

Abstract
Background
Hormonal ablation is the standard of treatment for advanced androgen-dependent prostate cancer. Although tumor regression is usually achieved at first, the cancer inevitably evolves toward androgen-independence, in part because of the development of mechanisms of resistance and in part because at the tissue level androgen withdrawal is not fully attained. Current research efforts are focused on new therapeutic strategies that will increase the effectiveness of androgen withdrawal and delay recurrence. We used a syngeneic pseudo-orthotropic mouse model of prostate cancer to test the efficacy of combining androgen withdrawal with FDA-approved COX-2 inhibitor celecoxib.
Methods
GFP-tagged TRAMP-C2 cells were co-implanted with prostate tissue in the dorsal chamber model and tumors were allowed to establish and vascularize. Tumor growth and angiogenesis were monitored in real-time using fluorescent intravital microscopy (IVM). Androgen withdrawal in mice was achieved using surgical castration or chemical hormonal ablation, alone or in combination with celecoxib (15 mg/kg, twice daily).
Results
Celecoxib alone decreased the growth of prostate tumors mostly by inducing mitotic failure, which resulted in increased apoptosis. Surprisingly, celecoxib did not possess significant angiostatic activity. Surgical or chemical castration prevented the growth of prostate tumors and this, on the other hand, was associated with disruption of the tumor vasculature. Finally, androgen withdrawal combined with celecoxib caused tumor regression through decreased angiogenesis and increased mitosis arrest and apoptosis.
Conclusion
Celecoxib, a relatively safe COX-2-selective anti-inflammatory drug, significantly increases the efficacy of androgen withdrawal in vivo and warrants further investigation as a complement therapy for advanced prostate cancer.
Keywords: Prostate cancer, androgen therapy, COX-2, intravital microscopy, Celecoxib, Celebrex
INTRODUCTION
Current therapeutic interventions for advanced prostate cancer are not curative. Although androgen ablation does initially deliver a response, the return of hormone-refractory tumors invariably prevents long-term patient survival. More effective strategies are needed to extend life expectancy and improve the quality of life for patients with advanced prostate cancer. New strategies may involve the combination of known effective treatments such as androgen withdrawal with drugs that have relatively minor side effects.
Cyclooxygenase (COX), the key regulatory enzyme for prostaglandin synthesis, is transcribed from two distinct genes. COX-1 is expressed constitutively in most tissues, while COX-2 expression is normally low and induced by a wide variety of stimuli (it was initially identified as an immediate-early growth response gene). Cyclooxygenases catalyze the formation of prostaglandins (PGs), which are involved in tumor initiation and/or progression. For example, COX-1 and COX-2 promote inflammation, which may directly contribute to the development of prostate cancer (1). In addition, COX-2-induced PGE2 activates cell signaling involved in proliferation and thereby directly promotes tumor cell growth. COX-2 is overexpressed in prostate cancer and its level of expression correlates with Gleason score and cancer progression (2).
Nonsteroidal anti-inflammatory drugs (NSAIDs) that inhibit both COX-1 and COX-2, as well as COX-2 selective inhibitors, are currently being evaluated clinically for the prevention of major types of cancer because of their positive effects in epidemiological and animal studies. Indeed, a recent meta-analysis of epidemiological studies concluded that NSAIDs – whether or not they are selective for COX-2 – have a chemopreventive effect against cancer of the colon, breast, lung and prostate (3). In addition, COX-2 promotes angiogenesis and therefore COX-2 inhibitors may impair tumor growth by blocking angiogenesis (4). Whereas genetic ablation of COX-2 decreases tumor formation in mouse models, its overexpression favors transformation and cancer progression (reviewed in (2)). COX-2 is overexpressed in a number of malignancies and is associated with increased production of PGE2, which plays a role in the initiation and progression of tumors (2). It is now well accepted that COX-2 contributes to prostate cancer and the evidence that COX-2 inhibitor celecoxib may be beneficial to prostate cancer patients is mounting (5,6).
In animal models, celecoxib (alone or in combination with another drug) decreased the growth of androgen-independent PC3 xenograft (7) and suppressed the re-growth of LNCaP xenografts following androgen withdrawal (8). In addition, several clinical studies have started to evaluate the effect of celecoxib in various therapeutic settings. These trials show that celecoxib is safe, with a low cytotoxicity profile. Two studies have described neoadjuvant celecoxib prior to prostatectomy in men with clinically localized prostate cancer, showing measurable amounts of celecoxib in tumors (9) and measurable biological effects in prostate cancer tissue (10) but lacking clear clinical benefit. Other studies have examined the effect of celecoxib in combination with chemotherapy for the treatment of advanced, hormone-independent prostate cancer, without much success (11,12). A larger trial is underway (13), which will provide further information regarding the potential efficacy of celecoxib for advanced prostate cancer. Finally, the efficiency of celecoxib was assessed in patients with recurrent prostate cancer following radiation therapy or radical prostatectomy. A decline or stabilization of PSA levels was observed in both trials, indicative of biological activity and suggesting that the drug may delay the growth of recurrent tumors and extend time before hormone-deprivation therapy (14,15). Of note, there has been no clinical trial so far to assess whether celecoxib may delay the progression of prostate cancer toward androgen-independence in patients undergoing hormone-deprivation therapy. Thus, more studies are warranted to discover the best use for COX-2 inhibitors and examine the efficacy of various strategies.
We have previously described a syngeneic pseudo-orthotropic model to study prostate cancer progression in vivo (16). This model is based on the dorsal skinfold chamber technique, in which a transparent chamber for microscopy is positioned in the dorsal skinfold of a mouse. Mouse cells derived from the prostate tumor of a TRAMP mouse, known as TRAMP-C2 cells (17), were implanted into mouse dorsal chambers. A H2B-GFP fusion protein was stably introduced into the TRAMP-C2 cells by retroviral transduction. As shown by Kanda et al., the H2B-GFP fusion protein is incorporated into chromatin without affecting cell cycle progression (18). Because the cells are stably transfected with fluorescent H2B-GFP, tumors can be visualized and imaged in real time using intravital fluorescence video-microscopy (IVM). IVM allows measuring tumor growth, vascular parameters and intratumoral mitotic and apoptotic indices. To create a pseudo-orthotropic microenvironment, prostate tissue from a donor mouse was co-implanted with TRAMP-C2-GFP in the chambers. We have shown previously that after 1-2 weeks post-implantation the prostate tissue grafted into the chamber was able to connect its vasculature to the skin vasculature of the recipient mouse, which in turn supported angiogenesis within the growing tumor (16).
The present study investigated the effect of celecoxib alone or in combination with surgical and chemical castration on the growth of TRAMP-C2-GFP tumors.
METHODS
Antibodies and reagents
Cell culture media, culture-grade PBS, L-Glutamine, Trypsin-EDTA, penicillin/streptomycin, and FBS (Fetal bovine Serum) were from Mediatech (Herndon, VA). G418 and Insulin-Selenium-Transferrin supplement (#41400-Gibco) was from Invitrogen (Carlsbad, CA). Celecoxib was from Toronto Research Chemicals (Ontario, Canada).
Antibodies against phospho-ERK1/2, PARP, and phospho-histone H3 (Ser10; Ab 6G3) were from Cell Signaling Technology (Danvers, MA). Antibodies to β-actin (AC-15) were from Sigma-Aldrich (St Louis, MO). Monoclonal anti-p27Kip1 antibodies were from BD Pharmingen (San Jose, CA). Alexa Fluor-488 goat anti-mouse antibodies were from Invitrogen (Carlsbad, CA).
Cell culture
H2B-GFP/TRAMP-C2 (TRAMP-C2-GFP) cells that stably express histone H2B-GFP fusion protein (18) were generated as described (19). TRAMP-C2-GFP cells were grown in RPMI containing 10% FBS, 2mM L-glutamine, 100 U/ml penicillin/100 μg/ml streptomycin, insulin-selenium-transferrin (10 μg/ml insulin), and DHT 10−8M final. G418 (100 μg/ml) was added to maintain stable expression of H2B-GFP. Androgen withdrawal was achieved by keeping the cells in phenol red-free RPMI medium containing 10% charcoal-treated FBS and the same supplements as in the normal medium except for DHT.
Human prostate cancer cells DU145 and PC3 were grown in RPMI containing 10% FBS, 2mM L-glutamine, 100 U/ml penicillin/100 μg/ml streptomycin. Cells were maintained in a humidified incubator at 37°C and 5% CO2.
Measurement of cell growth in vitro
Cell growth was monitored by direct counting. Cells in 12-well plates were washed once with PBS, detached using Trypsin, and transferred to a suspension vial in a final volume of 10ml PBS. Cells were counted using a COULTER™ Multisizer II instrument (Beckman Coulter Inc., Hialeah, FL) gated for the appropriate cell size and corrected for particulate debris. Each experiment was performed in biological replicates and each vial was counted twice.
Flow cytometry quantification of mitosis
Cells were treated with celecoxib for 24 hrs, suspended using trypsin, fixed and permeabilized using BD cytofix/cytoperm solution (BD Pharmingen, San Jose, CA) according to instructions. Cells were incubated with antibodies to phospho-histone H3 for 30 min, washed three times in BD perm/wash buffer, and stained with Alexa Fluor-488 anti-mouse antibodies for 20 min followed by three more washes. The cells were resuspended at a density of approximately 106 cells/0.5ml in BD perm/wash buffer containing 50 μg/ml DNase-free RNase A, and 50 μg/ml propidium iodine. Fluorescence of single cells was recorded using a Facscan flow cytometer (BD Pharmingen, San Jose, CA). FlowJo™ Software was used for data analysis.
Western-blot analysis of protein expression
Cells treated with 40 μM celecoxib for the indicated times were lysed on ice in the presence of phosphatase and protease inhibitors. Lysates were clarified by centrifugation and the protein concentration in each sample was measured using a BCA assay (Pierce, Rockford, IL). Lysates were submitted to SDS-PAGE electrophoresis. Proteins were transferred to Immobilon-P® membranes (Millipore, Billerica, MA), which were incubated with a blocking buffer for 20 min (Pierce, Rockford, IL). The first antibody was incubated overnight. Peroxidase-conjugated antibodies (Amersham Biosciences, Piscataway, NJ) were added for 45min. Proteins were revealed using a chromogenic stabilized substrate from Promega (Madison, WI). When appropriate, membranes were stripped using Restore™ Stripping Buffer (Pierce) for 15min and reprobed.
Animal model and surgical techniques
Animal experiments have been approved by our Institutional IACUC and were conducted in accordance to NIH guidelines. The dorsal skinfold chambers were prepared as described previously (16,19). Briefly, male C57/bl6 mice (25-30 g body weight) were anesthetized and placed on a heating pad. Two symmetrical titanium frames were implanted into the dorsal skinfold. A circular layer was excised from one of the skin layers. The underlying muscle and subcutaneous tissues were covered with a glass coverslip incorporated in one of the frames. After a recovery period of 2-3 days, prostate tissue and tumor cells were carefully placed in the chamber.
Preparation of tumor spheroids
TRAMP-C2-GFP cells were trypsinized and adjusted to a concentration of 250,000 cells/ml. Cell suspensions were then overlaid into 96-well round bottom plates coated with 1% agarose (100ul cell suspension/well). Cell spheroids were allowed to compact for 48 hours and were washed in serum-free medium before implantation into the mouse chambers.
Implantation of prostate tissue and cancer cells
Anterior prostate tissue was excised from a normal C57/BL6 mouse, minced into small pieces (< 1 mm2), and implanted into a chamber. A small indentation was made in the center of the prostate tissue, in which a pre-formed tumor spheroid was placed. The prostate tissue and tumor spheroid were allowed to re-vascularize prior to experimentation.
Surgical Castration
Mice were anesthetized with 7.3 mg ketamine hydrochloride and 2.3 mg xylazine /100 g body weight, i.p. A lateral incision across the scrotum was made and the testes were individually ligated and excised. The wound was cauterized. The incision was then sutured and sealed with Nexaband® acrylic.
Chemical Castration
The mice were chemically castrated through oral administration of Cyproterone acetate twice daily (0.5mg/kg) and injection of Leuprolide acetate daily (0.07mg/kg).
Celecoxib Treatment
Celecoxib was administered orally twice daily (15mg/kg/administration).
Intravital microscopy
Fluorescence microscopy, image analysis, measurement of tumor growth and vascular parameters, calculation of mitotic and apoptotic indices have been carefully detailed in our previous study (16).
RESULTS
TRAMP-C2 cells were derived from the prostate tumor of a TRAMP mouse and were shown previously to have lost the viral SV40-T antigen and to be tumorigenic in vivo (17). The GFP-tagged cells (TRAMP-C2-GFP) display the same growth characteristics as parental cells and are androgen-dependent in vivo (16), and in vitro (Supplemental figure 1).
We first tested the sensitivity of TRAMP-C2-GFP cells to celecoxib in vitro. As shown in figure 1, a concentration of 20μM reduced cell growth by more than 50%, whereas 50μM completely inhibited cell growth. It should be noted that these concentrations are within the physiological range. Indeed, average plasma levels achieved by the administration of 800 mg Celecoxib (FDA-approved dose for the treatment of familial adenomatous polyposis) are 8 μM within 24 hrs of administration and can reach up to 40 μM, whereas even higher peak levels are reached 3 hrs post-administration (20-22).
Figure 1. Effect of celecoxib on the growth of prostate cancer cells in vitro.

Mouse TRAMP-C2 cells stably transfected with H2B-GFP (top panel), as well as human prostate cancer cells PC3 (middle panel) and DU145 (bottom panel), were treated with increasing concentrations of celecoxib for 48 hrs. Cells were counted using a Cell Coulter Multisizer II as described in Methods. Results are expressed relative to untreated cells and are means ± SEM of 3 separate experiments, each done in biological duplicates.
Relevance to human cancer was assessed by measuring the growth of human prostate cancer cells following treatment with celecoxib in similar conditions (Figure 1). A concentration of 40μM was needed to reduce the growth of DU145 and PC3 cells by 50%. Thus, aggressive human prostate cancer cells are also sensitive to celecoxib-induced toxicity, as previously shown by others (8,23-25).
The morphology of celecoxib-treated TRAMP-C2-GFP cells was assessed by fluorescent and bright-field microscopy (figure 2A). Celecoxib at 10 μM did not alter the morphology of most cells, although a few cells were observed that contained enlarged nuclei. Some dead cells were observed (thin arrow). The number of cells in mitosis (thick arrows) was higher compared to the control condition. At 20 μM, celecoxib induced a dramatic change in cell morphology. Most cells had become flat, large cells with enlarged and abnormal nuclei. At 50 μM, most cells were visibly dying. These observations are consistent with the hypothesis that celecoxib induces growth arrest of TRAMP-C2-GFP cells by impairing mitosis, which is eventually followed by mitotic catastrophe and cell death.
Figure 2. Effect of celecoxib in TRAMP-C2-GFP prostate cancer cells.
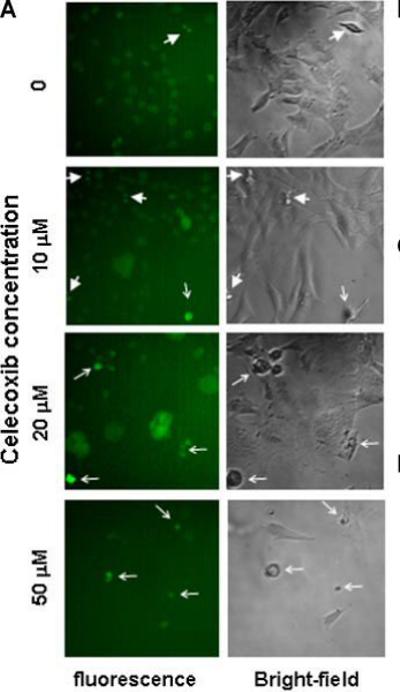

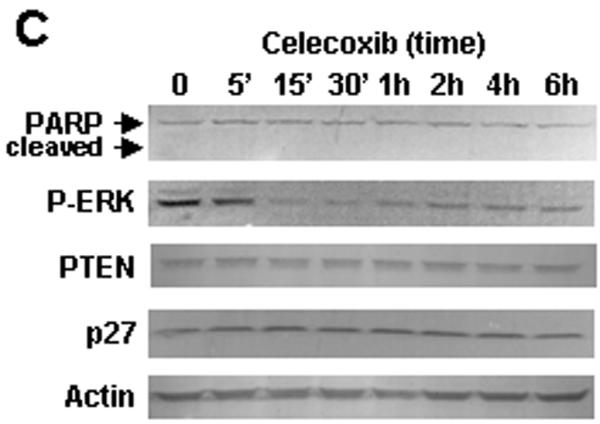

Panel A: Bright field microscopy (right) and fluorescence microscopy (left) of TRAMP-C2-GFP cells treated with increasing doses of Celecoxib for 48 hrs. Thick arrows point to mitotic cells; thin arrows point to dead cells. Panel B: Cells were treated with the indicated concentrations of celecoxib for 24 hrs. Cells were detached using trypsin, fixed, and co-stained for phospho-H3 (alexa-fluor 488) and DNA content (propidium iodide). The graph shows the proportion of cells in mitosis as compared to control, determined by flow cytometry on Facscan (BD Biosciences). Panel C: TRAMP-C2-GFP cells were treated with 40 μM celecoxib for the indicated times. Cells were lysed and protein expression was analyzed by western blot. Blot membranes were stripped and reprobed using the indicated antibodies. P-ERK: phosphorylation-specific antibodies to ERK. Panel D: TRAMP-C2-GFP cells were incubated in medium with or without androgen and treated with increasing concentrations of celecoxib for 48 hrs before cell counting. Results are expressed relative to untreated cells grown in medium containing androgen, and are means ± SEM of 3 separate experiments, each done in biological triplicate.
The effect of celecoxib on mitosis was further validated by flow cytometry analysis. Thus, cells treated with celecoxib were fixed and incubated with antibodies to phospho-histone H3. Phosphorylation of histone H3 on serine 10 is restricted to mitosis and therefore allows to specifically stain mitotic cells. DNA content was measured in parallel by PI staining. The flow cytometry profiles of control (untreated) and cells treated with two doses of celecoxib are shown in Supplemental Figure S2. Celecoxib increased the proportion of mitotic cells by 4-fold, an effect that was maximum at the lowest concentration tested (figure 2B).
A time-course of celecoxib was performed in TRAMP-C2-GFP cells, and the expression or phosphorylation levels of several proteins were examined by Western-Blot (figure 2C). At these early times of treatment (up to 6 hrs), celecoxib had no effect on PARP integrity, and did not visibly alter the expression of p27Kip1. No change in the phosphorylation of Akt was seen, although the signal was very weak (data not shown). Low levels of constitutive Akt phosphorylation are not unexpected, since there is no know alteration of the PTEN/PI3-kinase pathway in these cells. However, the cells do exhibit constitutive ERK phosphorylation (figure 2C), which was inhibited by celecoxib within 15 min of treatment.
We next examined the hypothesis that hormonal ablation, which is the standard of treatment for androgen-dependent prostate cancer, may be more efficacious when combined with celecoxib. As shown in figure 2D, androgen deprivation alone decreased cell proliferation by 56±11%, whereas the combination of androgen deprivation and 20 μM celecoxib inhibited cell proliferation by 88±6%. We conclude that in vitro, the combination treatment was more efficient than either treatment alone, although the combined effect was less than additive.
The mouse dorsal chamber was used to evaluate the effect of this combination therapy on tumor growth in vivo. TRAMP-C2-GFP cell spheroids were co-implanted in the dorsal chambers of mice with prostate tissue obtained from a donor mouse. The implanted prostate tissue and tumor cells were allowed to vascularize for two weeks. Once the prostate tissue and the tumor were established, surgical castration was used to induce androgen deprivation (considered day 0 of treatment). Four treatment groups were studied: control untreated mice, castrated mice, celecoxib 15 mg/kg twice daily, celecoxib 15 mg/kg twice daily combined with castration. Figure 3A illustrates the effects of celecoxib and castration on tumor growth in our pseudo-orthotropic model, whereas figures 3B-C depict the quantification of tumor growth parameters measured by fluorescent intravital microscopy as described in (16). The growth of tumors was apparent between day 14 and day 21 in the control mice, with a 4-fold increase in both tumor area and relative tumor intensity at day 21. Celecoxib caused a significant slowing of tumor growth, since only a 2-fold increase in both tumor area and tumor intensity was observed after 21 days. In agreement with our previous report (16), castration completely prevented the growth of TRAMP-C2-GFP tumors. However, none of these treatments alone resulted in the regression of established tumors. In contrast, the combination of surgical castration with celecoxib caused a 3 to 4-fold tumor regression. Indeed, a decrease of 80% in tumor area (figure 3B), and a decrease of 65% in tumor intensity (figure 3C) were observed compared to untreated mice.
Figure 3. Effect of celecoxib and/or surgical castration on prostate tumor growth in vivo.



TRAMP-C2-GFP cell spheroids were co-implanted with prostate tissue and allowed to vascularize. When there was proper blood flow within the growing tumors, the mice were surgically castrated (Day 0) and Celecoxib treatment (15 mg/kg/administration) was started by oral administration twice daily. Tumors were imaged by intravital microscopy once a week. Panel A: a representative collage of tumor growth in the four treatment groups. Bar ~ 500μm. Panel B-C: graphic representation of relative tumor areas (B) and relative tumor intensities (C) calculated from intravital microscopy data (log scale).
The apoptotic and mitotic index of each tumor in this experiment were measured using a higher microscopy magnification (shown in figure 4). The initial rate of apoptosis within the tumors decreased 5-fold in the control mice, indicative of a high cell survival rate when the implanted cells start growing into a tumor. In contrast, the rate of apoptosis remained constant, or was somewhat increased, within the tumors of the treated mice.
Figure 4. Mitotic and apoptotic index.


Graphic representation of the mean apoptotic index (Panel A) and the mean mitotic index (Panel B) within the tumors, calculated from intravital microscopy data. The animal experiments are the same as described in figure 2.
On the other hand, the mitotic index was stable in the growing tumors of control mice, indicating that the ratio of cells undergoing mitosis remained constant within the cell population. Treatment with celecoxib alone increased the mitotic index 4-fold, whereas celecoxib combined with castration caused a 5-fold increase in the intratumoral mitotic index, suggesting that many cells arrested in mitosis (figure 4B). Close examination of tumor cells nuclei over time (using H2B-GFP fluorescence) allowed us to visualize the onset of mitosis (figure 5, panels A-B). Two days later we observed that cell division failed (panel C) and the nuclei eventually became pycnotic (panel D). These observations suggest that celecoxib induced mitotic arrest, leading to mitotic failure and apoptosis.
Figure 5. Intravital microscopy at high magnification of celecoxib-treated TRAMP-C2-GFP tumors.

Tumors from celecoxib-treated mice (shown in figure 2) were imaged by intravital microscopy at high magnification. Panels A and B: H2B-GFP fluorescence of TRAMP-C2-GFP tumors showing the onset of mitosis at day 3. Bar ~25μ (A); ~10μ (B). Panels C and D: failed mitosis (C) with the nuclei becoming pycnotic at day 5 (D). Bar ~25μ (C, D).
It had been reported previously that COX-2 inhibitors alter tumor growth in part through an anti-angiogenic activity (26). Surprisingly, the vascular parameters measured from our experiments showed no difference between control and celecoxib-treated mice, suggesting that celecoxib did not possess angiostatic activity in this mouse model (figure 6). The combination of celecoxib and castration decreased both the mean vascular area and the density of the vasculature.
Figure 6. Effect of celecoxib treatment on intra-tumoral angiogenesis.

Tumors were imaged by intravital microscopy and vascular parameters were calculated. Panel A: Graphic representation of vascular parameters (area, diameter and density) for control, celecoxib-treated and celecoxib + castrated animals.
In prostate cancer patients, androgen withdrawal is achieved chemically. To mimic the clinical setting, chemical castration was combined with COX-2 inhibition. TRAMP-C2-GFP cell spheroids were implanted with orthotropic prostate stroma in the dorsal chambers of mice. All the mice were treated with cyproterone acetate and leuprolide to induce androgen deprivation. One group of chemically castrated mice was treated with celecoxib 15 mg/kg (twice daily) whereas the other group received sham treatment (figure 7). As can be seen from comparing figures 3 and 7, chemical castration and surgical castration had very similar effects on tumor growth. Combining celecoxib treatment with chemical castration caused tumor regression, similarly to the combination of celecoxib and surgical castration. The rate of mitosis increased significantly in tumors of mice treated with combination therapy compared to androgen deprivation alone (figure 7E). Our experiments demonstrate that surgical and chemical castration have similar effect on tumor regression when combined with celecoxib treatment.
Figure 7. Effect of combining celecoxib and hormonal ablation by chemical castration on the growth of tumors.





TRAMP-C2-GFP spheroids were co-implanted with prostrate tissue and allowed to vascularize. When there was proper flow within the tumors, mice were chemically castrated by oral administration of cyproterone acetate twice daily and injection of leuprolide acetate daily, starting at Day 0. Celecoxib was administered orally twice daily, also starting at Day 0. Panel A: Representative collage of tumor growth. Bar ~ 500μm. Panels B-C: Graphic representation of relative tumor area and intensity calculated from intravital microscopy data. Panels D-E: Apoptotic and Mitotic Index.
In this model of the clinical condition, we observed a deep regression of angiogenesis within only 2 days, as shown in figure 8. Androgen deprivation combined with celecoxib caused the vasculature to shrink, as measured by the vascular area and the vascular diameter (panel B).
Figure 8. Effect of hormonal ablation by chemical castration on intra-tumoral angiogenesis.


Tumors from the mice treated with the combination therapy as described in figure 6, were imaged by intravital microscopy and vascular parameters were calculated. Panel A: Phase contrast representative images of tumor vasculature at day 0 and day 2 post-castration. Bar~ A,B 500μm, C,D 50μm. Panel B: Graphic representation of the vascular area and the mean diameter of tumor vasculature calculated from intravital microscopy data.
In conclusion, celecoxib alone decreased tumor growth by causing cell cycle arrest and mitotic failure. It had no measurable effect on vascular parameters in our model. Castration, which directly inhibited the proliferation of prostate cancer cells in vitro (Suppl figure 1), blocked tumor growth in vivo but did not result in regression. The combination of celecoxib and androgen withdrawal, however, resulted in tumor regression and was associated with rapid shrinkage of the vasculature.
DISCUSSION
Animal models are crucial to our understanding of the mechanisms underlying tumor progression and growth. Current rodent models such as xenograft human tumors in immunodeficient mice do not sufficiently represent relevant clinical cancer models, especially with regard to angiogenesis and drug sensitivity. Transgenic animals, on the other hand, do not permit the direct measurement of tumor growth, and time-dependent observations can be made only by inference after killing mice at various time points. The dorsal skinfold chamber allows repeated observations in the same animal over extended time periods. Thus, evaluation of vascular responses to treatment can be done in real-time. The use of TRAMP-C2 cells transfected with H2B-GFP also allows us to measure increases or decreases in tumor growth and to assess other underlying mechanisms (mitosis or apoptosis) that influence tumor progression.
We have used this model to examine the effect of COX-2 inhibitor celecoxib in combination with androgen withdrawal for the treatment of prostate cancer. Surgical castration combined with celecoxib caused tumor regression, which was not observed with castration or celecoxib alone.
These results are in line with the recent finding that a combination of celecoxib and androgen withdrawal delayed the acquisition of androgen-independence in a xenograft model using human LNCaP cells (8), which suggests that these observations may be relevant to human disease.
Observation of various tumor parameters indicated that regression was caused by a combination of decreased vascularization due to androgen withdrawal, together with tumor cells growth arrest due mostly to celecoxib treatment. Thus, the efficacy of the combination was much better in vivo than in vitro, because of the separate effects of each treatment on distinct biological compartments (vasculature, stroma, possibly inflammatory cells) that are not represented in the culture of cell lines.
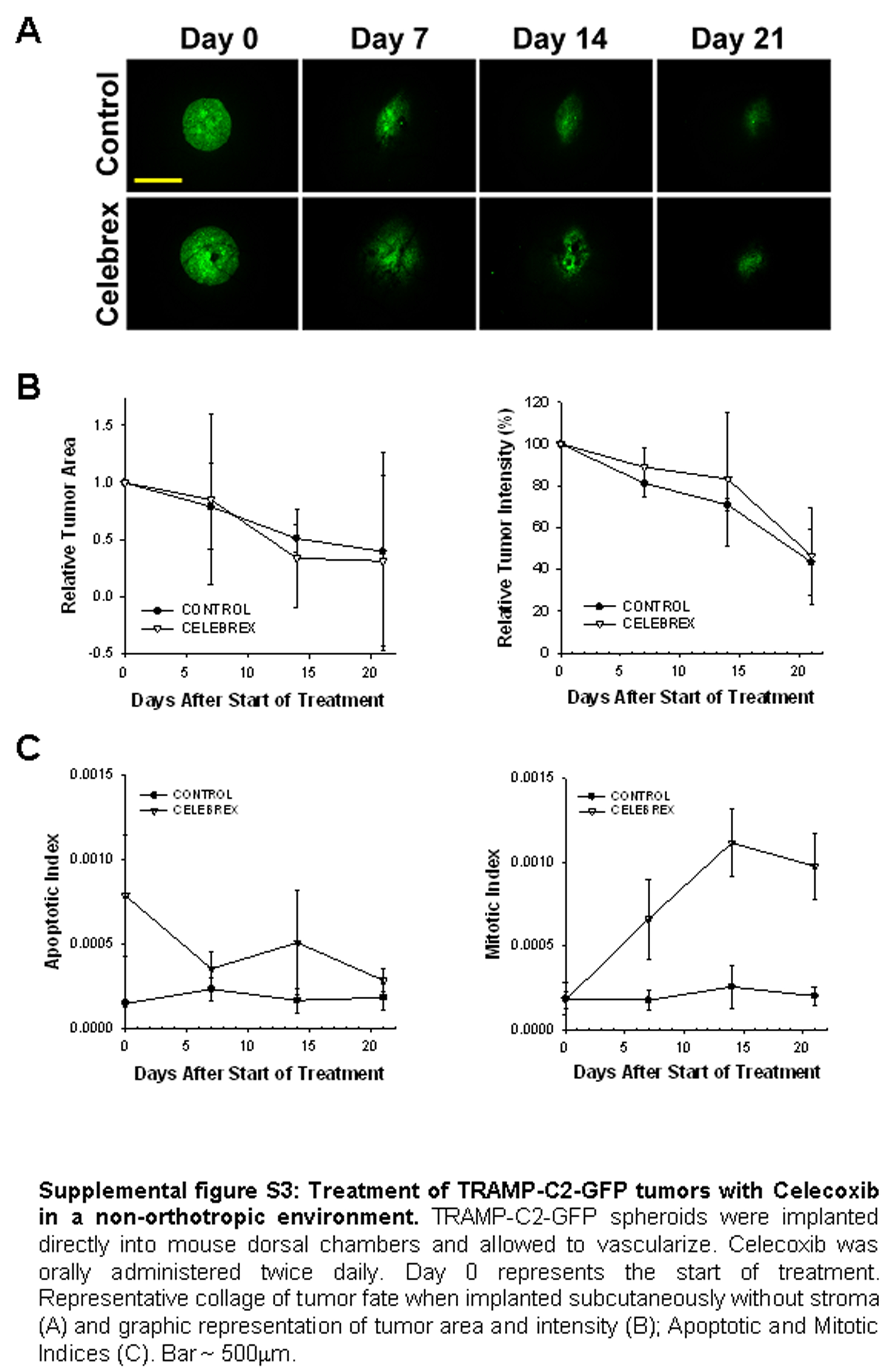
A critical aspect in understanding and treating cancer progression is the relationship between the tumor and the “soil” that supports its growth and progression. It is well known that stromal-epithelial interactions are very important for androgen dependent prostate cancer (27). Thus, co-implanting TRAMP-C2 cells with prostate stroma obtained from a donor mouse provides the tumor cells with an environment which closely resembles orthotropic implantation. When cancer cells were implanted in the chambers in the absence of prostate tissue, they did not grow into tumors, and treatment with celecoxib did not alter tumor parameters (supplemental figure 3), exemplifying the importance of the orthotropic milieu in assessing treatment parameters.
The in vivo mechanism by which COX-2 affects tumor growth is still not completely understood. It has been suggested that COX-2 inhibitors induce cell cycle arrest and apoptosis in cancer cells through a mechanism that is fundamentally different from the apoptosis caused by cancer chemotherapeutic agents. Our results showing that celecoxib caused growth arrest and mitotic failure, characterized by deep alterations of nuclei morphology, and followed by mitotic catastrophe and cell death, are in agreement with this hypothesis.
Celecoxib did not alter protein levels of p53 or PARP, which are involved at early stages of apoptosis. Although in some cell lines celecoxib inhibits the PI3-kinase pathway and decreases the phosphorylation of Akt (28), we did not detect changes in phospho-Akt. However, the PI3-kinase pathway is not constitutively active in the TRAMP-C2 cells, therefore levels of phospho-Akt are barely detectable and probably not amenable to celecoxib alteration. In contrast, ERK phosphorylation was constitutively high and was inhibited by celecoxib, a result consistent with previous observations (reviewed in (29)).
We conclude that COX-2 selective inhibitor celecoxib, through COX-2 dependent and independent mechanisms, significantly increases the efficacy of androgen withdrawal in a mouse model of prostate cancer. This combination warrants further investigation as a complement therapy for aggressive prostate cancer.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We thank Dale Winger for his excellent assistance in the preparation of the dorsal skinfold chambers and Reeti Bandyopadhyay for technical assistance. We also thank Dr. David Chambers (Salk Insitute, San Diego) for his help with Flow Cytometry.
This work was supported by funding from Pfizer, contract COXAV-V0190-010 (Dr. Per Borgström); NIH/NCI grant RO1-CA102688 (Dr. Véronique Baron); NIH/NCI grant R21CA133638 (Dr. John Welsh); DOD-CDMRP grant W81XWH-09-1-0280 and UC/CBCRP 151B-0133 (Dr. Per Borgström and Dr. John Welsh); Fellowship CTS07:1 from Carl Tryggers Stiftelse for Scientific Research, Stockholm (Dr. Parisa Abedinpour).
REFERENCES
- 1.Stock D, Groome PA, Siemens DR. Inflammation and prostate cancer: a future target for prevention and therapy? The Urologic clinics of North America. 2008;35(1):117–130. vii. doi: 10.1016/j.ucl.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Furstenberger G, Krieg P, Muller-Decker K, Habenicht AJ. What are cyclooxygenases and lipoxygenases doing in the driver’s seat of carcinogenesis? International journal of cancer. 2006;119(10):2247–2254. doi: 10.1002/ijc.22153. [DOI] [PubMed] [Google Scholar]
- 3.Harris RE. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology. 2009;17(2):55–67. doi: 10.1007/s10787-009-8049-8. [DOI] [PubMed] [Google Scholar]
- 4.Sahin M, Sahin E, Gumuslu S. Cyclooxygenase-2 in cancer and angiogenesis. Angiology. 2009;60(2):242–253. doi: 10.1177/0003319708318378. [DOI] [PubMed] [Google Scholar]
- 5.Pruthi RS, Wallen EM. Cyclooxygenase-2: a therapeutic target for prostate cancer. Clinical genitourinary cancer. 2005;4(3):203–211. doi: 10.3816/CGC.2005.n.034. [DOI] [PubMed] [Google Scholar]
- 6.Sooriakumaran P, Langley SE, Laing RW, Coley HM. COX-2 inhibition: a possible role in the management of prostate cancer? Journal of chemotherapy (Florence, Italy) 2007;19(1):21–32. doi: 10.1179/joc.2007.19.1.21. [DOI] [PubMed] [Google Scholar]
- 7.Zheng X, Cui XX, Avila GE, Huang MT, Liu Y, Patel J, Kong AN, Paulino R, Shih WJ, Lin Y, Rabson AB, Reddy BS, Conney AH. Atorvastatin and celecoxib inhibit prostate PC-3 tumors in immunodeficient mice. Clin Cancer Res. 2007;13(18 Pt 1):5480–5487. doi: 10.1158/1078-0432.CCR-07-0242. [DOI] [PubMed] [Google Scholar]
- 8.Zheng X, Cui XX, Gao Z, Zhao Y, Lin Y, Shih WJ, Huang MT, Liu Y, Rabson A, Reddy B, Yang CS, Conney AH. Atorvastatin and celecoxib in combination inhibits the progression of androgen-dependent LNCaP xenograft prostate tumors to androgen independence. Cancer prevention research (Philadelphia, Pa. 2010;3(1):114–124. doi: 10.1158/1940-6207.CAPR-09-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antonarakis ES, Heath EI, Walczak JR, Nelson WG, Fedor H, De Marzo AM, Zahurak ML, Piantadosi S, Dannenberg AJ, Gurganus RT, Baker SD, Parnes HL, DeWeese TL, Partin AW, Carducci MA. Phase II, randomized, placebo-controlled trial of neoadjuvant celecoxib in men with clinically localized prostate cancer: evaluation of drug-specific biomarkers. J Clin Oncol. 2009;27(30):4986–4993. doi: 10.1200/JCO.2009.21.9410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sooriakumaran P, Coley HM, Fox SB, Macanas-Pirard P, Lovell DP, Henderson A, Eden CG, Miller PD, Langley SE, Laing RW. A randomized controlled trial investigating the effects of celecoxib in patients with localized prostate cancer. Anticancer Res. 2009;29(5):1483–1488. [PubMed] [Google Scholar]
- 11.Carles J, Font A, Mellado B, Domenech M, Gallardo E, Gonzalez-Larriba JL, Catalan G, Alfaro J, Del Alba A Gonzalez, Nogue M, Lianes P, Tello JM. Weekly administration of docetaxel in combination with estramustine and celecoxib in patients with advanced hormone-refractory prostate cancer: final results from a phase II study. Br J Cancer. 2007;97(9):1206–1210. doi: 10.1038/sj.bjc.6604030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontana A, Galli L, Fioravanti A, Orlandi P, Galli C, Landi L, Bursi S, Allegrini G, Fontana E, Di Marsico R, Antonuzzo A, D’Arcangelo M, Danesi R, Del Tacca M, Falcone A, Bocci G. Clinical and pharmacodynamic evaluation of metronomic cyclophosphamide, celecoxib, and dexamethasone in advanced hormone-refractory prostate cancer. Clin Cancer Res. 2009;15(15):4954–4962. doi: 10.1158/1078-0432.CCR-08-3317. [DOI] [PubMed] [Google Scholar]
- 13.James ND, Sydes MR, Clarke NW, Mason MD, Dearnaley DP, Anderson J, Popert RJ, Sanders K, Morgan RC, Stansfeld J, Dwyer J, Masters J, Parmar MK. Systemic therapy for advancing or metastatic prostate cancer (STAMPEDE): a multi-arm, multistage randomized controlled trial. BJU international. 2009;103(4):464–469. doi: 10.1111/j.1464-410X.2008.08034.x. [DOI] [PubMed] [Google Scholar]
- 14.Pruthi RS, Derksen JE, Moore D, Carson CC, Grigson G, Watkins C, Wallen E. Phase II trial of celecoxib in prostate-specific antigen recurrent prostate cancer after definitive radiation therapy or radical prostatectomy. Clin Cancer Res. 2006;12(7 Pt 1):2172–2177. doi: 10.1158/1078-0432.CCR-05-2067. [DOI] [PubMed] [Google Scholar]
- 15.Smith MR, Manola J, Kaufman DS, Oh WK, Bubley GJ, Kantoff PW. Celecoxib versus placebo for men with prostate cancer and a rising serum prostate-specific antigen after radical prostatectomy and/or radiation therapy. J Clin Oncol. 2006;24(18):2723–2728. doi: 10.1200/JCO.2005.03.7804. [DOI] [PubMed] [Google Scholar]
- 16.Frost GI, Lustgarten J, Dudouet B, Nyberg L, Hartley-Asp B, Borgstrom P. Novel syngeneic pseudo-orthotopic prostate cancer model: vascular, mitotic and apoptotic responses to castration. Microvascular research. 2005;69(1-2):1–9. doi: 10.1016/j.mvr.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer research. 1997;57(16):3325–3330. [PubMed] [Google Scholar]
- 18.Kanda T, Sullivan KF, Wahl GM. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr Biol. 1998;8(7):377–385. doi: 10.1016/s0960-9822(98)70156-3. [DOI] [PubMed] [Google Scholar]
- 19.Frost GI, Borgstrom P. Real time in vivo quantitation of tumor angiogenesis. Methods in molecular medicine. 2003;85:65–78. doi: 10.1385/1-59259-380-1:65. [DOI] [PubMed] [Google Scholar]
- 20.Brautigam L, Vetter G, Tegeder I, Heinkele G, Geisslinger G. Determination of celecoxib in human plasma and rat microdialysis samples by liquid chromatography tandem mass spectrometry. Journal of chromatography. 2001;761(2):203–212. doi: 10.1016/s0378-4347(01)00333-4. [DOI] [PubMed] [Google Scholar]
- 21.Chow HH, Anavy N, Salazar D, Frank DH, Alberts DS. Determination of celecoxib in human plasma using solid-phase extraction and high-performance liquid chromatography. Journal of pharmaceutical and biomedical analysis. 2004;34(1):167–174. doi: 10.1016/j.japna.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 22.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999;96(1):272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu W, Tinsley HN, Keeton A, Qu Z, Piazza GA, Li Y. Suppression of Wnt/beta-catenin signaling inhibits prostate cancer cell proliferation. European journal of pharmacology. 2009;602(1):8–14. doi: 10.1016/j.ejphar.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narayanan NK, Narayanan BA, Reddy BS. A combination of docosahexaenoic acid and celecoxib prevents prostate cancer cell growth in vitro and is associated with modulation of nuclear factor-kappaB, and steroid hormone receptors. Int J Oncol. 2005;26(3):785–792. [PubMed] [Google Scholar]
- 25.Patel MI, Subbaramaiah K, Du B, Chang M, Yang P, Newman RA, Cordon-Cardo C, Thaler HT, Dannenberg AJ. Celecoxib inhibits prostate cancer growth: evidence of a cyclooxygenase-2-independent mechanism. Clin Cancer Res. 2005;11(5):1999–2007. doi: 10.1158/1078-0432.CCR-04-1877. [DOI] [PubMed] [Google Scholar]
- 26.Masferrer JL, Leahy KM, Koki AT, Zweifel BS, Settle SL, Woerner BM, Edwards DA, Flickinger AG, Moore RJ, Seibert K. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer research. 2000;60(5):1306–1311. [PubMed] [Google Scholar]
- 27.Cunha GR, Hayward SW, Wang YZ, Ricke WA. Role of the stromal microenvironment in carcinogenesis of the prostate. International journal of cancer. 2003;107(1):1–10. doi: 10.1002/ijc.11335. [DOI] [PubMed] [Google Scholar]
- 28.Kulp SK, Yang YT, Hung CC, Chen KF, Lai JP, Tseng PH, Fowble JW, Ward PJ, Chen CS. 3-phosphoinositide-dependent protein kinase-1/Akt signaling represents a major cyclooxygenase-2-independent target for celecoxib in prostate cancer cells. Cancer research. 2004;64(4):1444–1451. doi: 10.1158/0008-5472.can-03-2396. [DOI] [PubMed] [Google Scholar]
- 29.Grosch S, Maier TJ, Schiffmann S, Geisslinger G. Cyclooxygenase-2 (COX-2)-independent anticarcinogenic effects of selective COX-2 inhibitors. Journal of the National Cancer Institute. 2006;98(11):736–747. doi: 10.1093/jnci/djj206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.