set gene. Mol Cell Biol. 1992;12:3346–55. doi: 10.1128/mcb.12.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Tanimukai H, Grundke-Iqbal I, Iqbal K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am J Pathol. 2005;166:1761–71. doi: 10.1016/S0002-9440(10)62486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen S, Grundke-Iqbal I, Iqbal K. I1PP2A and I2PP2A affect tau phosphorylation via association with the catalytic subunit of protein phosphatase 2A. Alzheimer's & Dementia. 2006;2:S471. doi: 10.1074/jbc.M709852200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Alonso AD, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci USA. 2001;98:6923–8. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25:59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang JZ, Grundke-Iqbal I, Iqbal K. Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer's disease. Nat Med. 1996;2:871–5. doi: 10.1038/nm0896-871. [DOI] [PubMed] [Google Scholar]
- 137.Liu F, Zaidi T, Iqbal K, Grundke-Iqbal I, Merkle RK, Gong CX. Role of glycosylation in hyperphosphorylation of tau in Alzheimer's disease. FEBS Lett. 2002;512:101–6. doi: 10.1016/s0014-5793(02)02228-7. [DOI] [PubMed] [Google Scholar]
- 138.Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3beta. FEBS Lett. 2002;530:209–14. doi: 10.1016/s0014-5793(02)03487-7. [DOI] [PubMed] [Google Scholar]
- 139.Liu F, Zaidi T, Iqbal K, Grundke-Iqbal I, Gong CX. Aberrant glycosylation modulates phosphorylation of tau by protein kinase A and dephosphorylation of tau by protein phosphatase 2A and 5. Neuroscience. 2002;115:829–37. doi: 10.1016/s0306-4522(02)00510-9. [DOI] [PubMed] [Google Scholar]
- 140.Arnold CS, Johnson GV, Cole RN, Dong DL, Lee M, Hart GW. The microtubule-associated protein tau is extensively modified with O-linked N-acetylglu-cosamine. J Biol Chem. 1996;271:28741–4. doi: 10.1074/jbc.271.46.28741. [DOI] [PubMed] [Google Scholar]
- 141.Hart GW, Kreppel LK, Comer FI, Arnold CS, Snow DM, Ye Z, Cheng X, DellaManna D, Caine DS, Earles BJ, Akimoto Y, Cole RN, Hayes BK. O-GlcNAcylation of key nuclear and cytoskeletal proteins: reciprocity with O-phosphorylation and putative roles in protein multimerization. Glycobiology. 1996;6:711–6. doi: 10.1093/glycob/6.7.711. [DOI] [PubMed] [Google Scholar]
- 142.Hart GW. Dynamic O-linked glycosylation of nuclear and cytoskeletal proteins. Annu Rev Biochem. 1997;66:315–35. doi: 10.1146/annurev.biochem.66.1.315. [DOI] [PubMed] [Google Scholar]
- 143.Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci USA. 2004;101:10804–9. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 145.Mizutani T, Sakata M, Snook ME. Pathological heterogeneity of Alzheimer type dementia. In: Iqbal K, Winblad B, Nishimura T, Takeda M, Wisniewski HM, editors. Alzheimer Disease: Biology, Diagnosis and Therapeutics. Chichester, UK: John Wiley & Sons Ltd; 1997. pp. 247–55. [Google Scholar]
- 146.Schott JM, Price SL, Frost C, Whitwell JL, Rossor MN, Fox NC. Measuring atrophy in Alzheimer diseaseL a serial MRI study over 6 and 12 months. Neurology. 2005;65:119–24. doi: 10.1212/01.wnl.0000167542.89697.0f. [DOI] [PubMed] [Google Scholar]
- 147.Karas GB, Scheltens P, Rombouts SA, Visser PJ, Van Schijndel RA, Fox NC, Barkhof F. Global and local gray matter loss in mild cognitive impairment and Alzheimer's disease. Neuroimage. 2004;23:708–16. doi: 10.1016/j.neuroimage.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 148.Best JD, Jay MT, Otu F, Churcher I, Reilly M, Morentin-Gutierrez P, Pattison C, Harrison T, Shearman MS, Atack JR. In vivo characterization of Abeta(40) changes in brain and cerebrospinal fluid using the novel gamma-secretase inhibitor N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl) cyclohexyl]-1,1, 1-trifluoromethanesulfonamide (MRK-560) in the rat. J Pharmacol Exp Ther. 2006;317:786–90. doi: 10.1124/jpet.105.100271. [DOI] [PubMed] [Google Scholar]
- 149.El Mouedden M, Vandermeeren M, Meert T, Mercken M. Reduction of Abeta levels in the Sprague Dawley rat after oral administration of the functional gamma-secretase inhibitor, DAPT:a novel non-transgenic model for Abeta production inhibitors. Curr Pharm Des. 2006;12:671–6. doi: 10.2174/138161206775474233. [DOI] [PubMed] [Google Scholar]
- 150.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 151.Clark CM, Xie S, Chittams J, Ewbank D, Peskind E, Galasko D, Morris JC, McKeel DW, Jr, Farlow M, Weitlauf SL, Quinn J, Kaye J, Knopman D, Arai H, Doody RS, DeCarli C, Leight S, Lee VM, Trojanowski JQ. Cerebrospinal fluid tau and betaamyloid: how well do these biomarkers reflect autopsy-confirmed dementia diagnoses? Arch Neurol. 2003;60:1696–702. doi: 10.1001/archneur.60.12.1696. [DOI] [PubMed] [Google Scholar]
- 152.Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, DeBernardis J, Kerkman D, McCulloch C, Soininen H, Hampel H. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer's disease. Brain. 2006;129:3035–41. doi: 10.1093/brain/awl269. [DOI] [PubMed] [Google Scholar]
- 153.Zemlan FP, Rosenberg WS, Luebbe PA, Campbell TA, Dean GE, Weiner NE, Cohen JA, Rudick RA, Woo D. Quantification of axonal damage in traumatic brain injury: affinity purification and characterization of cerebrospinal fluid tau proteins. J Neurochem. 1999;72:741–50. doi: 10.1046/j.1471-4159.1999.0720741.x. [DOI] [PubMed] [Google Scholar]
- 154.Iqbal K, Flory M, Khatoon S, Soininen H, Pirttila T, Lehtovirta M, Alafuzoff I, Blennow K, Andreasen N, Vanmechelen E, Grundke-Iqbal I. Subgroups of Alzheimer's disease based on cerebrospinal fluid molecular markers. Ann Neurol. 2005;58:748–57. doi: 10.1002/ana.20639. [DOI] [PubMed] [Google Scholar]
- 155.Anonymous. Consensus report of the Working Group on: “Molecular and Biochemical Markers of Alzheimer's Disease”. The Ronald and Nancy Reagan Research Institute of the Alzheimer's Association and the National Institute on Aging Working Group. Neurobiol Aging. 1998;19:109–16. [PubMed] [Google Scholar]
- 156.Chohan MO, Khatoon S, Iqbal IG, Iqbal K. Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett. 2006;580:3973–9. doi: 10.1016/j.febslet.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 157.Gunnarsson M, Kilander L, Sudelof J, Basun H, Lannfelt L. Reduction of hyperphosphorylated tau during memantine treatment of Alzheimer's disease. Alzheimer's & Dementia. 2006;2:63–4. [Google Scholar]
- 158.Winblad B, Poritis N. Memantine in severe dementia: results of the 9M-Best Study (Benefit and efficacy in severely demented patients during treatment with memantine) Int J Geriatr Psychiatry. 1999;14:135–46. doi: 10.1002/(sici)1099-1166(199902)14:2<135::aid-gps906>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 159.Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333–41. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 160.Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005;280:37755–62. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- 161.Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Jr, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–58. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM, Trojanowski JQ. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci USA. 2005;102:227–31. doi: 10.1073/pnas.0406361102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–29. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Michaelis ML, Ansar S, Chen Y, Reiff ER, Seyb KI, Himes RH, Audus KL, Georg GI. {beta}-Amyloid-induced neurodegeneration and protection by structurally diverse microtubule-stabilizing agents. J Pharmacol Exp Ther. 2005;312:659–68. doi: 10.1124/jpet.104.074450. [DOI] [PubMed] [Google Scholar]
- 165.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–7. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 166.Roses AD. On the discovery of the genetic association of Apolipoprotein E genotypes and common late-onset Alzheimer disease. J Alzheimers Dis. 2006;9:361–6. doi: 10.3233/jad-2006-9s340. [DOI] [PubMed] [Google Scholar]