

Abstract
Energy production presents a formidable challenge to axons as their mitochondria are synthesized and degraded in neuronal cell bodies. To meet the energy demands of nerve conduction, small mitochondria are transported to and enriched at mitochondrial stationary sites located throughout the axon. In this study, we investigated whether size and motility of mitochondria in small myelinated CNS axons are differentially regulated at nodes, and whether mitochondrial distribution and motility are modulated by axonal electrical activity. The size/volume of mitochondrial stationary sites was significantly larger in juxtaparanodal/internodal axoplasm than in nodal/paranodal axoplasm. With three-dimensional electron microscopy, we observed that axonal mitochondrial stationary sites were composed of multiple mitochondria of varying length, except at nodes where mitochondria were uniformly short and frequently absent altogether. Mitochondrial transport speed was significantly reduced in nodal axoplasm compared with internodal axoplasm. Increased axonal electrical activity decreased mitochondrial transport and increased the size of mitochondrial stationary sites in nodal/paranodal axoplasm. Decreased axonal electrical activity had the opposite effect. In cerebellar axons of the myelin-deficient rat, which contain voltage-gated Na+ channel clusters but lack paranodal specializations, axonal mitochondrial motility and stationary site size were similar at Na+ channel clusters and other axonal regions. These results demonstrate juxtaparanodal/internodal enrichment of stationary mitochondria and neuronal activity-dependent dynamic modulation of mitochondrial distribution and transport in nodal axoplasm. In addition, the modulation of mitochondrial distribution and motility requires oligodendrocyte–axon interactions at paranodal specializations.
Introduction
Myelination affects the function and morphology of axons. By confining voltage-gated Na+ channels in the nodal axolemma, myelination facilitates rapid nerve conduction in an energy efficient manner (Ritchie, 1995; Waxman, 1995). Myelinated fibers can be divided into four morphologically and biochemically distinct segments (nodes, paranodes, juxtaparanodes, and internodes) (Poliak and Peles, 2003; Trapp and Kidd, 2004). Nodes are the short, unmyelinated axonal segments enriched for voltage-gated Na+ channels, located between adjacent myelin internodes. Nodes are flanked by paranodes, where adhesion molecules form septate-like junctions that tether the myelin sheath to the axon (Peles and Salzer, 2000; Bhat, 2003). Juxtaparanodal axolemma is the ∼15 μm region adjacent to the paranodes and is enriched for Na+/K+ ATPases and voltage-gated K+ channels (Mata et al., 1991; Waxman, 1995; Young et al., 2008). The remaining internodal region includes the majority of the myelinated fiber and also contains Na+/K+ ATPase. Following activation of nodal Na+ channels, the Na+/K+ ATPases rapidly exchange axoplasmic Na+ for extracellular K+ in an energy-dependent manner to permit repetitive firing of axons. Based on the presence of Na+/K+ ATPases and their close proximity to nodes (Young et al., 2008), juxtaparanodal regions of CNS myelinated fibers appear to have the greatest potential for energy utilization during saltatory nerve conduction.
Mitochondria are the major source of axonal ATP required for repetitive propagation of action potentials by saltatory conduction. Mammalian axons can extend a meter or more from their neuronal cell bodies where the bulk of mitochondria are formed and degraded (Chang and Reynolds, 2006). The bulk of axonal mitochondria are located at stationary sites, axoplasmic areas occupied by single or multiple mitochondria that do not move during the timeframe of imaging studies (Morris and Hollenbeck, 1995; Miller and Sheetz, 2004; Misgeld et al., 2007; Kiryu-Seo et al., 2010). The stationary sites are distributed throughout the axon and are thought to be enriched at areas of high energy consumption (Miller and Sheetz, 2004; Hollenbeck and Saxton, 2005; Chang and Reynolds, 2006). A less abundant pool of small motile mitochondria is transported to and from the neuronal cell body and stationary sites. Motile mitochondria may fuse with stationary mitochondria and facilitate stationary mitochondrial growth and/or turnover. Older dysfunctional segments of stationary mitochondria are removed by fission and degraded (Chang and Reynolds, 2006; Twig et al., 2008). To meet increased energy demands of nerve conduction, the size of stationary sites and the speed of mitochondria transport are significantly increased following demyelination of axons (Kiryu-Seo et al., 2010). In addition, the speed of mitochondrial transport in amphibian peripheral nervous system (PNS) nodal axoplasm is decreased by neuronal activity in a Ca2+-mediated manner (Zhang et al., 2010). However, knowledge of mitochondrial distribution, motility, and dynamics in myelinated axons is limited and whether stationary mitochondria are enriched at nodes of Ranvier and/or modulated by electrical activity remains to be established.
We used mitochondria-targeted DsRed2 and confocal time-lapse imaging in organotypic slice cultures to examine mitochondrial behavior in myelinated Purkinje cell axons under basal, enhanced, or inhibited neuronal activity. We also investigated the three-dimensional distribution and ultrastructure of mitochondria in cerebellar and optic nerve myelinated axons in vivo. Our results establish a juxtaparanodal–internodal enrichment of mitochondrial stationary sites and a neuronal activity-dependent modulation of mitochondrial distribution and transport in nodal axoplasm. In addition, focal modulation of mitochondrial distribution and transport was absent from axons containing voltage-gated Na+ channel clusters but lacking paranodal specializations.
Materials and Methods
Slice culture preparation, transfection of lentiviral vectors.
Lentivirus with mitochondrial-targeted DsRed2 and GCaMP3 was prepared using a pLenti6/V5 directional TOPO cloning kit (Invitrogen) according to the manufacturer's protocol with slight modifications (Hirai, 2008). Briefly, 293FT cells were transfected with pLentiV5/6 lentiviral vector including Mito-DsRed2 (Clontech) or GCaMP3 (Addgene) and packaging plasmids. The medium was changed 4 h after the transfection. Viral supernatants were collected 24 h after the transfection, filtered with 0.22 μm PVDF membrane (Millipore), and centrifuged at 100,000 g for 2 h. The viral pellet was resuspended in 1/100 volume of PBS. Cerebellar organotypic slice cultures from male 10 d postnatal (P10) Sprague Dawley and myelin-deficient (md) rats were cut at a thickness of 150–200 μm, as described previously (Stoppini et al., 1991). Slices from P10 pups were chosen because Purkinje cells survive and their axons undergo robust myelination (Dusart et al., 1997). The slices were cultured on Millicell organotypic culture inserts (Millipore) in six-well plates containing 50% minimal essential medium, 25% horse serum, 25% HBSS, 6 mg/ml d-glucose, 1× Glutamax (Invitrogen), and penicillin/streptomycin. Two hours after plating, the lentiviral vectors were injected into the Purkinje cell layer using a glass microinjection needle (Kasri et al., 2008).
Time-lapse imaging by confocal microscopy.
Purkinje cell axons with DsRed2-positive mitochondria or cell bodies with GCaMP3 were imaged by confocal microscopy as reported previously (Kumada et al., 2006; Kiryu-Seo et al., 2010). Glass-bottom dishes with culture medium and pieces of Millicell membrane with attached slices were transferred into the chamber of a micro-incubator (PDMI-2; Medical System) attached to an inverted laser-scanning confocal microscope (TCS SP; Leica). The slices were illuminated with 488 or 543 nm light through a 63× oil-immersion objective (NA, 1.32; Leica) and transmitted light and fluorescence signals were detected. Images of mitochondria were collected at 1024 × 1024 pixel resolution every 6 s. Imaging at 6 s intervals did not induce mitochondrial fragmentation, acute mitochondrial swelling, or cessation of axonal transport in >50 experiments and cell bodies and axons remained healthy 3 d after imaging. We also restricted total observation time to <20 min to minimize the possibility of phototoxic damage to the cells. In some experiments, bath application of 1 μm tetrodotoxin (TTX; Sigma) or 10 μm bicuculline methobromide (BCC; Alexis Biochemicals) was applied 15 min before live imaging.
Kymographs of mitochondria in time lapse images were produced using ImageJ, as described previously (Miller and Sheetz, 2004). Motile mitochondria were identified as moving objects in movies and diagonal lines in the kymographs. Mean velocity of motile mitochondria was measured by dividing total displacement distance by total time in nodal (±10 μm from nodes) and internodal (20-∼40 μm from nodes) axoplasm. Within this 20-μm-long axoplasmic region, peak velocity of a mitochondrion was defined as maximum velocity for specific 5-μm-long regions, and turn frequency was measured as described previously (Morris and Hollenbeck, 1993; Wang and Schwarz, 2009). Stationary mitochondria were identified as profiles without any displacement during the timeframe of our in vitro imaging studies and as vertical lines on the kymographs. The pixel areas and lengths of DsRed2-positive profiles were measured with ImageJ software after thresholding. In measurements of stationary mitochondria, one representative frame was used after excluding out-of-focus regions and motile profiles. Variations in mitochondria stationary sites in nodal/paranodal, juxtaparanodal, and internodal axoplasm were established for individual axons by normalizing mitochondria size to the average mitochondria size for that axon. Caspr immunostaining established the location of the paranodal/nodal axoplasm.
For GCaMP3 imaging analyses, the time-lapse changes of fluorescent intensity in Purkinje cell bodies before and after drug treatments were measured using ImageJ with Time Series Analyzer V2.0 (http://rsbweb.nih.gov/ij/plugins/time-series.html). The cell bodies were traced and mean fluorescent intensity in the cell bodies was measured. Frequency of Ca2+ transients was defined as the number of increase of fluorescent intensity (ΔF/F > 0.2), and amplitude of Ca2+ transients was defined as peak ΔF/F of each Ca2+ transients.
Immunohistochemistry.
For direct correlation of time-lapse images and immunostaining for nodal/paranodal markers, the observed slices were fixed with 4% paraformaldehyde in 0.1 m phosphate buffer for 3 h immediately after live imaging. The slices were immunostained as previously described (Kiryu-Seo et al., 2010) with the following antibodies: rat anti-proteolipid protein (anti-PLP; Agmed), mouse anti-pan Nav (Sigma), mouse anti-Caspr1 (a gift from James S. Trimmer, University of California, Davis, California). Immunostaining was visualized with secondary antibodies conjugated with Alexa 488 or 647 (Invitrogen) and by confocal microscopy (SP5; Leica).
Mitochondrial sizes in node–paranodal regions in fixed tissues.
Slice cultures at 14 d in vitro (DIV) with DsRed-positive Purkinje cells were treated with TTX, BCC, and BCC in combination with 10 μm BAPTA-AM, 5 mm EGTA, or 1 μm ω-conotoxin MVIIC for 1 h, fixed with 4% paraformaldehyde for 3 h, and immunostained for Caspr and PLP as described above. Caspr immunostaining defined the boundaries of the paranodal/nodal axoplasm and nodal–paranodal axoplasmic area and their DsRed profiles were measured in stacked images.
Serial ion ablation scanning electron microscopy.
Tissue preparation and imaging were performed as previously described (Knott et al., 2008; Kidd et al., 2010). Rats and mice were perfused at postnatal day 30 with cacodylate-buffered 2.5% glutaraldehyde and 4% paraformaldehyde. Rat cerebellar white matter (three rats) was removed, postfixed with 1% tannic acid, and stained with buffered 0.4% OsO4 followed by aqueous uranyl acetate, then embedded in Durcupan resin. Mouse optic nerves were treated with OsO4-ferricyanide, followed sequentially by thiocarbohydrazine, aqueous OsO4, aqueous uranyl acetate, and Walton's lead aspartate stain, then embedded in Durcupan resin (Deerinck et al., 2010). Blocks were trimmed in a Leica ultramicrotome and longitudinally oriented white matter tracts were identified in 1-μm-thick sections. Samples were imaged using either Nova-200 Nanolab or Helios 650 dual beam FIB (FEI) systems using G2 or G3 Slice&View operating software, and serial images of the block face was generated by repeated cycles of milling (i.e., sample surface ablation) and imaging. Milling was performed with a 1 nA ion beam current. Images were acquired using a 2 kV (21–84 pA) beam in high-resolution mode (immersion lens) using the through lens detector in backscattered electron mode. Sets of 250–500 images at ∼40 nm steps (i.e., milling depth) were obtained either at 6.5 or 17 nm/pixel resolution, producing images ∼13 × 17 μm or 35 × 45 μm and 8–20 μm deep. These imaging conditions were chosen to maximize axon sample length while still discerning individual mitochondria. At the lower resolution (17 nm/pixel), individual mitochondria were identified as oval or cigar-shaped organelles (∼0.1 μm or greater in diameter) with stained matrix and unstained cristae cisternae. Each mitochondrion was present in several serial sections, which greatly enhanced their ultrastructural identification. At high resolution (6.5 nm/pixel), classic mitochondrial membranes and structure were evident in all sections. Images were registered, corrected for aspect ratio, and derivative stacks were generated using ImageJ/FIJI software. Mitochondria and axons were traced and analyzed using Reconstruct software (Fiala, 2005), with paranodal and nodal axoplasm traced separately to provide accurate volumes. Where necessary, compensation for slice thickness disparity was made using the method of Fiala and Harris (2001). Distances and distributions were measured directly or calculated using 3D distance formulae. Axonal diameters were calculated from length and volume data assuming axons, nodal, and paranodal axoplasm were cylinders, and mitochondria were cylinders with a hemisphere at either end (cigar-shaped). Montage images were constructed from multiple single slices through the same axon (Kidd and Heath, 1988).
Statistical analysis.
Statistical analysis was performed using SigmaStat (Aspire Software International). Data were presented as mean + SEM or median with first and third quartile. Comparisons were made using the Mann–Whitney U test for rates of motile mitochondria, mitochondrial volume, mitochondrial/axonal volume ratio, and length of stationary mitochondria, and two-tailed Student's t test for number of motile mitochondria, sizes of DsRed2 profiles, and percentage of nodal–paranodal axoplasm area occupied by DsRed. Multiple comparisons were made with Bonferroni correction. Time-lapse images were obtained from at least 10 slices from five rats for each group. Data describing the percentage of nodal–paranodal axoplasm area occupied by DsRed were obtained from at least five slices from three rats for each group.
Results
Mitochondrial motility and distribution in myelinated axons
Mitochondrial distribution and motility were monitored in live myelinated rat Purkinje cell axons in cerebellar organotypic slice cultures obtained from P10 rat pups. The slices were transfected with lentiviral vectors carrying mitochondria-targeted DsRed2 and then maintained in vitro for 14 d (Fig. 1a). Mito-DsRed2 is a soluble red fluorescent DsRed2 protein fused with a cleavable mitochondria-targeting sequence from cytochrome c oxidase subunit VIII and targeted to the mitochondrial matrix. DsRed2 was colocalized with all axonal mitochondria immunopositive for cytochrome c oxidase subunit I (data not shown). By 10 DIV, white matter tracts, which contain myelinated Purkinje cell axons (Birgbauer et al., 2004), were macroscopically visible in the slice cultures (Fig. 1b). The lentivirus specifically infected a small percentage of Purkinje cells (Fig. 1d, arrowhead), which project single DsRed2-positive axons into the white matter tracts (Fig. 1d, arrows). To identify the direction of mitochondrial movement, we followed Purkinje cell axons from the soma and time-lapse images of DsRed2 distribution and motility were acquired. In these axons, 82% of the DsRed2-positive mitochondrial profiles were stationary. Motile mitochondrial movement was bidirectional and characterized by velocity changes and occasional cessation of transport (Hollenbeck, 1996). The median velocity of mitochondrial movement was 0.25 μm/s (mean ± SEM, 0.36 ± 0.4) in the anterograde direction and 0.23 μm/s (mean ± SEM, 0.33 ± 0.4) in the retrograde direction, comparable to axonal transport rates reported previously in unmyelinated CNS axons (Morris and Hollenbeck, 1993; Wang and Schwarz, 2009) and myelinated PNS axons (Kiryu-Seo et al., 2010). Multiple motile mitochondria often stopped or slowed at single axonal sites.
Figure 1.
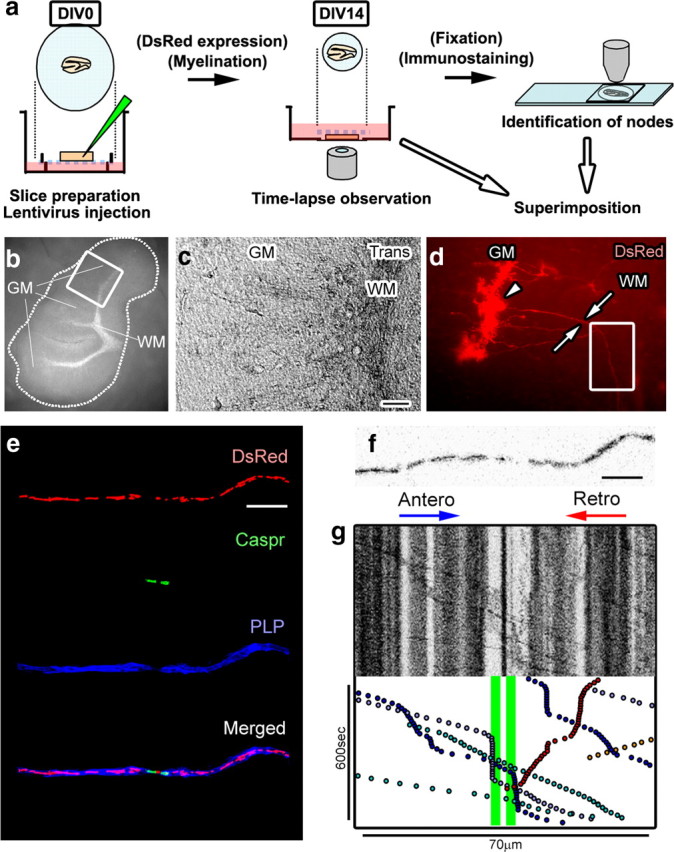
Time-lapse imaging of axonal mitochondria in cerebellar slice cultures. a, Schematic summary of experimental approach. b, c, P10 cerebellar slices are myelinated by 14DIV with microscopically apparent white matter (WM) and gray matter (GM). d, Purkinje cell soma are transfected with Mito-DsRed (arrowhead) and project DsRed-positive myelinated axons (arrows) into white matter. e, Time-lapse images of DsRed-labeled axonal mitochondria were obtained (d, box) and superimposed on images of the same axon immunostained for the paranodal protein Caspr and the compact myelin protein PLP. f, g, A single frame from the time-lapse imaging (f) and a kymographic summary (g) of stationary (vertical black lines) and motile (diagonal lines) mitochondria within different regions of this myelinated axon. Paranodal loops are colored green in the kymograph (g). Scale bars: c, 50 μm; e, f, 10 μm. Antero, anterograde; Retro, retrograde.
To identify the site of slowed mitochondrial motility, kymographs of mitochondrial distribution and motility were obtained by time-lapse imaging. Slices were then fixed and immunostained for proteins exclusively localized to nodal (pan-Na+ channel: Nav), paranodal (contactin-associated protein: Caspr), and internodal (PLP) segments of myelinated fibers. Time-lapse images of mitochondrial distribution and movement were coregistered with nodal, paranodal, juxtaparanodal, and internodal segments of the myelinated axons (Fig. 1a,e–g). These analyses established that motile axonal mitochondria stopped or slowed down preferentially in node–paranodal axoplasm (i.e., ±10 μm from nodes) of myelinated CNS fibers (Fig. 1g), and that the mean velocity in node–paranodal axoplasm was reduced ∼60% compared with that in internodal axoplasm (i.e., ±20–40 μm away from nodes) (Fig. 2a). Peak velocity and turn frequency were similar in nodal and internodal regions (data not shown).
Figure 2.

Mitochondrial transport velocity and mitochondrial stationary site sizes are decreased in nodal axoplasm. a, Mean anterograde and retrograde mitochondrial transport velocities are significantly decreased in nodal (±10 μm from nodes) compared with internodal (20-∼40 μm from nodes) axoplasm (**p < 0.001). b, b′, Mitochondrial stationary site size and length were measured at different axonal sites defined by length of and distance from Caspr staining. c, d, The normalized size (c) and length (d) of mitochondrial stationary sites were significantly smaller in nodal–paranodal axoplasm (PNP) than in juxtaparanodal (Juxta) or internodal (Inter) axoplasm and mitochondrial stationary site size was greater in juxtaparanodal compared with internodal axoplasm (c). *p < 0.02, **p < 0.008. n shows number of mitochondria in a and d. n = 12-∼15 axons in b and c. b′, c, Bars, Mean + SEM. a, d, Boxes, Median with first and third quartiles. Scale bar, 10 μm.
We next compared the area occupied by mitochondrial stationary sites and the length of each stationary site in node–paranodal, juxtaparanodal, and internodal axoplasm (Fig. 2b). The size of stationary mitochondrial sites in juxtaparanodal and internodal axoplasm were similar at proximal and distal sides of the node (Fig. 2b′). In contrast, stationary mitochondrial sites in node–paranodal axoplasm had approximately half the volume (Fig. 2c) and were ∼30% shorter (Fig. 2d) than those in juxtaparanodal and internodal axoplasm. In addition, the volume of mitochondrial stationary sites was greater in juxtaparanodal axoplasm than in internodal axoplasm (Fig. 2c). These results demonstrate juxtaparanodal and internodal enrichment of axonal stationary mitochondrial sites and slowing of mitochondria transport in node–paranodal axoplasm.
Ultrastructural distribution of mitochondria in vivo
To investigate mitochondrial distribution in cerebellar axons in vivo, we used serial ion ablation scanning electron microscopy [three-dimension electron microscopy (3D-EM)] to study P30 rat cerebellar white matter axons (Knott et al., 2008; Kidd et al., 2010; Kiryu-Seo et al., 2010). We selected P30 because the extent of myelination at this age in vivo is comparable to that in organotypic slice culture at DIV14 used in time-lapse imaging. Three-dimensional reconstructions were generated for 54 axons (0.6–1.7 μm diameter, ∼35 μm long segments), including 21 internodal (Fig. 3a,b) and 33 nodal (Fig. 3c,d) regions and 624 mitochondria from three rats. Axonal mitochondria were typically elongated; they varied in length from 0.3 to 13 μm (Fig. 3e) and had diameters of ∼0.1–0.4 μm. Most mitochondria (57%, Fig. 3e) were shorter than 1.5 μm, but longer mitochondria accounted for the majority of the total axonal mitochondrial volume (68%) (Fig. 3f), with the longest 10% representing >30% of axonal mitochondrial volume. Mitochondrial sizes were not normally distributed. In adjacent oligodendrocyte and astrocyte somata, mitochondria often formed multilobed, branching arrangements (data not shown), as described in other cell types (Scheffler, 2008). In contrast, <0.5% of axonal mitochondria branched. Axonal mitochondria were not evenly distributed along myelinated axons. Within internodal axoplasm, mitochondria often had overlapping distributions within the same axon territory (Fig. 3b,d). When quantified, 85.7% of mitochondria were present in clusters containing 2–11 mitochondria; 14.3% (±2.1 SEM) of mitochondria had non-overlapping distributions (Fig. 3g). The average length of axonal domains occupied by mitochondria, both clustered and isolated, was 3.75 μm (±0.21 SEM), which was similar to the lengths of stationary sites measured in internodal regions in slice cultures imaged by confocal microscopy (3.2 μm, ±0.20 SEM). Stationary sites in vitro occupied 63% of the axonal length, compared with 62.7% of the axon in 3D reconstructions in vivo. Mitochondria with overlapping distributions rarely contacted each other, indicating that mitochondrial clustering at stationary sites does not require mitochondria–mitochondria contacts.
Figure 3.
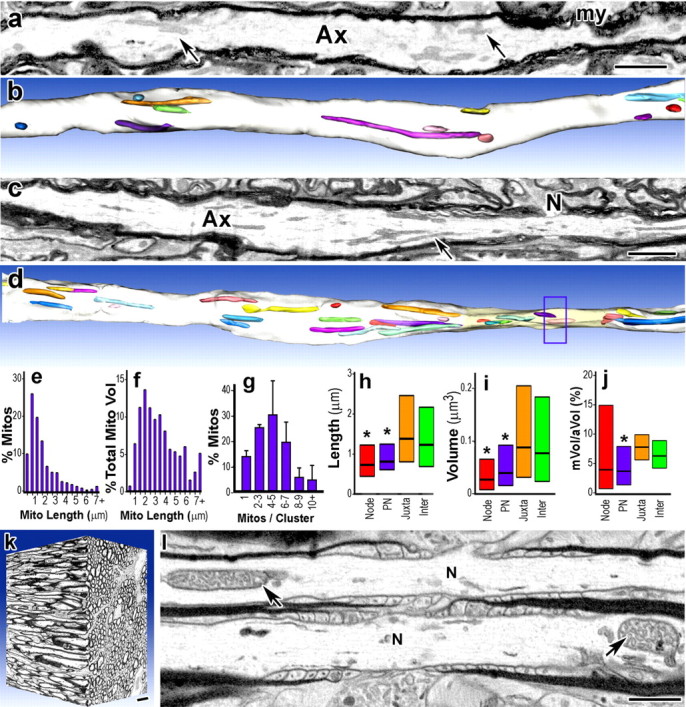
Mitochondria are not enriched in nodal axoplasm and stationary sites usually contain multiple mitochondria. a–l, The three-dimensional distribution of axonal mitochondrial was determined by serial-section electron microscopy of rat cerebellar white matter (a–i) and mouse optic nerve (j–l). a–d, Single sections (a, c) and 3D reconstructions of internodal (b) and nodal (d) regions of cerebellar myelinated axons (Ax). A node (c, N) is marked in the 3D reconstruction (d, box). e, f, Axonal mitochondrial lengths vary from <1 μm to >7 μm (e). Axonal mitochondria longer than 4 μm represent <10% of the total axonal mitochondria (e), but >25% of the total axonal mitochondrial volume (f). g, Internodal mitochondria (a, c, arrows; b, d, colored areas) frequently overlap and 86% are present in clusters of 2–11 mitochondria (Mito). h, i, Mitochondria in nodal and paranodal (PN) axoplasm are shorter (h) and smaller (i) than mitochondria in juxtaparanodal (Juxta) and internodal (Inter) axoplasm. j, The ratio of mitochondrial volume to axonal volume (mVol/aVol, expressed as a percentage) was highly variable in nodal axoplasm, and many nodes (39%) lacked mitochondria. k, Mitochondria in mouse optic nerve axons were distributed similarly to those in myelinated cerebellar axons. l, Internodal mitochondria (arrows) were abundant and 67% of nodes lacked mitochondria. g, Bars, Mean + SEM. h–j, Boxes, Median with first and third quartiles. *p < 0.001 Mann–Whitney U tests. a, c, Images montaged from serial slices. Scale bars, 1 μm.
Substantial differences were observed between mitochondrial sizes in the node–paranodal axoplasm and juxtaparanodal/internodal axoplasm. Internodal and juxtaparanodal axoplasm contained the full range of mitochondrial sizes, with lengths varying from ∼0.3 to 13.2 μm (median 1.4 μm) (Fig. 3h), and volumes from 0.008 to 1.652 μm3 (median 0.088 μm3) (Fig. 3i). In nodal axoplasm, the largest mitochondrion was only 1.9 μm long. Both nodal and paranodal mitochondria were substantially shorter and smaller in volume than those in juxtaparanodal/internodal axonal regions (p < 0.001) (Figs. 3h,i). Nodal mitochondria were also slightly smaller in diameter (median 0.21 μm in node vs 0.28 in juxtanode/internode, p < 0.001 Mann–Whitney U test). Mitochondrial presence in nodal axoplasm was highly variable; 39% contained no mitochondria, 40% had a single mitochondrion, and 21% had 2–5 mitochondria (n = 33 axons). Ratios of mitochondrial volume (mVol) to axonal volume (aVol) have been frequently used to compare the concentration of mitochondria in different axonal regions. In P30 cerebellar nodal axoplasm, the percentage of axonal volume occupied by mitochondria (mVol/aVol) values varied extensively (median 3.5%) (Fig. 3j), which was expected considering that 39% of nodes had no mitochondria and 21% had multiple mitochondria. In addition, the reduced axonal volume at the node skewed the ratios for the 61% of nodes containing mitochondria toward higher mean values compare with juxtaparanodal and internodal axoplasm. Because of the large variability in nodal mVol/aVol values (median 3.5%), they were not significantly different from internodal values (Fig. 3j). Paranodal axoplasm mVol/aVol values (median 3.3%), however, showed less variation and were significantly reduced compared with juxtaparanodal (median, 7.4%) and internodal (median, 5.7%) regions (p < 0.001, Mann–Whitney U test) (Fig. 3j). These results indicate that mitochondrial size and presence are differently regulated in nodal and paranodal axoplasm compared with juxtaparanodal and internodal axoplasm.
To test whether these results were due to species-specific or tract-specific properties of cerebellar white matter axons, we also examined P30 mouse optic nerve axons, using a staining regime that enhances membrane contrast. As illustrated in Figure 3k, optic nerve axons at P30 were of a similar dimension to those in cerebellar white matter (0.6–1.6 μm diameter). Optic nerve mitochondria were longer (median length, 1.54 vs 1.27 μm in cerebellum; p < 0.016, n = 173 vs 624) and larger (median volume, 0.12 vs 0.08 μm3; p < 0.003) than those in cerebellum, but the differences in the two populations were not extensive. Internodal mitochondria had overlapping distributions that were similar in length to mitochondrial clusters in cerebellar axons, indicating that stationary sites were also common in optic nerve axons. A survey of 21 optic nerve nodes indicated that only a third had mitochondria at nodal axoplasm (Fig. 3l), which is consistent with a previous stereological study that reported a lack of mitochondria in the majority of nodes in P60 mouse optic nerve (Edgar et al., 2008).
Neuronal activity modulates mitochondria in nodal axoplasm
Previous studies suggested that neuronal activation reduces mitochondrial motility and recruits mitochondria to metabolically active regions, such as presynaptic terminals (Li et al., 2004; Macaskill et al., 2009). In addition, increased electrical activity slows mitochondrial motility in nodal axoplasm of amphibian myelinated PNS axons (Zhang et al., 2010). To investigate whether axonal electrical activity modulates the distribution or motility of mitochondria in myelinated CNS axons, we treated slice cultures with either TTX, which blocks voltage-dependent Na+ channels and eliminates axonal action potentials; or BCC, which blocks GABAA receptors, suppresses IPSCs and increases electrical activity of Purkinje cells in slice cultures (Dupont et al., 2006; Gähwiler, 1975). To confirm whether these drugs modulate Purkinje cell electrical activity, we transfected Purkinje cells (DIV0) with a lentiviral vector carrying the protein-based Ca2+ indicator GCaMP3 (Tian et al., 2009) (Fig. 4a) and measured the frequency and amplitude of Ca2+ transients (ΔF/F > 0.2) (Fig. 4b–d) before and after treatment with TTX (Fig. 4e) or BCC (Fig. 4f) at DIV14. TTX treatment decreased and BCC treatment increased the frequency (Fig. 4g) and amplitude (data not shown) of Purkinje cell Ca2+ transients, confirming previous studies that reported decreased and increased axonal firing following the treatment of cerebellar slices with TTX and BCC, respectively (Khaliq and Raman, 2005).
Figure 4.
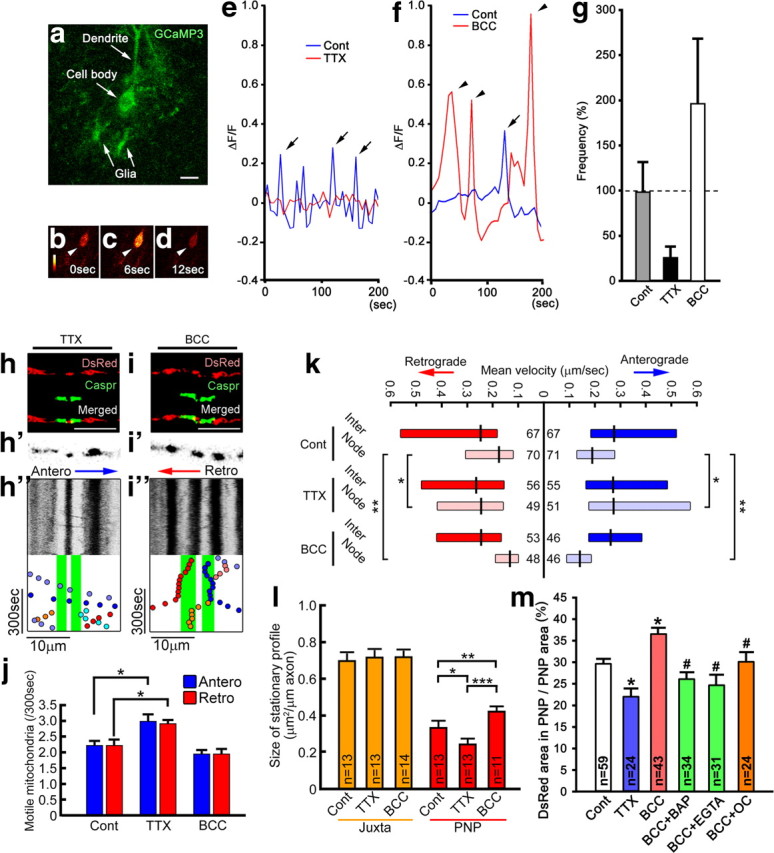
Electrical activity and Ca2+ increase the size of mitochondrial stationary sites and decrease mitochondrial motility in nodal axoplasm. Mitochondrial transport speed and distribution were investigated following TTX and BCC treatment. a–g, In a Purkinje cell body expressing GCaMP3 (a), increase (b to c, arrowheads) and return (c to d, arrowheads) of fluorescent intensity show a Ca2+ transient. The frequency (and amplitude) of Ca2+ transients are decreased threefold by TTX (e) and increased twofold (f) by BCC (g). h–j, Kymographs of nodal axoplasm after TTX (h–h″) and BCC (i–i″) show the number of motile mitochondria is increased by TTX and unchanged by BCC (j). *p < 0.01; n = 18 axons for each group. k, The speed of mitochondrial transport was significantly increased by TTX and significantly decreased by BCC in nodal axoplasm but not in internodal axoplasm. *p = 0.03, **p < 0.001; n, number of mitochondria marked on each bar. l, TTX decreased and BCC increased mitochondrial stationary site size in nodal axoplasm, but not in juxtaparanodal axoplasm. *p = 0.02, **p = 0.01, ***p < 0.001. m, TTX also decreased and BCC increased the percentage nodal axoplasm area occupied by mitochondria. Ca2+ chelaters [EGTA, BAPTA-AM (BAP)] or a Ca2+ channel blocker [ω-conotoxin MVIIC (OC)] eliminated the increase of nodal mitochondria size by BCC. #p < 0.05 compared with BCC alone, *p < 0.001; n = number of fibers (l) or nodes (m) examined. g, j, l, m, Bars, Mean + SEM. k, Boxes, Median with first and third quartiles. Cont, control; PNP, nodal–paranodal; Inter, internodal; Juxta, juxtaparanodal; Antero, anterograde; Retro, retrograde.
Following 15 min of TTX treatment, motile mitochondria stopped or slowed less frequently in node–paranodal axoplasm (Fig. 4h–h″). As a result, the number of motile mitochondria in node–paranodal axoplasm increased and the mean mitochondrial speeds were significantly greater in electrically silent than in electrically active control cultures (Fig. 4j,k). In addition, TTX treatment significantly decreased the total size of mitochondria stationary sites in node–paranodal axoplasm (Fig. 4l). Conversely, following 15 min of BCC treatment, the number of motile mitochondria and the mean velocity of mitochondrial transport were decreased in nodal axoplasm, but not in internodal axoplasm (Fig. 4i–i″,j,k). Furthermore, BCC treatment increased the total size of mitochondrial stationary sites in node–paranodal axoplasm, but not in juxtaparanodal (Fig. 4l) or internodal axoplasm (data not shown). These studies support the concept that increased axonal firing focally increases mitochondrial stationary site size and decreases mitochondrial motility in node–paranodal axoplasm.
The recruitment and function of mitochondria at synaptic terminals depends on Ca2+ (Li et al., 2004; Pardo et al., 2006; Macaskill et al., 2009), which regulates the electrical activity-dependent decrease of mitochondrial motility in nodal axoplasm of amphibian PNS myelinated fibers (Zhang et al., 2010). Since it is most likely that stationary mitochondria are the major source of axonal ATP, we examined whether neuronal activity-dependent changes of mitochondrial stationary site size in node–paranodal axoplasm are modulated by Ca2+. In these analyses, we measured the percentage of axonal area occupied by mitochondria in node–paranodes to exclude the possibility that the differences of mitochondrial sizes are simply due to possible variance of node–paranodal axonal volume. Following TTX treatment, the percentage of node–paranodal axonal area occupied by mitochondria was decreased (Fig. 4m). Conversely, following BCC treatment, the percentage of node–paranodal axonal area occupied by mitochondria was significantly increased (Fig. 4m). When slices were treated with BCC in combination with BAPTA-AM (intracellular Ca2+ chelator), EGTA (Ca2+ chelator), or ω-conotoxin MVIIC (P/Q- and N-type Ca2+ channel blocker), mitochondrial stationary site sizes in node–paranodal axoplasm were significantly smaller compared with BCC treatment alone (Fig. 4m). The mitochondrial stationary site sizes in node–paranodal axoplasm following TTX and ω-conotoxin treatment were similar to those following TTX treatment alone (data not shown). Given that voltage-dependent Ca2+ channels are expressed in myelinated axons (Hillman et al., 1991; Alix et al., 2008), these results establish that increased axoplasmic Ca2+ mediates increased stationary mitochondrial size in node–paranodal axoplasm upon neuronal activation.
Mitochondrial motility and distribution in myelin-deficient rats
To investigate the role of oligodendrocyte–axon interactions in axonal mitochondrial behavior, we established cerebellar organotypic slice cultures from P10 md rats and transfected Purkinje cells with DsRed2 targeted to mitochondria. In md rats, oligodendrocytes ensheath axons but fail to form normal compact myelin due to a point mutation in the PLP gene (Csiza and de Lahunta A., 1979; Boison and Stoffel, 1989). md rat oligodendrocyte processes, however, ensheath axons and concentrate voltage-gated Na+ channel in the axolemma. Because of the lack of myelin, paranodal loops do not form and the distribution of the paranodal septate junction molecules Caspr and contactin, as well as saltatory conduction, are significantly altered (Waxman et al., 1990; Utzschneider et al., 1994; Arroyo et al., 2002). In md rat cerebellar organotypic slice cultures, voltage-gated Na+ channels were clustered at discrete axonal locations despite a paucity of PLP-positive ensheathment of Purkinje cell axons. In these axons, the mean velocity of axonal mitochondrial transport (Fig. 5a–c) was similar at regions of Na+ channel clusters (20-μm-long segment centered by the Na+-channel clusters) and in internodal axoplasm (20–40 μms from the Na+-channel clusters) (Fig. 5d). In addition, mean velocities of axonal mitochondrial transport were similar in node-like axoplasm and other axonal regions following 15 min of BCC treatment (data not shown). We next examined the distribution of axonal mitochondria in md rats. Consistent with a previous report (Dentinger et al., 1985), mitochondrial stationary sites in md rat axons were larger than those found in wild-type myelinated axons, including total (p < 0.05) and node-like (p < 0.001) axoplasm (Fig. 5e). In addition, stationary sites were similar in volume and length in node-like and other axonal regions of md axons (Fig. 5f,g). The focal modulation of mitochondrial transport and distribution in node–paranodal axoplasm appear to require oligodendrocyte–axon interaction at paranodal specializations.
Figure 5.

Oligodendrocyte–axon interactions affect distribution and transport of axonal mitochondria. a–c, The distribution and transport rates of axonal mitochondria were compared in slice cultures of wild-type (WT) and myelin-deficient (MD) rats; myelin-deficient axons have Na+ channel clusters, but lack paranodal loops and compact myelin. Vertical bar, 300 s; horizontal bar, 10 μm. d, In contrast to wild-type myelinated axons, mean mitochondrial transport speed was identical in internodal and nodal-like axoplasm of myelin-deficient fibers. e–g, The mean size (e), normalized size (f), and length (g) of axonal mitochondria were similar in node-like (NL) and internodal axoplasm of myelin-deficient fibers. Mean mitochondria size in nodal-like and all axoplasm were significantly larger than mean mitochondrial size in wild-type axons. *p = 0.05, **p < 0.001; n, number of mitochondria (d, g) or axons (e, f). e, f, Bars, Mean + SEM. d, g, Boxes, Median with first and third quartiles. PNP, nodal–paranodal; Inter, internodal; Juxta, juxtaparanodal; Antero, anterograde; Retro, retrograde.
Discussion
The present study describes fundamental properties of mitochondrial distribution and motility in myelinated CNS axons. Time-lapse imaging in vitro and three-dimensional ultrastructural reconstructions in vivo establish that >90% of axonal mitochondrial are present in internodal stationary sites, which consist of multiple mitochondria that vary in length but not in diameter. In contrast, mitochondria in nodal and paranodal axoplasm were consistently small. In myelinated axons from rat cerebellum and mouse optic nerves, mitochondria were not enriched in nodal axoplasm and were absent from 39% (cerebellum) and 66% (optic nerve) of nodes examined. Increased axonal firing, however, increased mitochondrial stationary site size and decreased the transport speed of mitochondrial transport in nodal/paranodal but not internodal axoplasm. In these small diameter fibers, mitochondria are not enriched nor do they appear to be an absolute requirement for the nodal axoplasm. The presence of mitochondria in nodal axoplasm, however, is dynamic and increases in response to axonal electrical activity coupled to increased axoplasmic Ca2+.
Cerebellar organotypic slice cultures provide several advantages for investigating mitochondrial behavior in myelinated axons. As described previously (Ross et al., 1962; Birgbauer et al., 2004), myelination is abundant; we show here that nodes of Ranvier can be reliably coregistered in kymographs of living tissues and immunostained fixed images. Purkinje cells can be selectively transfected with lentiviral vectors and their electrophysiological properties, including their response to TTX and BCC, have been previously described in detail (Gähwiler, 1975; Dupont et al., 2006; Kessler et al., 2008). The size and distribution of mitochondrial stationary sites in myelinated axons were consistent with previous studies (Morris and Hollenbeck, 1993; Hollenbeck, 1996; Miller and Sheetz, 2004; Wang and Schwarz, 2009; Zhang et al., 2010) and stationary sites in myelinated axons in these slice cultures were comparable to those measured in vivo by 3D electron microscopy and previously in PNS myelinated cultures (Kiryu-Seo et al., 2010). These results validate the cerebellar slice system for the examination of mitochondrial behavior in CNS myelinated axons.
The juxtaparanodal–internodal enrichment of axonal mitochondria was unexpected in light of the commonly stated view that mitochondria are enriched in nodal axoplasm (Hollenbeck and Saxton, 2005; Chang and Reynolds, 2006; Chen and Chan, 2006). Based on our 3D-EM analysis, 39% of nodes in the cerebellum and 68% of nodes in the optic nerve lacked axonal mitochondria. Similar results have been reported previously: 36% (3 of 8) of cat ventral root nodes and 44% (4 of 9) of dorsal root nodes lacked mitochondria (Berthold et al., 1993); in rat spinal roots, “it was common to find nodes with no mitochondria present and just as common to observe nodes with several mitochondria in the compact axoplasmic region” (Perkins and Ellisman, 2011); the majority of paranodal/nodal complexes in P60 mouse optic nerve and spinal cord dorsal columns were devoid of mitochondria (Edgar et al., 2008); and 30% (3 of 10) of nodes in the cat spinal cord lacked mitochondria (Fabricius et al., 1993). Collectively, all EM studies show that mitochondria are not obligatory or essential components of the nodal axoplasm. The concept of nodal enrichment of mitochondria was largely based on comparisons involving the ratio of mitochondrial volume to axonal volume. Because axonal diameters are constricted at the node (i.e., nodal axoplasm volume is small), the mVol/aVol value is high for those fibers that contained nodal mitochondria and zero when nodal mitochondria are not present. Nodal mVol/aVol values are thus are not normally distributed and, when averaged across multiple axons, their mean values may be very misleading. For example, in the present study, the average nodal mVol/aVol (10.2%) was 1.3 times the internodal average, whereas the median value (a more appropriate nonparametric measure of centrality) was 0.6 times that of internodal regions (Fig. 3j). Although mitochondria are not enriched in nodal axoplasm, many nodes do contain mitochondria. Their presence may depend on multiple factors, including electrical activity and fiber tract location. As demonstrated by our cerebellum–optic nerve comparisons, the percentage of nodes containing mitochondria can vary in different CNS fiber tracts and may also differ between CNS and PNS axons (Perkins and Ellisman, 2011).
The bulk of mitochondrial volume resides in the longer mitochondrial stationary sites in internodes, suggesting that most ATP within axons is generated there. We have recently reported that axonal Na+-K+-ATPases are absent from nodal and paranodal axolemmae and are enriched in juxtaparanodal and internodal axolemmae (Young et al., 2008). The juxtaparanodal enrichment of mitochondrial stationary sites, therefore, is likely to reflect the energy demands of saltatory conduction by myelinated axons. Since mitochondria are the major source of ATP, the internodal enrichment of stationary mitochondria helps facilitate axonal transport, which is also energy-dependent. Due to the rapid repolarization of the axolemma, it is unlikely that internodal Na+-K+-ATPases and mitochondria play a significant role in repolarizing the axolemma during saltatory conduction. In support of this hypothesis, manipulation of axonal electrical activity in myelinated axons had no effect on stationary mitochondrial site size or transport speed of motile mitochondria in internodal segments of myelinated axons. This stands in contrast to demyelination, which increased both the speed of axonal transport and the size of stationary mitochondrial sites in all regions of formerly myelinated axons (Kiryu-Seo et al., 2010). These mitochondrial changes are likely to result from the diffuse distribution of voltage-gated Na+ channels on demyelinated axolemmae and the increased energy demands of nonsaltatory axonal conduction (Trapp and Stys, 2009).
Internodal stationary sites contained multiple and often large mitochondria, which collectively account for >90% of the mitochondrial volume in axons. Larger axonal mitochondria were not mobile in our imaging studies. In contrast to internodal mitochondria, our results demonstrate that mitochondria in nodal axoplasm are dynamically regulated by voltage-gated Na+ channel activity. This is apparent at basal firing rates of Purkinje cell axons in slice cultures as mitochondrial motility is significantly decreased in nodal/paranodal axoplasm compared with juxtaparanodal/internodal axoplasm. It remains to be determined whether the decreased mean velocity in nodal/paranodal axoplasm is caused by an actual decrease in transport speed and/or an increase in transitory transport stops that cannot be detected by in vitro imaging at 6 s intervals. A similar decrease in mitochondrial motility in nodal axoplasm was reported in amphibian PNS myelinated internodes (Zhang et al., 2010). We confirm and extend this observation by establishing that increased electrical activity not only decreases the speed of motile mitochondria, but also increases the size of mitochondrial stationary sites in node–paranodal axoplasm, but not internodal axoplasm, of myelinated CNS axons. Furthermore, we show that reduced or abolished axonal activity in TTX-treated cultures increases mitochondrial motility and decreases the size of mitochondrial stationary sites in node–paranodal axoplasm. Although these alterations should be confirmed in vivo, our observations establish a connection between transport rates of motile mitochondria and stationary mitochondrial site size in vitro. Increased electrical activity and Na+ channel activation slows mitochondrial transport and increases mitochondrial stationary site size. It is likely, therefore, that Na+ channel activity regulates mitochondrial transport and/or mitochondrial fusion/fission.
A possible link between mitochondrial transport and mitochondrial stationary site size at nodes of Ranvier is increased axoplasmic Ca2+, which has been implicated in activity-dependent mitochondrial recruitment to synaptic terminals in neurons (Li et al., 2004; Macaskill et al., 2009). A recent study in frog sciatic nerve correlated increased axonal firing, elevated axoplasmic Ca2+, and reduced mitochondrial transport in nodal axoplasm of amphibian peripheral nerves (Zhang et al., 2010). We extend these observations by demonstrating that removal of extracellular Ca2+ by EGTA or pharmacologically blocking P/Q- and N-type axonal Ca2+ channels mitigates the effects of increased axonal electrical activity in reducing mitochondrial transport and increasing the size of mitochondrial stationary sites in node–paranodal axoplasm. The functions of axoplasmic Ca2+ are diverse, as it can directly modify mitochondrial-related proteins or indirectly affect mitochondria by activating kinases and phosphatases. Ca2+ can enhance mitochondrial energy production by modulating mitochondrial aspartate–glutamate carriers and multiple dehydrogenases, including α-ketoglutarate dehydrogenase (Pardo et al., 2006; Gellerich et al., 2010). Ca2+ can reduce mitochondrial transport by binding to the mitochondrial Rho GTPase (Miro) and regulating Miro's interaction with the microtubule motor kinesin-1 (Saotome et al., 2008; Macaskill et al., 2009; Wang and Schwarz, 2009). Ca2+ can also shorten mitochondria by augmenting fission (Han et al., 2008). It remains to be determined whether mitochondria that accumulate in the node–paranodal axoplasm in response to increased electrical activity fuse to form larger stationary mitochondria and/or represent accumulation of motile mitochondria.
Axonal degeneration is the major cause of permanent neurological disability in primary diseases of myelin (Trapp and Nave, 2008). Myelination affects axons in many ways, including modulation of axonal cytoskeleton. In addition, axonal pathologies that precede axonal degeneration in inherited diseases of myelin are manifested first and most abundantly in paranodal regions (Nave and Trapp, 2008). These paranodal pathologies include decreased phosphorylation of neurofilaments and reduced axonal diameters in MAG-null mice (Yin et al., 1998) and altered axonal transport in CNP- and PLP-null mice (Lappe-Siefke et al., 2003; Edgar et al., 2004). It is likely, therefore, that mitochondrial transport and distribution are modulated by oligodendrocyte–axon interactions at paranodal loops. In support of this hypothesis, mutations of the septate junction-specific protein Caspr cause impaired paranodal junctions and abnormal distribution and morphology of mitochondria in paranodal axoplasm (Garcia-Fresco et al., 2006; Sun et al., 2009). Our studies of md rat cerebellar axons further support this hypothesis and establish that voltage-gated Na+ channel clusters are not sufficient to generate node-like behavior of axonal mitochondrial transport or distribution. The studies described in this report therefore set the stage for investigating posttranslational modifications of axonal molecules that are modulated by paranodal loops and activity of voltage-gated Na+ channels.
Footnotes
This work was supported by National Institutes of Health Grant NS38186 (to B.D.T.) and a National Multiple Sclerosis Society Postdoctoral Fellowship (to N.O.). We thank Christopher Nelson for careful editorial assistance and Ansi Chang, Xinghua Yin, Danielle Klein, Erin Petrovic, and Nan Avishai for technical assistance. We also thank Dr. Loren L. Looger for information of GCaMP3.
References
- Alix JJ, Dolphin AC, Fern R. Vesicular apparatus, including functional calcium channels, are present in developing rodent optic nerve axons and are required for normal node of Ranvier formation. J Physiol. 2008;586:4069–4089. doi: 10.1113/jphysiol.2008.155077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo EJ, Xu T, Grinspan J, Lambert S, Levinson SR, Brophy PJ, Peles E, Scherer SS. Genetic dysmyelination alters the molecular architecture of the nodal region. J Neurosci. 2002;22:1726–1737. doi: 10.1523/JNEUROSCI.22-05-01726.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthold CH, Fabricius C, Rydmark M, Andersén B. Axoplasmic organelles at nodes of Ranvier. I. Occurrence and distribution in large myelinated spinal root axons of the adult cat. J Neurocytol. 1993;22:925–940. doi: 10.1007/BF01218351. [DOI] [PubMed] [Google Scholar]
- Bhat MA. Molecular organization of axo-glial junctions. Curr Opin Neurobiol. 2003;13:552–559. doi: 10.1016/j.conb.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Birgbauer E, Rao TS, Webb M. Lysolecithin induces demyelination in vitro in a cerebellar slice culture system. J Neurosci Res. 2004;78:157–166. doi: 10.1002/jnr.20248. [DOI] [PubMed] [Google Scholar]
- Boison D, Stoffel W. Myelin-deficient rat: a point mutation in exon III (A—-C, Thr75—-Pro) of the myelin proteolipid protein causes dysmyelination and oligodendrocyte death. EMBO J. 1989;8:3295–3302. doi: 10.1002/j.1460-2075.1989.tb08490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Reynolds IJ. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog Neurobiol. 2006;80:241–268. doi: 10.1016/j.pneurobio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. Critical dependence of neurons on mitochondrial dynamics. Curr Opin Cell Biol. 2006;18:453–459. doi: 10.1016/j.ceb.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Csiza CK, de Lahunta A. Myelin deficiency (md): a neurologic mutant in the Wistar rat. Am J Pathol. 1979;95:215–224. [PMC free article] [PubMed] [Google Scholar]
- Deerinck TJ, Bushong EA, Lev-Ram V, Shu X, Tsien RY, Ellisman MH. Enhancing serial block-face scanning electron microscopy to enable high resolution 3-D nanohistology of cells and tissues. Microsc Microanal. 2010;16:1138–1139. [Google Scholar]
- Dentinger MP, Barron KD, Csiza CK. Glial and axonal development in optic nerve of myelin deficient rat mutant. Brain Res. 1985;344:255–266. doi: 10.1016/0006-8993(85)90803-0. [DOI] [PubMed] [Google Scholar]
- Dupont JL, Fourcaudot E, Beekenkamp H, Poulain B, Bossu JL. Synaptic organization of the mouse cerebellar cortex in organotypic slice cultures. Cerebellum. 2006;5:243–256. doi: 10.1080/14734220600905317. [DOI] [PubMed] [Google Scholar]
- Dusart I, Airaksinen MS, Sotelo C. Purkinje cell survival and axonal regeneration are age dependent: an in vitro study. J Neurosci. 1997;17:3710–3726. doi: 10.1523/JNEUROSCI.17-10-03710.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler JH, Montague P, Barrie JA, McCulloch MC, Duncan ID, Garbern J, Nave KA, Griffiths IR. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol. 2004;166:121–131. doi: 10.1083/jcb.200312012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JM, McCulloch MC, Thomson CE, Griffiths IR. Distribution of mitochondria along small-diameter myelinated central nervous system axons. J Neurosci Res. 2008;86:2250–2257. doi: 10.1002/jnr.21672. [DOI] [PubMed] [Google Scholar]
- Fabricius C, Berthold CH, Rydmark M. Axoplasmic organelles at nodes of Ranvier. II. Occurrence and distribution in large myelinated spinal cord axons of the adult cat. J Neurocytol. 1993;22:941–954. doi: 10.1007/BF01218352. [DOI] [PubMed] [Google Scholar]
- Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Harris KM. Extending unbiased stereology of brain ultrastructure to three-dimensional volumes. J Am Med Inform Assoc. 2001;8:1–16. doi: 10.1136/jamia.2001.0080001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gähwiler BH. The effects of GABA, picrotoxin and bicuculline on the spontaneous bioelectric activity of cultured cerebellar Purkinje cells. Brain Res. 1975;99:85–95. doi: 10.1016/0006-8993(75)90610-1. [DOI] [PubMed] [Google Scholar]
- Garcia-Fresco GP, Sousa AD, Pillai AM, Moy SS, Crawley JN, Tessarollo L, Dupree JL, Bhat MA. Disruption of axo-glial junctions causes cytoskeletal disorganization and degeneration of Purkinje neuron axons. Proc Natl Acad Sci U S A. 2006;103:5137–5142. doi: 10.1073/pnas.0601082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellerich FN, Gizatullina Z, Trumbeckaite S, Nguyen HP, Pallas T, Arandarcikaite O, Vielhaber S, Seppet E, Striggow F. The regulation of OXPHOS by extramitochondrial calcium. Biochim Biophys Acta. 2010;1797:1018–1027. doi: 10.1016/j.bbabio.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman D, Chen S, Aung TT, Cherksey B, Sugimori M, Llinás RR. Localization of P-type calcium channels in the central nervous system. Proc Natl Acad Sci U S A. 1991;88:7076–7080. doi: 10.1073/pnas.88.16.7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H. Progress in transduction of cerebellar Purkinje cells in vivo using viral vectors. Cerebellum. 2008;7:273–278. doi: 10.1007/s12311-008-0012-5. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ. The pattern and mechanism of mitochondrial transport in axons. Front Biosci. 1996;1:d91–102. doi: 10.2741/a118. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM. The axonal transport of mitochondria. J Cell Sci. 2005;118:5411–5419. doi: 10.1242/jcs.02745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasri NN, Govek EE, Van Aelst L. Characterization of oligophrenin-1, a RhoGAP lost in patients affected with mental retardation: lentiviral injection in organotypic brain slice cultures. Methods Enzymol. 2008;439:255–266. doi: 10.1016/S0076-6879(07)00419-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler M, Kiliman B, Humes C, Arai AC. Spontaneous activity in Purkinje cells: multi-electrode recording from organotypic cerebellar slice cultures. Brain Res. 2008;1218:54–69. doi: 10.1016/j.brainres.2008.04.063. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM. Axonal propagation of simple and complex spikes in cerebellar Purkinje neurons. J Neurosci. 2005;25:454–463. doi: 10.1523/JNEUROSCI.3045-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd GJ, Heath JW. Double myelination of axons in the sympathetic nervous system of the mouse. II. Mechanisms of formation. J Neurocytol. 1988;17:263–276. doi: 10.1007/BF01674212. [DOI] [PubMed] [Google Scholar]
- Kidd GJ, Avishai A, Yin X, Trapp BD. Three-dimensional analysis of optic nerve axons using a focused ion beam-based approach. Microscopy Today. 2010;18:18–22. [Google Scholar]
- Kiryu-Seo S, Ohno N, Kidd GJ, Komuro H, Trapp BD. Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochondrial transport. J Neurosci. 2010;30:6658–6666. doi: 10.1523/JNEUROSCI.5265-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott G, Marchman H, Wall D, Lich B. Serial section scanning electron microscopy of adult brain tissue using focused ion beam milling. J Neurosci. 2008;28:2959–2964. doi: 10.1523/JNEUROSCI.3189-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumada T, Lakshmana MK, Komuro H. Reversal of neuronal migration in a mouse model of fetal alcohol syndrome by controlling second-messenger signalings. J Neurosci. 2006;26:742–756. doi: 10.1523/JNEUROSCI.4478-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata M, Fink DJ, Ernst SA, Siegel GJ. Immunocytochemical demonstration of Na+,K(+)-ATPase in internodal axolemma of myelinated fibers of rat sciatic and optic nerves. J Neurochem. 1991;57:184–192. doi: 10.1111/j.1471-4159.1991.tb02114.x. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW. Imaging axonal transport of mitochondria in vivo. Nat Methods. 2007;4:559–561. doi: 10.1038/nmeth1055. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104:917–927. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol. 1995;131:1315–1326. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- Pardo B, Contreras L, Serrano A, Ramos M, Kobayashi K, Iijima M, Saheki T, Satrústegui J. Essential role of aralar in the transduction of small Ca2+ signals to neuronal mitochondria. J Biol Chem. 2006;281:1039–1047. doi: 10.1074/jbc.M507270200. [DOI] [PubMed] [Google Scholar]
- Peles E, Salzer JL. Molecular domains of myelinated axons. Curr Opin Neurobiol. 2000;10:558–565. doi: 10.1016/s0959-4388(00)00122-7. [DOI] [PubMed] [Google Scholar]
- Perkins GA, Ellisman MH. Mitochondrial configurations in peripheral nerve suggest differential ATP production. J Struct Biol. 2011;173:117–127. doi: 10.1016/j.jsb.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Peles E. The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci. 2003;4:968–980. doi: 10.1038/nrn1253. [DOI] [PubMed] [Google Scholar]
- Ritchie JM. Physiology of axons. In: Waxman SG, Kocsis JD, Stys PK, editors. The axon. New York: Oxford UP; 1995. pp. 68–96. [Google Scholar]
- Ross LL, Bornstein MB, Lehrer GM. Electron microscopic observations of rat and mouse cerebellum in tissue culture. J Cell Biol. 1962;14:19–30. doi: 10.1083/jcb.14.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A. 2008;105:20728–20733. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffler IE. Mitochondria. Hoboken, NJ: Wiley; 2008. [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Sun XY, Takagishi Y, Okabe E, Chishima Y, Kanou Y, Murase S, Mizumura K, Inaba M, Komatsu Y, Hayashi Y, Peles E, Oda S, Murata Y. A novel Caspr mutation causes the shambling mouse phenotype by disrupting axoglial interactions of myelinated nerves. J Neuropathol Exp Neurol. 2009;68:1207–1218. doi: 10.1097/NEN.0b013e3181be2e96. [DOI] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Kidd GJ. Structure of the myelinated axon. In: Lazzarini R, editor. Myelin biology and disorders. San Diego: Elsevier; 2004. pp. 3–25. [Google Scholar]
- Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009;8:280–291. doi: 10.1016/S1474-4422(09)70043-2. [DOI] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DA, Archer DR, Kocsis JD, Waxman SG, Duncan ID. Transplantation of glial cells enhances action potential conduction of amyelinated spinal cord axons in the myelin-deficient rat. Proc Natl Acad Sci U S A. 1994;91:53–57. doi: 10.1073/pnas.91.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell. 2009;136:163–174. doi: 10.1016/j.cell.2008.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG. Voltage-gated ion channels in axons: localization, function, and development. In: Waxman SG, Kocsis JD, Stys PK, editors. The axon. New York: Oxford UP; 1995. pp. 218–243. [Google Scholar]
- Waxman SG, Black JA, Duncan ID, Ransom BR. Macromolecular structure of axon membrane and action potential conduction in myelin deficient and myelin deficient heterozygote rat optic nerves. J Neurocytol. 1990;19:11–28. doi: 10.1007/BF01188436. [DOI] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu P, Lee VM, Li C, Roder J, Trapp BD. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young EA, Fowler CD, Kidd GJ, Chang A, Rudick R, Fisher E, Trapp BD. Imaging correlates of decreased axonal Na+/K+ ATPase in chronic multiple sclerosis lesions. Ann Neurol. 2008;63:428–435. doi: 10.1002/ana.21381. [DOI] [PubMed] [Google Scholar]
- Zhang CL, Ho PL, Kintner DB, Sun D, Chiu SY. Activity-dependent regulation of mitochondrial motility by calcium and Na/K-ATPase at nodes of Ranvier of myelinated nerves. J Neurosci. 2010;30:3555–3566. doi: 10.1523/JNEUROSCI.4551-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]