

Abstract
The folding mechanisms for β-barrel membrane proteins present unique challenges because acquisition of both secondary and tertiary structure is coupled with insertion into the bilayer. For the porins in Escherichia coli outer membrane, the assembly pathway also includes association into homotrimers. We study the folding pathway for purified LamB protein in detergent and observe extreme hysteresis in unfolding and refolding, as indicated by the shift in intrinsic fluorescence. The strong hysteresis is not seen in unfolding and refolding a mutant LamB protein lacking the disulfide bond, as it unfolds at much lower denaturant concentrations than wild type LamB protein. The disulfide bond is proposed to stabilize the structure of LamB protein by clasping together the two sides of Loop 1 as it lines the inner cavity of the barrel. In addition we find that low pH promotes dissociation of the LamB trimer to folded monomers, which run at about one third the size of the native trimer during SDS PAGE and are much more resistant to trypsin than the unfolded protein. We postulate the loss at low pH of two salt bridges between Loop 2 of the neighboring subunit and the inner wall of the monomer barrel destabilizes the quaternary structure.
Keywords: folding β-barrel proteins, outer membrane, LamB protein, maltoporin, disulfide bond, oligomerization
1. Introduction
The outer membranes of Gram negative bacteria provide important barriers that protect cells from harmful agents while allowing uptake of nutrients by transport through β-barrel proteins [1]. In addition to transporters, outer membrane β-barrel proteins function in adhesion and virulence and act as enzymes. In Gram negative bacteria the known β-barrel proteins are found exclusively in the outer membranes, yet β-barrels are predicted to make up 2-3% of the Gram negative proteome [2]. They are also found in Mycobacteria and in the mitochondria and chloroplasts of eukaryotes [1].
Porins are a class of β-barrel proteins that provide solute diffusion channels of varying selectivity. They are homotrimers, with a channel in each monomer. The most abundant porins in Escherichia coli, the OmpF and OmpC porins, are called general porins: they allow passage of hydrophilic solutes up to a size limit that is due to constrictions of their channels evident in the crystal structures [3, 4]. The LamB protein is a well-characterized example of a specific porin: it is called maltoporin because it is required for growth on limiting concentrations of maltose and it facilitates passage of larger maltodextrins [5, 6]. Enhanced uptake of starch breakdown products through the LamB channel is likely to give the enteric bacteria a competitive advantage. A large assortment of lamB mutants has been used to probe the roles of different parts of the protein [7, 8, 9]. Once the x-ray structure provided details of its β-barrel topology and the residues lining the “greasy slide” that confer specificity to the pore [10, 11, 12], genetic analysis of the architecture of LamB protein could be targeted to determining structure-function relationships of various loops and pore residues [13, 14].
Biogenesis of outer membrane porins is complicated by their location and their structure. Because of their destined location in the outer membrane, newly synthesized porins must cross the cytoplasmic membrane and periplasmic space, thus their biogenesis involves several stages [15]. Recognition of their signal sequences as they emerge from the ribosome targets the nascent peptides for export across the cytoplasmic membrane via a complex called the SecYEG/SecA translocon in E. coli [16]. Upon entry to the periplasm, nascent porins interact with chaperones such as Skp and DegP, which maintain their unfolded state [17], and SurA, which may begin their folding process [18, 19]. Incorporation into the outer membrane utilizes a second transport complex called Bam (for β-barrel assembly machinery) that consists of a major pore in the outer membrane formed by the β-barrel BamA (or YaeT, a homolog of Omp85) and several accessory lipoproteins [20]. Just how the Bam complex facilitates outer membrane insertion is an active field of research [21].
Biogenesis of porins is also complicated by their structure: unlike α-helical membrane proteins, the β-barrel cannot assemble from previously inserted transmembrane segments because the cost of insertion into the hydrophobic core of the bilayer of individual β-strands lacking hydrogen bonds is prohibitive [22]. Rather, membrane insertion must be coupled with acquisition of both secondary and tertiary structure to produce the contiguous membrane-spanning barrel held by interstrand hydrogen bonds. Furthermore, the porins must additionally attain their quaternary structure, presumably by associations formed in the membrane. Therefore the β-barrel membrane proteins present interesting challenges to the protein folding problem. Fortunately, folding and assembly of these β-barrel proteins can be probed in vitro because they are capable of assembling directly into lipid bilayers with little or no assistance.
The most extensive folding studies of outer membrane β-barrel proteins have examined the OmpA protein, in particular the barrel domain of OmpA consisting of residues 172-325 of the peptide chain [reviewed in 23, 24]. Unfolded OmpA barrel domain that has been purified in urea or SDS folds when diluted into a variety of detergents or phospholipids. The native state of refolded OmpA has been demonstrated with various spectroscopic methods as well as conductance measurements and phage binding. In early studies, acquisition of tertiary structure inferred from faster mobility on SDS PAGE was found to be synchronous with formation of secondary structure observed by CD. Kinetic assays based on the shift in electrophoretic mobility showed that OmpA folding follows a single-step second-order rate law (pseudo-first order at high lipid concentrations) and depends on the nature of the amphiphile (head group and chain length of phospholipid) as well as the size of the lipid vesicles. The use of depth-dependent fluorescence quenchers as well as single-Tryptophan mutants allowed characterization of intermediates along the folding pathway as four Trp residues move across the bilayer and the fifth stays near the insertion site. Recently, intramolecular quenching of the individual Trp residues by nitroxides on engineered Cys residues determined when pairs of β-strands converged and demonstrated that they form the OmpA barrel concurrent with insertion into the membrane [25].
How much more complex is the assembly pathway of an oligomeric β-barrel, such as the OmpF and LamB porins of Escherichia coli? In an early study, OmpF purified in a nonionic detergent, n-octyl (polydisperse)oligo-oxyethylene (octylPOE), was denatured by heating in 6 M guanidinium chloride (GdmCl) and resolubilized in SDS [26]. Unfolded OmpF lost the characteristic minimum at 217 nm in its CD spectrum that typifies β structure, appeared as a monomer on SDS PAGE, and was digested by trypsin. Once refolded by either dialysis or dilution into octylPOE-lecithin mixed micelles in the presence of SDS, from 40 to 80% appeared as trimer on SDS PAGE, the trimer band was resistant to trypsin, and the reconstituted refolded OmpF gave conductance in lipid bilayers characteristic of native OmpF pores.
For a direct comparison of the in vitro assembly of OmpA and OmpF, OmpF was purified using chaotropic agents (both GdmCl and urea) and alcohol precipitation with no detergent [27]. This procedure produced protein in 8 M urea that was unfolded according to its CD and fluorescence spectra. Refolding by dilution to 20 mM urea produced a very low yield of OmpF trimer with addition of DMPC alone, but with a 1:1 mixture of DMPC:dodecylmaltoside (DM) over 70% of the OmpF was trimers and another 25% dimers. Both trimers and dimers were resistant to trypsin, and the refolded OmpF had characteristic CD and fluorescence spectra as well as conductance properties of native OmpF. The kinetics of assembly from aqueous solutions of OmpA or OmpF in urea exhibited a striking difference. The appearance of trypsin-resistant OmpF trimer was much slower (15 % after 3 hours in DMPC and 73% after 5 hrs in DMPC + DM) than appearance of folded OmpA (70% after three hours in DMPC). The slower assembly of OmpF was attributed to the oligomerization step.
Using the ability to refold β-barrel membrane proteins from aqueous solutions, a recent study expanded this approach to compare the folding efficiencies of nine outer membrane proteins that were overexpressed and solubilized in urea [28]. The “heat modifiability” of these proteins produces different bands of folded and unfolded proteins on SDS PAGE and thus is used to quantitate their folding. This allowed determination of the effects of many different bilayer compositions on the folding process. Although the outer membrane proteins tested vary in their ability to fold in any single bilayer environment, they generally folded better in thinner bilayers, in bilayers with increased curvatures, and at higher pH values. Interestingly, only four of the nine outer membrane proteins could fold into small vesicles prepared with E. coli lipid extracts. The one trimeric porin included, OmpF, folded only with phosphatidyl choline with unnaturally short acyl chains. OmpF also does not conform to the “heat modifiability” that is the basis of the SDS-PAGE assay for folding. Rather than two bands corresponding to folded and unfolded species, OmpF exhibits three bands on SDS PAGE: folded trimers, unfolded monomers, and presumed dimers.
The folding and assembly of porins is a significant aspect of outer membrane biogenesis, given their critical role in nutrient transport. Folding studies can also reveal new information about the effects of mutations on porin structure. While numerous studies have been reported on other outer membrane proteins, there has been no previous information about the in vitro folding of LamB protein. Such investigations can take advantage of the wealth of lamB mutants to ultimately probe many aspects of its assembly. In the approach we have taken, intrinsic fluorescence of purified LamB protein in detergent is used to monitor unfolding and refolding of both the wild type protein and a mutant protein lacking the disulfide bond. We have also tested the folding state of a compact monomer observed at low pH on SDS PAGE as a means to assay trimer dissociation separate from denaturation.
2. Materials and Methods
2.1 Materials
N-octyl (polydisperse)oligo-oxyethylene (octylPOE) was purchased from Alexis and guanidium chloride (8 M solution) purchased from Pierce. Highly-purified SDS was purchased from Gallard-Schlesinger. Trypsin from bovine pancreas and soybean trypsin inhibitor were obtained from Calbiochem. Low Molecular Weight Protein Standards, including Prestained standards, were purchased from BioRad. Phosphorylase A was purchased from Boehringer Mannheim, Catalase, Fumarase and Glycerolaldehydephosphatase from Boehringer, and Cytochrome C from Fluka. Carboanhydrase B, dioleophosphatidyl choline and egg phosphatidyl choline were purchased from Sigma. All chemicals were reagent grade.
2.2 Protein purification
LamB protein was purified from octylPOE extracts of whole envelopes of E. coli strain MCR106 harboring high expression plasmids for wild type or for the C22S mutant [29] using starch affinity chromatography as described in reference 30. Purified protein in 20 mM Hepes (N-2-Hydroxypiperazine-N-ethanesulfonic acid), 0.5% octylPOE and 0.02% NaN3 was stored in glass vials at 4°C. Protein concentration was determined by the Lowry assay [31] with 0.1% SDS added to samples containing octylPOE [32].
2.3 Gel electrophoresis
SDS PAGE was carried out on an 11% resolving gel as previously described [29] with BioRad low molecular weight standards, except where indicated on a 10% resolving gel with a noncommercial mixture of standards composed of Phosphorylase A (95 kDa), Catalase (57.5 kDa), Fumarase (49 kDa), Glycerolaldehydephosphatase (36 kDa), Carboanhydrase B (29 kDa), and Cytochrome C (11.7 kDa). Every gel included additional standards containing 2 μg samples of LamB protein with and without heat treatment (100° C for 10 min). For electrophoresis of unfolded and refolded samples containing significant levels of GdmCl, precipitates that formed in the gel samples were removed by suctioning the gel lanes. The amount of SDS in the sample buffer was varied from the concentration in the running buffer (0.05%) to the concentration typically used for sample solubilization (2%) to see the effect of SDS concentration on the formation of compact monomer. For elution of the compact monomer from unstained gels, BioRad prestained standards were used. Horizontal strips cut from the region near 37 kDa were immersed in buffers containing 200 mM phosphate with 200 mM glycine or citrate and gently agitated overnight. The pH of the elution step was varied from 2.9 to 6.0 and the eluate examined by SDS PAGE for presence of the compact monomer. Ninety-seven percent of the eluted material was compact monomer when elution was carried out in 200 mM phosphate-200 mM glycine, pH 3.8.
2.4 Unfolding and Refolding
For unfolding studies, samples of LamB protein in 20 mM Hepes pH 7.4, 0.5% octylPOE, 0.02% NaN3, were mixed with the same buffer containing 7.3 M GdmCl to give 45 μg/ml of protein in 0 to 7 M final concentrations of denaturant and stored at 25°C for 24 hours.
For refolding, LamB protein (∼2 mg/ml) was incubated in 6.8 M GdmCl 24 hr at 25°C, then diluted into buffers containing varying amounts of GdmCl and allowed to refold overnight at 25°C. These steady state measurements exhibited hysteresis between the unfolding and refolding processes (see Fig. 3A in section 3.2), preventing thermodynamic analysis.
Fig. 3.
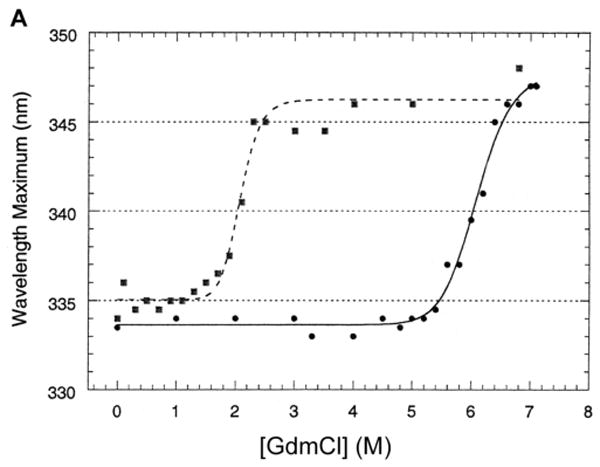
Unfolding and refolding assayed by the shift of fluorescence emission maximum.
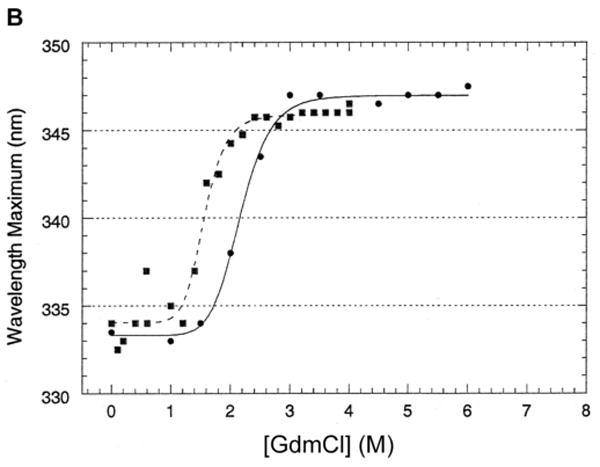
A. To study refolding, LamB protein at high concentration (1.9 mg/ml) was incubated in 6.8 M GdmCl for 24 hr at 25°C to unfold, then diluted into buffers containing varying amounts of GdmCl and allowed to refold (■) to steady state overnight at 25°C. For direct comparison, data for unfolding (●) is included for samples prepared at the same time.
B. The same experiments were carried out with a mutant LamB protein lacking the disulfide bond. LamB protein containing the C22S point mutation was treated with GdmCl for unfolding (●) and refolding (■) studies as described in Figures 1B and 3A. Compared to studies with the wild type protein, the unfolding occurs at much lower denaturant concentrations and the hysteresis is greatly reduced.
2.5 Fluorescence
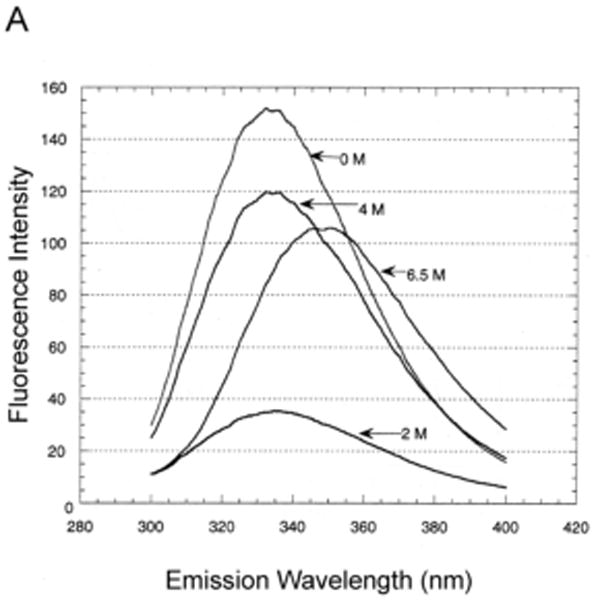
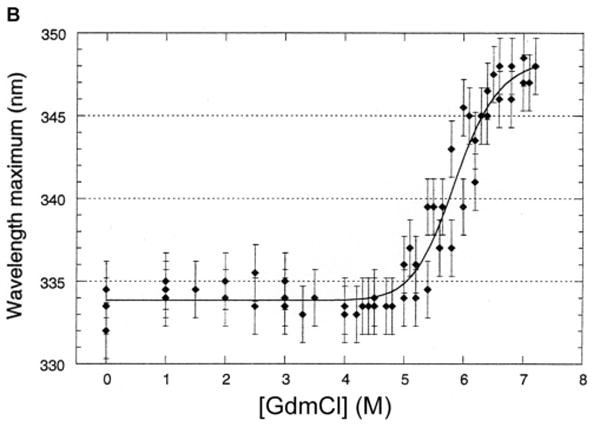
Fluorescence was measured at 25°C at a steady state obtained after sample incubation for 24 hr using a Proton Technology International Quantum Master spectrofluorometer, with a Xenon lamp at 75 watts and slit widths of 5 nm (excitation) and 1 nm (emission). Following excitation at 290 nm, emission spectra were collected from 300 to 400 nm and corrected using the PTI Felix software to subtract the buffer background. In all unfolding studies, a sharp decrease in fluorescence intensity occurred at low levels of denaturant but did not persist in samples of higher concentration of GdmCl, suggesting that it resulted from interaction of detergent with the denaturant (at low concentrations) rather than from conformational changes of the protein. For this reason the fluorescence intensity was not used to determine the fraction unfolded, and the unfolding is determined from the shift in λmax. A similar problem did not occur in the refolding process, indicating that dilution from a high concentration of denaturant avoided the interaction. For consistency, both unfolding and refolding are given as the change in λmax. The large number of aromatic residues in LamB protein (including 19 Trp per monomer) produces a strong but broad peak of intrinsic fluorescence. Because of the breadth of the peaks, many samples were done in triplicate and the standard deviation of the values was used to provide the error bars in Fig. 1B in section 3.1. Given this clear result, the data were fit to third order polynomials for this and all following unfolding and refolding curves.
Fig. 1.
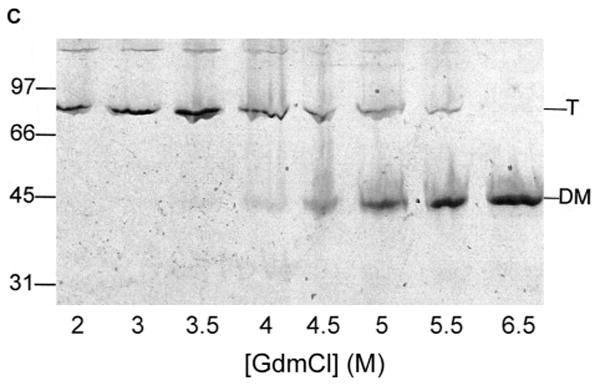
Unfolding of purified LamB protein in guanidium chloride.
Samples containing 45 μg/ml LamB protein in 20 mM Hepes, 0.5% octylPOE and 0.02% NaN3 were incubated in the indicated concentrations of guanidium chloride for 24 hours at 25°C prior to fluorescence scanning and gel electrophoresis.
A. Fluorescence was measured at 25°C after excitation at 290 nm. Representative fluorescence emission spectra are shown to illustrate the highly reproducible drop in intensity at low concentrations (e.g. 2 M) of GdmCl. The loss of intensity did not occur at higher concentrations (e.g. 4 M and 6.5 M). The same drop in intensity was also observed when the C22S LamB protein was unfolded.
B. Steady state unfolding of LamB protein is indicated by the shift in emission maximum of the intrinsic fluorescence as the concentration of GdmCl increases. The data from several experiments were fit to a polynomial curve, and error bars were standardized using a 1.7 nm deviation calculated from those denaturant levels with multiple scans.
C. Unfolding is accompanied by a mobility shift on SDS PAGE. Samples containing 5 μg protein were removed from the fluorescence samples, added to sample buffer with a final concentration of 2% SDS, and electrophoresed without heat treatment. The mobility of trimer (T) and denatured monomer (DM) and molecular weight standards are indicated.
2.6 Circular Dichroism
Circular dichroism spectra were obtained on an Aviv 410 circular dichroism spectrophotometer. The spectra were measured in a 0.1 nm cuvette at 25°C scanned over a range of 210 to 300 nm in steps of 1 nm. Each scan was repeated and the average of the two scans was corrected by subtracting the appropriate buffer blank. Each curve was terminated when the diode became excessively high (above 600). For CD experiments, samples containing 0.5 mg purified LamB protein were precipitated with ethanol overnight at -20°C and dissolved in 20 mM phosphate, pH 7.0, 0.5% octylPOE, 0.02% NaN3, with and without 6.0 M GdmCl.
The compact monomer of LamB was eluted from an unstained SDS gel in 200 mM phosphate - 200 mM glycine, pH 4.0, and diluted ten-fold for CD measurements.
2.7 Transport activity
The liposome swelling assay was used to measure transport activity of refolded LamB protein [6]. Multilamellar proteoliposomes were prepared and assayed as described in reference 6, except the lipids used were dioleophosphatidyl choline and egg phosphatidyl choline, in a 2:1 ratio by weight. Because high levels of GdmCl interfered with liposome formation, unfolded LamB protein was not tested, and refolded LamB protein was dialyzed to 0.1M GdmCl before incorporation into liposomes. Concurrent batches of control liposomes contained no protein and 2 μg untreated LamB protein. Uptake of the sugars maltose, glucose, lactose, and sucrose were tested after determining the isotonic concentration with raffinose. Specificity of the uptake channel was confirmed by very low uptake of lactose and sucrose. Transport activity was determined from initial rates of uptake of maltose measured in triplicate and given as relative permeability based on the rate of swelling of the liposomes, derived from d(1/OD500/dt where OD500 is the optical density at 500 nm [6].
2.8 Protease treatment
Trypsin was added to LamB samples at a ratio of 1:100 by weight (trypsin:LamB), incubated at 37°C for 30 min and then inactivated with trypsin inhibitor at a ratio of 1:3 (trypsin:trypsin inhibitor) by weight. Trypsin-treated samples were solubilized in sample buffer containing 2% SDS and analyzed by SDS PAGE without heat treatment. Protease sensitivity of the compact monomer was determined with the same protocol using LamB protein that was stored in 20 mM phosphate-20 mM glycine pH 2.5, 0.5% octylPOE, at 4°C and then dialyzed back to pH 7.2 in 20 mM phosphate-20 mM glycine, 0.5% octylPOE.
3. Results
3.1 Unfolding of purified LamB protein in detergent
Like other β-barrel proteins, purified LamB protein is very stable in mild detergent, such as Triton X-100 or octylPOE, and resists denaturation by SDS and urea in the absence of heat treatment. Samples of LamB protein that have not been boiled show a mobility corresponding to ca. 95 kDa on SDS PAGE, much lower than the Mr of 142 kDa, which indicates the trimers are compact, i.e. folded, in sample buffer containing 2% SDS. Concentrations of urea as high as 9 M do not unfold LamB protein at room temperature or at 70°C, although the protein does unfold in higher urea concentrations (9.4 M) at 70°C [29].
We were able to unfold LamB protein in detergent (octylPOE) at 25°C with ≥6 M guanidinium chloride (GdmCl), observing a shift in the fluorescence emission maximum from 334 nm (native) to 346 nm (unfolded) (Fig, 1). Intensity of fluorescence dropped markedly at low concentrations of denaturant but not at higher concentrations (Fig. 1A). Therefore the initial intensity decrease did not reflect unfolding of the protein and must be due instead to some interactions of detergent and denaturant at the low concentrations. However, the unfolding process is well characterized by the increase in λmax as a function of the concentration of GdmCl, which shows a steep increase between 4.5 M and 7 M denaturant (Fig. 1B). Consistent with these results, observation of these samples on SDS PAGE revealed the shift from trimer (folded) to monomer (unfolded) started at 4.5 M and was complete at 6.5 M GdmCl (Fig. 1C). Also the circular dichroism spectrum of a sample in 6 M GdmCl showed no secondary structure, in contrast to the readily apparent β structure seen in the CD spectrum of native LamB protein (Fig. 2). When purified LamB protein was first reconstituted into phospholipids, even these very high levels of denaturants did not unfold it.
Fig. 2.
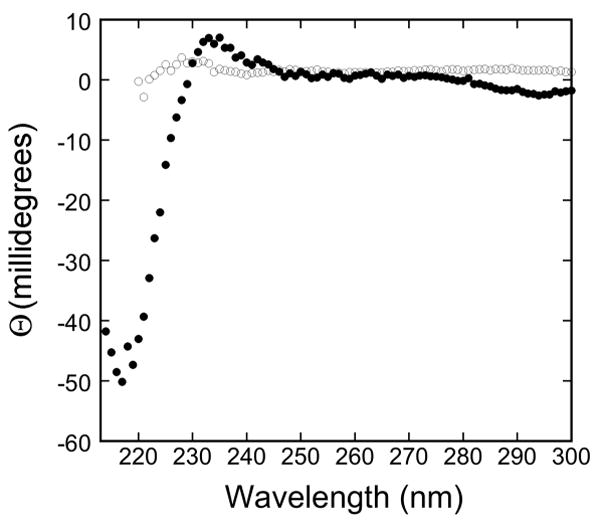
Circular dichroism spectra for native and unfolded LamB protein.
The circular dichroism spectra of purified LamB protein in 20 mM phosphate, pH 7.0, 0.5% oPOE, 0.02% NaN3, with no additions (●) and with 6 M GdmCl (○). The minimum at 217 nm observed in the protein with no denaturant added is characteristic of β structure.
3.2 Refolding of unfolded LamB protein
After LamB protein was unfolded by overnight incubation in 6.8 M GdmCl, it refolded upon dilution. Refolding, as assayed by the shift of fluorescence emission maximum, occurred only when concentrations of GdmCl around 2M had been reached, revealing extreme hysteresis between unfolding and refolding at steady state (Fig. 3A). Refolding also occurred after overnight dialysis to reduce the GdmCl (not shown) but less consistently, as dialysis sometimes produced precipitation.
Transport activity of refolded LamB protein was measured by the liposome swelling assay, which gives only relative values of permeability, not the absolute values of permeability coefficients [6]. Because 1-2 M GdmCl was incompatible with liposome formation, the refolded protein was further dialyzed to 0.1 M GdmCl. Refolded LamB protein exhibited the expected specificity, with very rapid uptake of maltose and glucose and very slow uptake of lactose and sucrose. Quantitative rates of uptake of maltose in proteoliposomes derived from averages of triplicate samples were 1.157 +/- 0.004 for refolded LamB protein dialyzed to 0.1 M GdmCl; 1.014 +/- 0.072 for refolded LamB protein after dialysis and storage at 4°C for one week; and 1.357 +/- 0.027 for control LamB protein (untreated native protein). The transport activity of refolded LamB protein reconstituted into liposomes corresponded to the proportion of trimer that appeared on SDS PAGE, both indicating that ca. 80% was refolded (Fig. 4A). Furthermore, the refolded LamB protein was resistant to trypsin (Fig. 4B), as expected for folded LamB trimer and not the denatured monomer [33].
Fig. 4.
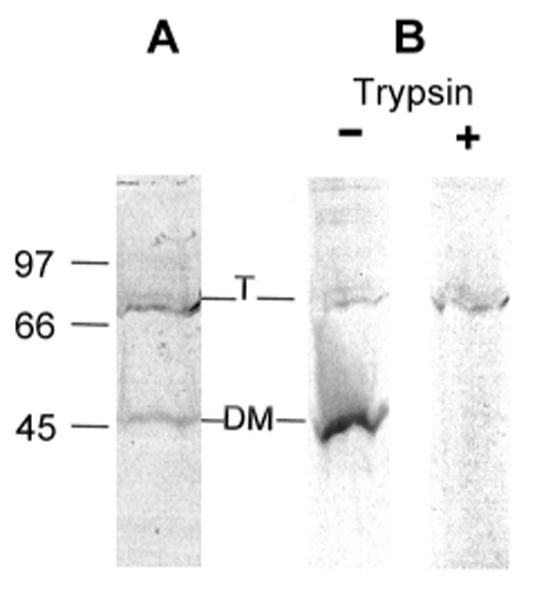
Characterization of refolded LamB protein.
The refolded LamB protein was analyzed by SDS PAGE without heat treatment. Note that residual GdmCl causes the bands to be wavy.
A. Purified LamB protein (0.8 μg/ml) in 0.5% octylPOE was unfolded overnight in 7.4 M GdmCl and confirmed as unfolded by fluorescence. Then it was diluted to 1.25 M GdmCl and sampled for SDS PAGE without further incubation. Both trimer and denatured monomer are visible on the gel.
B. Trypsin resistance of refolded LamB protein was determined after unfolded LamB protein (incubated in 6.8 M GdmCl overnight) was diluted to give only 20% refolded. When this material is treated with trypsin for 30 min at 37°C, the denatured monomer is completely digested, while the refolded trimer is resistant to digestion.
3.3 Unfolding and refolding of mutant LamB protein lacking the disulfide bond
The purified LamB C22S mutant protein, which lacks the intrasubunit disulfide bond of the wild type protein, has been shown to have decreased thermal stability [29]. When unfolded with GdmCl under the same conditions as the wild type LamB protein, it unfolded between 2 and 3 M GdmCl (Fig. 3B). This is markedly different from unfolding the wild type protein. Since it refolded after dilution to under 2M GdmCl (Fig. 3B), the C22S mutant protein exhibits much less hysteresis between unfolding and refolding curves than that of the wild type LamB protein.
3.4 Reversible dissociation of LamB protein trimer observed on SDS PAGE
We have reported the appearance of two types of monomers apparent on SDS PAGE when purified LamB protein was treated with HCl. The process is reversible, as neutralization with NaOH restored the trimer [29]. One of the monomers corresponds to the denatured monomer routinely seen at a position of Mr 47 kDa. The other is a compact monomer with a higher mobility, appearing at 37 kDa on an 11% resolving gel and 35 kDa on a 10% resolving gel. This apparent size makes it approximately one third of the apparent size of the folded trimer on SDS PAGE. We show here that the compact monomer is likely a folded monomer, and therefore the observed gel shift is indicative of dissociation of the LamB trimer without unfolding.
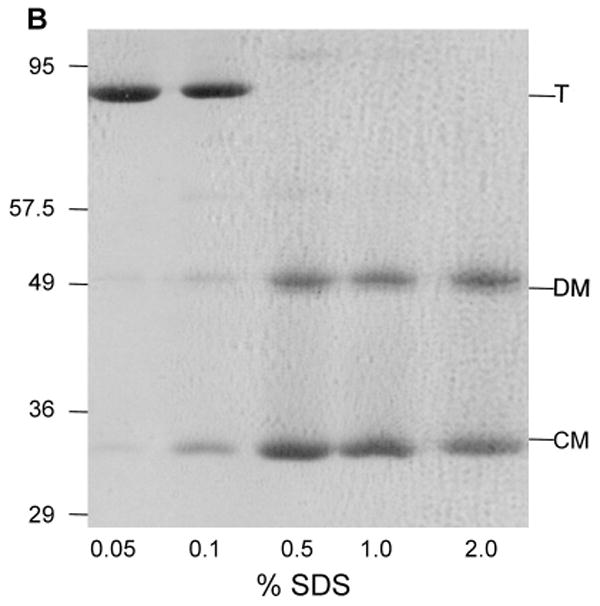
LamB trimer dissociates to compact monomer during dialysis to pH in the range of 2.5-3 (Fig. 5A). At pH above 4.5 the trimer did not dissociate, and at pH ∼2 the protein denatured (not shown). Increased ionic strength (1 M NaCl) or addition of reducing agents (β-mercaptoethanol, dithiothreitol, or p-chloromercuribenzoic acid) had no effect on either trimer dissociation or reassociation upon neutraliation (not shown). However, the presence of ionic detergent proved to be critical, as shown by varying the concentration of SDS in the sample buffer prior to PAGE. At 2% SDS in the electrophoresis sample, the dissociation of trimer to a mixture of compact monomer and denatured monomer was observed; decreasing SDS concentrations to 0.1% or less prevented the dissociation of LamB trimer (Fig. 5B).
Fig. 5.
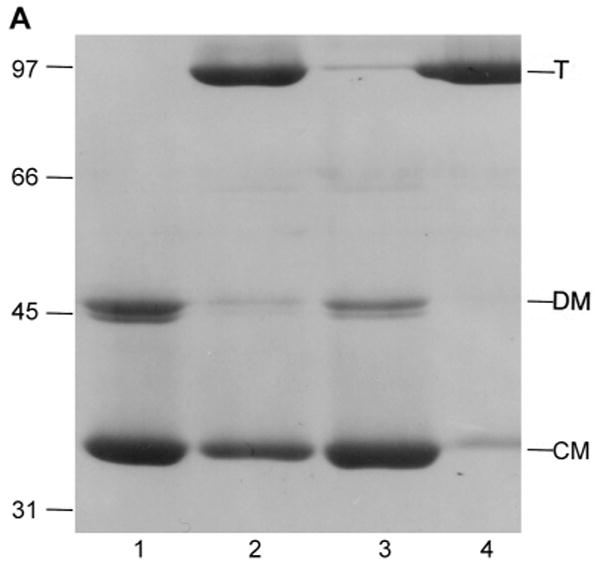
Characterization of low pH LamB monomer by SDS PAGE.
A. Dissociation of LamB protein trimer by dialysis at low pH is reversed by neutralization. Purified LamB protein (0.5 mg) was dialyzed overnight against 100 ml of 20 mM phosphate - 20 mM glycine, pH 2.5, 1% octylPOE or against 100 ml of 20 mM phosphate - 20 mM citrate, pH 3.5, 1% octylPOE. Aliquots were withdrawn and neutralized by addition of NaOH where indicated. All samples were loaded on to the gel without heat treatment. Lane 1, LamB dialyzed to pH 2.5; Lane 2, LamB neutralized after dialysis at pH 2.5; Lane 3, LamB dialyzed to pH 3.5; Lane 4, LamB neutralized after dialysis at pH 3.5. Mr of standards are indicated in kDa, and the positions of trimer (T), denatured monomer (DM) and compact monomer (CM) are marked.
B. Dissociation of LamB protein was shown to be a function of SDS concentration during SDS PAGE by varying the percent SDS in the sample buffer. The resolving gel in this case is 10% acrylamide. Samples containing 12 μg of purified LamB protein were treated with 33 mM HCl, then added to an equal volume of sample buffer containing no reducing agent and a varying amount of SDS. The concentrations of SDS in the sample are: Lane 1, 0.05%; Lane 2, 0.1%; Lane 3, 0.5%; Lane 4, 1.0%; and Lane 5, 2.0%. All samples were loaded on to the gel without heat treatment. The positions of molecular weight standards from a noncommercial mixture (see section 2.3) are shown. In addition, trimer (T), denatured monomer (DM) and compact monomer (CM) are indicated.
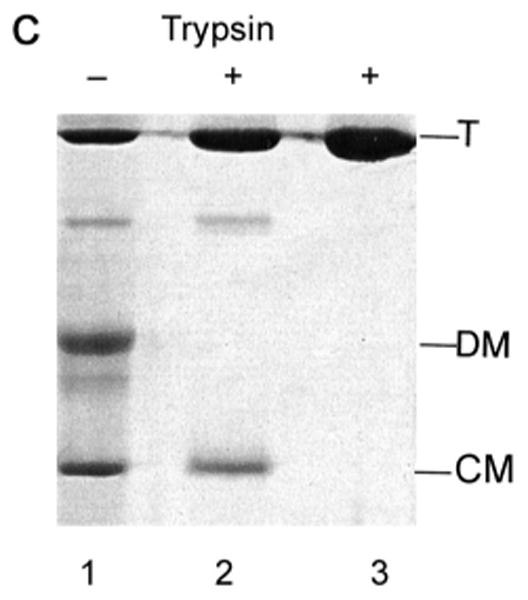
C. The resistance of the compact monomer to digestion by trypsin was determined with a stored sample of purified LamB protein that had been dialyzed overnight into 20 mM phosphate-20 mM glycine buffer, pH 2.6, with 0.5% oPOE. After storage at 4°C, it was again dialyzed to 10 mM phosphate-10 mM glycine, pH 7.2, 0.5% oPOE. Both samples, each containing 42 μg of LamB protein, along with untreated LamB protein (pH 7.2) were treated with 1 μg trypsin 30 min at 37°C and then 3 μg of trypsin inhibitor was added. SDS PAGE was carried out without heat treatment. Lane 1 and 2 are samples dialyzed back to pH 7.2 after storage with and without trypsin. Lane 3 is native LamB with trypsin. The positions of trimer (T), denatured monomer (DM) and compact monomer (CM) are shown.
While SDS stabilizes the compact monomer during gel electrophoresis, it did not stabilize it in solution, where as little as 0.1% SDS caused the protein to precipitate. Without SDS the sample either stayed a trimer or denatured at low pH during purification by gravity gel filtration or high pressure gel filtration (not shown). Similarly, analytical ultracentrifugation of a sample of LamB protein dialyzed to pH 2.5 in 1% octylPOE in the absence of SDS gave S20 of 6.2, corresponding to a Mr of 142 kDa and indicative of a trimer. Again the presence of SDS interfered, since adding 1% SDS precipitated LamB protein dialyzed to low pH. After addition of 0.05% SDS, the low pH sample did not resolve on the analytical untracentrifuge.
In order to determine the secondary structure of the compact monomer from its CD spectrum a sufficient quantity of the compact monomer was eluted at pH 3.8 from all lanes of an unstained SDS gel. The eluted material was unfortunately quite dilute, but it exhibited the minimum at 217 nm typical of β structure by circular dichroism of folded LamB protein (not shown).
Trypsin treatment was used to test the folded state of the compact monomer. When acid-treated LamB protein that had been stored at 4°C was dialyzed back to pH 7.2 it did not completely reassemble to trimers, allowing observation on SDS PAGE of the four bands corresponding to trimer, presumed dimer, denatured monomer and compact monomer. Treatment of this mixture with trypsin resulted in complete digestion of denatured monomer, while the trimer was resistant, as expected (Fig. 5C). The compact monomer was quite resistant to trypsin, being one third digested compared to the trimer.
4. Discussion and Conclusions
We report here the first in vitro folding studies of LamB protein, a porin that has been extensively characterized using the tools of biochemistry, genetics, and structural biology. Unfolding of LamB protein in nonionic detergent requires very high concentrations of denaturant, ≥ 6 M GdmCl, as shown in Fig. 1. Clearly some of the 19 Trp residues per LamB monomer become exposed to a more polar environment upon unfolding, shifting the fluorescence emission maximum from 334 nm to 348 nm. Similar shifts have been reported for OmpA and OmpF: OmpA, with 5 Trp residues, exhibits shifts from 330 nm (folded) to 348 nm (unfolded) [34] and OmpF, with only 2 Trp/monomer, exhibits shifts from 332 nm (folded) to 350 nm (unfolded) [27]. The unfolding of LamB protein was confirmed by loss of the minimum at 217 nm in its circular dichroism spectrum and appearance as denatured monomer on SDS PAGE. Refolding upon dilution did not occur until the level of GdmCl reached about 2 M. The refolded protein was trimeric, resistant to trypsin, and active in proteoliposomes.
The extreme hysteresis between unfolding and refolding observed in wild type LamB protein was not observed in the LamB mutant C22S that lacks the disulfide bond. Rather, the purified C22S protein unfolds at 2-3 M GdmCl (Fig. 3). The disulfide bond between Cys22 and Cys38 is located in Loop 1, a long loop that folds into the interior of the β-barrel [10]. The unfolding of the C22S mutant protein at such low levels of GdmCl is consistent with its loss of thermal stability [29] and indicates an important role for the disulfide bond in the stability of the wild type protein.
It is unusual to find disulfide bonds in outer membrane proteins. Other porins in E. coli lack Cys residues, and yet they are highly stable molecules due to the extensive hydrogen bond network between the β strands. So why does the lack of the disulfide bond in LamB protein lead to loss of stability? The crystal structure of the LamB monomer provides clues to this matter [10]. Loop 1, which contains the disulfide bond, is one of three long loops that fold inward to fill much of the channel of the LamB monomer. The disulfide bond forms a clasp on the two sides of Loop 1, apparently holding them together as they start to cross the lumen along one wall of the monomer interior. As Loop 1 reaches the far wall of the barrel a number of its polar residues, namely Thr25, Thr26, Gln29, Ser30 and Tyr32, make very close contact with the inner wall of the β-barrel. Unclasping Loop 1 by the absence of the disulfide bond in the C22S mutant would allow its two sides to separate, making it less constrained as it traverses the channel. Given its close fit in the lumen in the native structure, it is likely that unclasped Loop 1 in the mutant would more readily interfere with other nearby loops, such as Loop 6, part of which folds into the channel and part of which contributes to the protective structure over the channel entrance, and Loop 2, which hooks into the adjacent subunit. The crowding of loops could allow denaturants or heat to more easily disrupt the barrel of LamB protein.
Trimer assembly, an important step in porin biogenesis, is difficult to isolate in in vitro folding studies since trimer dissociation is normally accompanied by denaturation. We use low pH to dissociate the LamB trimer to a compact or folded monomer, detected by its increased electrophoretic mobility in samples with no heat treatment prior to SDS PAGE. Although the Mr of the LamB trimer is 142 kDa, it appears as a band at ∼95 kDa on SDS PAGE, indicating it is compact or folded. The compact monomer runs as a 37 kDa band in 11% acrylamide and 35 kDa in 10% acrylamide (Fig. 5B), roughly a third of the size of the folded trimer. We find the appearance of the compact monomer requires SDS concentrations higher than 0.1% in the electrophoresis sample. Including SDS during dialysis to low pH causes LamB protein to aggregate, thus the compact monomer of LamB protein appears to be unstable in solution. However, its detection on SDS PAGE allows separation of the dissociation step from denaturation of LamB in electrophoretic mobility assays.
A strong indicator of whether the compact monomer observed on SDS PAGE is folded is its resistance to trypsin digestion. Trypsin resistance has been used to indicate folding of electrophoresis samples of other outer membrane proteins, including both OmpF and OmpA [27, 35]. In the case of OmpF, where three bands are visible, the monomer is denatured and is digested by trypsin, while both trimers and dimers are trypsin resistant. A compact or folded monomer of OmpF has never been reported and cannot be observed at low pH because the protein is unstable in SDS below pH 4.5 [27]. The significant trypsin resistance of the compact monomer of LamB protein (Fig. 5C) contrasts with the complete digestion of the denatured monomer. This result strongly suggests the compact monomer is folded, which indicates that the low pH treatment has dissociated the trimer into folded monomers.
How does acidic pH destabilize the quaternary structure of LamB protein? The crystal structure reveals nonpolar surfaces between monomers [10]. The only acidic residue whose side chain points into the interface between subunits is Asp78, at the end of Loop 2. Loop 2 plays a special role in quaternary structure, in that it hooks into the β-barrel of a neighboring subunit. One residue at the tip of this loop, Trp74, actually forms part of the “greasy slide” lining the pore of the adjacent subunit [10]. Trp74 is flanked by two acidic residues that can form salt links with residues of the barrel wall in the neighboring subunit: Asp73 is 2.59 Å from Arg82 on the adjacent chain, and Glu75 is 4.06 Å from Lys104 in the adjacent chain (determined with PyMol). Since this pattern is repeated three times in the trimer, it makes a significant contribution to trimer stability. Protonation at pH∼3 would disrupt these salt links and thus destabilize the interaction between the hook and the inner wall of the barrel of the adjacent subunit. While OmpF has a similar hook made by Loop 2, which also contains two acidic residues [3], it cannot be determined whether they play a similar role in trimer stabilization since lowering the pH completely denatures the OmpF protein [27].
When electrophoretic mobility is used to monitor the folding reactions of oligomers, it is important to determine the species each band represents. In samples of purified LamB protein that were stored at low pH prior to neutralization, four species of the protein are seen on SDS PAGE, and all four were proven to be LamB protein by Cleveland analysis [29]. Thus the faint band below the trimer is likely to be a dimer and is partially trypsin-resistant (see Fig. 5C). Trypsin-resistant dimers of OmpF have also been observed on SDS PAGE [27]. Do dimers represent assembly intermediates? Like the compact monomer of LamB, dimers have only been observed on SDS PAGE. Dimers were not observed in experiments of LamB assembly performed in cells [35]. These studies followed the appearance of newly synthesized LamB protein using immunoprecipitation with trimer-specific and monomer-specific antibodies. While monomer-specific antibodies do not react with LamB trimer and thus would not be expected to react with dimer, the trimer-specific antibodies do pull down some monomers and could possibly detect dimers. The precursor to trimers in the in vivo pathway for LamB assembly was not dimers but metastable trimers, detected as trimers that dissociate to monomers with heat treatments at 50-60°C before SDS PAGE. A lamB mutant that was defective in trimer assembly at the restrictive temperature (42°C) accumulated LamB monomers that were degraded in cells. This mutant would be a good candidate for future folding studies, as it would be especially interesting to see if the mutant protein purified from cells grown at 37°C formed a more stable compact monomer.
These folding studies of LamB protein revealed two properties that are not observed in folding studies of other OMPs. First, starting with native protein in detergent, conventional unfolding and refolding studies reveal a clear effect of the disulfide bond on protein stability. Second, the ability to detect on SDS PAGE a folded monomer separates the dissociation of the trimer from unfolding. Given the large number of lamB mutants that have been characterized in several laboratories, application of these methods to altered LamB proteins is likely to reveal more about the assembly process of these β-barrel trimers,
Research highlights
Unfolding and refolding LamB protein in detergent exhibits extreme hysteresis.
Without its disulfide bond, LamB protein unfolds in low denaturant concentrations.
The disulfide bond clasps the sides of L1 at the inner wall of the β-barrel.
Low pH dissociates the LamB trimer to folded monomers, observable on SDS PAGE.
Low pH disrupts salt bridges between L2 of the adjacent subunit and the β- barrel.
Acknowledgments
We thank Lin Yuan for help with production of purified LamB protein and Kai Hui for early contributions to fluorescence experiments at SFSU. We also thank Jim Keck and Rachel Bernstein in the laboratory of Prof. Susan Marqusee at the University of California, Berkeley, for circular dichroism measurements and Ariel Lustig of the University of Basel for analytical ultracentrifugation. The work was supported by NIH Grant #R15 GM54310 to ML.
The abbreviations used are
- octylPOE
n-octyl (polydisperse)oligo-oxyethylene
- SDS
sodium dodecyl sulfate
- DM
dodecylmaltoside
- GdmCl
guanidinium chloride
- DTT
dithiothreitol
- Hepes
N-2-Hydroxypiperazine-N-ethanesulfonic acid
- CD
circular dichroism
- PAGE
polyacrylamide gel electrophoresis
- DMPC
dimyristoylphosphatidylcholine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nikaido H. Molecular Basis of Bacterial Outer Membrane Permeability Revisited, Microbiol. Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gromiha MM, Suwa M. Current developments on β-barrel membrane proteins. Curr Protein Pept Sci. 2007;8:580–599. doi: 10.2174/138920307783018712. [DOI] [PubMed] [Google Scholar]
- 3.Cowan SW, Schirmer T, Rummel G, Steiert M, Ghosh R, Pauptit RA, Jansonius JN, Rosenbusch JP. Crystal structures explain functional properties of two E. coli porins. Nature. 1992;358:727–733. doi: 10.1038/358727a0. [DOI] [PubMed] [Google Scholar]
- 4.Basle A, Rummel G, Storici P, Rosenbusch JP, Schirmer T. Crystal structure of osmoporin OmpC from E. coli at 2.0 Å. J Mol Biol. 2006;362:933–942. doi: 10.1016/j.jmb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 5.Szmelcman S, Hofnung M. Maltose transport in Escherichia coli K-12: involvement of the bacteriophage lambda receptor. J Bacteriol. 1975;124:112–118. doi: 10.1128/jb.124.1.112-118.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luckey M, Nikaido H. Specificity of diffusion channels produced by λ phage receptor protein of Escherichia coli. Proc Nat Acad Sci US. 1980;77:167–171. doi: 10.1073/pnas.77.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Charbit A, Gehring K, Nikaido H, Ferenci T, Hofnung M. Maltose transport and starch binding in phage-resistant point mutations of maltoporin. J Mol Biol. 1988;201:487–496. doi: 10.1016/0022-2836(88)90630-4. [DOI] [PubMed] [Google Scholar]
- 8.Rancis G, Brennan L, Stretton S, Ferenci T. Genetic mapping of starch- and Lambda-receptor sites in maltoporin. Mol Microbiol. 1991;5:2293–2301. doi: 10.1111/j.1365-2958.1991.tb02160.x. [DOI] [PubMed] [Google Scholar]
- 9.Charbit A, Wang J, Michel V, Hofnung M. A cluster of charged and aromatic residues in the C-terminal portion of maltoporin participates in sugar binding and uptake. Mol Gen Genet. 1998;260:185–192. doi: 10.1007/s004380050884. [DOI] [PubMed] [Google Scholar]
- 10.Schirmer T, Keller TF, Wang YF, Rosenbusch JP. Structural basis for sugar translocation through maltoporin channels at 3.0 Å resolution. Science. 1995;267:512–514. doi: 10.1126/science.7824948. [DOI] [PubMed] [Google Scholar]
- 11.Wang YF, Dutzler R, Rizkallah PJ, Rosenbusch JP, Schirmer T. Channel specificity: Structural basis for sugar discrimination and differential flux rates in maltoporin. J Mol Biol. 1997;272:56–63. doi: 10.1006/jmbi.1997.1224. [DOI] [PubMed] [Google Scholar]
- 12.Dutzler R, Wang YF, Rizkallah PJ, Rosenbusch JP, Schirmer T. Crystal structures of various maltooligosaccharides bound to maltoporin reveal a specific sugar translocation pathway Structure. 1996;4:127–134. doi: 10.1016/s0969-2126(96)00016-0. [DOI] [PubMed] [Google Scholar]
- 13.Charbit A, Andersen J, Wang J, Schiffler B, Michel B, Benz R, Hofnung M. In vivo and in vitro studies of transmembrane beta-strand deletion, insertion or substitution mutants of the Escherichia coli K-12 maltoporin. Mol Micribiol. 2000;35:777–790. doi: 10.1046/j.1365-2958.2000.01748.x. [DOI] [PubMed] [Google Scholar]
- 14.Denker K, Orlik F, Schiffler B, Benz R. Site-directed mutagenesis of the greasy slide aromatic residues withing the LamB (maltoporin) channel of Escherichia coli. J Mol Biol. 2005;352:534–550. doi: 10.1016/j.jmb.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 15.Walther DM, Rapaport D, Tommassen J. Biogenesis of β-barrel membrane proteins in bacteria and eukaryotes: Evolution conservation and divergence. Cell Mol Life Sci. 2009;66:2789–2804. doi: 10.1007/s00018-009-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Papanikou E, Karamanou S, Economou A. Bacterial protein secretion through the translocase nanomachines. Nat Rev Microbiol. 2007;5:839–851. doi: 10.1038/nrmicro1771. [DOI] [PubMed] [Google Scholar]
- 17.Patel G, Gehrens-Kneip S, Holst O, Kleinschmidt JH. The Periplasmic Chaperone Skp Facilitates Targeting, Insertion and Folding of OmpA into Lipid Membranes with a Negative Membrane Surface Potential. Biochemistry. 2009;48:10235–10245. doi: 10.1021/bi901403c. [DOI] [PubMed] [Google Scholar]
- 18.Rouvierre PE, Gross CA. SurA, a periplasmic protein with peptidyl-prolyl isomerase activity, participates in the assembly of outer membrane proteins. Genes Dev. 1966;10:3170–3182. doi: 10.1101/gad.10.24.3170. [DOI] [PubMed] [Google Scholar]
- 19.Sklar JG, Wu T, Kahne D, Silhavy TJ. Defining the roles of the periplasmic chaperones Sur A, Skp, and DegP in Escherichia coli. Genes Dev. 2007;21:2473–2484. doi: 10.1101/gad.1581007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell. 2005;121:235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 21.Knowles TJ, Scott-Tucker A, Overduin M, Henderson IR. Membrane protein architects: the role of the BAM complex in outer membrane assembly. Nature Rev Microbiol. 2009;7:206–214. doi: 10.1038/nrmicro2069. [DOI] [PubMed] [Google Scholar]
- 22.White SH, Wimley WC. Hydrophobic interactions of peptides with membrane surfaces. Biochim Biophys Acta. 1998;1376:339–352. doi: 10.1016/s0304-4157(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 23.Tamm LK, Hong H, Liang B. Folding and assembly of β-barrel membrane proteins. Biochim Biophys Acta. 2004;1666:250–263. doi: 10.1016/j.bbamem.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 24.Kleinschmidt JH. Folding kinetics of the outer membrane proteins OmpA and FomA into phospholipid bilayers. Chem Phys of Lipids. 2006;141:30–47. doi: 10.1016/j.chemphyslip.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Kleinschmidt JH, Bulieris PV, Qu J, Dogterom M, den Blaauwen T. Association of neighboring β-strands of Outer Membrane Protein A in lipid bilayers revealed by site-directed fluorescence quenching. J Mol Biol. 2011;407:316–332. doi: 10.1016/j.jmb.2011.01.021. [DOI] [PubMed] [Google Scholar]
- 26.Eisele JL, Rosenbusch JP. In vitro folding and oligomerization of a membrane protein. J Biol Chem. 1990;265:10217–10220. [PubMed] [Google Scholar]
- 27.Surrey T, Schmid A, Jahnig F. Folding and membrane insertion of the trimeric β-barrel protein. OmpF, Biochemistry. 1996;35:2283–2288. doi: 10.1021/bi951216u. [DOI] [PubMed] [Google Scholar]
- 28.Burgess NK, Dao TP, Stanley AM, Fleming KG. β-Barrel Proteins that reside in the Escherichia coli Outer Membrane in vivo demonstrate varied folding behavior in vitro. J Biol Chem. 2008;283:26748–26758. doi: 10.1074/jbc.M802754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luckey M, Ling R, Dose A, Malloy B. Role of a disulfide bond in the thermal stability of the LamB Protein Trimer in the Escherichia coli Outer Membrane. J Biol Chem. 1991;266:1866–1871. [PubMed] [Google Scholar]
- 30.Keller TA, Ferenci T, Prilipov A, Rosenbusch JP. Crystallization of monodisperse maltoporin from wild-type and mutant strains of various Enterobacteriaceae. Biochem Biophys Res Commun. 1994;199:767–771. doi: 10.1006/bbrc.1994.1295. [DOI] [PubMed] [Google Scholar]
- 31.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 32.Sandermann H, Strominger JL. Purification and properties of C55-isoprenoid alcohol phophopkinase from Staphylococcus aureus. J Biol Chem. 1972;247:5123–5131. [PubMed] [Google Scholar]
- 33.Schenkman S, Tsugita A, Schwartz M, Rosenbusch JP. Topology of phage λ receptor protein. J Biol Chem. 1984;259:7570–7576. [PubMed] [Google Scholar]
- 34.Surrey T, Jahnig F. Refolding and oriented insertion of a membrane protein into a lipid bilayer. Proc Nat Acad Sci US. 1992;89:7457–7461. doi: 10.1073/pnas.89.16.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misra R, Peterson A, Ferenci T, Silhavy TJ. A genetic approach for analyzing the pathway of LamB assembly into the outer membrane of Escherichia coli. J Biol Chem. 1991;266:13592–13597. [PubMed] [Google Scholar]