

Abstract
Macrophages exert divergent effects in the injured CNS, causing either neurotoxicity or regeneration. The mechanisms regulating these divergent functions are not understood but can be attributed to the recruitment of distinct macrophage subsets and the activation of specific intracellular signaling pathways. Here, we show that impaired signaling via the chemokine receptor CX3CR1 promotes recovery after traumatic spinal cord injury (SCI) in mice. Deficient CX3CR1 signaling in intraspinal microglia and monocyte-derived macrophages (MDMs) attenuates their ability to synthesize and release inflammatory cytokines and oxidative metabolites. Also, impaired CX3CR1 signaling abrogates the recruitment or maturation of MDMs with presumed neurotoxic effects after SCI. Indeed, in wild-type mice, Ly6Clo/iNOS+/MHCII+/CD11c− MDMs dominate the lesion site, whereas CCR2+/Ly6Chi/MHCII−/CD11c+ monocytes predominate in the injured spinal cord of CX3CR1-deficient mice. Replacement of wild-type MDMs with those unable to signal via CX3CR1 resulted in anatomical and functional improvements after SCI. Thus, blockade of CX3CR1 signaling represents a selective anti-inflammatory therapy that is able to promote neuroprotection, in part by reducing inflammatory signaling in microglia and MDMs and recruitment of a novel monocyte subset.
Introduction
Clinical and experimental spinal cord injury (SCI) elicits an inflammatory response comprised mostly of activated microglia and monocyte-derived macrophages (MDMs). Within the lesion, these cells, collectively referred to as CNS macrophages, adopt unique molecular phenotypes with divergent effects on neuron survival and axon growth (Kigerl et al., 2009). The mechanisms controlling functional diversity in CNS macrophages are poorly defined but undoubtedly include differential signaling via chemokines, cytokines, and other cues present in the lesion microenvironment.
In the healthy CNS, fractalkine (CX3CL1), a membrane-bound chemokine, is expressed by neurons, while its receptor (CX3CR1) is highly expressed on microglia (Harrison et al., 1998; Mildner et al., 2007). The interaction between CX3CL1 and CX3CR1 is important for maintenance of microglial function in health and disease (Zujovic et al., 2000; Mizuno et al., 2003). CX3CL1 also is upregulated on activated endothelia (Fraticelli et al., 2001; Tarozzo et al., 2002), where it acts as an adhesion molecule to regulate recruitment of circulating leukocytes to sites of inflammation (Imai et al., 1997). Interestingly, CX3CL1 is not induced in CNS vasculature during experimental autoimmune encephalomyelitis (EAE), suggesting disease-specific regulation of CX3CL1/CX3CR1 signaling (Sunnemark et al., 2005). The relative expression of CX3CR1 defines two distinct monocyte subsets in the blood (Geissmann et al., 2003; Sunderkötter et al., 2004; Ancuta et al., 2006, 2009). Thus, SCI-induced changes in CX3CL1 or CX3CR1 could influence how microglia and MDMs affect recovery after SCI.
To date, the effects of manipulating CX3CR1 signaling after traumatic SCI have not been evaluated. However, other model systems have shown that loss of signaling via this chemokine receptor significantly impacts the onset and progression of neurological disease. In experimental paradigms where microglia are the predominant CNS macrophages (e.g., Parkinson's disease and ALS), deficient CX3CR1 signaling enhances their neurotoxic potential (Cardona et al., 2006). In contrast, in stroke models, deficient CX3CR1 signaling reduces infarct size and improves neurological function, in part through reducing neuroinflammation (Soriano et al., 2002; Dénes et al., 2008). In experimental models of autoimmune inflammation, CX3CR1 deficiency exacerbates pathology and neurological dysfunction (Huang et al., 2006), an effect related to impaired recruitment of NK cells.
Here we show that CX3CL1 and CX3CR1 are dynamically regulated after SCI. When CX3CR1 signaling is abolished, neurological recovery is improved, spinal cord pathology is reduced, and the phenotype and function of microglia and monocytes is functionally altered from that normally present after SCI. The present data show that loss of CX3CR1 signaling attenuates the inflammatory and oxidative capacity of microglia and MDMs. Moreover, the dynamics of monocyte recruitment and/or the differentiation of newly recruited monocyte subsets are altered after SCI in CX3CR1-deficient mice. Collectively, these data identify CX3CR1 as a putative therapeutic target for selectively reducing neurodestructive inflammatory cascades after SCI.
Materials and Methods
CX3CR1-deficient mice.
Breeding pairs of CX3CR1GFP/GFP mice were established from Dan Littman's original colony as described previously (Jung et al., 2000; Cardona et al., 2006). Knock-outs were generated by replacing the second exon of the CX3CR1 gene with the enhanced green fluorescent protein (GFP) reporter gene (Jung et al., 2000). Throughout this document, homozygous knock-out mice are labeled as CX3CR1GFP/GFP. Mice were backcrossed to a C57BL/6 background for a minimum of 10 generations. Wild-type C57BL/6 mice were obtained from Taconic.
Generation of chimeric mice.
To minimize or eliminate the nonspecific effects of irradiation on myeloid cell recruitment into the uninjured spinal cord, preliminary dosing studies were completed. Briefly, wild-type recipient mice were exposed to 5, 7.5, or 10 Gy of x-irradiation, after which they received intravenous injections of 10 × 106 bone marrow cells obtained from actin-GFP mice (The Jackson Laboratory). Recipients were maintained in sterile housing and received prophylactic Ringer's solution (subcutaneously) for 1 week and acidified water supplemented with antibiotics (Baytril; 20 mg/L) for 1 month. Eight weeks later, the level of chimerism was established by cytofluorometric analysis of circulating blood leukocytes and the number of GFP+CD11b+ cells was quantified in longitudinal sections of the thoracic spinal cord. As shown in Figure 6, the 5 Gy dose caused minimal infiltration of GFP+ cells into the naive spinal cord. This dose was used in subsequent experiments designed to test the effects of CX3CR1 deletion on monocytes after SCI. Chimeras were prepared by subjecting 6- to 8-week-old wild-type mice to 5 Gy whole-body irradiation followed by intravenous injection of 10 × 106 bone marrow cells harvested from wild-type or CX3CR1GFP/GFP donor mice.
Figure 6.

Improved recovery of function with neuroprotection after SCI in bone marrow chimeric mice with CX3CR1-deficient monocyte-derived macrophages. A–C, Longitudinal thoracic spinal cord sections of uninjured actin-GFP→WT chimeric mice reveal an effect of irradiation dose on accumulation of bone-marrow cells in spinal cord. Nearly complete bone marrow chimerization (> 96%; data not shown) was achieved with each dose (5, 7.5, and 10 Gy); however, minimal GFP+ cell infiltration was observed into intact spinal cord with low-dose (5 Gy) irradiation (effect of dose was p = 0.0317; D). An irradiation dose of 5 Gy was used subsequently to create CX3CR1GFP/GFP→WT (GFP→WT) or wild-type→wild-type chimeras (WT→WT). After SCI, locomotor recovery (BMS and BMS subscore) was improved in GFP→WT mice (effect of genotype was p = 0.0009 and p = 0.0121, respectively; E, F). This was accompanied by increased sparing of white matter at and nearby the impact site (effect of genotype was p = 0.0006; G). H, I, Representative cross-sections of injured spinal cord at 42 dpi showing increased myelin (MBP staining) and axonal (NF staining) sparing in GFP→WT versus WT→WT chimeras. Inset in H and I is zoomed image of white box shown. J, Representative cross-section of injured spinal cord of GFP→WT chimera at 42 dpi showing that the majority of monocytes (GFP+) accumulate within the core of the contusion lesion, especially in the axon-deficient penumbra (box in J); minimal accumulation occurs in spared white matter. K, High magnification of box shown in J; arrows delineate interface between lesion core and surrounding astroglial scar. Data were analyzed by two-way ANOVA followed by Bonferroni's post hoc test. n = 5–6 mice/genotype/time. *p < 0.05. Scale bars: C (for A–C), 20 μm; J (for H–J), 100 μm; K, 20 μm.
Spinal cord injury.
All housing, surgical, and postoperative care procedures were performed in accordance with The Ohio State University Institutional Animal Care and Use Committee and have been described previously (Jakeman et al., 2000; Kigerl et al., 2006). For SCI, 10- to 12- (14–16 for chimeras) week-old male and female mice received a laminectomy at T9–10 under ketamine (80 mg/kg, i.p.) and xylazine (40 mg/kg, i.p.) anesthesia followed by a moderate spinal cord contusion at this level using either The Ohio State University electromechanical device (0.5 mm over 30 ms) (see Figs. 1, 2) or the Infinite Horizons device (75 kdyn; Precision Systems and Instrumentation) (see Figs. 3, 5–8). Sham controls received laminectomy only. Postoperatively, mice received prophylactic antibiotics (1 mg/kg Gentacin, s.c.). To prevent dehydration, mice were injected with Ringer's solution (subcutaneously). Bladders were voided manually twice a day for the duration of the study. Urine pH was monitored weekly and mice were observed daily for signs of infection or abnormal wound healing at the site of surgery. No signs of infection were evident in any mice.
Figure 1.

Postinjury regulation of CX3CL1 and CX3CR1 mRNA and protein. Changes in CX3CL1 and CX3CR1 mRNA were quantified using real-time PCR (A and B, respectively). Semiquantitative analyses of CX3CL1 and CX3CR1 protein were performed using Western blot (C; representative of n = 3 blots). Densitometric analysis of protein expression (D) revealed a decrease in CX3CL1 expression during the first 7 dpi. Similarly, the 50 kDa isoform of CX3CR1 decreased slightly by 3 dpi and then remained below levels found in sham-injured mice through 42 dpi. The 55 kDa CX3CR1 isoform was decreased below the limits of detection through 3 dpi and then increased by 7 dpi, returning toward baseline levels by 42 dpi. Data in A, B, and D were analyzed via one-way ANOVA with Dunnett's post hoc test. In D, * indicates significance for CX3CL1 and ∧ indicates significance for the 50 kDa CX3CR1. *,∧p < 0.05, **,∧∧p < 0.01, ***p < 0.001 versus sham. Gene expression was normalized to GAPDH; protein expression was normalized to β-actin and then expressed relative to values obtained in sham-injured mice. n = 5 mice for mRNA analysis, n = 3 mice for protein analysis.
Figure 2.

Locomotor function is improved and lesion pathology is reduced after SCI in CX3CR1GFP/GFP mice. Overground locomotion (BMS score) was improved in CX3CR1GFP/GFP compared to wild-type mice (A). CX3CR1GFP/GFP mice have more spared myelin at and nearby the site of injury (B) and significantly shorter contusion lesions (C, D). Note the marked attenuation of tissue pathology within 0.4 mm of the injury site in CX3CR1GFP/GFP mice (D). Immunofluorescent double labeling for myelin basic protein (MBP) and neurofilament confirms the increased myelin and axon sparing in CX3CR1GFP/GFP mice (E, F). Inset in E and F is zoomed image of white box shown. Data are average of two independent studies using n = 4–6 mice/genotype. Data were analyzed via repeated-measures two-way ANOVA (A) and unpaired t tests (B, C). *p < 0.05, **p < 0.01, ***p < 0.001 versus wild-type. Scale bars, 100 μm.
Figure 3.
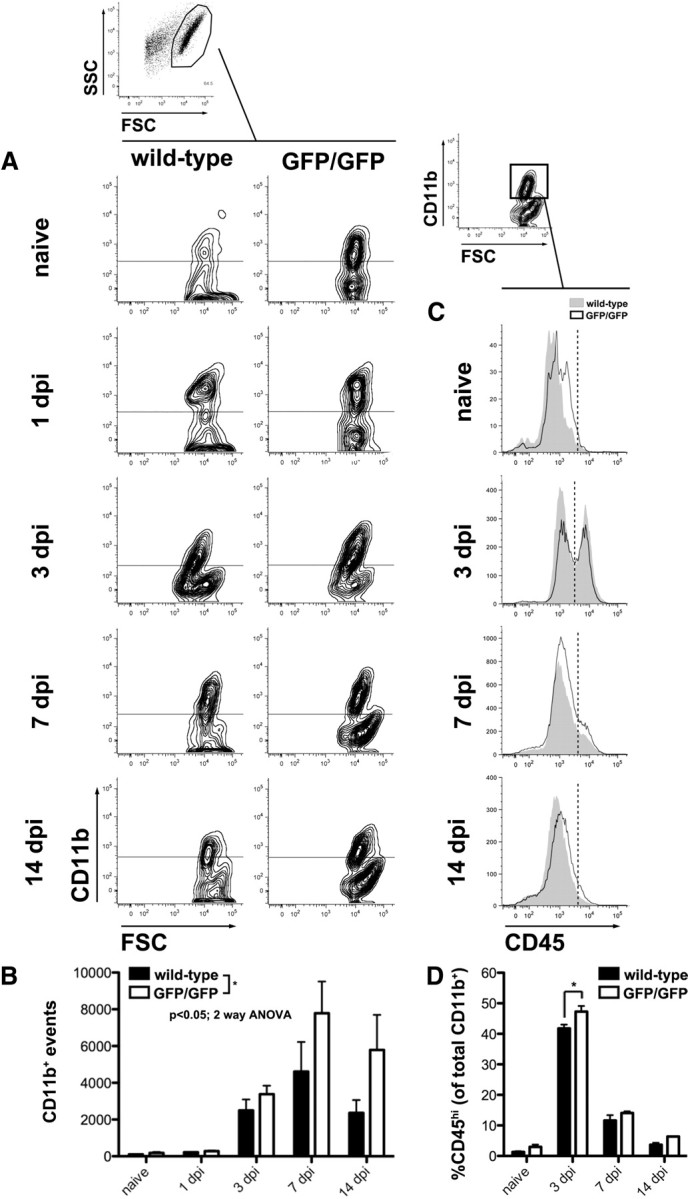
Intraspinal accumulation of monocyte-derived macrophages is increased in CX3CR1GFP/GFP mice. All CNS macrophages (microglia+MDMs) were identified by gating on CD11b+ events identified in forward versus side scatter plots (FSC vs SSC, respectively). A, Representative contour plots (outliers removed for clarity) show CD11b+ populations in wild-type and CX3CR1GFP/GFP mice as a function of time after injury [0 (naive), 1, 3, 7, and 14 dpi]. Horizontal lines indicate positive expression defined with isotype controls. B, Quantification of plots in A reveal that the CNS macrophage response is modestly increased in CX3CR1GFP/GFP mice, especially at later times after injury when MDMs are expected to infiltrate the spinal cord (significant main effect of time, p < 0.0001 and genotype, p = 0.0286). C, Representative histograms show CD45 high (MDMs) and low (microglia) populations from naive and SCI wild-type and CX3CR1GFP/GFP. Relative expression of CD45 was analyzed from the CD11b+ gate (one example is shown). Vertical dotted lines indicate distinction between CD45 high and low expression. D, Entry of CD45hi MDMs is maximal at 3 dpi and is greater in CX3CR1GFP/GFP mice (significant effect of time, p < 0.0001, and genotype, *p = 0.0017). Data were analyzed by two-way ANOVA followed by Bonferroni's post hoc test. n = 3–4 mice/genotype/time.
Figure 5.

Intraspinal macrophages in CX3CR1-deficient mice do not express iNOS. iNOS+ macrophages were identified by gating on CD11b+ events (CD11b vs FSC). A, Representative flow cytometry contour plots (outliers removed for clarity) show iNOS+ macrophages isolated from naive and injured spinal cord of wild-type and CX3CR1GFP/GFP mice. Mean numbers (events) of iNOS+ macrophages are shown in each plot. Horizontal lines indicate positive expression defined with isotype controls. B, In both genotypes, iNOS+ macrophages increased at 1 dpi and then decreased at 3 dpi. At 7 and 14 dpi, corresponding with the period immediately after maximal monocyte-derived macrophage accumulation (see Fig. 3), the percentage of iNOS+ macrophages increased in wild-type but not CX3CR1GFP/GFP mice. Significant main effects of time (p < 0.0001) and genotype (p < 0.0001) were noted, as was a time × genotype interaction (p = 0.0016). The difference in iNOS expression between genotypes was significant at 14 dpi. Data were analyzed by two-way ANOVA followed by Bonferroni's post hoc test. n = 4 mice/genotype/time. ***p < 0.001.
Figure 7.

Phenotype of CD11b+/Ly6Chi intraspinal macrophages after SCI. Ly6Chi macrophages were identified by gating on CD11b+ events (as in Fig. 5). Subsequently, iNOS, CCR2, MHCII, and CD11c expression was analyzed within the Ly6Chi gate (one example is shown). Representative histograms of postinjury iNOS, CCR2, MHCII, and CD11c expression are shown for CX3CR1GFP/GFP (white) and wild-type (shaded) mice. Mean fluorescent intensity is indicated within each representative histogram for each genotype. Vertical lines indicate positive expression defined with isotype controls. Quantification of the number of events in the Ly6Chi/CD11b+ gate is graphed below each column. There was a nonsignificant trend for reduced numbers of iNOS+/Ly6Chi/CD11b+ macrophages but a significant increase (effect of genotype was p = 0.0054) in numbers of CCR2+/Ly6Chi/CD11b+ macrophages in CX3CR1GFP/GFP mice. MHCII+/Ly6Chi/CD11b+ and CD11c+/Ly6Chi/CD11b+ macrophages were not significantly affected by CX3CR1 deletion. Data were analyzed by two-way ANOVA followed by Bonferroni's post hoc test. n = 4 mice/genotype/time.
Figure 8.

Phenotype of CD11b+/Ly6Clo intraspinal macrophages after SCI. Ly6Clo macrophages were identified by gating on CD11b+ events (as in Fig. 5). Subsequently, iNOS, CCR2, MHCII, and CD11c expression was analyzed within the Ly6Clo gate (one example is shown). Representative histograms of postinjury iNOS, CCR2, MHCII, and CD11c expression are shown for CX3CR1GFP/GFP (white) and wild-type (shaded) mice. Mean fluorescent intensity is indicated within each representative histogram for each genotype and significant differences are indicated with an asterisk(s). Vertical lines indicate positive expression defined with isotype controls. Quantification of the number of events in the Ly6Clo/CD11b+ gate is graphed below each column. Decreased numbers of iNOS+/Ly6Clo/CD11b+ macrophages were found in CX3CR1-deficient mice (effect of genotype was p = 0.0016). The interaction of time and genotype was also significant (p = 0.0057). Conversely, CCR2+/Ly6Clo/CD11b+ macrophages were significantly increased in CX3CR1-deficient mice (effect of genotype was p = 0.0006). MHCII+/Ly6Clo/CD11b+ macrophages were decreased in CX3CR1-deficient mice (effect of genotype was p = 0.0133). The interaction was also significant (p = 0.0131). The effect of genotype alone on CD11c+/Ly6Clo/CD11b+ macrophage numbers was not significant, but there was a significant interaction (p = 0.0473), suggesting that the onset of CD11c+ macrophage accumulation is a late occurrence, but still altered by CX3CR1 deletion. Data were analyzed by two-way ANOVA followed by Bonferroni's post hoc test. n = 4 mice/genotype/time. **p < 0.01, ***p < 0.001.
Open field locomotor testing.
Motor recovery was assessed using a standardized open field locomotor rating scale (the Basso Mouse Scale, BMS) that was developed from operational definitions of hindlimb movement, paw placement, and coordination across four strains of mice recovering from SCI (Basso et al., 2006). Before SCI, mice were acclimated to handling and to the testing environment for two 10 min sessions. SCI mice were scored at regular intervals for a testing period of 4 min by a pair of raters blinded to genotype. Testing occurred at 0, 1, 5, 7, 10, and 14 dpi and then weekly until 42 dpi. BMS subscores were obtained for chimeras in Figure 6 (Basso et al., 2006).
Tissue processing.
Mice designated for histology were anesthetized (as above) and then perfused intracardially with PBS (0.1 m, pH 7.4) followed by 4% paraformaldehyde in 0.1 m PBS. Perfused spinal cords were removed and postfixed for 2 h and then rinsed and stored overnight in 0.2 m phosphate buffer, pH 7.4. Tissues were cryoprotected by immersion in 30% sucrose. Spinal cord tissue blocks were prepared over a rostral–caudal extent of 1 cm centered on the injury site and rapidly frozen in OCT compound. Transverse serial sections (10 μm) were cut through each block on a Microm cryostat (HM 505E), mounted on slides, and then stored at −20°C before staining. To ensure uniformity of staining between groups, sections from different genotypes were randomized onto slides. Mice designated for RNA analysis were perfused with sterile PBS as above; tissues were rapidly dissected, snap frozen in 2-methylbutane, and then stored at −80°C until analyses were completed.
Spinal cord lesion analysis.
To visualize intact myelin and distinguish it from gray matter and lesioned tissue, sections were stained with eriochrome cyanine (EC) as described previously (Kigerl et al., 2006). To calculate the area of spared myelin and lesion length, images of EC-stained sections were digitized using a Zeiss Axioplan 2 Imaging microscope (Carl Zeiss) and an MCID Elite image analysis system (Imaging Research). The total cross-sectional area and the lesioned area were quantified at 0.2 mm intervals using either the Cavalieri method (Fig. 2) (Howard and Reed, 1998) or digital image analysis with the MCID system [see Fig. 6; similar to that reported previously for macrophage quantitation (Donnelly et al., 2009)]. Lesion length was determined by calculating the distance between the rostral-most and caudal-most sections devoid of visible lesion or abnormal tissue architecture.
Immunohistochemistry.
Spinal cord sections were dried at room temperature for 1 h and then at 37°C for an additional hour, followed by rinsing in 0.1 m PBS. Fluorescent (GFP+) detection of CX3CR1 expression is facilitated by the transgenic approach described above (Jung et al., 2000). For Iba-1 staining (to label all microglia and macrophages), blocking serum was applied for 1 h at room temperature and then sections were overlaid overnight at 4°C with anti-Iba-1 primary antibody (1:1000; Wako). Antibody specificity has been previously confirmed by Western blot and immunoprecipitation assays. The next day, slides were rinsed in 0.1 m PBS and Alexa Fluor 546 dye-conjugated goat anti-rabbit secondary antibody was applied for 40 min at room temperature. For MBP/NF staining, a complex of anti-NF primary antibody (Developmental Studies Hybridoma Bank; 1:500) and Alexa Fluor 546 dye-conjugated goat anti-mouse (1:500) secondary antibody was first incubated for 1 h at 37°C. This antibody complex was then mixed with normal mouse serum and incubated for 1 h at 37°C, followed by incubation on ice for an additional hour. Blocking serum was applied for 1 h at room temperature and then sections were overlaid overnight at 4°C with the antibody complex and also anti-MBP primary antibody (Aves; 1:500). The next day, slides were rinsed in 0.1 m PBS and Alexa Fluor 633 dye-conjugated goat anti-chicken secondary antibody was applied for 40 min at room temperature. After rinsing, all slides were coverslipped with Immu-Mount (Thermo Scientific). Fluorescent images were taken using an Olympus FluoView FV1000 laser scanning confocal microscope (Olympus).
RNA isolation, reverse transcription, and real-time PCR.
Macrophages (5 × 105 to 6 × 106 cells), microglia (1 × 105 to 3 × 105 cells), or spinal cord tissue (5 mm centered at the impact site or at T9/10 for uninjured tissue) were homogenized in Trizol (Invitrogen). RNA was isolated according to the manufacturer's protocol and then treated with DNase I (1 μg/μl) to eliminate genomic DNA contamination (Invitrogen). One to two micrograms of DNase-treated RNA were primed with random hexamers (1 μm, Applied Biosystems) and reverse transcribed using SuperScript II reverse transcriptase (Applied Biosystems). Primer pairs were designed using ABI Prism Primer Express 2.0. Primer sequence specificity was confirmed by performing BLAST analysis for highly similar sequences against known sequence databases. Primer sequences included the following: CX3CR1-F-5′-CAGCATCGACCGGTACCTT-3′, CX3CR1-R-5′-GCTGCACTGTCCGGTTGTT-3′; CX3CL1-F-5′-CCGCGTTCTTCCATTTGTGT-3′, CX3CL1-R-5′-GCACATGATTTCGCATTTCG-3′; GAPDH-F-5′-GCTTAAGAGACAGCCGCATCT-3′, GAPDH-R-5′-CGACCTTCACCATTTTGTCTACA-3′; IGF1-F-5′-TGACATGCCCAAGACTCAGAAG-3′, IGF1-R-5′-GCGGTGATGTGGCATTTTC-3′; IL1b-F-5′-CAGGCTCCGAGATGAACAAC-3′, IL1b-R-5′-GGTGGAGAGCTTTCAGCTCATAT-3′; IL6-F-5′-GCCTTCTTGGGACTGATGCT-3′, IL6-R-5′-AGTCTCCTCTCCGGACTTGTG-3′; iNOS-F-5′-CCCTTCAATGGTTGGTACATGG-3′, iNOS-R-5′-ACATTGATCTCCGTGACAGCC-3′; TNFa-F-5′-GTGATCGGTCCCCAAAGG, and TNFa-R-5′ GGTCTGGGCCATAGAACTGATG. RNA profiles were analyzed using primers specific to the gene of interest (above) and SYBR Green master mix (Applied Biosystems). All PCR was performed using an Applied Biosystems 7300 system. Melting point analyses were performed for each reaction to confirm single amplified products. Gene expression was extrapolated from standard curves generated concurrently for each gene using a control cDNA dilution series. Expression was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for each sample.
Flow cytometry.
Mice designated for flow cytometry were anesthetized (as above) and then perfused intracardially with Krebs' Ringer's buffer (KRB; 121 mm NaCl, 4.8 mm KCl, 1.2 mm KH2PO4, 25 mm NaHCO3, and 14 mm d-glucose) containing 0.2% heparin. Whole spinal cords were removed and placed in Petri dishes containing KRB/BSA (KRB with 0.09% BSA). The cords were then minced with a razor blade, then resuspended in a solution of KRB/BSA with 0.03% trypsin and incubated on a shaker (175 rpm) at 37°C for 30 min. The cell suspensions were then incubated and triturated repeatedly with different concentrations of trypsin inhibitor and DNase I, type II until no solids were visible. The cells were then resuspended in 70% Percoll in KRB, and underlaid into a tube already containing KRB previously underlaid with 35% Percoll in KRB. Gradients were then centrifuged at 1250 × g for 20 min. Cells were harvested from the lower interface and washed.
All cells isolated from spinal cord were stained. Briefly, cells were first incubated in blocking solution (anti-CD16/32 antibody; eBioscience). Samples were then split into two for staining. Panel 1 was stained with the following antibodies: V450-conjugated anti-CD11b (BD Biosciences), PerCP-Cy5.5-conjugated anti-Ly6C (BD Biosciences), and anti-CCR2 (Abcam) for 30 min. After washing, the samples were stained with APC-conjugated anti-goat antibody (to label anti-CCR2) for 20 min. Panel 2 was stained with the following antibodies: V450-conjugated anti-CD11b (BD Biosciences), PerCP-Cy5.5-conjugated anti-Ly6C (BD Biosciences), PE-conjugated anti-MHCII (I-A/I-E; BD Biosciences), and APC-conjugated anti-CD11c (BD Biosciences) for 30 min. Cells from both panels were then fixed and permeabilized with BD Cytofix/Cytoperm. Afterward, cells were incubated in normal mouse serum (eBioscience) for 15 min. Then cells in panel 1 were stained with PE-conjugated anti-iNOS antibody (Santa Cruz Biotechnology) for 30 min. In a separate set of experiments, cells from injured spinal cord were stained with V450-conjugated anti-CD11b and APC-conjugated anti-CD45 (BD Biosciences) for 30 min.
After all staining was completed, cells were washed and analyzed using a FACSCanto II flow cytometer and FACSDiva software (BD Biosciences). Subsequent data analyses were completed using FlowJo software (version 9.2; Tree Star). CNS macrophages were gated based on forward versus side scatter and CD11b expression. Total numbers of CD11b+ events were quantified from the total cell isolate from each spinal cord. iNOS, CD45, and Ly6C expression were analyzed from the CD11b+ gate. For analysis of Ly6Chi and Ly6Clo populations, iNOS, CCR2, MHC II, and CD11c were analyzed from the Ly6Chi/lo gate. Isotype control antibodies (BD Biosciences) were matched for fluorochrome and used for cursor placement.
Microglia and bone marrow-derived macrophage cultures.
Primary cultures of microglia were prepared as described previously (McCarthy and de Vellis, 1980; Le et al., 2001). Brains were harvested from P0–P3 wild-type and CX3CR1GFP/GFP mice, minced in 0.25% trypsin/EDTA (Invitrogen), and then incubated in enzyme for 30 min at 37°C with occasional agitation. The enzyme reaction was halted by adding media (RPMI + 10% FBS (Hyclone) + 1% penicillin/streptomycin (P/S; Invitrogen), and tissue was briefly triturated. Cellular material was allowed to settle and supernatant was collected and passed through a 100 μm filter. This was repeated until tissue was fully dissociated. The cell suspension was washed and then resuspended and plated at a density of 1–2 brains/T75 flask. Cultures were maintained for 14–21 d with media changes at day 1 and then every 3–4 d. To isolate microglia, flasks were shaken for 1–2 h at 37°C at 200 rpm. Detached cells were collected and replated. Cells were >99% positive for CD11b as assessed by immunostaining.
Bone marrow-derived macrophages (BMDMs) from adult mice were obtained as described previously (Longbrake et al., 2007) and then were cultured in RPMI 1640 supplemented with 1% P/S, 1% HEPES, 0.001% β-mercaptoethanol, 10% FBS, and 20% supernatant from M-CSF secreting L929 cells for 6–10 d before use (sL929; gift from Dr. Caroline Whitacre, The Ohio State University, Columbus, OH; all other reagents from Invitrogen).
The BV-2 cell line (gift from Dr. Jonathan Godbout, The Ohio State University, Columbus, OH) was derived from immortalized C57BL/6 microglia (Blasi et al., 1990). Cells were cultured in DMEM + 10% FBS, 1% P/S, 1% GlutaMax, and 1% sodium pyruvate and split twice a week (all reagents from Invitrogen).
One day before stimulation, microglia and BMDMs were replated in media appropriate to the assay (RPMI + 10% FBS) and then were acclimated overnight. Lipopolysaccharide (LPS) (100 ng/ml) or serum (20%) was applied for 6 (RNA) or 24 [nitric oxide (NO) production] h at 37°C. Serum was obtained from naive C57BL/6 mice, and the efficacy of the 20% dilution was defined empirically based on iNOS mRNA induction in control microglia. In a subset of experiments, wild-type BMDMs or microglia were activated in the presence of a neutralizing polyclonal anti-CX3CR1 antibody (Torrey Pines Biolabs).
Nitric oxide assay.
NO production was assessed using the Griess reaction. BMDMs (5 × 105/well) or BV-2 cells (3 × 105/well) were plated in a 24-well plate and allowed to adhere overnight. Stimuli were then applied for 24 h. After that time, 150 μl of supernatant was combined with 130 μl of dH2O and 20 μl of Griess reagent (Invitrogen), incubated at room temperature for 30 min and read at 548 nm (SpectraMax 190, Molecular Devices). Absorbance readings were converted to nitrite concentrations based on a sodium nitrite standard curve.
Western blotting.
Spinal cord segments (5 mm centered at the impact site or at T9/10 for uninjured tissue) were digested using T-PER Tissue Protein Extraction Reagent (Pierce) and then protein concentrations were determined using Coomassie Plus Protein Assay Kit (Pierce). Samples (20 mg each) were diluted in NuPAGE LDS sample buffer and then separated on NuPAGE 4–12% Bis-Tris gels (45 min at 200 V). Proteins were then transferred to a nitrocellulose membrane for 90 min at 30 V. The membrane was blocked in 5% bovine serum albumin and incubated with antibodies against either CX3CL1 (1:250, Torrey Pines TP233) or CX3CR1 (1:400, Abcam ab8021) overnight at 4°C. Detection of β-actin was done to assess equal loading. HRP-conjugated secondary antibodies were then incubated with the membrane for 1 h at room temperature. Blots were developed using a chemiluminescent HRP substrate kit (Pierce) and visualized on a Kodak Image Station 4000MM PRO. A total of three Western blots were performed using different animal tissue for each. Densitometric analysis of CX3CL1 and CX3CR1 expression was done using the MCID Elite system (InterFocus Imaging).
Statistics.
Real-time PCR data were analyzed by ANOVA followed by Dunnett's post hoc test and/or unpaired t tests as indicated in the figure legends. Behavioral data were analyzed using repeated-measures two-way ANOVA followed by Bonferroni's post hoc test, or by unpaired t tests as indicated. Recovery of coordination was assessed with a χ2 analysis. Immunohistochemical data were evaluated using unpaired t tests. Flow cytometry data were evaluated by two-way ANOVA followed by Bonferroni's post hoc test. Results were considered statistically significant at p < 0.05. All data points represent group mean ± SEM.
Results
CX3CL1 and CX3CR1 are regulated by SCI
Expression of CX3CL1 mRNA is reduced 1–7 d postinjury (dpi), returning to preinjury levels by 14 dpi (Fig. 1A). This acute decline corresponded with a loss of CX3CL1 protein at 7 dpi (Fig. 1C,D). In contrast, expression of CX3CR1 mRNA increased, beginning at 3 dpi, reaching levels 8–11× higher than sham-injured mice by 7–28 dpi (Fig. 1B). Western blots revealed dynamic regulation of the 50 and 55 kDa isoforms of CX3CR1 (Fig. 1C,D). Both were reduced after SCI, but only the 50 kDa isoform remained below preinjury levels out to 42 dpi. Conversely, the 55 kDa isoform was reduced below the limits of detection through 3 dpi and then increased above preinjury levels beginning at 7 dpi.
CX3CR1 deficiency is neuroprotective and promotes functional recovery after SCI
CX3CL1 and CX3CR1 exhibit a monogamous relationship (Imai et al., 1997). Therefore, the functional significance of postinjury regulation of this ligand/receptor pair can be inferred using mice deficient in either protein. We analyzed spontaneous motor recovery after SCI in wild-type C57BL/6 mice and mice in which the enhanced green fluorescent protein (GFP) gene was inserted into the coding region of Cx3cr1. Thus, signaling via CX3CR1 is abolished in CX3CR1GFP/GFP mice and cells expressing the receptor are GFP+ (Jung et al., 2000).
Before injury, wild-type and CX3CR1GFP/GFP mice displayed identical patterns of overground locomotion, and immediately after SCI mice in both groups were equally impaired (Fig. 2A). However, by 5 dpi, recovery of hindlimb function improved in CX3CR1GFP/GFP mice relative to wild-type mice. Functional improvements continued over the ensuing weeks. Importantly, 62% of CX3CR1GFP/GFP mice reestablished forelimb/hindlimb coordination (BMS score ≥ 6) by 28 dpi, while only 13% of wild-type mice achieved this functional milestone (χ2 analysis; p = 0.03). This difference was sustained at 35 dpi and was reduced slightly (62% vs 25%) at 42 dpi. Improved recovery in CX3CR1GFP/GFP mice was accompanied by a significant reduction in myelin and axon loss at and beyond the site of injury (Fig. 2B–F).
CX3CR1 and accumulation of CNS macrophages after SCI
CX3CR1 deficiency could attenuate tissue damage and improve recovery of function by reducing the recruitment and/or the activation of microglia and MDMs (Lesnik et al., 2003; Nanki et al., 2004; Dénes et al., 2008; Oh et al., 2008). To test this hypothesis, macrophages were isolated from injured spinal cord of wild-type and CX3CR1GFP/GFP mice and then CD11b+ macrophages were quantified using flow cytometry. Counter to our hypothesis, a modest but significant increase in CD11b+ macrophages was detected in the injured spinal cords of CX3CR1GFP/GFP mice (vs wild-type) (Fig. 3A,B).
To determine whether this was a result of enhanced microglial activation or increased monocyte recruitment, CD45 expression was quantified on cells in the CD11b gate (Fig. 3C,D). The relative expression of CD45 has been used to differentiate resident microglia (CD45lo) from recruited monocytes (CD45hi) (Sedgwick et al., 1991). In naive spinal cord of both genotypes, CD45lo/CD11b+ cells (microglia) predominate. However, by 3 dpi, more CD45hi cells accumulate in the spinal cord of CX3CR1GFP/GFP mice (Fig. 3C,D). Consistent with previous reports, numbers of CD45hi cells decrease by 7–14 dpi in both groups (Stirling and Yong, 2008; Saiwai et al., 2010), perhaps because monocyte recruitment is attenuated and CD45 expression is reduced on microglia and MDMs within the lesion (Ponomarev et al., 2011).
CX3CR1 deletion impairs inflammatory signaling in CNS macrophages
To determine whether changes in MDM recruitment described above (Fig. 3) are associated with changes in the expression of inflammatory mediators produced by activated macrophages and microglia, real-time PCR was used to screen injured spinal cord homogenates for differences in cytokine and iNOS mRNA. Consistent with previous reports from our laboratory and others (Pineau and Lacroix, 2007; Lee et al., 2009), expression of a subset of inflammatory cytokines (e.g., IL-6, IL-1β) and iNOS mRNA was increased after SCI, regardless of genotype (data not shown). However, postinjury expression of IL-6 and iNOS mRNA was markedly reduced in CX3CR1GFP/GFP mice (data not shown). To determine whether deficient CX3CR1 signaling in intraspinal macrophages could account for these mRNA expression changes, a series of controlled in vitro experiments and ex vivo flow cytometry assays was completed.
Microglia isolated from wild-type or CX3CR1GFP/GFP mice were stimulated with diluted preparations of mouse serum. Serum stimulation is relevant in the context of SCI since blood proteins that accumulate in the injured CNS parenchyma can elicit inflammatory signaling (Sakurai et al., 1997; Okamura et al., 2001; Beg, 2002). As shown in Figure 4A, serum increased IL-6 and iNOS mRNA expression in wild-type but not CX3CR1GFP/GFP microglia. There was minimal effect on the expression of IL-1β or TNF-α mRNA.
Figure 4.

Impaired CX3CR1 signaling in CNS macrophages renders them less responsive to inflammatory stimuli. A, Mouse serum (20%) induces IL-6 and iNOS mRNA expression in wild-type but not CX3CR1GFP/GFP primary microglia. B, In response to LPS (100 ng/ml; 24 h stimulation), CX3CR1GFP/GFP BMDMs produce significantly less nitrite than wild-type BMDMs. C, D, This effect was replicated in wild-type primary BMDMs (C) and BV-2 microglia (D) treated with a neutralizing CX3CR1 antibody. The inhibitory effects of the antibody could be titrated (2.5–10 μg/ml). LPS-triggered nitrite release was unaffected by different concentrations of nonspecific IgG indicating that the effects of the blocking antibody were specific to CX3CR1. Data are representative of 2–3 independent experiments for each graph. Real-time PCR data were obtained from duplicate or triplicate wells and mRNA expression was normalized to GAPDH and to unstimulated controls (dashed line). Data were analyzed by unpaired t tests (A, B) or one-way ANOVA followed by Bonferroni's post hoc test (C, D). *p < 0.05, **p < 0.01, ***p < 0.001 compared to wild-type (A, B) or to LPS alone (C, D).
Next, we expanded our in vitro models to include BMDMs. MDMs that infiltrate the injured spinal cord are derived from the bone marrow (Longbrake et al., 2007). Instead of using serum as a stimulus and mRNA expression as the sole readout of activation, we stimulated cells with LPS, a canonical inflammatory stimulus (Hoffmann et al., 1999), and then measured release of nitrite, the metabolite produced as a consequence of iNOS activation. As with primary microglia, CX3CR1GFP/GFP BMDMs produced nitrite but significantly less than wild-type BMDMs (Fig. 4B). To ensure that impaired inflammatory signaling in CX3CR1-deficient cells was not caused by a functional defect that arises during development of the genetically modified cells, nitrite production was measured from wild-type BMDMs and BV-2 microglia that were stimulated in the presence of a neutralizing anti-CX3CR1 antibody. In both BMDMs and BV-2 cells, the anti-CX3CR1 antibody reduced nitrite production in a concentration-dependent manner (Fig. 4C,D).
Finally, microglia and macrophages were isolated from injured spinal cords of wild-type and CX3CR1GFP/GFP mice and then iNOS+/CD11b+ cells were quantified using flow cytometry. Using this approach, a biphasic increase of iNOS+ cells was observed after SCI in wild-type mice, peaking initially at 1 dpi with a secondary increase evident at 7–14 dpi (Fig. 5). The acute changes are presumably mediated by activated glia and perhaps early infiltrating neutrophils, while later changes correspond with the accumulation of MDMs (Stirling and Yong, 2008; Saiwai et al., 2010). This biphasic response was noticeably absent from the injured spinal cord of CX3CR1GFP/GFP mice. Numbers of iNOS+/CD11b+ cells peaked at 1 dpi, but cells with this phenotype fail to accumulate at later stages of recovery (>7 dpi) in CX3CR1GFP/GFP mice.
Loss of CX3CR1 signaling in monocyte-derived macrophages promotes recovery from SCI
Collectively, the above data indicate that serum and perhaps other lesion-derived factors enhance inflammatory and oxidative functions in microglia and MDMs and that signaling via CX3CR1 regulates these functions. Based on the current data and past work from our group, we predicted that the neuroprotection and enhanced recovery of function observed after SCI in CX3CR1GFP/GFP mice was primarily a result of altered function in MDMs. Indeed, improved functional recovery becomes evident in CX3CR1GFP/GFP mice by 3 dpi (Fig. 2), a time that coincides with an increase in intraspinal CX3CR1 expression (Fig. 1) and accumulation of iNOS+/CD11b+ macrophages. Also, the onset of monocyte recruitment occurs at 3 dpi (Popovich and Hickey, 2001). To test this hypothesis and differentiate between the functional contributions of microglia and MDMs, we generated bone marrow chimeric mice.
A dose–response study indicated that a single 5 Gy irradiation dose was optimal for our experimental design. Indeed, >90% chimerization of transplanted bone marrow cells was observed 4 weeks after transplantation with limited recruitment of bone-marrow cells into the intact spinal cord (Fig. 6A–D). Next, bone marrow of C57BL/6 wild-type mice was sublethally irradiated (5 Gy) and then reconstituted with bone marrow cells from wild-type (WT→WT) or CX3CR1GFP/GFP (GFP→WT) mice. Thus, any adverse (or priming) effects of irradiation on the CNS are controlled for in both groups (Mildner et al., 2007), but CX3CR1 signaling is impaired only in MDMs of GFP→WT mice. All mice received a SCI 8 weeks after reconstitution of irradiated bone marrow.
As shown in Figure 6, patterns of spontaneous behavioral recovery were similar in SCI WT→WT and GFP→WT mice for at least 2–3 weeks after injury, after which recovery was significantly improved in GFP→WT mice. Corresponding with the improved recovery was a significant increase in myelin and axon sparing at the impact site of GFP→WT mice. Consistent with data in Figure 3, deficient CX3CR1 signaling did not impair recruitment of MDMs to the injury site; quantification of Iba-1+ macrophages was identical between groups (data not shown). Moreover, the spatial distribution of MDMs was consistent with our previous data in SCI rats showing preferential recruitment to the lesion epicenter with fewer cells distributing within the lesion penumbra or spared white matter (Fig. 6J,K) (Popovich and Hickey, 2001). The kinetics of improved recovery in GFP→WT mice does not coincide with that observed in CX3CR1GFP/GFP mice, suggesting that deficient signaling in microglia imparts an additional neuroprotective benefit in these latter mice. However, the anatomical and behavioral improvements with sustained MDM recruitment in GFP→WT mice indicate that the onset of deleterious functions in monocytes after SCI is dependent on CX3CR1 signaling.
CX3CR1 deficiency alters the phenotype of MDMs that infiltrate the injured spinal cord
Reduced inflammatory signaling in CX3CR1GFP/GFP MDMs attenuates their neurotoxic effects after SCI; however, the types of MDMs that are recruited to the injury site may also be affected. Relative expression of CX3CR1 defines phenotypically distinct monocyte subsets: CCR2+/Ly6Chi/CX3CR1lo “inflammatory” monocytes and CCR2−/Ly6CloCX3CR1hi “resident” monocytes (Geissmann et al., 2003; Nahrendorf et al., 2007). Recently, it was shown that spontaneous recovery after SCI could be improved by boosting the recruitment of Ly6Chi/CCR2+/CD11c+ (CX3CR1lo) monocytes (Shechter et al., 2009). Although these monocytes were found to express high levels of Ly6C antigen, they were designated as being “anti-inflammatory.”
Here, we used flow cytometry to determine the phenotype of monocytes recruited to the injured spinal cord of wild-type and CX3CR1GFP/GFP mice. CNS macrophages isolated from injured spinal cords were analyzed via five-color flow cytometry. The relative expression of iNOS, CCR2, MHCII and CD11c was quantified on CD11b+/Ly6Chi and CD11b+/Ly6Clo macrophages (Figs. 7, 8). Consistent with data in Figure 5, the number and relative expression of iNOS+ CNS macrophages increased after SCI in wild-type mice compared to CX3CR1GFP/GFP mice, especially at 7–14 dpi in the CD11b+/Ly6Clo population (Fig. 8). This latter population of cells was scarce in CX3CR1GFP/GFP mice and was replaced by the recruitment of CCR2+ cells. Increased numbers of CCR2+ macrophages were found in both the Ly6Chi and Ly6Clo macrophage subsets, but consistent with their predilection for migrating to inflammatory sites, the relative expression of CCR2 was consistently higher on Ly6Chi macrophages (compare MFI values in Figs. 7 and 8).
During the first week after injury, CD11b+/Ly6Chi and CD11b+/Ly6Clo macrophages express minimal MHC class II or CD11c, regardless of genotype (Figs. 7, 8). By 14 dpi, the number and relative expression of MHCII+ and CD11c+ macrophages were predictive of genotype; cell number and the relative expression of MHCII increased on a per-cell basis in wild-type mice, while the number of CD11c+ macrophages (and expression of this antigen) increased in CX3CR1GFP/GFP mice during a similar time course.
Thus, the MDM responses (> 3 dpi) of wild-type and CX3CR1GFP/GFP mice after SCI are distinct; a CD11b+/Ly6Clo/iNOS+/MHCII+/CD11c− population dominates the injury site of wild-type mice, while CD11b+/CCR2+/Ly6Chi/MHCII−/CD11c+ cells predominate in spinal cord of CX3CR1GFP/GFP mice. These latter macrophages also expressed some iNOS, albeit at low levels compared to those found in wild-type mice.
Discussion
Spinal cord injury elicits a protracted macrophage response comprised of resident microglia and MDMs. Injurious and repair functions have been attributed to microglia and macrophages; however, the mechanisms that regulate these opposing functions remain poorly defined.
Fractalkine (CX3CL1), a unique chemokine with profound effects on healthy and injured/diseased tissues (Soriano et al., 2002; Lesnik et al., 2003; Nanki et al., 2004; Cardona et al., 2006; Oh et al., 2008), is synthesized as a membrane-spanning adhesion molecule. In healthy CNS, CX3CL1 is expressed by neurons, while its receptor, CX3CR1, is expressed on microglia (Harrison et al., 1998; Mildner et al., 2007). In the presence of TNF-α-converting enzyme (TACE, ADAM17) or disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), membrane-bound CX3CL1 can be released by proteolysis (Garton et al., 2001; Hundhausen et al., 2003). Soluble CX3CL1 acts as a conventional chemokine, enhancing the recruitment and activation of CX3CR1-expressing leukocytes, mostly monocytes. Here we show that intraspinal CX3CL1 and CX3CR1 are dynamically regulated after SCI and that abolishing CX3CR1 signaling confers neuroprotection and promotes recovery of function. These benefits are associated with suppression of inflammatory signaling in microglia and MDMs and reduced accumulation of Ly6Clo/iNOS+ monocytes.
After SCI, CX3CR1 signaling regulates two distinct phases of intraspinal inflammation. The first phase occurs within 72 h after injury and is associated with a modest loss of CX3CL1 and CX3CR1. These changes are likely to cause dysregulation of microglia homeostasis with little or no effect on monocytes, which are not recruited until ∼3 dpi. The delayed increase in CX3CR1 after 3 dpi represents the second phase and coincides with monocyte entry into the lesion (Popovich and Hickey, 2001). In CX3CR1-deficient mice, recovery of function is accelerated during this second phase, despite enhanced intraspinal infiltration by monocytes (Fig. 3), suggesting that the injurious effects of MDMs in wild-type mice are controlled in part by CX3CR1.
In wild-type mice, despite a delayed increase in mRNA synthesis, CX3CL1 protein is unchanged 1–6 weeks after injury. This could be explained by enhanced proteolytic cleavage of CX3CL1 from the membrane with immediate binding to CX3CR1 during the second phase of intraspinal inflammation (Chapman et al., 2000; Garton et al., 2001). Thus, measurable changes in soluble CX3CL1 would be transient and below the levels of detection by Western blot (Fig. 1C,D). Regardless, an increase in soluble CX3CL1 could potentiate inflammatory signaling in CX3CR1-expressing microglia and macrophages, resulting in the enhanced release of toxic cytokines and oxidative metabolites. This would help explain why deletion of CX3CL1 or CX3CR1 confers neuroprotection after experimental stroke (Soriano et al., 2002; Dénes et al., 2008) and, in the present study, SCI. Common to each of these models is activation of macrophages via toll-like receptor 4 (TLR4). After an ischemic or traumatic CNS injury, vascular damage is substantial and serum proteins and necrotic tissue debris elicit inflammatory signaling in microglia and monocytes via TLR4 (Beg, 2002; Yang et al., 2008; Ransohoff and Perry, 2009). Activation via TLR4 is known to regulate CX3CL1 and CX3CR1 expression (Boddeke et al., 1999; Pachot et al., 2008). Since activation of CX3CR1 can also increase NFκB-dependent transcription (Ishida et al., 2008), signaling via this receptor could potentiate the inflammatory effects of TLR4 activation, while loss of CX3CR1 signaling would have the opposite effect. This function of CX3CR1 signaling appears to extend beyond the CNS. In accordance with our data, CX3CR1GFP/GFP macrophages produce less iNOS and inflammatory cytokines than wild-type cells in models of septic peritonitis and acute colitis (Ishida et al., 2008; Kostadinova et al., 2010).
A decrease in the number of CD11b+/Ly6Clo/iNOS+ macrophages after 7 dpi in CX3CR1-deficient mice was associated with reduced neuropathology and enhanced functional recovery, suggesting that in wild-type mice, these cells cause pathology or inhibit repair. The deleterious effects of wild-type monocytes were confirmed using chimeric mice in which wild-type monocytes were replaced with CX3CR1-deficient cells. Any confounding effects of irradiation-induced priming were controlled for using wild-type chimeras (Ajami et al., 2007; Mildner et al., 2007)). These data, when considered together with previous reports describing enhanced recovery and reduced pathology in iNOS-deficient mice (Isaksson et al., 2005; Genovese et al., 2006), indicate that CD11b+/Ly6Clo/iNOS+ macrophages are pathological effector cells after SCI.
In parallel with its ability to regulate intracellular signaling, the relative expression of CX3CR1 dictates patterns of monocyte recruitment. Two subsets of circulating monocytes with varying levels of surface CX3CR1 have been described (Geissmann et al., 2003; Sunderkötter et al., 2004; Nahrendorf et al., 2007). Ly6Chi/CX3CR1lo monocytes have a short half-life and express CCR2, a chemokine receptor that facilitates recruitment to sites of inflammation. Conversely, Ly6Clo/CX3CR1hi monocytes are CCR2−, have a comparatively longer half-life, and are dependent on CX3CR1 for recruitment (Nahrendorf et al., 2007; Combadière et al., 2008). Whether these monocyte subsets are distinct cells or the same cells at different stages of maturation is controversial (Geissmann et al., 2003; Sunderkötter et al., 2004; Nahrendorf et al., 2007). What is agreed upon is that phenotypically distinct monocyte populations are associated with tissue pathology and repair in various forms of disease (Robbins and Swirski, 2010).
In a model of myocardial infarction, effective wound resolution and tissue repair requires sequential recruitment of Ly6Chi and then Ly6Clo monocytes (Nahrendorf et al., 2007). Since Ly6Chi monocytes have high proteolytic activity and degrade injured and intact tissues (Sunderkötter et al., 2004; Zhu et al., 2007; Ancuta et al., 2009; King et al., 2009; Mildner et al., 2009), we predicted that a bias in the recruitment of Ly6Chi (vs Ly6Clo) monocytes could contribute to lesion pathology and loss of function after SCI. Indeed, Ly6Chi monocytes are thought to give rise to M1 macrophages in vivo (Lin et al., 2009), and recent data indicate that intraspinal macrophages acquire a neurotoxic “classically activated” M1 phenotype after SCI (Kigerl et al., 2009). Contrary to our original hypothesis, we find that a novel Ly6Clo/iNOS+ monocyte population predominates at later stages of recovery in SCI wild-type mice and that these cells are responsible for impaired recovery. This delayed accumulation of Ly6Clo monocytes may be specific to SCI, as Ly6Clo monocytes are excluded from the CNS in models of autoimmune inflammation (EAE) and acute viral encephalitis (Gaupp et al., 2003; Huang et al., 2006; King et al., 2009). This disparity underscores the controversial nature of the role of CX3CR1 in monocyte recruitment and also the potential for disease-specific regulation of monocyte phenotype.
Recently, impaired recruitment of CCR2+/Ly6Chi/CD11c+ monocytes was implicated as a mechanism underlying poor recovery after SCI (Shechter et al., 2009). Injection of large numbers of Ly6Chi monocytes into the circulation enhanced recovery after SCI. Also, vaccination-enhanced recruitment of Ly6Chi monocytes after SCI improved recovery, an effect that was abrogated by preinjecting mice with anti-CCR2 depleting antibodies. Thus, selective depletion of subsets of monocyte/macrophages may be developed as a therapy after SCI. If CCR2+/Ly6Chi/CD11c+ monocytes have anti-inflammatory effects, it would be unwise to deplete them (Lu et al., 2011). Fate-mapping studies are needed to determine whether these cells differentiate into CCR2−/Ly6Clo/CX3CR1hi/CD11c− monocytes and if so, whether they maintain anti-inflammatory functions indefinitely (Sunderkötter et al., 2004; Arnold et al., 2007). Although the monocytes that accumulate after SCI in CX3CR1GFP/GFP mice are similar in phenotype to those described by Shechter et al. (2009), we do not know whether they have anti-inflammatory functions. Instead, their phenotype (CCR2+/Ly6Chi/MHCII−/ CD11c+) is reminiscent of immature myeloid precursor cells, and expression of CX3CR1 has been shown to be important for myeloid precursors to develop into mature monocytes (Sunderkötter et al., 2004; Auffray et al., 2009). It will be interesting to see whether prolongation of an immature monocyte phenotype has neuroprotective effects in SCI and other neuroinflammatory diseases.
There are human CD16+ and CD16− monocyte subsets that correspond with Ly6Chi and Ly6Clo monocytes in mice (Geissmann et al., 2003; Urra et al., 2009). Low and high levels of CX3CR1 also define these distinct monocyte populations, respectively. Accordingly, a greater understanding of the role played by CX3CR1 in regulating post-SCI inflammation will yield novel opportunities for therapeutic intervention. Because CX3CR1 signaling has been implicated in the development of neuropathic pain, malignancies, and progression of inflammatory disease (e.g., atherosclerosis, rheumatoid arthritis), drugs that target CX3CR1 signaling are under development (Davis et al., 2004; Inoue et al., 2005; Streit et al., 2005; Charo and Ransohoff, 2006; Dorgham et al., 2009). These drugs should be tested in models of CNS injury. It also will be useful to determine whether CX3CR1 polymorphisms affect recovery after SCI in people. Data suggest that common variants of the cx3cr1 gene can lead to blunted CX3CR1 signaling and confer resistance or alleviate pathology in cardiovascular disease, age-related macular degeneration, and brain cancer (Combadière et al., 2007; Rodero et al., 2008; Apostolakis et al., 2009). Perhaps prospective genetic analysis of CX3CR1 could be of prognostic value in predicting functional recovery or the onset of systemic inflammation and neuropathic pain after SCI (Gris et al., 2004; Gris et al., 2009; Popovich and McTigue, 2009; Austin and Moalem-Taylor, 2010).
Footnotes
This work was funded by grants from the National Institutes of Health: NIH R01 NS37846 (P.G.P.), NIH F31 NS064725 (D.J.D.), and P30-NS045758. We thank Zhen Guan, Ming Wang, Ping Wei, Violeta McGaughy, and Pat Walters for technical assistance.
The authors declare no competing financial interests.
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–1543. doi: 10.1038/nn2014. [DOI] [PubMed] [Google Scholar]
- Ancuta P, Wang J, Gabuzda D. CD16+ monocytes produce IL-6, CCL2, and matrix metalloproteinase-9 upon interaction with CX3CL1-expressing endothelial cells. J Leukoc Biol. 2006;80:1156–1164. doi: 10.1189/jlb.0206125. [DOI] [PubMed] [Google Scholar]
- Ancuta P, Liu KY, Misra V, Wacleche VS, Gosselin A, Zhou X, Gabuzda D. Transcriptional profiling reveals developmental relationship and distinct biological functions of CD16+ and CD16- monocyte subsets. BMC Genomics. 2009;10:403. doi: 10.1186/1471-2164-10-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolakis S, Amanatidou V, Papadakis EG, Spandidos DA. Genetic diversity of CX3CR1 gene and coronary artery disease: new insights through a meta-analysis. Atherosclerosis. 2009;207:8–15. doi: 10.1016/j.atherosclerosis.2009.03.044. [DOI] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol. 2010;229:26–50. doi: 10.1016/j.jneuroim.2010.08.013. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Beg AA. Endogenous ligands of Toll-like receptors: implications for regulating inflammatory and immune responses. Trends Immunol. 2002;23:509–512. doi: 10.1016/s1471-4906(02)02317-7. [DOI] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Boddeke EW, Meigel I, Frentzel S, Biber K, Renn LQ, Gebicke-Härter P. Functional expression of the fractalkine (CX3C) receptor and its regulation by lipopolysaccharide in rat microglia. Eur J Pharmacol. 1999;374:309–313. doi: 10.1016/s0014-2999(99)00307-6. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci. 2000;20(RC87) doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Combadière C, Feumi C, Raoul W, Keller N, Rodéro M, Pézard A, Lavalette S, Houssier M, Jonet L, Picard E, Debré P, Sirinyan M, Deterre P, Ferroukhi T, Cohen SY, Chauvaud D, Jeanny JC, Chemtob S, Behar-Cohen F, Sennlaub F. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–2928. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117:1649–1657. doi: 10.1161/CIRCULATIONAHA.107.745091. [DOI] [PubMed] [Google Scholar]
- Davis CN, Zujovic V, Harrison JK. Viral macrophage inflammatory protein-II and fractalkine (CX3CL1) chimeras identify molecular determinants of affinity, efficacy, and selectivity at CX3CR1. Mol Pharmacol. 2004;66:1431–1439. doi: 10.1124/mol.104.003277. [DOI] [PubMed] [Google Scholar]
- Dénes A, Ferenczi S, Halász J, Környei Z, Kovács KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Gensel JC, Ankeny DP, van Rooijen N, Popovich PG. An efficient and reproducible method for quantifying macrophages in different experimental models of central nervous system pathology. J Neurosci Methods. 2009;181:36–44. doi: 10.1016/j.jneumeth.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorgham K, Ghadiri A, Hermand P, Rodero M, Poupel L, Iga M, Hartley O, Gorochov G, Combadière C, Deterre P. An engineered CX3CR1 antagonist endowed with anti-inflammatory activity. J Leukoc Biol. 2009;86:903–911. doi: 10.1189/jlb.0308158. [DOI] [PubMed] [Google Scholar]
- Fraticelli P, Sironi M, Bianchi G, D'Ambrosio D, Albanesi C, Stoppacciaro A, Chieppa M, Allavena P, Ruco L, Girolomoni G, Sinigaglia F, Vecchi A, Mantovani A. Fractalkine (CX3CL1) as an amplification circuit of polarized Th1 responses. J Clin Invest. 2001;107:1173–1181. doi: 10.1172/JCI11517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garton KJ, Gough PJ, Blobel CP, Murphy G, Greaves DR, Dempsey PJ, Raines EW. Tumor necrosis factor-alpha-converting enzyme (ADAM17) mediates the cleavage and shedding of fractalkine (CX3CL1) J Biol Chem. 2001;276:37993–38001. doi: 10.1074/jbc.M106434200. [DOI] [PubMed] [Google Scholar]
- Gaupp S, Pitt D, Kuziel WA, Cannella B, Raine CS. Experimental autoimmune encephalomyelitis (EAE) in CCR2(-/-) mice: susceptibility in multiple strains. Am J Pathol. 2003;162:139–150. doi: 10.1016/S0002-9440(10)63805-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Genovese T, Mazzon E, Mariotto S, Menegazzi M, Cardali S, Conti A, Suzuki H, Bramanti P, Cuzzocrea S. Modulation of nitric oxide homeostasis in a mouse model of spinal cord injury. J Neurosurg Spine. 2006;4:145–153. doi: 10.3171/spi.2006.4.2.145. [DOI] [PubMed] [Google Scholar]
- Gris D, Marsh DR, Oatway MA, Chen Y, Hamilton EF, Dekaban GA, Weaver LC. Transient blockade of the CD11d/CD18 integrin reduces secondary damage after spinal cord injury, improving sensory, autonomic, and motor function. J Neurosci. 2004;24:4043–4051. doi: 10.1523/JNEUROSCI.5343-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gris P, Tighe A, Thawer S, Hemphill A, Oatway M, Weaver L, Dekaban GA, Brown A. Gene expression profiling in anti-CD11d mAb-treated spinal cord-injured rats. J Neuroimmunol. 2009;209:104–113. doi: 10.1016/j.jneuroim.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann JA, Kafatos FC, Janeway CA, Ezekowitz RA. Phylogenetic perspectives in innate immunity. Science. 1999;284:1313–1318. doi: 10.1126/science.284.5418.1313. [DOI] [PubMed] [Google Scholar]
- Howard CV, Reed MG. New York: Springer; 1998. Unbiased stereology: three-dimensional measurement in microscopy. [Google Scholar]
- Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar-Mather TP, He TT, Weaver JT, Ljunggren HG, Biron CA, Littman DR, Ransohoff RM. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 2006;20:896–905. doi: 10.1096/fj.05-5465com. [DOI] [PubMed] [Google Scholar]
- Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102:1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- Inoue A, Hasegawa H, Kohno M, Ito MR, Terada M, Imai T, Yoshie O, Nose M, Fujita S. Antagonist of fractalkine (CX3CL1) delays the initiation and ameliorates the progression of lupus nephritis in MRL/lpr mice. Arthritis Rheum. 2005;52:1522–1533. doi: 10.1002/art.21007. [DOI] [PubMed] [Google Scholar]
- Isaksson J, Farooque M, Olsson Y. Improved functional outcome after spinal cord injury in iNOS-deficient mice. Spinal Cord. 2005;43:167–170. doi: 10.1038/sj.sc.3101672. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Hayashi T, Goto T, Kimura A, Akimoto S, Mukaida N, Kondo T. Essential involvement of CX3CR1-mediated signals in the bactericidal host defense during septic peritonitis. J Immunol. 2008;181:4208–4218. doi: 10.4049/jimmunol.181.6.4208. [DOI] [PubMed] [Google Scholar]
- Jakeman LB, Guan Z, Wei P, Ponnappan R, Dzwonczyk R, Popovich PG, Stokes BT. Traumatic spinal cord injury produced by controlled contusion in mouse. J Neurotrauma. 2000;17:299–319. doi: 10.1089/neu.2000.17.299. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, McGaughy VM, Popovich PG. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neurol. 2006;494:578–594. doi: 10.1002/cne.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. 2009;113:3190–3197. doi: 10.1182/blood-2008-07-168575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostadinova FI, Baba T, Ishida Y, Kondo T, Popivanova BK, Mukaida N. Crucial involvement of the CX3CR1-CX3CL1 axis in dextran sulfate sodium-mediated acute colitis in mice. J Leukoc Biol. 2010;88:133–143. doi: 10.1189/jlb.1109768. [DOI] [PubMed] [Google Scholar]
- Le W, Rowe D, Xie W, Ortiz I, He Y, Appel SH. Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson's disease. J Neurosci. 2001;21:8447–8455. doi: 10.1523/JNEUROSCI.21-21-08447.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MY, Chen L, Toborek M. Nicotine attenuates iNOS expression and contributes to neuroprotection in a compressive model of spinal cord injury. J Neurosci Res. 2009;87:937–947. doi: 10.1002/jnr.21901. [DOI] [PubMed] [Google Scholar]
- Lesnik P, Haskell CA, Charo IF. Decreased atherosclerosis in CX3CR1-/- mice reveals a role for fractalkine in atherogenesis. J Clin Invest. 2003;111:333–340. doi: 10.1172/JCI15555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SL, Castaño AP, Nowlin BT, Lupher ML, Jr, Duffield JS. Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol. 2009;183:6733–6743. doi: 10.4049/jimmunol.0901473. [DOI] [PubMed] [Google Scholar]
- Longbrake EE, Lai W, Ankeny DP, Popovich PG. Characterization and modeling of monocyte-derived macrophages after spinal cord injury. J Neurochem. 2007;102:1083–1094. doi: 10.1111/j.1471-4159.2007.04617.x. [DOI] [PubMed] [Google Scholar]
- Lu H, Huang D, Saederup N, Charo IF, Ransohoff RM, Zhou L. Macrophages recruited via CCR2 produce insulin-like growth factor-1 to repair acute skeletal muscle injury. FASEB J. 2011;25:358–369. doi: 10.1096/fj.10-171579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Brück W, Priller J, Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- Mildner A, Mack M, Schmidt H, Brück W, Djukic M, Zabel MD, Hille A, Priller J, Prinz M. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. 2009;132:2487–2500. doi: 10.1093/brain/awp144. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003;979:65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanki T, Urasaki Y, Imai T, Nishimura M, Muramoto K, Kubota T, Miyasaka N. Inhibition of fractalkine ameliorates murine collagen-induced arthritis. J Immunol. 2004;173:7010–7016. doi: 10.4049/jimmunol.173.11.7010. [DOI] [PubMed] [Google Scholar]
- Oh DJ, Dursun B, He Z, Lu L, Hoke TS, Ljubanovic D, Faubel S, Edelstein CL. Fractalkine receptor (CX3CR1) inhibition is protective against ischemic acute renal failure in mice. Am J Physiol Renal Physiol. 2008;294:F264–271. doi: 10.1152/ajprenal.00204.2007. [DOI] [PubMed] [Google Scholar]
- Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF., 3rd The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- Pachot A, Cazalis MA, Venet F, Turrel F, Faudot C, Voirin N, Diasparra J, Bourgoin N, Poitevin F, Mougin B, Lepape A, Monneret G. Decreased expression of the fractalkine receptor CX3CR1 on circulating monocytes as new feature of sepsis-induced immunosuppression. J Immunol. 2008;180:6421–6429. doi: 10.4049/jimmunol.180.9.6421. [DOI] [PubMed] [Google Scholar]
- Pineau I, Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol. 2007;500:267–285. doi: 10.1002/cne.21149. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med. 2011;17:64–70. doi: 10.1038/nm.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich P, McTigue D. Damage control in the nervous system: beware the immune system in spinal cord injury. Nat Med. 2009;15:736–737. doi: 10.1038/nm0709-736. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Hickey WF. Bone marrow chimeric rats reveal the unique distribution of resident and recruited macrophages in the contused rat spinal cord. J Neuropathol Exp Neurol. 2001;60:676–685. doi: 10.1093/jnen/60.7.676. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Robbins CS, Swirski FK. The multiple roles of monocyte subsets in steady state and inflammation. Cell Mol Life Sci. 2010;67:2685–2693. doi: 10.1007/s00018-010-0375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero M, Marie Y, Coudert M, Blondet E, Mokhtari K, Rousseau A, Raoul W, Carpentier C, Sennlaub F, Deterre P, Delattre JY, Debré P, Sanson M, Combadière C. Polymorphism in the microglial cell-mobilizing CX3CR1 gene is associated with survival in patients with glioblastoma. J Clin Oncol. 2008;26:5957–5964. doi: 10.1200/JCO.2008.17.2833. [DOI] [PubMed] [Google Scholar]
- Saiwai H, Ohkawa Y, Yamada H, Kumamaru H, Harada A, Okano H, Yokomizo T, Iwamoto Y, Okada S. The LTB4-BLT1 axis mediates neutrophil infiltration and secondary injury in experimental spinal cord injury. Am J Pathol. 2010;176:2352–2366. doi: 10.2353/ajpath.2010.090839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Kaise T, Yadomae T, Matsubara C. Different role of serum components and cytokines on alveolar macrophage activation by soluble fungal (1–>3)-beta-D-glucan. Eur J Pharmacol. 1997;334:255–263. doi: 10.1016/s0014-2999(97)01191-6. [DOI] [PubMed] [Google Scholar]
- Sedgwick JD, Schwender S, Imrich H, Dörries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A. 1991;88:7438–7442. doi: 10.1073/pnas.88.16.7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. 2009;6:e1000113. doi: 10.1371/journal.pmed.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano SG, Amaravadi LS, Wang YF, Zhou H, Yu GX, Tonra JR, Fairchild-Huntress V, Fang Q, Dunmore JH, Huszar D, Pan Y. Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J Neuroimmunol. 2002;125:59–65. doi: 10.1016/s0165-5728(02)00033-4. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Yong VW. Dynamics of the inflammatory response after murine spinal cord injury revealed by flow cytometry. J Neurosci Res. 2008;86:1944–1958. doi: 10.1002/jnr.21659. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Davis CN, Harrison JK. Role of fractalkine (CX3CL1) in regulating neuron-microglia interactions: development of viral-based CX3CR1 antagonists. Curr Alzheimer Res. 2005;2:187–189. doi: 10.2174/1567205053585765. [DOI] [PubMed] [Google Scholar]
- Sunderkötter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, Leenen PJ. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- Sunnemark D, Eltayeb S, Nilsson M, Wallström E, Lassmann H, Olsson T, Berg AL, Ericsson-Dahlstrand A. CX3CL1 (fractalkine) and CX3CR1 expression in myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis: kinetics and cellular origin. J Neuroinflammation. 2005;2:17. doi: 10.1186/1742-2094-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarozzo G, Campanella M, Ghiani M, Bulfone A, Beltramo M. Expression of fractalkine and its receptor, CX3CR1, in response to ischaemia-reperfusion brain injury in the rat. Eur J Neurosci. 2002;15:1663–1668. doi: 10.1046/j.1460-9568.2002.02007.x. [DOI] [PubMed] [Google Scholar]
- Urra X, Villamor N, Amaro S, Gómez-Choco M, Obach V, Oleaga L, Planas AM, Chamorro A. Monocyte subtypes predict clinical course and prognosis in human stroke. J Cereb Blood Flow Metab. 2009;29:994–1002. doi: 10.1038/jcbfm.2009.25. [DOI] [PubMed] [Google Scholar]
- Yang QW, Li JC, Lu FL, Wen AQ, Xiang J, Zhang LL, Huang ZY, Wang JZ. Upregulated expression of toll-like receptor 4 in monocytes correlates with severity of acute cerebral infarction. J Cereb Blood Flow Metab. 2008;28:1588–1596. doi: 10.1038/jcbfm.2008.50. [DOI] [PubMed] [Google Scholar]
- Zhu B, Bando Y, Xiao S, Yang K, Anderson AC, Kuchroo VK, Khoury SJ. CD11b+Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J Immunol. 2007;179:5228–5237. doi: 10.4049/jimmunol.179.8.5228. [DOI] [PubMed] [Google Scholar]
- Zujovic V, Benavides J, Vigé X, Carter C, Taupin V. Fractalkine modulates TNF-alpha secretion and neurotoxicity induced by microglial activation. Glia. 2000;29:305–315. [PubMed] [Google Scholar]