

Abstract
The function of the endogenous angiogenesis inhibitor thrombospondin-1 (TSP-1) in tissue repair has remained controversial. We established transgenic mice with targeted overexpression of TSP-1 in the skin, using a keratin 14 expression cassette. TSP-1 transgenic mice were healthy and fertile, and did not show any major abnormalities of normal skin vascularity, cutaneous vascular architecture, or microvascular permeability. However, healing of full-thickness skin wounds was greatly delayed in TSP-1 transgenic mice and was associated with reduced granulation tissue formation and highly diminished wound angiogenesis. Moreover, TSP-1 potently inhibited fibroblast migration in vivo and in vitro. These findings demonstrate that TSP-1 preferentially interfered with wound healing-associated angiogenesis, rather than with the angiogenesis associated with normal development and skin homeostasis, and suggest that therapeutic application of angiogenesis inhibitors might potentially be associated with impaired wound vascularization and tissue repair.
Keywords: blood vessels/skin/transgenic mice/wound healing
Introduction
Wound healing is characterized by the formation of a richly vascularized, hyperpermeable granulation tissue, supporting the increased nutritional needs of rapidly proliferating and migrating epidermal keratinocytes, fibroblasts and leukocytes (Dvorak, 1986). Several angiogenesis factors have been found to be upregulated in healing wounds, including vascular endothelial growth factor (VEGF) expressed by epidermal keratinocytes (Brown et al., 1992). Impaired wound healing in genetically diabetic mice is associated with diminished expression of VEGF by epidermal keratinocytes (Frank et al., 1995), as well as reduced expression of platelet-derived growth factor-A (PDGF-A), PDGF-B and of the B-type PDGF receptor (Beer et al., 1997). Moreover, mice deficient for basic fibroblast growth factor were characterized by delayed wound healing (Ortega et al., 1998). While these data strongly suggest that impaired wound healing may be partially caused by insufficient stimulation of blood vessels, due to decreased activity of angiogenesis factors, the biological role of endogenous inhibitors of angiogenesis during tissue repair has remained controversial.
Several naturally occurring angiogenesis inhibitors have been identified, including thrombospondin-1 (TSP-1) (Iruela-Arispe et al., 1991), TSP-2 (Volpert et al., 1995), angiostatin (O’Reilly et al., 1994), endostatin (O’Reilly et al., 1997) and vasostatin (Pike et al., 1998). TSP-1 is a 450 kDa homotrimeric matricellular glycoprotein that regulates attachment, proliferation, migration and differentiation of various cell types (for review see Bornstein, 1995). TSP-1 interacts with several cell surface receptors, including the integrins αvβ3, α3β1, α4β1, α5β1, the integrin-associated protein CD47, heparan sulfate proteoglycans and CD36 (Adams, 1997). TSP-1 inhibits proliferation and migration of vascular endothelial cells in vitro and inhibits neovascularization and tumor growth in vivo (Tolsma et al., 1993; Boukamp et al., 1997; Streit et al., 1999a). In human skin, TSP-1 is secreted by basal epidermal keratinocytes and is deposited in the basement membrane area (Wight et al., 1985), contributing to the normal anti-angiogenic barrier that separates the avascular epidermis from the vascularized dermis (Detmar, 1996). The role of TSP-1 during tissue repair has remained controversial. Increased amounts of TSP-1 have been detected in the extracellular matrix of early skin wounds and around vessels during the late stages of wound closure, possibly contributing to the normalization of vessel numbers at this stage (Raugi et al., 1987; Reed et al., 1993; DiPietro et al., 1996; Roth et al., 1998). In contrast, antisense inhibition of TSP-1 expression was reported to cause delayed cutaneous wound healing, and TSP-1 has been proposed as an important mediator of tissue repair (DiPietro et al., 1996).
To characterize the biological role of TSP-1 for tissue repair in vivo, and to investigate further the physiological role of TSP-1 for vascular development and function, we established a transgenic mouse model for the targeted overexpression of TSP-1 in the basal epidermis of the skin and in the outer root sheath keratinocytes of the hair follicle, using a keratin 14 expression cassette (Vassar et al., 1989; Detmar et al., 1998). Here, we report that chronic overexpression of TSP-1 in the skin of transgenic mice potently inhibited cutaneous tissue repair, granulation tissue formation and wound angiogenesis.
Results
Targeted overexpression of TSP-1 in the skin of transgenic mice
We used a keratin 14 promoter cassette (Figure 1A) to target TSP-1 transgene expression selectively to basal epidermal keratinocytes. Southern blot analysis of genomic DNA revealed transgene incorporation in eight founder mice, and three independent transgenic lines with copy numbers between 8 and ∼20 were established on the FVB background. No major phenotypical differences between these lines were observed. In all three transgenic lines, northern blot analysis confirmed transgenic TSP-1 mRNA expression in both dorsal and ventral skin of TSP-1 transgenic mice (Figure 1B), whereas TSP-1 mRNA expression was not significantly increased above baseline levels in the liver, the heart, or the kidneys of transgenic mice, demonstrating selective transgene expression in the skin. Enhanced TSP-1 mRNA expression in the skin of TSP-1 transgenic mice resulted in increased amounts of TSP-1 protein, as evaluated by western blot analysis of skin lysates (Figure 1C). In situ hybridizations confirmed targeted TSP-1 mRNA expression in basal keratinocytes of the epidermis and in outer root sheath keratinocytes of hair follicles (Figure 2E and F), whereas little TSP-1 mRNA expression was detected in wild-type skin (Figure 2C and D), mainly restricted to follicular keratinocytes and to blood vessels that were also labeled in transgenic skin.

Fig. 1. (A) Schematic representation of the K14–TSP-1 transgene construct. A 3.6 kb human TSP-1 cDNA fragment was ligated to the BamHI restriction site of the keratin 14 promoter cassette. (B) Overexpression of TSP-1 mRNA in the dorsal and ventral skin of 3-week-old K14–TSP-1 transgenic mice was confirmed by northern blot analysis. TSP-1 mRNA expression was not increased above wild-type (WT) levels in the liver of TSP-1 transgenic mice. Hybridization with a murine β-actin probe served as a control for equal loading. (C) Western blot analysis of skin lysates demonstrates the presence of the intact, 180 kDa TSP-1 protein in TSP-1 transgenic skin. TSP-1, natural human TSP-1. (D) Strong TSP-1 mRNA expression levels in keratinocytes isolated from TSP-1 transgenic mice. TSP-1 expression levels were higher under low Ca2+ conditions than under high Ca2+ conditions that favor keratinocyte differentiation. (E) Western blot analysis confirmed efficient secretion of transgenic TSP-1 protein into culture supernatants and increased protein expression in cell lysates obtained from transgenic keratinocytes, as compared with wild-type (WT) controls.

Fig. 2. (A and B) Twelve-day-old TSP-1 transgenic mice (right) were smaller than their wild-type littermates and showed a delayed first hair cycle. TSP-1 transgenic mice were phenotypically indistinguishable from their wild-type littermates after 4 weeks of age. Little TSP-1 mRNA expression was detected by in situ hybridization in wild-type skin (C and D), mainly restricted to follicular keratinocytes of the outer root sheath and to blood vessels. (E and F) Targeted overexpression of the K14–TSP-1 transgene in the basal keratinocyte layer (arrowheads) and in the outer root sheath keratinocytes of hair follicles (arrows) was confirmed by in situ hybridization. Bar: 250 µm. Giemsa staining of the tail skin of adult 7-week-old TSP-1 transgenic mice (H) showed no differences in skin structure as compared with wild-type mice (G). Bar: 250 µm.
The efficient expression of transgenic TSP-1 was further confirmed in primary keratinocyte cultures from neonatal wild-type and transgenic mice. Whereas wild-type keratinocytes expressed little TSP-1 mRNA, strong TSP-1 mRNA expression was detected in transgenic keratinocytes (Figure 1D). In both types of keratinocytes, TSP-1 expression levels were consistently higher under low Ca2+ conditions than under high Ca2+ conditions that favor keratinocyte differentiation (Hennings et al., 1980). Western blot analyses confirmed the efficient secretion of transgenic TSP-1 protein into culture supernatants (Figure 1E). Highly increased levels of TSP-1 protein were also detected in lysates obtained from transgenic keratinocytes.
Normal skin structure, vascularization and microvascular permeability in adult TSP-1 transgenic mice
TSP-1 transgenic mice were smaller than their wild-type littermates during the first 3 weeks post-partum and showed a delayed first hair cycle (Figure 2A and B). No other obvious alterations were observed, and transgenic mice were phenotypically indistinguishable from their wild-type littermates after 4 weeks of age. Histological analysis of the skin of adult TSP-1 transgenic mice showed no major differences in the thickness and morphology of the epidermis and dermis (Figure 2G and H), whereas some minor differences in skin architecture during the first 3 weeks post-partum were mainly due to the delayed first hair cycle (data not shown). Vascular perfusions did not reveal any obvious differences in the branching pattern and structure of blood vessels in the ear skin of TSP-1 transgenic mice (Figure 3A and B). Using computer-assisted morphometric analysis of CD31-stained sections, no significant differences in vascular density and average vessel size were observed in the skin of adult transgenic mice (74 ± 12 vessels/mm2; 141 ± 26 µm2), as compared with wild-type skin (67 ± 9 vessels/mm2; 154 ± 21 µm2). Moreover, cutaneous microvascular permeability was unaltered in TSP-1 transgenic mice, as demonstrated in a modified Miles assay. Comparable vascular leakage was observed in both transgenic and wild-type mice after intradermal injection of 50 and 100 ng of VEGF, and baseline permeability levels were not different (Figure 3C). In addition, bleeding times were comparable in wild-type and TSP-1 transgenic mice (data not shown).

Fig. 3. Comparable branching pattern and structure of cutaneous blood vessels in adult wild-type mice (A) and TSP-1 transgenic mice (B), visualized in whole-mount preparations of mouse ears after perfusion with fluorescein-labeled L.esculentum lectin. Bar: 1 mm. (C) Microvascular permeability of skin vessels was not altered in TSP-1 transgenic mice (right panels) when compared with wild-type littermates (left panels) in a modified Miles assay. Comparable extravasation of intravenously injected Evans blue was observed after intradermal injection of recombinant VEGF (50 and 100 ng/ml). Injection of PBS served as a control for baseline permeability levels.
Impaired wound healing and granulation tissue formation in TSP-1 transgenic mice
Excisional full-thickness wounds were created on the backs of 8-week-old TSP-1 transgenic mice and wild-type littermates. At 24 h after wounding, all wounds were covered with a dry scab that remained adherent until days 5–7 in control mice and until day 10 in TSP-1 transgenic mice (Figure 4A). Three days after wounding, wound areas were reduced by >50% in control mice, and wounds were completely closed after 8 days. In contrast, no significant wound closure was observed in TSP-1 transgenic mice during the first 4 days after wounding (Figure 4). At day 9, the original wound size was reduced by >50%, and complete wound closure occurred on day 14. Thus, wound closure was delayed by 6 days (p <0.001) in TSP-1 transgenic mice. Comparable results were obtained in three independent wound-healing studies.
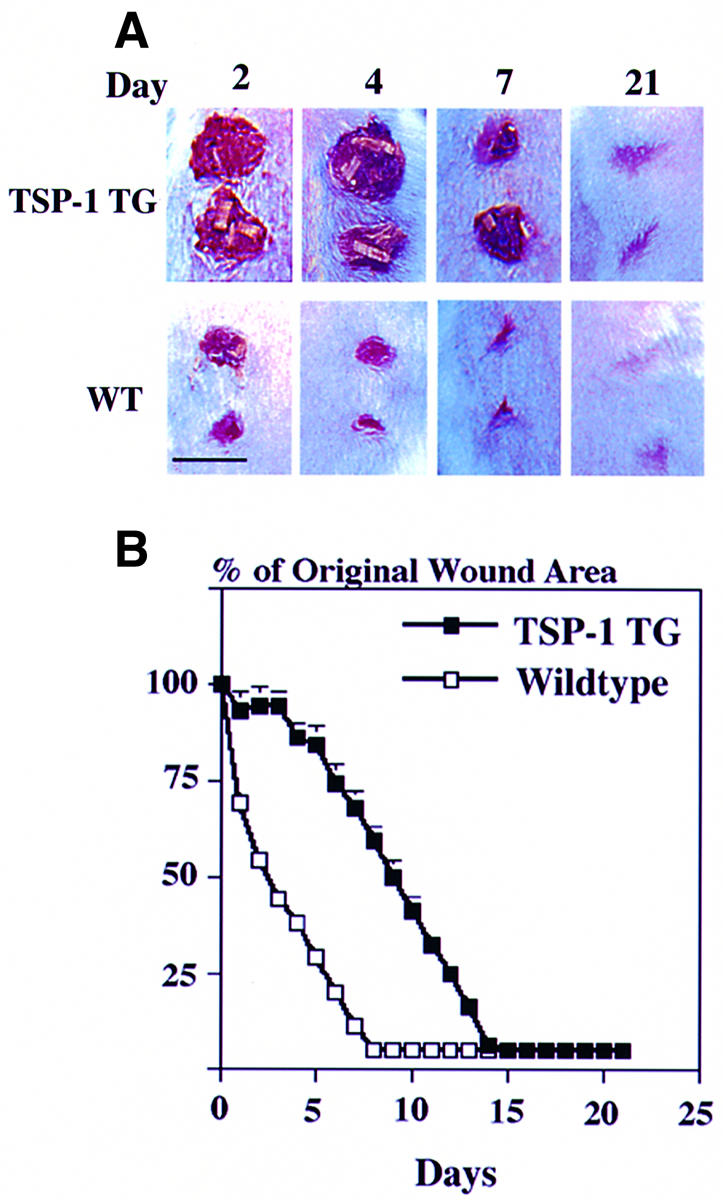
Fig. 4. Delayed closure of full-thickness wounds in TSP-1 transgenic mice as compared with wild-type (WT) littermates. (A) At 24 h after injury, wounds were covered by a dry scab, which remained adherent until day 10 in TSP-1 transgenic mice and until day 5–7 in control mice. Bar: 6 mm. (B) A >50% reduction in wound area was observed 3 days after wounding in wild-type mice (n = 30) but only after 9 days in TSP-1 transgenic mice (n = 30) (p <0.001). Complete wound closure occurred after 8 days in wild-type mice and after 14 days in TSP-1 transgenic mice.
In wild-type mice, invasion of granulation tissue from the wound margins into the wound bed was observed 2 days after injury (data not shown). At day 3, the wound bed was completely filled with granulation tissue (Figure 5A), and at day 7 the granulation tissue was completely covered by newly formed epidermis (Figure 5C). In contrast, granulation tissue formation was delayed in TSP-1 transgenic mice, with only sparse granulation tissue formation after 3 days (Figure 5B). After 7 days, the wound bed was still not completely filled with granulation tissue (Figure 5D), and the formation of neoepidermis was only completed after 10 days. No major differences in the number and cell type of inflammatory cells infiltrating the wounds were found in TSP-1 transgenic mice when compared with wild-type controls, as evaluated by hematoxylin–eosin stains and by immunostains for CD45 and CD11b (data not shown). In situ hybridization studies confirmed potently increased TSP-1 mRNA expression in transgenic epidermal keratinocytes during wound healing (Figure 5F and H), as compared with wild-type mice (Figure 5E and G). Immunofluorescence studies at 7 days after wounding demonstrated strong TSP-1 protein expression in the neoepidermis overlying the granulation tissue in TSP-1 transgenic mice, whereas only little TSP-1 expression was seen in wild-type epidermis (Figure 5I and J). We did not detect significant amounts of TSP-1 protein in non-resident cells in the granulation tissue. Comparable expression levels of VEGF mRNA were detected in the epidermis at the wound edge of transgenic and wild-type mice (data not shown).

Fig. 5. Impaired granulation tissue formation in cutaneous wounds of TSP-1 transgenic mice. In wild-type mice, the wound bed was completely filled with granulation tissue 3 days after wounding (A), and was completely covered by newly formed epidermis after 7 days (C). In contrast, only minor granulation tissue infiltration from the wound margins was observed in TSP-1 transgenic mice after 3 days (B), and by day 7 the wound bed was still only partially filled with granulation tissue (D). Arrows indicate the front line of granulation tissue invading the wound bed. (A–D) Hematoxylin–eosin stain. Magnification: 96×. Bar: 250 µm. (E–H) In situ hybridization confirmed high levels of TSP-1 mRNA expression in transgenic epidermal keratinocytes at day 7 after wounding (F and H), as compared with low TSP-1 mRNA expression in wild-type mice (E and G). Dots indicate the epidermal–dermal junction. Magnification: 192×. Bar: 250 µm. (I and J) Immunofluorescence staining of TSP-1 in 7-day-old wounds reveals strong TSP-1 protein expression in the neoepidermis of TSP-1 transgenic mice (J), as compared with low levels of expression in wild-type wounds (I). Little or no staining of non-resident cells is visible. Vessels are depicted in red (CD31 stain). Magnification: 96×. Bar: 250 µm.
TSP-1 inhibits wound angiogenesis in transgenic mice
The vascular density and the average vessel sizes were quantitatively evaluated on CD31-stained frozen sections of wound tissues, and proliferating endothelial cells were visualized by double-fluorescence stainings for bromodeoxyuridine (BrdU) and CD31. In wild-type mice, vascular endothelial cell proliferation was already detected in the early granulation tissue of 2-day-old wounds (Figure 6A) with a subsequent increase in the number of proliferating endothelial cells after 4 days (Figure 6C), leading to a vessel-rich granulation tissue (Figure 6E). In contrast, little or no vascular proliferation was seen in 2-day-old wounds in TSP-1 transgenic mice, with only slight increases after 4 days (Figure 6B and D). Accordingly, 7-day-old wounds in transgenic mice were markedly less vascularized, as compared with wild-type controls (Figure 6E and F). Blood vessel density in the granulation tissue of wild-type mice increased rapidly to reach a peak of 259 ± 32 vessels/mm2 after 4 days, with subsequent reduction of vessel densities to the baseline levels observed in unwounded skin (Figure 6G). In contrast, the increase in vascular density was less pronounced in the granulation tissue of TSP-1 transgenic mice, reaching a peak of only 191 ± 43 vessels/mm2 after 7 days. Importantly, the changes of vessel size during wound healing in wild-type mice were much more pronounced than the increase in vessel numbers, with a >4-fold increase in vessel size after 7 and 14 days (729 ± 120 µm2), as compared with unwounded skin (165 ± 36 µm2). In contrast, no significant increase in vessel size was detected in granulation tissue of TSP-1 transgenic mice (p <0.001; Figure 6H). In accordance with these data, the relative wound area occupied by blood vessels in wild-type mice increased by >6-fold to reach 14 ± 2.5% after 7 days, as compared with only 6 ± 1.6% in TSP-1 transgenic mice (Figure 6I). After 3 weeks, blood vessel density and size in both wild-type and transgenic mice were comparable to those observed in unwounded skin. No differences in the smooth muscle cell actin staining of the new vessels were found in wounds (days 2, 4, 7 and 14) obtained from TSP-1 transgenic mice, as compared with their wild-type littermates (data not shown).

Fig. 6. Pronounced inhibition of vascular endothelial cell proliferation and neovascularization in granulation tissue of wounds from TSP-1 transgenic mice at 2 days (B) and 4 days (D) after injury, as compared with numerous proliferating endothelial cells in wild-type mice at day 2 (A) and day 4 (C). Immunofluorescence staining demonstrates CD31-stained blood vessels (red), BrdU-labeled proliferating non-endothelial cells (green) and CD31/BrdU double-stained proliferating vascular endothelial cells (yellow, arrows). Staining for CD31 demonstrated reduced neovascularization of 7-day-old granulation tissue in TSP-1 transgenic mice (F), as compared with wild-type mice (E). Bar: 250 µm. (G–I) Computer-assisted morphometric analysis of CD31-stained wound sections revealed a significantly (p <0.01) reduced microvascular density in granulation tissue of TSP-1 transgenic mice (G), as compared with wild-type (WT) controls. (H) Inhibition of vessel size increases through day 14 after injury in granulation tissue of TSP-1 transgenic mice. (I) A strong increase in the total area covered by blood vessels was detected in granulation tissue in wild-type mice, but not in TSP-1 transgenic mice (p <0.001). CD31-stained blood vessels were evaluated in granulation tissue in corresponding 10× fields of five different wounds per time point. Data are expressed as mean ± SEM. **p <0.01; ***p <0.001.
TSP-1 inhibits fibroblast but not keratinocyte migration
The percentage of proliferating, BrdU-positive epidermal keratinocytes was comparable in wild-type mice (unwounded skin: 7 ± 0.8; wounded skin, day 4: 24 ± 3; day 7: 9 ± 2) and in TSP-1 transgenic mice (unwounded skin: 6 ± 1; wounded skin, day 4: 30 ± 3; day 7: 9 ± 2). Moreover, despite marked differences in TSP-1 secretion (see Figure 1C and D), no significant differences in proliferation and migration were observed between cultured TSP-1 transgenic and wild-type keratinocytes (data not shown). Addition of 20 µg/ml exogenous TSP-1 to wild-type or TSP-1 transgenic keratinocytes for up to 6 days did not significantly influence keratinocyte proliferation (data not shown). In contrast, TSP-1 potently inhibited the proliferation of human dermal microvascular endothelial cells cultured in the presence of VEGF (Figure 7A). TSP-1 dose-dependently inhibited dermal fibroblast migration towards collagen type I (Figure 7C) and also inhibited migration towards fibronectin (Figure 7D), whereas fibroblast proliferation was not affected by TSP-1 (Figure 7B).

Fig. 7. (A) Dose-dependent inhibition of human dermal microvascular endothelial cell (HDMEC) proliferation by TSP-1 in the presence of 20 ng/ml VEGF. (B) Absence of fibroblast growth modulation by added TSP-1 (20 µg/ml), as compared with untreated controls. (C) TSP-1 dose-dependently inhibited in vitro migration of human dermal fibroblasts (p <0.001) towards a collagen type I matrix. (D) Comparable inhibition of fibroblast migration on collagen type I or fibronectin matrices. Transwell migration chamber assay; TSP-1 concentration: 20 µg/ml. Results are the mean ± SD of two independent experiments. **p <0.01; ***p <0.001.
Discussion
In this study, we have established a new transgenic model for the chronic, orthotopic overexpression of the endogenous angiogenesis inhibitor TSP-1. Taking advantage of a keratin 14-promoter expression cassette, we selectively directed TSP-1 expression to the basal epidermal keratinocyte layer of the skin and to the outer root sheath keratinocytes of hair follicles. Targeted expression of TSP-1 was confirmed by northern analysis, which demonstrated highly increased TSP-1 mRNA levels in transgenic skin extracts, but not in extracts from several other organs including heart, kidneys and liver, in accordance with the previously reported tissue-specific expression of the keratin 14 gene (Vassar et al., 1989). In situ hybridization studies revealed increased mRNA expression in basal epidermis and the hair follicle sheath, confirming that TSP-1 expression was induced orthotopically in cells that show TSP-1 expression in normal skin (Wight et al., 1985), and western analysis of skin lysates revealed enhanced amounts of TSP-1 protein in transgenic skin. Efficient secretion of transgenic TSP-1 was confirmed in primary keratinocyte cultures derived from neonatal skin, and western blot analyses of conditioned media revealed that transgenic TSP-1 protein was predominantly secreted as the intact, full-length protein with little evidence for degradation/cleavage products. Importantly, TSP-1 expression in wild-type keratinocyte cultures was downregulated by a switch from low Ca2+ to high Ca2+ culture conditions, which mediate a more differentiated keratinocyte phenotype (Hennings et al., 1980). These findings demonstrate an inverse relationship between keratinocyte differentiation and TSP-1 expression, which is in accordance with the preferred TSP-1 expression in the undifferentiated basal epidermal layer, but not in the differentiated suprabasal layers.
Skin-specific overexpression of transgenic TSP-1 resulted in a delayed first hair cycle. However, no obvious developmental abnormalities of cutaneous blood vessel formation, vascular density and vascular branching pattern were observed. Moreover, no major alterations of the skin structure and of acute, VEGF-induced microvascular permeability were seen in adult transgenic mice. Because hair growth during the anagen phase of the first hair cycle is associated with pronounced angiogenesis (K.Yano, L.F.Brown and M.Detmar, unpublished results), the growing hair follicle might represent a more suitable target for the anti-angiogenic action of transgenic TSP-1 than quiescent normal skin. Because keratin 14 expression is correlated with the proliferative activity of keratinocytes, these differences might, in part, also be due to different transgene expression levels in proliferative outer root sheath keratinocytes, as compared with keratinocytes in the differentiated interfollicular epidermis. In agreement with our findings in normal skin, we did not detect any major differences in the vascularization and histological structure of the tongue, the esophagus and the forestomach between transgenic and wild-type mice; these tissues also express keratin 14.
Early full-thickness wounds are characterized by marked wound contraction, ingrowth of fibroblast-rich granulation tissue from the deep wound sides into the tissue defect, and prominent neovascularization of the granulation tissue (Clark, 1993). Using computer-assisted image analysis of sequential wound sections stained for the endothelial junction molecule CD31, we found that in wild-type mice, angiogenesis was already observed 2 days after wounding, leading to high vascular density in the granulation tissue, which reached its peak after 4 days. Angiogenesis was associated with proliferation of vascular endothelial cells, as detected by double stainings for CD31 and BrdU. When we quantified the size of blood vessels during wound healing, we detected a pronounced, >3-fold increase in the average size of individual vessels, beginning at day 2 post-wounding and reaching a peak between day 7 and day 14. These findings demonstrate a biphasic regulation of wound angiogenesis, with an initial phase of sprouting angiogenesis with intense endothelial cell mitogenesis, leading to increased vascular densities and vessel sizes. The second phase is characterized by normalization of vessel numbers but persistent vessel enlargement.
The striking feature of K14/TSP-1 transgenic mice was the potent inhibition of cutaneous tissue repair, as investigated in an established model of cutaneous full-thickness wound healing (Werner et al., 1992, 1994). Importantly, the early phase of tissue repair was most severely affected, leading to a 6 day delay of wound closure in transgenic mice. TSP-1 overexpression markedly inhibited endothelial cell mitogenesis and angiogenesis during early wound healing. It is of interest that transgenic TSP-1 almost completely prevented vascular enlargement with a less pronounced reduction of vascular densities. These findings are in accordance with our previous results in human squamous cell carcinomas (Streit et al., 1999a), in which transfected TSP-1 exerted a more potent inhibitory effect on peritumoral vessel sizes than on vessel numbers. While the mechanisms for this preferential effect on vessel size remain unknown, the increased size of angiogenic vessels appears to be a more sensitive target for angiogenesis inhibition, since treatment of human tumor xenografts with a neutralizing anti-VEGF antibody also resulted predominantly in a reduction in tumor vessel diameter (Yuan et al., 1996). Importantly, TSP-1 exerted its inhibitory effect on wound angiogenesis even in the presence of high VEGF expression during the early phases of tissue repair, in accordance with our in vitro studies, which demonstrated that TSP-1 potently inhibited microvascular endothelial cell proliferation even in the presence of 20 ng/ml VEGF. The molecular mechanisms of TSP-1-mediated angiogenesis inhibition remain to be established. A previous analysis of proteolytic fragments of the TSP-1 molecule revealed anti-angiogenic activity of peptides derived from the procollagen-like domain, in particular the peptide NGVQYRN (Tolsma et al., 1993). In addition, specific sequences, contained within the properdin-like type I repeats, have been identified that interact with the CD36 receptor on endothelial cells (Tolsma et al., 1993; Volpert et al., 1995; Dawson et al., 1997).
The early phase of tissue repair involves wound contraction, granulation tissue invasion and fibroblast proliferation (Martin, 1997; Stadelmann et al., 1998; Yehualaeshet et al., 1999). The pronounced delay of early wound closure in TSP-1 transgenic mice suggested that TSP-1 might also affect fibroblast functions. Indeed, we detected a marked reduction in the number of proliferating non-endothelial cells, most likely representing fibroblasts, in early wounds in TSP-1 transgenic mice, and a delay of fibroblast migration into the fibrin-rich wound clot. To evaluate potential effects of TSP-1 on fibroblast functions, we treated cultured human dermal fibroblasts with TSP-1 isolated from human platelets. In contrast to previous reports that described a stimulatory effect of TSP-1 on fibroblast proliferation in vitro (Phan et al., 1989) and on the growth of aortic-culture-derived myofibroblasts (Nicosia and Tuszynski, 1994), we did not detect any effect of TSP-1 on fibroblast proliferation. However, fibroblast migration towards collagen type I and fibronectin, both highly expressed during early cutaneous wound healing (Stadelmann et al., 1998), was significantly inhibited. This effect was cell type specific since epidermal keratinocytes derived from the same K14/TSP-1 transgenic mice did not show any inhibition of proliferation or migration, despite high levels of TSP-1 secretion. Since fibroblast activation and migration are believed to be a rate-limiting step of granulation tissue induction (McClain et al., 1996), these in vitro data reveal a new mechanism of action of TSP-1 in tissue repair and strongly suggest that the inhibitory effect of transgenic TSP-1 on fibroblasts contributed to the impaired granulation tissue formation in the wound. These findings are in accordance with a previous preliminary report demonstrating increased and prolonged granulation tissue formation in TSP-1-deficient mice (Polverini et al., 1995) and with the impaired granulation tissue formation in polyvinyl-sponge implants by a 19 residue peptide derived from the type I repeats of TSP-1 (Tolsma et al., 1993). Together, they support the concept of interdependence of endothelial cell and fibroblast activation during granulation tissue formation (Arnold and West, 1991; Martin, 1997).
The potent inhibitory effect of TSP-1 on wound angiogenesis in K14/TSP-1 transgenic mice, in the absence of major alterations of the cutaneous vascular architecture and microvascular permeability in unwounded skin, demonstrates that skin-specific overexpression of TSP-1 preferentially interfered with wound tissue repair-associated angiogenesis, rather than with the angiogenesis associated with normal development and skin homeostasis. These findings suggest that therapeutic application of angiogenesis inhibitors might potentially be associated with impaired wound vascularization and tissue repair. To the best of our knowledge, the K14/TSP-1 transgenic mouse represents the first transgenic model for the chronic, orthotopic overexpression of an endogenous angiogenesis inhibitor and might also provide an attractive experimental system for studies on the biological role of TSP-1 in other angiogenesis-dependent processes including carcinogenesis and chronic inflammation.
Materials and methods
Generation of TSP-1 transgenic mice
A 3.644 kb human TSP-1 cDNA, comprising the full coding sequence, was obtained by RT–PCR of total RNA extracted from human dermal fibroblasts, using the following primers: 5′-AGCTCCACCATGGGGCTGGC-3′ (nucleotides 67–88 of DDBJ/EMBL/GenBank accession No. X04665) and reverse primer 5′-CAAGCACAGAAAAGAAGGAAGC-3′ (nucleotides 3710–3689). The TSP-1 cDNA was cloned into a pGEM-7Z(f) vector. After restriction digestion with SacI, SphI and PvuI, a 3.6 kb human TSP-1 cDNA fragment was blunted and ligated into the blunted BamHI site of a pGEM-3Z transgene vector containing a keratin 14 expression cassette (Vassar et al., 1989; kindly provided by Dr E.Fuchs, University of Chicago). The correct sequence and orientation of the TSP-1 insert were verified by restriction mapping and by direct sequencing using the Sanger dideoxy method. An 8.3 kb KpnI–HindIII fragment (Figure 1A) was used to generate transgenic mice as described (Detmar et al., 1998). Transgenic founders were detected by Southern blot analysis of BamHI-digested genomic tail DNA using a 32P-labeled, 3.6 kb, human TSP-1 cDNA probe. For rapid identification, genomic tail DNA was subjected to PCR as described previously (Detmar et al., 1998). Transgenic lines were established on the FVB genetic background. All animal studies were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care.
Cell cultures
Primary epidermal keratinocytes were isolated from the skin of neonatal TSP-1 transgenic mice and of wild-type littermates as described previously (Kamimura et al., 1997). Cells were resuspended in minimal essential medium (Life Technologies, Grand Island, NY), supplemented with 4% fetal calf serum and 10 ng/ml epidermal growth factor (Becton Dickinson, Bedford, MA) at low calcium concentrations (0.05 mM), and were plated onto collagen type I (Collagen Corp., Palo Alto, CA) coated culture dishes. Human dermal fibroblasts (IMR-91) were obtained from the National Institute of Aging (Camden, NJ), and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 4% fetal calf serum. Human dermal microvascular endothelial cells were isolated from human neonatal foreskins and were maintained in culture as described previously (Richard et al., 1998).
RNA isolation, northern blot and western blot analysis
Total cellular RNA was isolated from primary mouse keratinocytes and from tissues as described (Detmar et al., 1998), using the RNeasy kit (Qiagen, Chatsworth, CA). RNA (10 µg) was fractionated by electrophoresis on 1% agarose formaldehyde gels and transferred to Biotrans nylon supported membranes (ICN Pharmaceuticals, Costa Mesa, CA). 32P-radiolabeled cDNA probes were prepared with a random primed synthesis kit (Multiprime; Amersham, Arlington Heights, IL). mRNAs for TSP-1 were detected with a 3.6 kb human TSP-1 cDNA probe, and a 2.0 kb human β-actin cDNA probe (Clontech, Palo Alto, CA) was used as a control for equal RNA loading. Blots were washed at high stringency as described (Detmar et al., 1997) and exposed to X-OMAT film (Kodak, Rochester, NY). Conditioned media were obtained from confluent primary mouse keratinocytes grown for 48 h in serum-free MEM at low Ca2+ (0.05 mM) or at high Ca2+ concentrations (1 mM). TSP-1 was concentrated as described (Streit et al., 1999a). Total cell lysates were prepared as described (Streit et al., 1999b), and skin lysates were obtained by homogenization of abdominal skin of 8-week-old TSP-1 transgenic mice and wild-type littermates. Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA). Samples were boiled in denaturing sample buffer (Laemmli, 1970), and equal protein amounts of each sample were electrophoresed on polyacrylamide gels and blotted onto polyvinylidene difluoride membranes (Bio-Rad). To verify equal protein loading, membranes were stained with 0.1% Ponceau red (Sigma), diluted in 5% acetic acid. Membranes were incubated overnight in phosphate-buffered saline (PBS) containing 0.1% Tween-20 and 3% bovine serum albumin (BSA) to block non-specific binding. Membranes were then incubated with anti-human TSP-1 antibody (1:200 dilution; clone 133; Genzyme, Cambridge, MA), incubated with horseradish peroxidase-conjugated anti-rabbit IgG, and analyzed by the enhanced chemiluminescence system (Amersham).
In situ hybridization and immunohistochemistry
In situ hybridization was performed on 6 µm paraffin sections as described (Detmar et al., 1997). An RNA probe to human TSP-1 was transcribed from a pBluescript II KS+ vector containing a 240 bp PCR fragment of the coding region of human TSP-1. This probe recognizes both human and mouse TSP-1. Antisense and sense single-stranded 35S-labeled RNA probes for VEGF were prepared from a 393 bp PCR rat VEGF cDNA fragment, cloned into pGEM-3Zf(+). Immunohistochemical and immunofluorescence stainings were performed on 6 µm paraffin sections or cryostat sections as described previously (Skobe et al., 1997; Detmar et al., 2000), using monoclonal antibodies against human TSP-1 (Genzyme), mouse CD31, BrdU, mouse CD45 or mouse CD11b (PharMingen, San Diego, CA).
Vascular perfusions and computer-assisted morphometric analysis of blood vessels
Seven-week-old TSP-1 transgenic mice and wild-type littermates were anesthetized with a mixture of ketamine (800 µg/10 g body weight Ketaset™; Fort Dodge Laboratories, Fort Dodge, IA) and avertin (0.5 µg/10 g body weight 2,2,2-tribromoethanol in 2.5% t-amyl alcohol; Sigma), and perfusion was performed via the left ventricle as recently described (Thurston et al., 1998). Mice were perfused with fluorescein-labeled Lycopersicon esculentum lectin (Vector Laboratories, Burlingame, CA) at a constant pressure of 120 mm Hg, leading to lectin binding to N-acetyl-d-glucosamine residues on the luminal surface of vascular endothelial cells (Thurston et al., 1996). The architecture of the vasculature was analyzed in whole mounts of mouse ears in Vectashield mounting medium (Vector Laboratories), using a Nikon E-600 fluorescence microscope (Nikon, Melville, NY). Cryostat sections (6 µm thick) were stained with a monoclonal rat anti-mouse CD31 monoclonal antibody, and representative sections obtained from at least five different wounds for each time point were analyzed using a Nikon E-600 microscope. Images were captured with a Spot digital camera (Diagnostic Instruments, Sterling Heights, MI), and computer-assisted morphometric analyses of blood vessels within the granulation tissue of each wound were performed as described previously (Streit et al., 1999b), using the IP-Lab software (Scanalytics, Fairfax, VA). Three different fields were examined on each section at 10-fold magnification, and the number of vessels per square millimeter, the size distribution, the average and the total area of blood vessels were determined (Streit et al., 1999b). The two-sided unpaired Student’s t-test was used to analyze differences in vessel areas. In addition, bleeding times were determined as described (Kyriakides et al., 1998).
Miles vascular permeability assay
Miles assays were performed as described previously (Claffey et al., 1996). Evans blue (1% in PBS, 100 µl/mouse; Sigma) was injected into the tail vein of anesthetized mice. After 10 min, human VEGF165 (0–100 ng in 50 µl of PBS; Peprotech, Rocky Hill, NJ) was injected intradermally into the back skin. Leakage of protein-bound dye was detected as blue spots on the underside of the back skin surrounding the injection site.
Wound-healing studies
Three independent wound-healing experiments were performed in TSP-1 transgenic mice as described (Werner et al., 1992), using heterozygous F2 generation TSP-1 transgenic mice and their wild-type littermates, obtained by crossing heterozygous F1 TSP-1 transgenic mice with wild-type mice. Six full-thickness wounds (3–4 mm apart) were created on the back of each of ten 8-week-old female TSP-1 transgenic mice or wild-type littermates, using 6 mm skin biopsy punches. Wound closure was monitored daily in 30 wounds for each genotype by covering each wound with a transparent plastic sheet and tracing the area of the wound with a marker (Gross et al., 1995). Area measurements were made using the IP-LAB software. Wound closure was calculated as the percentage of the initial wound area. In an additional experiment, wounds from at least three animals per time point were harvested at 2, 3, 5, 7, 14 and 21 days after wounding. Two hours prior to killing, mice received intraperitoneal injections of BrdU (200 µg; Sigma). An area of 7–8 mm in diameter, including the complete epithelial margins, was excised, cut in half, and wound specimens were frozen in OCT-compound (Sakura, Torrance, CA) or fixed for 48 h in 4% paraformaldehyde and embedded in paraffin.
Cell migration and proliferation assays
Keratinocytes obtained from wild-type or TSP-1 transgenic mice were grown to 80% confluence and cultures were wounded as described (Ando and Jensen, 1996), using 1.75 cm cell scrapers. The wound edge was marked and cell migration was monitored daily for 7 days, using a Nikon Eclipse TE300 inverted microscope and a Spot digital camera. The assay was performed in triplicate dishes, and the distance covered by migrating keratinocytes was measured in four different fields per dish at 60-fold magnification. The effect of human TSP-1, isolated from platelets (kindly provided by Dr J.Lawler, Harvard Medical School, Boston, MA), on fibroblast migration was measured as described (Senger et al., 1996) using 8 µm pore size Transwell migration chambers (Costar, Cambridge, MA) coated on the underside with 10 µg/ml human collagen type I or human fibronectin (Sigma). Fibroblasts (1 × 105 cells) were added to the upper chamber in 300 µl of DMEM, supplemented with 10 mg/ml BSA and human TSP-1 (0–20 µg/ml), for 4 h. Images of three different 10× fields were captured from each membrane, and the number of migrating cells per square millimeter was determined by using the IP-LAB software. All assays were performed in triplicate. For proliferation studies, 5 × 105 primary keratinocytes were seeded onto collagen type I-coated 60 mm culture dishes (Nunc, Naperville, IL). After 24 h, cell proliferation was evaluated using the BrdU labeling and detection kit (Boehringer Mannheim, Germany) according to the manufacturer’s instructions. The results represent the mean ± SD of four dishes per group. Human dermal microvascular endothelial cells were seeded into quadruplicate wells of 24-well plates, and were cultured for 48 h in the presence of 20 ng/ml human VEGF165 (Peprotech, Rocky Hill, NJ) with or without addition of human TSP-1 (1–10 µg/ml). Proliferation was assessed as described previously (Detmar et al., 1995). Dermal fibroblasts (5 × 104 cells) were cultured in triplicate 60 mm dishes in the presence or absence of 20 µg/ml human TSP-1, and cells were counted every second day, using a Coulter Z1 counter (Coulter, Hialeah, FL). Culture medium and TSP-1 were replaced every other day. Statistical analysis was performed using the two-sided unpaired Student’s t-test.
Acknowledgments
Acknowledgements
The authors thank E.Fuchs for providing the keratin 14 expression cassette, G.Thurston and D.McDonald for advice on vascular perfusions, and J.Gross for helpful discussions. This work was supported by NIH/NCI grants CA69184 and CA 6410 (M.D.), by American Cancer Society Program Project Grant 99-23901 (M.D.), by Deutscher Akademischer Austauschdienst (M.S.), by the Deutsche Forschungsgemeinschaft (B.L.-A., T.H.), and by the Cutaneous Biology Research Center through the Massachusetts General Hospital/Shiseido Co. Ltd Agreement (M.D.).
References
- Adams J.C. (1997) Thrombospondin-1. Int. J. Biochem. Cell Biol., 29, 861–865. [DOI] [PubMed] [Google Scholar]
- Ando Y. and Jensen,P. (1996) Protein kinase C mediates up-regulation of urokinase and its receptor in the migrating keratinocytes of wounded cultures, but urokinase is not required for movement across a substratum in vitro. J. Cell Physiol., 167, 500–511. [DOI] [PubMed] [Google Scholar]
- Arnold F. and West,D.C. (1991) Angiogenesis in wound healing. Pharmacol. Ther., 52, 407–422. [DOI] [PubMed] [Google Scholar]
- Beer H.-D., Longaker,M.T. and Werner,S. (1997) Reduced expression of PDGF and PDGF receptors during impaired wound healing. J. Invest. Dermatol., 109, 132–138. [DOI] [PubMed] [Google Scholar]
- Border W.A. and Noble,N.A. (1994) Transforming growth factor β in tissue fibrosis. N. Engl. J. Med., 331, 1286–1292. [DOI] [PubMed] [Google Scholar]
- Bornstein P. (1995) Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J. Cell Biol., 130, 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukamp P., Bleuel,K., Popp,S., Vormwald,D.V. and Fusenig,N. (1997) Functional evidence for tumor-suppressor activity on chromosome 15 in human skin carcinoma cells and thrombospondin-1 as the potential suppressor. J. Cell Physiol., 173, 256–260. [DOI] [PubMed] [Google Scholar]
- Brown L.F., Yeo,K.T., Berse,B., Yeo,T.K., Senger,D.R., Dvorak,H.F. and Van De Water,L. (1992) Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J. Exp. Med., 176, 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claffey K.P., Brown,L.F., del Aguila,L.F., Tognazzi,K., Yeo,K.-T., Manseau,E.J. and Dvorak,H.F. (1996) Expression of vascular permeability factor/vascular endothelial growth factor by melanoma cells increases tumor growth, angiogenesis and experimental metastasis. Cancer Res., 56, 172–181. [PubMed] [Google Scholar]
- Clark R.A.F. (1993) Basics of cutaneous wound repair. J. Dermatol. Surg. Oncol., 19, 693–706. [DOI] [PubMed] [Google Scholar]
- Dawson D.W., Pearce,S.F., Zhong,R., Silverstein,R.L., Frazier,W.A. and Bouck,N.P. (1997) CD36 mediates the in vitro inhibitory effects of thrombospondin-1 on endothelial cells. J. Cell Biol., 138, 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmar M. (1996) Molecular regulation of angiogenesis in the skin. J. Invest. Dermatol., 106, 207–208. [DOI] [PubMed] [Google Scholar]
- Detmar M., Yeo,K.-T., Nagy,J.A., Van De Water,L., Brown,L.F., Berse,B., Elicker,B.M., Ledbetter,S. and Dvorak,H.F. (1995) Keratinocyte-derived vascular permeability factor (vascular endothelial growth factor) is a potent mitogen for dermal microvascular endothelial cells. J. Invest. Dermatol., 105, 44–50. [DOI] [PubMed] [Google Scholar]
- Detmar M., Brown,L.F., Berse,B., Jackman,R.W., Elicker,B.M., Dvorak,H.F. and Claffey,K.P. (1997) Hypoxia regulates the expression of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) and its receptors in human skin. J. Invest. Dermatol., 108, 263–268. [DOI] [PubMed] [Google Scholar]
- Detmar M. et al. (1998) Increased microvascular density and enhanced leukocyte rolling and adhesion in the skin of VEGF transgenic mice. J. Invest. Dermatol., 111, 1–6. [DOI] [PubMed] [Google Scholar]
- Detmar M., Velasco,P., Richard,L., Claffey,K.P., Streit,M., Riccardi,L., Skobe,M. and Brown,L.F. (2000) Expression of vascular endothelial growth factor induces an invasive phenotype in human squamous cell carcinomas. Am. J. Pathol., 156, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPietro L.A., Nissen,N.N., Gamelli,R.L., Koch,A.E., Pyle,J.M. and Polverini,P.J. (1996) Thrombospondin 1 synthesis and function in wound repair. Am. J. Pathol., 148, 1851–1860. [PMC free article] [PubMed] [Google Scholar]
- Dvorak H.F. (1986) Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med., 315, 1650–1659. [DOI] [PubMed] [Google Scholar]
- Frank S., Huebner,G., Breier,G., Longaker,M.T., Greenhalgh,D.G. and Werner,S. (1995) Regulation of vascular endothelial growth factor expression in cultured keratinocytes: implications for normal and impaired wound healing. J. Biol. Chem., 270, 12607–12613. [DOI] [PubMed] [Google Scholar]
- Gross J.P., Farinelli,W., Sadow,P., Anderson,R. and Bruns,R. (1995) On the mechanism of skin wound ‘contraction’: a granulation tissue ‘knockout’ with a normal phenotype. Proc. Natl Acad. Sci. USA, 92, 5982–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennings H., Michael,D., Cheng,C., Steinert,P., Holbrook,K. and Yuspa,S.H. (1980) Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell, 19, 245–254. [DOI] [PubMed] [Google Scholar]
- Iruela-Arispe M., Bornstein,P. and Sage,H. (1991) Thrombospondin exerts an antiangiogenic effect on cord formation by endothelial cells in vitro. Proc. Natl Acad. Sci. USA, 88, 5026–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura J., Lee,D., Baden,H.P., Brissette,J. and Dotto,G.P. (1997) Primary mouse keratinocyte cultures contain hair follicle progenitor cells with multiple differentiation potential. J. Invest. Dermatol., 109, 534–540. [DOI] [PubMed] [Google Scholar]
- Kyriakides T.R. et al. (1998) Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density and a bleeding diathesis. J. Cell Biol., 140, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Martin P. (1997) Wound healing—aiming for perfect skin regeneration. Science, 276, 75–81. [DOI] [PubMed] [Google Scholar]
- McClain S.A., Simon,M., Jones,E., Nandi,A., Gailit,J.O., Tonnesen,M.G., Newman,D. and Clark,R.A.F. (1996) Mesenchymal cell activation is the rate-limiting step of granulation tissue induction. Am. J. Pathol., 149, 1257–1270. [PMC free article] [PubMed] [Google Scholar]
- Nicosia R.F. and Tuszynski,G.P. (1994) Matrix-bound thrombospondin promotes angiogenesis in vitro. J. Cell Biol., 124, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly M.S. et al. (1994) Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell, 79, 315–328. [DOI] [PubMed] [Google Scholar]
- O’Reilly M.S. et al. (1997) Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell, 88, 277–285. [DOI] [PubMed] [Google Scholar]
- Ortega S., Ittmann,M., Tsang,S.H., Ehrlich,M. and Basilico,C. (1998) Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc. Natl Acad. Sci. USA, 95, 5672–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan S.H., Dillon,R.G., McGarry,B.M. and Dixit,V.M. (1989) Stimulation of fibroblast proliferation by thrombospondin. Biochem. Biophys. Res. Commun., 163, 56–63. [DOI] [PubMed] [Google Scholar]
- Pike S.E. et al. (1998) Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J. Exp. Med., 188, 2349–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polverini P.J., DiPietro,L.A., Dixit,V.M., Hynes,R.O. and Lawler,J. (1995) Thrombospondin 1 knock out mice show delayed organization and prolonged neovascularization of skin wounds. FASEB J., 9, A272. [Google Scholar]
- Raugi G.J., Olerud,J.E. and Gown,A.M. (1987) Thrombospondin in early human wound tissue. J. Invest. Dermatol., 89, 551–554. [DOI] [PubMed] [Google Scholar]
- Reed M.J., Puolakkainen,P., Lane,T.F., Dickerson,D., Bornstein,P. and Sage,E.H. (1993) Differential expression of SPARC and thrombospondin 1 in wound repair: immunolocalization and in situ hybridization. J. Histochem. Cytochem., 41, 1467–1477. [DOI] [PubMed] [Google Scholar]
- Richard L., Velasco,P. and Detmar,M. (1998) A simple immunomagnetic protocol for the selective isolation and long-term culture of human dermal microvascular endothelial cells. Exp. Cell Res., 240, 1–6. [DOI] [PubMed] [Google Scholar]
- Roth J.J., Albo,D., Rothman,V.L., Longaker,M.T., Granick,M.S., Long,C.D., Solomon,M.P. and Tuszynski,G.P. (1998) Thrombospondin-1 and its CSVTCG-specific receptor in wound healing and cancer. Ann. Plast. Surg., 40, 494–501. [DOI] [PubMed] [Google Scholar]
- Senger D.R., Ledbetter,S.R., Claffey,K.P., Papadopoulos-Sergiou,A., Perruzzi,C.A. and Detmar,M. (1996) Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the αvβ3 integrin, osteopontin and thrombin. Am. J. Pathol., 149, 293–305. [PMC free article] [PubMed] [Google Scholar]
- Skobe M., Rockwell,P., Goldstein,N., Vosseler,S. and Fusenig,N.E. (1997) Halting angiogenesis suppresses carcinoma cell invasion. Nature Med., 3, 1222–1227. [DOI] [PubMed] [Google Scholar]
- Stadelmann W.K., Digenis,A.G. and Tobin,G.R. (1998) Physiology and healing dynamics of chronic cutaneous wounds. Am. J. Surg., 176, 26–38. [DOI] [PubMed] [Google Scholar]
- Streit M., Velasco,P., Brown,L.F., Skobe,M., Richard,L., Riccardi,L., Lawler,J. and Detmar,M. (1999a) Overexpression of thrombospondin-1 decreases angiogenesis and inhibits the growth of human cutaneous squamous cell carcinomas. Am. J. Pathol., 155, 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit M., Riccardi,L., Velasco,P., Brown,L.F., Hawighorst,T., Bornstein,P. and Detmar,M. (1999b) Thrombospondin-2: a potent endogenous inhibitor of tumor growth and angiogenesis. Proc. Natl Acad. Sci. USA, 96, 14888–14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston G., Baluk,P., Hirata,A. and McDonald,D.M. (1996) Permeability-related changes revealed at endothelial cell borders in inflamed venules by lectin binding. Am. J. Physiol., 271, H2547–2562. [DOI] [PubMed] [Google Scholar]
- Thurston G., Murphy,T.J., Baluk,P., Lindsey,J.R. and McDonald,D.M. (1998) Angiogenesis in mice with chronic airway inflammation: strain-dependent differences. Am. J. Pathol., 153, 1099–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolsma S.S., Volpert,O.V., Good,D.J., Frazier,W.A., Polverini,P.J. and Bouck,N. (1993) Peptides derived from two separate domains of the matrix protein thrombospondin-1 have anti-angiogenic activity. J. Cell Biol., 122, 497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R., Rosenberg,M., Ross,S., Tyner,A. and Fuchs,E. (1989) Tissue-specific and differentiation-specific expression of a human K14 keratin gene in transgenic mice. Proc. Natl Acad. Sci. USA, 86, 1563–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpert O.V., Tolsma,S.S., Pellerin,S., Feige,J.J., Chen,H., Mosher,D.F. and Bouck,N. (1995) Inhibition of angiogenesis by thrombospondin-2. Biochem. Biophys. Res. Commun., 217, 326–332. [DOI] [PubMed] [Google Scholar]
- Werner S., Peters,K.G., Longaker,M.T., Fuller,P.F., Banda,M.J. and Williams,L.T. (1992) Large induction of keratinocyte growth factor expression in the dermis during wound healing. Proc. Natl Acad. Sci. USA, 89, 6896–6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner S., Smola,H., Liao,X., Longaker,M.T., Krieg,T., Hofschneider,P.H. and Williams,L.T. (1994) The function of KGF in morphogenesis of epithelium and re-epithelialization of wounds. Science, 266, 819–822. [DOI] [PubMed] [Google Scholar]
- Wight T.N., Raugi,G.J., Mumby,S.M. and Bornstein,P. (1985) Light microscopic immunolocation of thrombospondin in human tissues. J. Histochem. Cytochem., 33, 295–302. [DOI] [PubMed] [Google Scholar]
- Yehualaeshet T., O’Connor,R., Green,J.J., Mai,S., Silverstein,R., Murphy,U.J. and Khalil,N. (1999) Activation of rat alveolar macrophage-derived latent transforming growth factor β-1 by plasmin requires interaction with thrombospondin-1 and its cell surface receptor, CD36. Am. J. Pathol., 155, 841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan F., Chen,Y., Dellian,M., Safabakhsh,N., Ferrara,N. and Jain,R.K. (1996) Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc. Natl Acad. Sci. USA, 93, 14765–14770. [DOI] [PMC free article] [PubMed] [Google Scholar]