

Abstract
22q11 Deletion syndrome (22q11DS) is a common microdeletion syndrome with variable expression, including congenital and later onset conditions such as schizophrenia. Most studies indicate that expression does not appear to be related to length of the deletion but there is limited information on the endpoints of even the common deletion breakpoint regions in adults. We used a real-time quantitative PCR (qPCR) approach to fine map 22q11.2 deletions in 44 adults with 22q11DS, 22 with schizophrenia (SZ; 12 M, 10 F; mean age 35.7 SD 8.0 years) and 22 with no history of psychosis (NP; 8 M, 14 F; mean age 27.1 SD 8.6 years). QPCR data were consistent with clinical FISH results using the TUPLE1 or N25 probes. Two subjects (one SZ, one NP) negative for clinical FISH had atypical 22q11.2 deletions confirmed by FISH using the RP11-138C22 probe. Most (n = 34; 18 SZ, 16 NP) subjects shared a common 3 Mb hemizygous 22q11.2 deletion. However, eight subjects showed breakpoint variability: a more telomeric proximal breakpoint (n = 2), or more centromeric (n = 3) or more telomeric distal breakpoint (n = 3). One NP subject had a proximal nested 1.4 Mb deletion. COMT and TBX1 were deleted in all 44 subjects, and PRODH in 40 subjects (19 SZ, 21 NP). The results delineate proximal and distal breakpoint variants in 22q11DS. Neither deletion extent nor PRODH haploinsufficiency appeared to explain the clinical expression of schizophrenia in the present study. Further studies are needed to elucidate the molecular basis of schizophrenia and clinical heterogeneity in 22q11DS.
Introduction
22q11 deletion syndrome (22q11DS), also known as velocardiofacial or DiGeorge syndrome (MIM #188400/#192430), involves a hemizygous interstitial microdeletion at 22q11.2 which occurs in an estimated 1 in 4,000 live births, making 22q11DS the most common microdeletion syndrome (Devriendt et al. 1998). The deletion is clinically detected by fluorescence in situ hybridization (FISH) (Driscoll et al. 1993), usually using a TUPLE1 or N25 probe from the proximal commonly deleted region. The 22q11DS phenotype is highly variable, with over 40 common features, including congenital anomalies and later onset conditions, such as psychiatric disorders (Bassett et al. 2005). Individuals with 22q11DS have a 20 to 30-fold increased risk for developing schizophrenia over the general population rate (Bassett et al. 2005; Murphy et al. 1999), suggesting that hemizygosity of one or more genes mapping to the 22q11.2 region may underlie susceptibility to psychosis in 22q11DS.
The 22q11.2 genomic region is known to be polymorphic and to carry a number of low copy repeats (LCRs), which have been proposed to play an important role in the observed high frequency of 22q11.2 deletions. Further it has been proposed that the breakpoints of 22q11.2 deletions usually occur within these LCRs. However, this has only been demonstrated for a small number of 22q11.2 deletions. Although most individuals with 22q11DS have a similar “common 3 Mb deletion”, a minority have other overlapping and non-overlapping 22q11.2 deletions (Amati et al. 1999; Saitta et al. 2004). In other microdeletion syndromes such as Williams syndrome, some aspects of the phenotype appear related to the length of the deletion (Stock et al. 2003). In contrast, most studies of 22q11DS have found no evidence for such an association (Carlson et al. 1997; Kurahashi et al. 1997; Lindsay et al. 1995; Saitta et al. 2004). Some recent studies have reported possible genotype–phenotype correlations, but these were based on small samples of patients with uncommon deletions and congenital phenotypic features (Bartsch et al. 2003; Rauch et al. 2005). Few studies (Jacquet et al. 2002; Karayiorgou et al. 1995; McQuade et al. 1999) have included common late onset phenotypes such as schizophrenia and finely mapped breakpoint regions to help determine which genes would be included in deletions. For example, candidate genes for schizophrenia, COMT and PRODH coding for catechol-O-methyl transferase and proline dehydrogenase enzymes, respectively, would be assumed to be included in virtually all of the ~3 Mb 22q11.2 of the FISH deletions confirmed using conventional TUPLE1 or N25 probes, based on assumptions about common deletion breakpoint regions (Saitta et al. 2004).
In this study we used real-time quantitative polymerase chain reaction (qPCR) analysis involving SYBR Green chemistry (Weksberg et al. 2005) to map 22q11.2 deletions in 44 well characterized adults with 22q11DS, 22 with schizophrenia and 22 with no history of psychotic features. This method allowed us to detect several recurrent deletion breakpoint regions and delineate two atypical deletions not clinically detectable using standard FISH probes. We found no evidence for association of 22q11.2 deletion variants with expression of schizophrenia. TBX1 and COMT genes, but not PRODH, were contained within the 22q11.2 deletions of all 44 subjects.
Materials and methods
Sample and clinical assessment
We investigated 44 unrelated adults aged 20 years or older having clinical diagnoses of 22q11DS and either schizophrenia/schizoaffective disorder or no lifetime history of any psychotic disorder or symptoms, and who had sufficient qPCR data to determine the length of the 22q11.2 deletion. We ascertained subjects as previously described (Bassett et al. 2005), through the Toronto Congenital Cardiac Centre for Adults (Toronto General Hospital), Clinical Genetics Research Program, Centre for Addiction and Mental Health (CAMH) and other psychiatric-related sources, Clinical and Metabolic Genetics Clinic (The Hospital for Sick Children) and as transmitting parents. All subjects met clinical screening criteria for 22q11DS and had standard clinical FISH studies using a TUPLE1 (Vysis) or N25 (ONCOR) probe. One subject with schizophrenia was known to have inherited the 22q11.2 deletion. Informed consent was obtained in writing, and the study approved by the Research Ethics Boards of the University of Toronto, Centre for Addiction and Mental Health, and University Health Network.
As previously described, subjects had comprehensive medical and psychiatric assessments using standard methods (Bassett et al. 2005). Subgrouping of the sample was based on the presence of schizophrenia or schizoaffective disorder that met DSM-IV criteria, or the absence of any lifetime history of psychosis. Age at onset in the schizophrenia group was defined as age at onset of psychotic symptoms. Family history of a psychotic disorder was determined for first degree relatives of subjects.
Real-time quantitative PCR
Genomic DNA was obtained from fresh or cultured lymphocytes using standard procedures. Real-time quantitative PCR (qPCR) was performed using SYBR Green I PCR Master Mix (Applied Biosystems, Foster City, USA) and the ABI Prism 7900 high-throughput sequence detection system. Primer selection, conditions and optimization methods were as previously described for ten of the primer sets and six of the subjects with 22qDS (Weksberg et al. 2005). In brief, we used publicly available databases to design primers with unique sequences, free of intra or inter-chromosomal duplication or other trivial repeats, based on the July 2003 human reference sequence (UCSC version hg16, based on NCBI Build 34). QPCR experiments were then run in triplicate for each subject and copy-number alterations calculated as previously described to determine the copy number threshold values consistent with the presence of hemizygous 22q11.2 deletions (Weksberg et al. 2005). For raw data from each sample, reference primers (G6PDH, HEM3) were used to control for varying amounts of DNA and four control samples were used to normalize and calculate threshold cycle ratios, which were then translated into fold changes, using standard methods. Conclusive results were assigned when two of three findings fell in the same range of ±0.5 ΔCt, inside the value 0.24 standard deviation. The interval −0.75 to −1.35 was deemed consistent with loss of one copy (hemizygous microdeletion), and −0.35 to +0.35 to indicate two copies (no loss). QPCR experiments were repeated using re-extracted DNA samples for out-of-range uncorrelated results.
The initial screen of the 22q11.2 region used 21 primer sets, where possible selecting sequences from exonic regions of genes, with the goal of ~200 kb resolution. Eleven primer pairs were used to span the commonly deleted 3 Mb region (PRODH, DGCR2, DGCR14, TUPLE1, COMT, ZNF74, PIK4CA, CRKL, LZTR1, SLC7A4, D22S936), three primer pairs from the proximal flanking region (D22S181, TUBA8, USP18), and seven from the distal flanking region (HIC2, UBE2L3, G31074, D22S1248, VPREB1, rs2519503, G15854). Once deletions were determined, we selected and designed an additional 15 primer sets (total 36 qPCR primer sets) to better map breakpoint regions. For the proximal breakpoint region between TUPLE1 and COMT (genomic distance of ~639 Kb) of the two atypical deletions (ID 1 and 23, Fig. 1), we selected ten additional primers (RM40, UFD1L, CDC45L, BC042982, CF798466, SEPT5, GPIBB, AA195001, TBX1 and GNB1L). For the distal endpoint of the nested short deletion (ID 24, Fig. 1) between COMT and ZNF74 (genomic distance of ~767 Kb), we selected five additional markers (ARVCF, RANBP1, RTN4R, SHGC-173648, SHGC-172569).
Fig. 1.

22q11.2 deletion size and breakpoints in 44 adults with 22q11DS. Solid red arrowheads indicate the 21 qPCR screening markers used. Additional 15 qPCR markers were used to further delineate breakpoint regions in selected subjects (ID = 1, 23, 24). FISH probes used to validate deletion status are indicated by stars as follows: purple N25, blue Tuple1, and red RP11-138C22. Green boxes represent the four low copy repeat regions located in 22q11.2 (http://www.projects.tcag.ca/humandup/). Solid blue horizontal lines indicate the extent of 22q11.2 deletions tested by qPCR. Dotted blue lines represent the unknown interval between markers where the actual breakpoints reside. Broken red vertical lines show the positions of the seven breakpoint regions
FISH studies
All subjects had standard clinical FISH studies using a TUPLE1 (Vysis) or N25 (ONCOR) probe. For the two subjects with atypical deletions negative for the clinical probe, BAC probe RP11-138C22 containing the COMT primer set, was selected for FISH analysis to confirm qPCR results. The BAC originated from the RPCI library 11 of BAC clones (Osoegawa et al. 2001), obtained courtesy of the Centre for Applied Genomics at the Hospital for Sick Children (Toronto, Canada). Both ends of the RP11-138C22 clone (AQ383658 and AQ383661) are free of repeats and map with 100% identity only to cytoband 22q11.21 in the interval 18253741-18429812. RP11-138C22 is 176 Kb in length, contains the COMT gene and maps ~510 Kb telomeric to TUPLE1. Metaphase chromosome spreads were prepared for FISH analysis according to established cytogenetic and hybridization protocols (Beatty et al. 2002). BAC DNA from RP11-138C22 was extracted by standard alkaline lysis and phenol–chloroform extraction and directly labelled with Spectrum Green-dUPT by nick translation using the Vysis Nick Translation Kit (Abbott laboratories, Chicago, USA). Following hybridization the slides were counterstained with DAPI and mounted with Vectashield, an antifade mounting medium (Vector Laboratories, Burlingame, USA). Assessment of the BAC location and hybridization efficiency was carried out on lymphocytes prepared from a normal female donor. At least ten metaphase spreads were analyzed from each sample using the Vysis Quips FISH Imaging System (Vysis Inc., Chicago, USA).
Results
Clinical characteristics
All individuals with 22q11DS included in the study had standard phenotypic assessments (Bassett et al. 2005), and clinical FISH analysis. Forty-two of forty-four had confirmed a detectable 22q11.2 deletion using TUPLE1 (n = 16) or N25 (n = 28) clinical probes. There were no significant differences in sex distribution (χ2 = 1.47, df = 1, p = 0.23) or ethnicity (Fisher’s exact p = 0.11) between the 22 subjects in the schizophrenia group (12 male, 10 female; 18 Caucasian) and 22 in the non-psychotic group (8 male, 14 female; 22 Caucasian). Subjects with schizophrenia were significantly older than those in the non-psychotic group (mean age 35.7 SD 8.0 years and 27.1 SD 8.6 years, respectively; t = 3.42, p = 0.001). Median age at onset of psychosis was 19.5 years in the schizophrenia group. There was a family history of a psychotic disorder in one subject from the schizophrenia group and one from the non-psychotic group.
22q11.2 deletions
Figure 1 shows the qPCR 22q11.2 deletion results for the 44 subjects, which are summarized in Table 1. These data were consistent with the clinical deletion status ascertained by FISH analyses with the commonly used TUPLE1 or N25 probes. The results show that the deletion interval for the majority of subjects (n = 34, 77%) mapped proximally between USP18 (not deleted) and PRODH (deleted), and distally between D22S936 (deleted) and HIC2 (not deleted), spanning about 3.1 Mb. There were no significant differences in frequency of this common deletion between the two phenotypic subgroups (n = 18 with schizophrenia, n = 16 with no psychosis; χ2 = 0.51, df = 1, p = 0.47).
Table 1.
Summary of 22q11.2 deletions found in 44 adults with 22q11DS
| Subjects
|
Proximal breakpoints
|
Distal breakpoints
|
Deletion size (Mb) | ||||
|---|---|---|---|---|---|---|---|
| N | % | Deleted | Non-deleted | Deleted | Non-deleted | ||
| Tuple 1 deleted | 34 | 81 | USP18 | PRODH | D22S936 | HIC2 | 3.1 |
| 3 | 7.1 | USP18 | PRODH | SLC7A4 | D22S936 | 2.6 | |
| 2 | 4.7 | PRODH | DGCR2 | D22S936 | HIC2 | 2.8 | |
| 1 | 0.4 | USP18 | PRODH | HIC2 | UBE2L3 | 3.2 | |
| Tuple 1 non-deleted | 1 | 0.4 | CF798466 | SEPT5 | D22S936 | HIC2 | 2.1 |
| 1 | 0.4 | CF798466 | SEPT5 | HIC2 | UBE2L3 | 2.3 | |
Amongst the 10 (23%) subjects who did not have the common 3 Mb 22q11.2 deletion, there were six other deletion variants detected. In addition to the proximal and distal breakpoint region seen in common deletions, these six variant deletions included two proximal and three distal breakpoint variants (Table 1). These deletion variants involved one to three subjects each and appeared approximately evenly divided between the two phenotypic subgroups, however numbers were too small for statistical comparisons. Included in these variant deletions were those found in the two individuals (ID 1 and 23, Fig. 1) with negative clinical FISH results (Table 1). These “atypical” 22q11.2 deletions were confirmed by FISH analysis using the RP11-138C22 probe (Fig. 2). These deletions shared the same proximal breakpoint region centromeric to the SEPT5 locus but had differing distal breakpoint regions. One of these breakpoint regions mapped to the same location as that in the common 3 Mb deletion and the other was the same as the more distal breakpoint variant found in two other subjects (ID 41 and 42, Fig. 1).
Fig. 2.
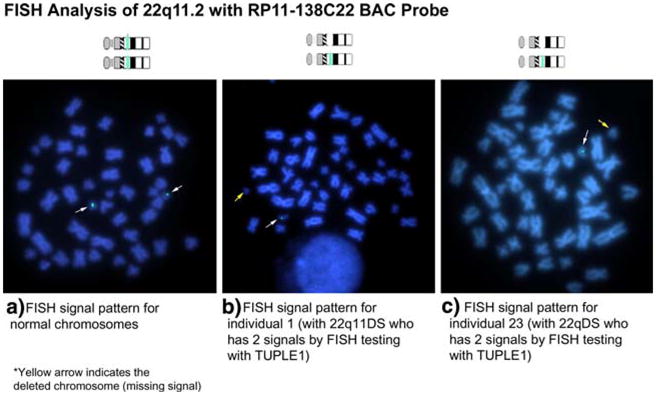
FISH analysis using the RP11-138C22 probe to confirm an atypical 22q11.2 deletion. Fluoresence in situ hybridization (FISH) validation of the RP11-138C22 probe to detect atypical 22q11DS deletions. A control individual (a) showing two copies of chromosome 22q11 using the RP11-138C22 probe. Individuals 1 (b) and 23 (c) are negative by FISH analysis for the TUPLE1 probe but are positive for the deletion using the RP11-138C22 probe due to the extent of their deletion, which was confirmed by qPCR analysis (Fig. 1)
The TBX1 and COMT genes were included in the deletions of all subjects assessed (Fig. 1). The PRODH gene was included in the deletions of 40 (91%) of the 44 subjects, including 19 (86%) with schizophrenia and 21 (95%) with no psychosis. In this sample of 44 subjects, all 22q11.2 deletions mapped shared a region spanning about 300 Kb that included proximal marker SEPT5, in the two atypical deletions, and distal marker ARVCF, in the proximal nested 1.4 Mb deletion; flanking markers spanned about 518 Kb (Table 1). The 22 subjects with schizophrenia shared a longer deletion region, extending from SEPT5, in the atypical deletion, to SLC7A4, in the centromeric distal variant 2.6 Mb deletion (Fig. 1, Table 1).
Discussion
Common deletion variants and recurrent breakpoint regions
Our results showed that the majority of the 44 adults with 22q11DS studied shared a common 3.1 Mb hemizygous deletion of 22q11.2, consistent with the literature (Carlson et al. 1997; Kurahashi et al. 1997; Lindsay et al. 1995; Saitta et al. 2004). We refined the proximal breakpoint location to a ~250 Kb segment between USP18 and PRODH, and the distal breakpoint to a ~350 Kb segment between D22S936 and HIC2. Although different methods, markers and maps have been used, this present study provides greater mapping density than most previous studies. Previously published analyses of the proximal breakpoint have been reported between D22S427 and D22S1638 (~400 kb) (Carlson et al. 1997; Kurahashi et al. 1997; Vittorini et al. 2001). However, the location of the telomeric breakpoint has not been conclusively mapped to one location as published studies implicate slightly different regions: D22S935/936 to D22S1709 (~850 Kb) (Carlson et al. 1997), D22S935 to D22S938 (~930 Kb) (Kurahashi et al. 1997), and D22S1709 to D22S306/308 (~1.1 Mb) (Vittorini et al. 2001). Our results are consistent with another study (Jacquet et al. 2002) that used a similar qPCR method with slightly less map coverage (n = 23 probes) in 14 patients with 22q11DS.
Nearly one quarter of the 44 individuals studied showed variations on the most common ~3 Mb 22q11.2 deletion, including three variants of this deletion, a short nested deletion, and two atypical deletions. All of these showed recurrent breakpoint regions within this sample (Table 1; Fig. 1), with the exception of the short nested 1.4 Mb deletion (ID 24), which had a distal breakpoint in a ~100 Kb segment between ARVCF and RANBP1. This deletion appears similar to the 1.5 Mb deletion previously described (Carlson et al. 1997; Kurahashi et al. 1997; Lindsay et al. 1995; Saitta et al. 2004), with breakpoints between D22S933 and ZNF74 (~706 Kb). The distal deletion breakpoint of these proximal nested deletions is commonly believed to be in LCR-B (Saitta et al. 2004), although this would not appear to be the case for the 1.4 Mb deletion we found (Fig. 1).
Improved resolution with the method we used allowed detection of three variants of the common 3 Mb deletions, in 17.1% of 41 subjects with 2.6–3.2 Mb deletions. These included breakpoint variants proximally between markers PRODH and DGCR2, a distance of ~209 Kb, and distally between markers SLC7A4 and D22S936 (~65 Kb) or between HIC2 and UBE2L3 (~115 Kb) (Fig. 1, Table 1). Variants of 3 Mb deletions have been reported previously (Matsuoka et al. 1998; Saitta et al. 2004; Urban et al. 2006). It appears likely though that, due to lower resolution methods, some of the “common 3 Mb deletions” reported in previous studies may have included both the most common 3.1 Mb deletion as well as the type variants found in our study.
Previous reports of “atypical variant” deletions support our current findings of recurrent breakpoint regions for deletions that fit the “common deletion sizes”. Even the two atypical deletions we found shared distal breakpoint regions with the common 3.1 Mb deletion or its longer variant (Fig. 1, Table 1). These atypical deletions also shared a common proximal breakpoint. These deletions and/or their breakpoint regions may be similar to atypical deletions previously reported (Amati et al. 1999; Levy et al. 1995; Lu et al. 2001; McQuade et al. 1999; Vittorini et al. 2001), further supporting the possibility of several recurring breakpoints in the 22q11.2 region.
Breakpoint position and repeat elements
The results of this study are consistent with those of many other studies indicating that the breakpoint regions for the common 3.1 Mb 22q11.2 deletion are in low-copy repeat (LCR) segments, specifically the 240 Kb LCRs termed LCR22-2 and LCR22-4 (Lindsay 2001), or A and D (Saitta et al. 2004). These LCRs are estimated to mediate ~90% of 22q11.2 deletions (Saitta et al. 2004). However, for variant ~3 Mb deletions, such as that with a telomeric distal breakpoint flanked by HIC2 and UBE2L3, it is unclear whether LCRs or other repeat elements are involved. The high density of LCRs and other repetitive sequences in 22q11.2 make this the region of the human genome most prone to rearrangements (Babcock et al. 2003). Breakpoints outside of LCRs may be mediated by other repeat elements present in the 22q11.2 region, likely including Alu elements which are known to play a role in modulating genomic architecture (Babcock et al. 2003; Shaw and Lupski 2005). Our results support the recurrence of breakpoints in specific regions of 22q11.2 but not necessarily in specific sites containing LCRs in 22q11. Other reported breakpoints outside of the LCRs include translocation breakpoints (ADU and GM00980), and interstitial deletion breakpoints (Carlson et al. 1997; Levy et al. 1995; McQuade et al. 1999); however, it is unknown whether or not these are recurrent and also which specific repeat elements are involved. Similar breakpoints are known to be associated with uncommon deletions in other microdeletion syndromes, and may have important implications for deletion mechanisms (Shaw and Lupski 2005).
22q11.2 deletions and schizophrenia in 22q11DS
Few previous studies have used flanking markers to determine the extent of 22q11.2 deletions in individuals with 22q11DS and schizophrenia (Jacquet et al. 2002; Karayiorgou et al. 1995; McQuade et al. 1999). In our study, the 22q11.2 region shared by all 22 adults with schizophrenia included COMT and most of the telomeric half of the 3.1 Mb common deletion, but not PRODH (Fig. 1). However, this telomeric region was not included in a proximal nested ~1.5 Mb deletion (Karayiorgou et al. 1996) or atypical deletions (Jacquet et al. 2002; McQuade et al. 1999) associated with schizophrenia in other cases, suggesting that longer deletions or hemizygosity of distal genes may not be necessary for expression of psychosis in 22q11DS. The COMT locus was included in the proximal nested deletion (Karayiorgou et al. 1996), and an atypical ~750 Kb interstitial deletion (McQuade et al. 1999). PRODH, but not COMT, was included in a ~350 Kb (PRODH-DGCR6) interstitial deletion in two sisters with schizophrenia and their unaffected mother (Jacquet et al. 2002), and in a 22pter-22q11.2 deletion associated with a translocation (11;22) in a woman (GM00980) and her adult daughter (AP1268) with schizophrenia spectrum disorder (Karayiorgou et al. 1996). Taken together, biallelic expression of the COMT and the PRODH loci would not appear to be essential for expression of schizophrenia in 22q11DS.
Phenotype and 22q11.2 deletion extent
Consistent with most studies of 22q11DS that primarily examined major congenital features (Carlson et al. 1997; Kurahashi et al. 1997; Saitta et al. 2004) we found no evidence for genotype–phenotype associations using a schizophrenia phenotype. Our study, like others with few individuals having any single variant deletion, had insufficient power to detect modest effects of deletion extent. However, especially considering the significant within family phenotypic variability of identical 22q11.2 deletions, there is little evidence to date for significant effects of deletion length on major phenotypes of 22q11DS (Yamagishi and Srivastava 2003).
The relatively small deletion region shared by all 44 adults with 22q11DS in our study includes TBX1 and COMT and would overlap with most, but not all, 22q11.2 deletions. Other “minimally deleted regions”, sometimes termed “single region of overlap” or “critical region”, have previously been reported in larger samples of patients (Carlson et al. 1997; Kurahashi et al. 1997). Consistent with the possibility of multiple 22q11.2 regions for 22q11DS (Amati et al. 1999) however, there are several 22q11.2 deletions associated with a 22q11DS phenotype, which would not overlap with our “minimally deleted region”, proximally (Amati et al. 1999; Karayiorgou et al. 1996; Lu et al. 2001) or distally (Kurahashi et al. 1997; Rauch et al. 2005; Saitta et al. 1999).
Identifying deletion variants has implications for the specific genes involved in expression of schizophrenia and other phenotypes. However, accumulating evidence supports the likelihood that several genes within the 22q11.2 region may be necessary for expression of major 22q11DS phenotypes and that genes from other areas of the genome can modify the effects of 22q11.2 hemizygosity (Guris et al. 2006; Yamagishi and Srivastava 2003). The common 3 Mb deletion contains more than 40 known genes from PRODH to D22S936 inclusive (UCSC Genome Browser, v128 http://www.genome.ucsc.edu). TBX1 has been shown to play an important role in congenital heart defects and other anomalies related to pharyngeal apparatus development in 22q11DS (Lindsay 2001). Interestingly, a recent study suggests TBX1 may play a role in behaviour as well (Paylor et al. 2006).
Advantages and limitations
Limitations of the current study include assay resolution, small sample size, ascertainment bias, and age difference between groups. The qPCR method used has advantages over many previous studies. For example, studies using polymorphic markers may be limited by low informativeness, density and span of markers. The flanking markers often used (e.g., D22S427, D22S306/308) would not be able to detect the breakpoint variants found in this study. We estimated deletion size (Table 1) conservatively, based on the genomic extent of those markers found to be hemizygous but deletions may extend close to flanking markers.
Most studies report infants and children with 22q11DS, particularly those with congenital heart defects and other congenital defects and there is limited information on later onset features of the syndrome or effects of ascertainment (Bassett et al. 2005). The current study of 22q11.2 deletions used the largest sample of adults with 22q11DS, or with 22q11DS-schizophrenia, yet reported. However, power was still insufficient for statistical analyses of individual deletion variants. Other 22q11.2 deletion variants, including deletions that do not overlap the common 3.1 Mb deletion (Rauch et al. 2005; Saitta et al. 1999), undoubtedly exist in the population but small sample size and ascertainment bias for deletions detectable by routine diagnostic testing would minimize the chance of their detection. We limited our study to the schizophrenia phenotype. There is a possibility that some non-psychotic subjects may develop schizophrenia in the future, which would have decreased power to find differences between phenotypic subgroups. We minimized this by using a sample older than the median age of onset of psychosis, but the frequency of the common 3 Mb deletion also makes it unlikely that this possibility would have had a substantial effect on the results.
In conclusion, the results demonstrate that qPCR can determine deletion span and breakpoint regions and identify 22q11DS patients with 22q11.2 deletions not detectable by standard clinical FISH testing. Although a 3.1 Mb deletion is clearly most common, there are several variants in length, position and precise breakpoints for 22q11.2 deletions. There was no correlation of SZ expression with deletion extent, and PRODH hemizygosity did not appear necessary for expression of psychosis in 22qDS in our sample. Further studies are needed to elucidate the molecular basis of schizophrenia in 22q11DS.
Acknowledgments
This research was supported by grants from the W. Garfield Weston Foundation, Ontario Mental Health Foundation, Medical Research Council of Canada (MOP-38099), and by a Young Investigator Award (EWCC) and Distinguished Investigator Awards (ASB and RW) from the National Alliance for Research on Schizophrenia and Depression, and Canada Research Chair in Schizophrenia Genetics (ASB). We also thank the National Cancer Institute of Canada (NCIC) with funds from the Canadian Cancer Society (JS and JB) for support. The authors thank the patients and their families for their participation, and many colleagues for referring patients, including Drs. Gary Webb and Michael Gatzoulis and the staff at the Toronto Congenital Cardiac Centre for Adults.
Footnotes
None of the authors have financial interests that might present a conflict of interest.
Contributor Information
Rosanna Weksberg, Division of Clinical and Metabolic Genetics, Department of Paediatrics, The Hospital for Sick Children, 555 University Ave., M5G 1X8 Toronto, ON, Canada, Department of Molecular and Medical Genetics, University of Toronto, Toronto, ON, Canada, The Research Institute, The Hospital for Sick Children, Toronto, ON, Canada, Institute of Medical Science, University of Toronto, Toronto, ON, Canada.
Andrea C. Stachon, The Research Institute, The Hospital for Sick Children, Toronto, ON, Canada, Institute of Medical Science, University of Toronto, Toronto, ON, Canada
Jeremy A. Squire, Ontario Cancer Institute and Department of Laboratory Medicine, Pathology and Medical Biophysics, University of Toronto, Toronto, ON, Canada
Laura Moldovan, The Research Institute, The Hospital for Sick Children, Toronto, ON, Canada.
Jane Bayani, Ontario Cancer Institute and Department of Laboratory Medicine, Pathology and Medical Biophysics, University of Toronto, Toronto, ON, Canada.
Stephen Meyn, Division of Clinical and Metabolic Genetics, Department of Paediatrics, The Hospital for Sick Children, 555 University Ave., M5G 1X8 Toronto, ON, Canada, Department of Molecular and Medical Genetics, University of Toronto, Toronto, ON, Canada, The Research Institute, The Hospital for Sick Children, Toronto, ON, Canada.
Eva Chow, Department of Psychiatry, University of Toronto, Toronto, ON, Canada, Clinical Genetics Research Program, Centre for Addiction and Mental Health, Toronto, ON, Canada.
Anne S. Bassett, Institute of Medical Science, University of Toronto, Toronto, ON, Canada, Department of Psychiatry, University of Toronto, Toronto, ON, Canada, Clinical Genetics Research Program, Centre for Addiction and Mental Health, Toronto, ON, Canada
References
- Amati F, Conti E, Novelli A, Bengala M, Diglio MC, Marino B, Giannotti A, Gabrielli O, Novelli G, Dallapiccola B. Atypical deletions suggest five 22q11.2 critical regions related to the DiGeorge/velo-cardio-facial syndrome. Eur J Hum Genet. 1999;7:903–909. doi: 10.1038/sj.ejhg.5200399. [DOI] [PubMed] [Google Scholar]
- Babcock M, Pavlicek A, Spiteri E, Kashork CD, Ioshikhes I, Shaffer LG, Jurka J, Morrow BE. Shuing of genes within low-copy repeats on 22q11 (LCR22) by Alu-mediated recombination events during evolution. Genome Res. 2003;13:2519–2532. doi: 10.1101/gr.1549503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch O, Nemeckova M, Kocarek E, Wagner A, Puchmajerova A, Poppe M, Ounap K, Goetz P. DiGeorge/velocardiofacial syndrome: FISH studies of chromosomes 22q11 and 10p14, and clinical reports on the proximal 22q11 deletion. Am J Med Genet A. 2003;117:1–5. doi: 10.1002/ajmg.a.10914. [DOI] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, Gatzoulis MA. Clinical features of 78 adults with 22q11 deletion syndrome. Am J Med Genet A. 2005;138:307–313. doi: 10.1002/ajmg.a.30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty B, Mai S, Squire J. FISH: a practical approach. Oxford University Press; Oxford, New York: 2002. [Google Scholar]
- Carlson C, Sirotkin H, Pandita R, Goldberg R, McKie J, Wadey R, Patanjali SR, Weissman SM, Anyane-Yeboa K, Warburton D, Scambler P, Shprintzen R, Kucherlapati R, Morrow BE. Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am J Hum Genet. 1997;61:620–629. doi: 10.1086/515508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devriendt K, Fryns JP, Mortier G, van Thienen MN, Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet. 1998;35:789–790. doi: 10.1136/jmg.35.9.789-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai EH, Emanuel BS. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counseling and prenatal diagnosis. J Med Genet. 1993;30:813–817. doi: 10.1136/jmg.30.10.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guris DL, Duester G, Papaioannou VE, Imamoto A. Dose–dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev Cell. 2006;10:81–92. doi: 10.1016/j.devcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Jacquet H, Raux G, Thibaut F, Hecketsweiler B, Houy E, Demilly C, Haouzir S, Allio G, Fouldrin G, Drouin V, Bou J, Petit M, Campion D, Frebourg T. PRODH mutations and hyperprolinemia in a subset of schizophrenic patients. Hum Mol Genet. 2002;11:2243–2249. doi: 10.1093/hmg/11.19.2243. [DOI] [PubMed] [Google Scholar]
- Karayiorgou M, Morris MA, Morrow B, Shprintzen RJ, Goldberg R, Borrow J, Gos A, Nestadt G, Wolyniec PS, Lasseter VK, et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci USA. 1995;92:7612–7616. doi: 10.1073/pnas.92.17.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayiorgou M, Gogos JA, Galke BL, Jeffery JA, Nestadt G, Wolyniec PS, Antonarakis SE, Kazazian HH, Housman DE, Driscoll DA, Pulver AE. Genotype and phenotype analysis at the 22q11 schizophrenia susceptibility locus. Cold Spring Harb Symp Quant Biol. 1996;61:835–843. [PubMed] [Google Scholar]
- Kurahashi H, Tsuda E, Kohama R, Nakayama T, Masuno M, Imaizumi K, Kamiya T, Sano T, Okada S, Nishisho I. Another critical region for deletion of 22q11: a study of 100 patients. Am J Med Genet. 1997;72:180–185. doi: 10.1002/(sici)1096-8628(19971017)72:2<180::aid-ajmg10>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Levy A, Demczuk S, Aurias A, Depetris D, Mattei MG, Philip N. Interstitial 22q11 microdeletion excluding the ADU breakpoint in a patient with DiGeorge syndrome. Hum Mol Genet. 1995;4:2417–2419. doi: 10.1093/hmg/4.12.2417. [DOI] [PubMed] [Google Scholar]
- Lindsay EA. Chromosomal microdeletions: dissecting del22q11 syndrome. Nat Rev Genet. 2001;2:858–868. doi: 10.1038/35098574. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Goldberg R, Jurecic V, Morrow B, Carlson C, Kucherlapati RS, Shprintzen RJ, Baldini A. Velo-cardio-facial syndrome: frequency and extent of 22q11 deletions. Am J Med Genet. 1995;57:514–522. doi: 10.1002/ajmg.1320570339. [DOI] [PubMed] [Google Scholar]
- Lu JH, Chung MY, Betau H, Chien HP, Lu JK. Molecular characterization of tetralogy of fallot within Digeorge critical region of the chromosome 22. Pediatr Cardiol. 2001;22:279–284. doi: 10.1007/s002460010230. [DOI] [PubMed] [Google Scholar]
- Matsuoka R, Kimura M, Scambler PJ, Morrow BE, Imamura S, Minoshima S, Shimizu N, Yamagishi H, Joh-o K, Watanabe S, Oyama K, Saji T, Ando M, Takao A, Momma K. Molecular and clinical study of 183 patients with conotruncal anomaly face syndrome. Hum Genet. 1998;103:70–80. doi: 10.1007/s004390050786. [DOI] [PubMed] [Google Scholar]
- McQuade L, Christodoulou J, Budarf M, Sachdev R, Wilson M, Emanuel B, Colley A. Patient with a 22q11.2 deletion with no overlap of the minimal DiGeorge syndrome critical region (MDGCR) Am J Med Genet. 1999;86:27–33. [PubMed] [Google Scholar]
- Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry. 1999;56:940–945. doi: 10.1001/archpsyc.56.10.940. [DOI] [PubMed] [Google Scholar]
- Osoegawa K, Mammoser AG, Wu C, Frengen E, Zeng C, Catanese JJ, de Jong PJ. A bacterial artificial chromosome library for sequencing the complete human genome. Genome Res. 2001;11:483–496. doi: 10.1101/gr.169601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paylor R, Glaser B, Mupo A, Ataliotis P, Spencer C, Sobotka A, Sparks C, Choi CH, Oghalai J, Curran S, Murphy KC, Monks S, Williams N, O’Donovan MC, Owen MJ, Scambler PJ, Lindsay E. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci USA. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Zink S, Zweier C, Thiel CT, Koch A, Rauch R, Lascorz J, Huffmeier U, Weyand M, Singer H, Hofbeck M. Systematic assessment of atypical deletions reveals genotype-phenotype correlation in 22q11.2. J Med Genet. 2005;42:871–876. doi: 10.1136/jmg.2004.030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitta SC, McGrath JM, Mensch H, Shaikh TH, Zackai EH, Emanuel BS. A 22q11.2 deletion that excludes UFD1L and CDC45L in a patient with conotruncal and craniofacial defects. Am J Hum Genet. 1999;65:562–566. doi: 10.1086/302514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitta SC, Harris SE, Gaeth AP, Driscoll DA, McDonald-McGinn DM, Maisenbacher MK, Yersak JM, Chakraborty PK, Hacker AM, Zackai EH, Ashley T, Emanuel BS. Aberrant interchromosomal exchanges are the predominant cause of the 22q11.2 deletion. Hum Mol Genet. 2004;13:417–428. doi: 10.1093/hmg/ddh041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw CJ, Lupski JR. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet. 2005;116:1–7. doi: 10.1007/s00439-004-1204-9. [DOI] [PubMed] [Google Scholar]
- Stock AD, Spallone PA, Dennis TR, Netski D, Morris CA, Mervis CB, Hobart HH. Heat shock protein 27 gene: chromosomal and molecular location and relationship to Williams syndrome. Am J Med Genet A. 2003;120:320–325. doi: 10.1002/ajmg.a.20055. [DOI] [PubMed] [Google Scholar]
- Urban AE, Korbel JO, Selzer R, Richmond T, Hacker A, Popescu GV, Cubells JF, Green R, Emanuel BS, Gerstein MB, Weissman SM, Snyder M. High-resolution mapping of DNA copy alterations in human chromosome 22 using high-density tiling oligonucleotide arrays. Proc Natl Acad Sci USA. 2006;103:4534–4539. doi: 10.1073/pnas.0511340103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vittorini S, Sacchelli M, Iascone MR, Collavoli A, Storti S, Giusti A, Andreani G, Botto N, Biagini A, Clerico A. Molecular characterization of chromosome 22 deletions by short tandem repeat polymorphism (STRP) in patients with conotruncal heart defects. Clin Chem Lab Med. 2001;39:1249–1258. doi: 10.1515/CCLM.2001.201. [DOI] [PubMed] [Google Scholar]
- Weksberg R, Hughes S, Moldovan L, Bassett AS, Chow EW, Squire JA. A method for accurate detection of genomic microdeletions using real-time quantitative PCR. BMC Genomics. 2005;6:180. doi: 10.1186/1471-2164-6-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi H, Srivastava D. Unraveling the genetic and developmental mysteries of 22q11 deletion syndrome. Trends Mol Med. 2003;9:383–389. doi: 10.1016/s1471-4914(03)00141-2. [DOI] [PubMed] [Google Scholar]