

Abstract
Objective
Heme oxygenase-1 induction (HO-1) elicits chronic weight loss in several rodent models of obesity. Despite these findings, the mechanism by which HO-1 induction reduces body weight is unclear. Chronic HO-1 induction does not alter food intake suggesting other mechanisms such as increases in metabolism and activity may be responsible for the observed reduction of body weight. In this study, we investigated the mechanism of weight loss elicited by chronic HO-1 induction in a model of genetic obesity due to melanocortin-4 receptor (MC4R) deficiency.
Design
Experiments were performed on loxTB MC4R deficient mice as well as lean controls. Mice were administered cobalt protoporphyrin (CoPP, 5 mg/kg), an inducer of HO-1, once weekly from 4 to 23 weeks of age. Body weights were measured weekly and fasted blood glucose and insulin as well as food intake were determined at 18 weeks of age. O2 consumption, CO2 production, activity, and body heat production were measured at 20 weeks of age.
Results
Chronic CoPP treatment resulted in a significant decrease in body weight from 5 weeks on in loxTB mice. Chronic CoPP treatment resulted in a significant decrease in fasted blood glucose levels, plasma insulin, and a significant increase in plasma adiponectin levels in MC4R deficient mice. Chronic CoPP treatment increased O2 consumption (47 ± 4 vs. 38 ± 3 ml/kg/min, P<0.05) and CO2 production (44 ± 7 vs. 34 ± 4 ml/kg/min, P<0.05) in treated versus non-treated, MC4R deficient mice (n=4). Heat production (10%) and activity (18%) were also significantly (P<0.05) increased in CoPP treated MC4R deficient mice.
Conclusion
Our results suggest that chronic HO-1 induction with CoPP induction elicits weight loss by increasing metabolism and activity by an MC4R independent pathway.
Keywords: Heme oxygenase, adiponectin, metabolism, gene knockout, diabetes, leptin
Introduction
Cobalt protoporphyrin (CoPP), an inducer of heme oxygenase (HO)-1, has been reported to elicit chronic weight loss in a variety of species including dogs, chickens, rats, and mice (1). This effect of HO induction on body weight has been replicated with other systemic inducers of HO-1 such as D-4F which is an apolipoprotein A1 mimetic peptide (2;3). While the affects of chronic HO induction on body weight have been established, the mechanism leading to weight loss has not been determined. Previous studies have demonstrated that direct injection of CoPP into either the lateral ventricles or directly into the medial nuclei of the hypothalamus results in acute decreases in food intake but chronic decreases in body weight (4;5). Central CoPP administration has also been associated with decreases in the acute feeding response to neuropeptide Y (NPY) as well as in neuronal nitric oxide synthase (nNOS) activity but increases nNOS message and protein (6-8). Despite exerting only acute effects on appetite, the effects of CoPP on body weight are persistent suggesting a mechanism independent of caloric intake mediates the chronic weight loss elicited by CoPP treatment.
Leptin, a hormone released from adipocytes, regulates appetite by stimulating the release of α-melanocyte-stimulating hormone (α-MSH) from pro-opiomelanocortin (POMC) neurons of the hypothalamus which in turn binds to melanocortin-3 and 4 receptors (MC3/4R) to mediate the metabolic and cardiovascular effects of leptin (9). Two mouse models of MC4R deficiency have been developed to examine the mechanism by which loss of MC4R receptors leads to obesity. The first model is a traditional gene-targeted deletion of the endogenous MC4R receptor (10). In these mice, most of the MC4R receptor coding region which is contained in a single exon has been eliminated. In the second model of MC4R deficiency, a transcriptional blocker flanked by loxP sites was placed between the transcription start site and the initiation ATG of the MC4R gene which causes disruption of the transcription of the gene resulting in MC4R deficiency (loxTB mice) (11). In loxTB mice, function of the endogenous MC4R can be “rescued” by selective expression of the Cre recombinase which deletes the transcription blocker and restores endogenous expression of the MC4R gene. Both models develop obesity, hyperphagia, hyperglycemia, and hyperinsulinemia resulting from disruption of the MC4R gene (10;11). Loss of the MC4R is associated with decreases in oxygen consumption (VO2) suggesting that a metabolic defect may also responsible for the development of obesity in both of these models (11;12).
Mutations in MC4Rs are the most prevalent cause of genetic obesity in humans (13-15). These findings underscore the important role for MC4R activation in the regulation of body weight and metabolism. Obese loxTB MC4R deficient mice were utilized to specifically examine the importance of the central MC4R system in mediating weight loss elicited by chronic HO-1 induction. We also directly measured VO2, carbon dioxide production (VCO2), locomotor activity and heat production to determine the role of metabolism in the chronic weight loss elicited by HO-1 induction. Mice were treated weekly with CoPP for 19 weeks and then housed in specific metabolic cages for the measurement of oxygen consumption (VO2), carbon dioxide production (VCO2), locomotor activity and heat production.
Methods
Mice
Experiments were performed on male C57BL/6J (Jackson Labs, Bar Harbor, ME, USA), loxTB MC4R deficient mice which were obtained from initial stocks kindly provided by Dr. Bradford B. Lowell, Harvard Medical School and MC4R gene knockout (KO) mice which were obtained from initial stocks kindly provided by Dr. Roger D. Cone, Vanderbilt University (10;11). LoxTB MC4R deficient mice were bred at the University of Mississippi Medical Center and maintained on a mixed C57BL/6 and 129Sv background. MC4R KO mice were also bred at the University of Mississippi Medical Center and maintained on a C57BL/6 genetic background. Mice were treated with once weekly injections of CoPP (5 mg/kg, s.c., Frontier Scientific, Logan, UT, USA) from 4 weeks to 23 weeks of age. The mice were fed a standard laboratory diet and were provided water ad libitum. All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Mississippi Medical Center and performed in accordance with the Guide for the Care and Use of Laboratory Animals of the U.S. National Institutes of Health. Mice were housed together during the experimental study and were weighed weekly for measurement of body weight. Food intake was determined in individually housed mice at 18 weeks of age by daily weighing of food provided to the mice over a 5 day period. O2 consumption, CO2 production, activity, and body heat production were measured at 20 weeks of age in mice placed in specially designed cages as described below. Mice were housed in rooms with an ambient temperature of 74°F with 12 hours of light (6AM-6PM) and 13 hours of dark (6PM-6AM). Mice were euthanized at 30 weeks at which time organs were collected and weighed.
Plasma Measurements
Blood was obtained from mice at 19 weeks of age by retro-orbital bleed under light isoflurane anesthesia. All mice were fasted overnight prior to blood sample collections. Whole blood samples were collected into EDTA containing tubes and centrifuged at 3,000 g for 5 min at 4°C. Plasma was then collected and stored at -20°C until use. Blood glucose concentration was immediately determined from a 5 ul blood sample with the Accuchek Advantage glucometer (Roche, Madison, WI, USA). Plasma insulin levels were measured by ELISA with the Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem, Inc., Downers Grove, IL, USA). Plasma adiponectin levels were also measured by ELISA with the Mouse Adiponectin ELSIA Kit (Millipore, Billerica, MA, USA).
Oxygen consumption, carbon dioxide & heat production, respiratory quotient motor activity
At 20 weeks of age, mice were placed individually in metabolic cages (AccuScan Instruments Inc, Columbus, OH) equipped with oxygen sensors to measure oxygen consumption (VO2), carbon dioxide sensors to measure carbon dioxide production (VCO2), and infrared beams to determine motor activity. VO2 was measured for 2-min at 10-minute intervals continuously 24-hours a day using a Zirconia oxygen sensor. Respiratory quotient (RQ) was automatically calculated by the software and expressed as VCO2/VO2. Motor activity was determined using infrared light beams mounted in the cages in X, Y and Z axes. Heat production was derived from the measurement of VO2 and VCO2 which were used to compute the calorific value and then applied to the volume of gases exchanged to compute heat production. After the mice were acclimatized to the new environment for 3 days, VO2, VCO2, activity and heat production were recorded for 3 consecutive days. The values for each of these parameters were then averaged for each individual mouse.
Western Blots
Western Blots were performed on lysates prepared from adipose tissue, liver, and muscle. Samples of 30 μg of protein were boiled in Laemmli sample buffer (Bio-Rad, Hercules, CA) for 5 min, electrophoresed on 12 or 10% SDS-polyacrylamide gels, and blotted onto nitrocellulose membrane. Membranes were blocked with Odyssey blocking buffer (LI-COR, Lincoln, NE) for 2 hours at room temperature, then incubated with the following antibodies: mouse anti-HO-1 antibody (StressGen, Vancouver, Canada, 1:2000), rabbit anti-pAMPK (Cell Signaling Technology, Danvers, MA, 1:1000), rabbit anti-pAKT (Cell Signaling Technology, Danvers, MA, 1:1000), mouse anti-glucose transporter 4 (Glut-4) (Abcam Cambridge, MA, 1:2,500), mouse anti-GAPDH monoclonal antibody (Abcam, 1:20,000), rabbit or mouse anti-β-actin antibody (Abcam, 1:5,000) overnight at 4°C. The membranes were then incubated with Alexa 680 goat anti-mouse IgG (Invitrogen, Carlsbad, CA 1:5,000) and IRDye 800 goat anti-rabbit IgG (Rockland, Gilbertsville, PA, 1:5,000) or Alexa 680 donkey anti-rabbit IgG (Invitrogen, 1:5,000) or IRDye 800 donkey anti-mouse IgG (Rockland, 1:5,000) for 1 hour at room temperature. The membranes were then visualized using an Odyssey infrared imager (Li-COR, Lincoln, NE). Densitometric analysis was performed using Odyssey software (LI-COR). Protein levels are expressed as a ratio to the level of β-actin in adipose and liver and to the level of GAPDH in muscle to normalize for protein loading.
Statistical analyses
All data are presented as mean ± s.e.m. Differences between treatment groups were determined using unpaired T-Tests or one-way analysis of variance with a post hoc test (Dunnett’s). A P<0.05 was considered to be significant. All analyses were performed with SigmaStat (Systat software, Inc., Richmond, CA, USA).
Results
Effect of chronic CoPP treatment on body weight, food intake, and fat distribution
Chronic treatment with CoPP (5 mg/kg/wk) significantly attenuated body weight gain in loxTB mice while having no effect on body weight- gain in control C57BL/6J mice (Figure 1). The effect of chronic CoPP treatment on body weight was also confirmed in MC4R KO mice (Figure 1). CoPP treated MC4R KO mice exhibited body weights similar to control mice. LoxTB mice exhibited greater obesity from 5 weeks of treatment as compared to MC4R KO mice. CoPP treatment in loxTB significantly attenuated weight gain which resembled the pattern of weight gain displayed in MC4R KO mice (Figure 1). Food intake, at 18 weeks of age, was significantly increased in both models of MC4R deficiency as compared to control mice averaging 3.5 ± 0.2, vs. 5.0 ± 0.2, vs. 4.7 ± 0.3 grams/day in control, MC4R KO, and loxTB mice, respectively (P<0.05 vs. control). CoPP treatment had no effect on food intake in any of the groups studied with food intake averaging 3.7 ± 0.1, vs. 5.0 ± 0.3, vs. 4.7 ± 0.2 grams/day in control, MC4R KO, and loxTB mice, respectively. Chronic CoPP treatment had a significant effect on fat distribution in all groups studied. CoPP treatment significantly decreased epidydimal fat in control and MC4R KO mice but had no effect in loxTB mice while significantly decreasing visceral fat in MC4R KO and loxTB mice but not in control mice (Figure 2). CoPP treatment resulted in a significant decrease in total fat in all groups (Figure 2).
Figure 1.
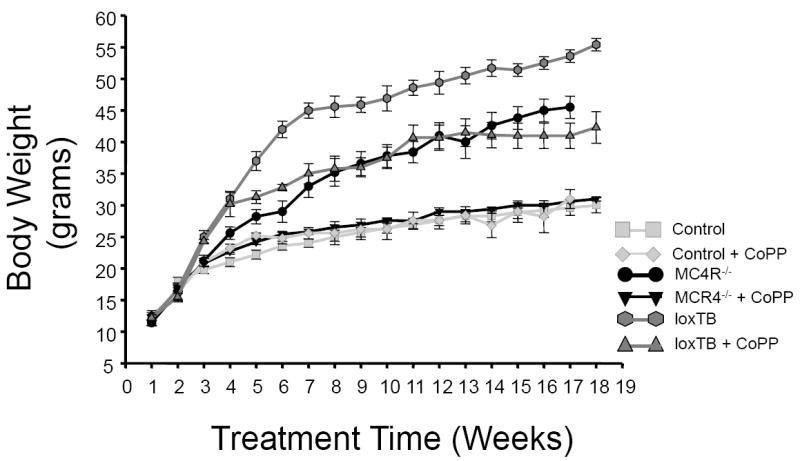
Effect of chronic CoPP treatment on body weight in control C57 mice, melanocortin-4 receptor knockout mice (MC4R-/-), and melanocortin 4 receptor transcriptional blocker (loxTB) mice. CoPP was administered once weekly. n=6/group.
Figure 2.
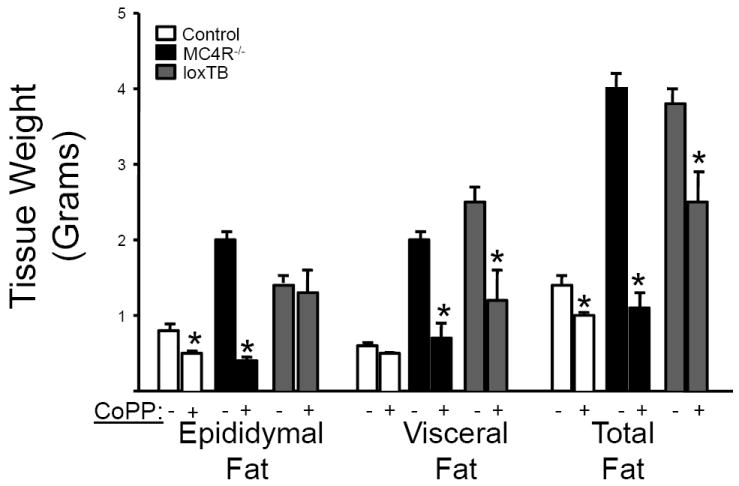
Effect of chronic CoPP treatment on distribution of fat in Control, MC4R-/-, and loxTB mice, n=6/group. Fat pads were weighted from 23 week old mice. *= p<0.05 as compared to corresponding value in non-treated.
Effect of chronic CoPP treatment on blood glucose, insulin and adiponectin levels
Fasting blood glucose levels were significantly increased in both loxTB and MC4R KO mice as compared to control mice. Chronic CoPP treatment equivalently lowered fasting blood glucose levels in both loxTB and MC4R KO mice but had no effect on fasting blood glucose levels in control mice (Figure 3A). Both models of MC4R deficiency exhibited increased fasting insulin levels as compared with control mice (Figure 3B). However, plasma insulin levels were significantly higher in loxTB mice as compared to MC4R KO mice. CoPP treatment significantly decreased fasting insulin levels in both models of MC4R deficiency. Plasma adiponectin levels in loxTB mice were significantly decreased as compared to control mice under basal conditions (Figure 3C). This finding was also confirmed in MC4R KO mice (Figure 3C). CoPP treatment significantly increased plasma adiponectin levels in all groups; however, the increase in the MC4R deficient mice was close to 4 fold as compared to a 70% increase exhibited in control mice (Figure 3C).
Figure 3.
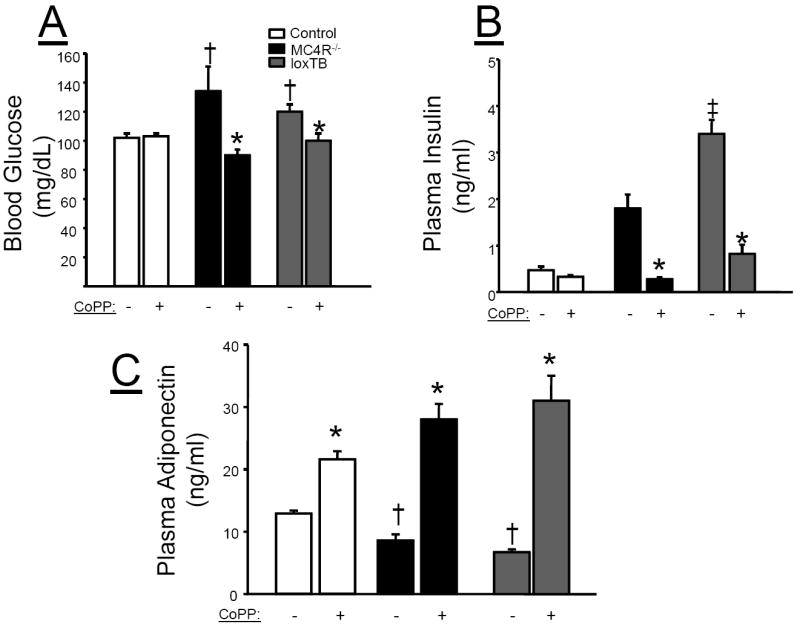
Plasma measurements in control, MC4R-/-, and loxTB mice. Blood samples were collected at 19 weeks of age as described in the Methods. A) Blood Glucose, B) Plasma Insulin, C) Plasma adiponectin. *=P<0.05 as compared to corresponding value in non-CoPP treated group. †=P<0.05 as compared to corresponding value in control mice. ‡ =P<0.05 as compared to corresponding value in MC4R-/- mice. n=6/group
Effect of chronic CoPP treatment on oxygen (O2) consumption and carbon dioxide (CO2) production, and respiratory quotient (RQ)
Basal O2 consumption was significantly decreased in MC4R deficient loxTB mice as compared to control mice when normalized for total body mass (Figure 4A, left panel). However, Basal O2 consumption when not normalized to body weight was not different in MC4R deficient loxTB mice as compared to control mice (Figure 4A, right panel). Chronic CoPP treatment significantly increased O2 consumption by 20% in control mice and 28% in loxTB mice irrespective normalization (Figure 4A). Basal CO2 production was significantly decreases in loxTB mice as compared to control mice when normalized for total body mass (Figure 4B, left panel). Basal CO2 production when not normalized to body weight was significantly increased in loxTB mice as compared to control mice (Figure 4B, right panel). Chronic CoPP treatment significantly increased CO2 production by 17% in control mice and 28% in loxTB mice and this effect was even more dramatic in loxTB mice when not normalized to body mass (Figure 4B). Chronic CoPP treatment had no effect on 24 hour RQ in loxTB which averaged 0.9 + .02 vs. 0.88 + .04 VO2/VCO2 in loxTB vs. CoPP treated loxTB mice. There was a trend for a decrease in RQ during the day time in CoPP treated loxTB mice with RQ averaging .87 + .01 vs. .82 + .02 VO2/VCO2 in each group respectively. CoPP treatment had no effect on 24 hour RQ in control mice with RQ values averaging 0.83 + .01 VO2/VCO2 in both each group.
Figure 4.
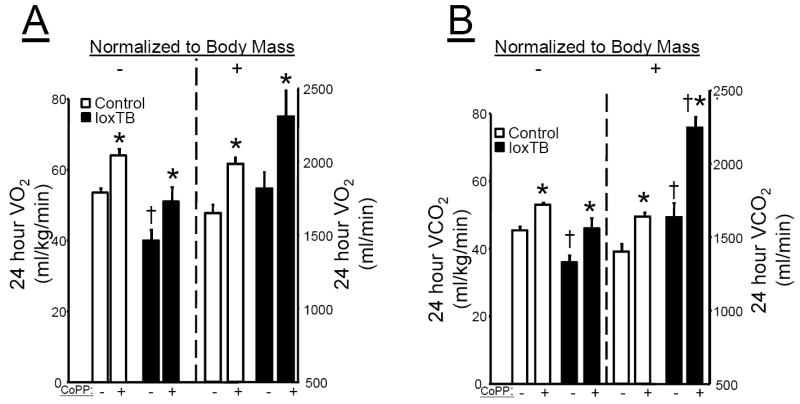
A) Effect of chronic CoPP treatment on 24 hour O2 consumption in control C57 and loxTB mice normalized to body mass in the left panel and not normalized in the right panel. B) Effect of chornic CoPP treatment on 24 hour CO2 production in control C57 and loxTB mice normalized to body mass in the left panel and not normalized in the right panel. * =P< 0.05 as compared to corresponding value in non-CoPP treated mice. † = P<0.05 as compared to corresponding value in non-CoPP treated control mice, n=4/group.
Effect of chronic CoPP treatment on heat production and activity
Chronic CoPP treatment had a similar effect to increase heat production in both control and loxTB treated mice (Figure 5A). CoPP treatment had no significant effect on locomotor activity in control mice; however, it did significantly increase locomotor activity in MC4R deficient loxTB mice (Figure 5B).
Figure 5.
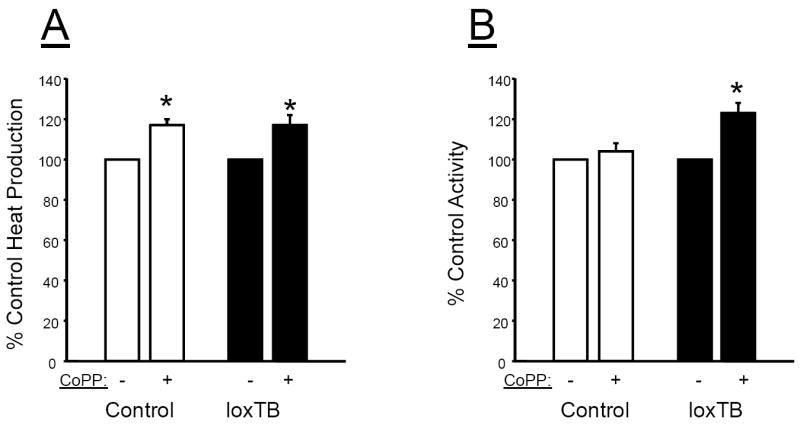
Effect of chronic CoPP treatment on A) 24 hour heat production and B) 24 hour locomotor activity in control C57 and loxTB mice. * =P< 0.05 as compared to corresponding value in non-CoPP treated mice, n=4/group.
Effect of chronic CoPP treatment on metabolic pathways in adipose tissue, liver and muscle
Chronic CoPP treatment induced HO-1 protein in the adipose tissue of loxTB mice (Figure 6A & B). The induction of HO-1 in the adipose tissue was also associated with increases in pAKT (Figure 6A &C), pAMPK (Figure 6A & D), pMTOR (Figure 6A & E), and Glut 4 (Figure 6A &F). Chronic CoPP treatment induced HO-1 protein in the liver of loxTB mice (Figure 7A &B). However, induction of HO-1 in the liver was not associated with changes in the levels of pAKT (Figure 7A &C), pAMPK (Figure 7A & D), or Glut 4 (Figure 7A & F). Hepatic induction of HO-1 was associated with a decrease in the levels of pMTOR (Figure 7A &E) in loxTB treated mice. Chronic CoPP treatment resulted in significant HO-1 induction in gastrocnemius muscle of loxTB mice (Figures 8A & B). Induction of HO-1 in the muscle was associated with no changes in the levels of pAMPK (Figures 8A & C) but resulted in a significant decrease in the levels of Glut 4 (Figures 8A & D).
Figure 6.
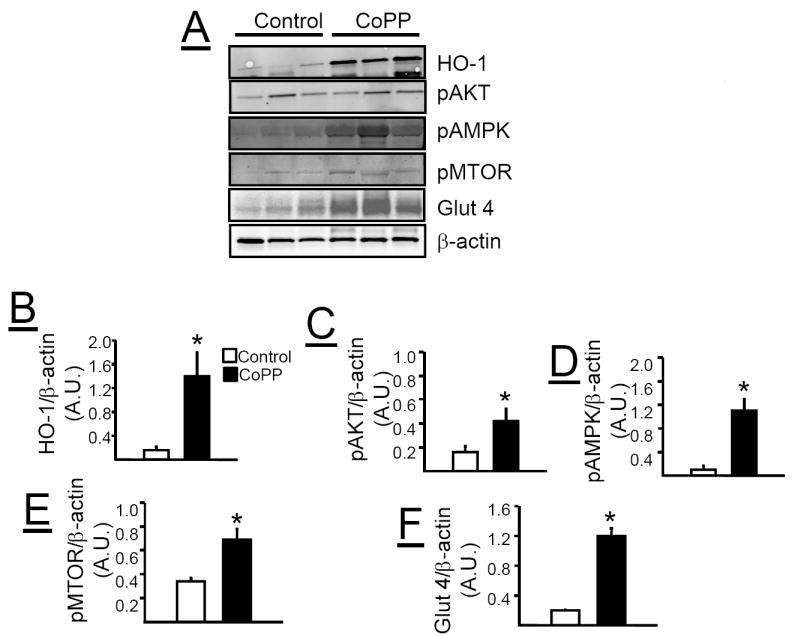
A) Representative Western blots from adipose tissue of control and CoPP treated loxTB mice. B) Levels of HO-1 protein. C) Levels of pAKT. D) Levels of pAMPK. E) Levels of pMTOR. F) Levels of glucose transporter 4 (Glut 4). * =P< 0.05 as compared to corresponding value in non-CoPP treated mice, n=6/group.
Figure 7.
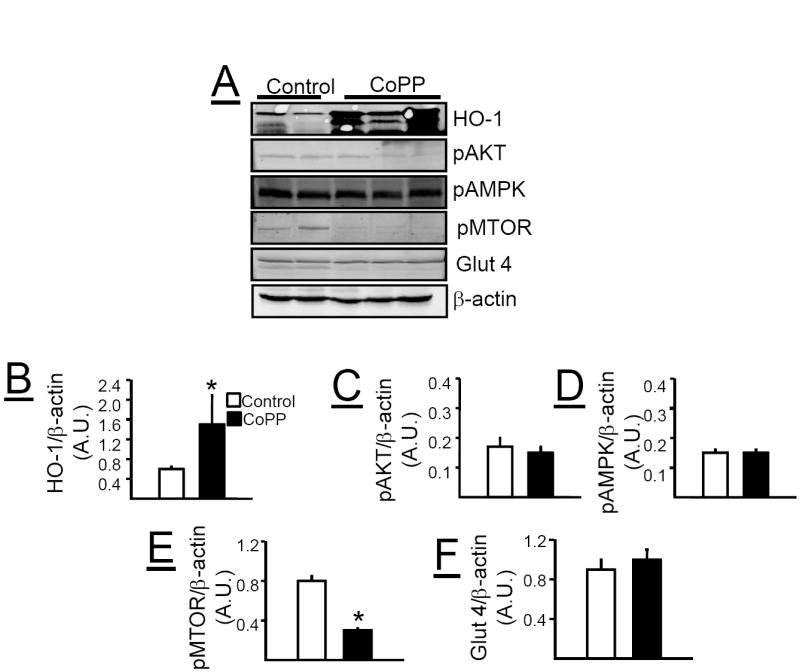
A) Representative Western blots from liver of control and CoPP treated loxTB mice. B) Levels of HO-1 protein. C) Levels of pAKT. D) Levels of pAMPK. E) Levels of pMTOR. F) Levels of glucose transporter 4 (Glut 4). * =P< 0.05 as compared to corresponding value in non-CoPP treated mice, n=4-6/group.
Figure 8.
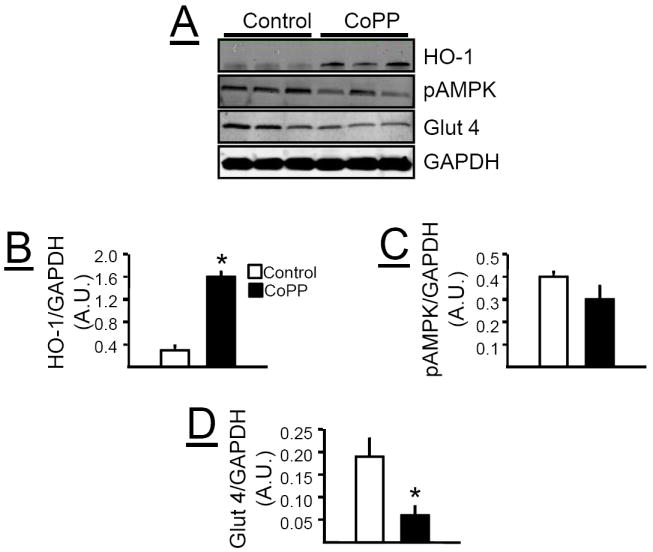
A) Representative Western blots from muscle of control and CoPP treated loxTB mice. B) Levels of HO-1 protein. C) Levels of pAMPK. D) Levels of glucose transporter 4 (Glut 4). * =P< 0.05 as compared to corresponding value in non-CoPP treated mice, n=6/group.
Discussion
We directly examined the effects of chronic HO-1 induction with CoPP on O2 consumption, CO2 and heat production, and locomotor activity in a mouse model of obesity resulting from MC4R deficiency. Chronic CoPP treatment increased in both O2 consumption and CO2 production and lowered body weight in obese loxTB MC4R deficient mice. An underlying decrease in metabolism in MC4R deficient mice is believed to be the initial driving force behind the obesity as hyperphagia in this model is not observed until after 14 weeks of age (12). Our results in the present study are consistent with these findings as we observed a significant increase in food intake in the obese MC4R deficient mice at 18 weeks of age as compared to control lean mice. Also consistent with previous studies (4;5), we did not observe any effect of chronic CoPP treatment on food intake at this later time point of administration. However, since food intake was not measured throughout the entire study, the potential importance of any initial decrease in food intake to the chronic weight reduction in CoPP treated MC4R deficient mice cannot be excluded. It is possible that early decreases in food intake in the MC4R deficient mice promote weight loss and this is maintained through increases in activity which maintain the initial weight loss. While this is possible scenario for the action of CoPP in MC4R deficient mice, several observations would argue against this as the mechanism for sustained weight loss in this model. First, decreases in food intake are generally associated with decreases in metabolism which were not observed in CoPP treated MC4R deficient or control lean mice. Next, while CoPP acted to increase O2 consumption, heat production, and adiponectin levels in both lean and obese mice, only obese MC4R deficient exhibited increases in activity which were most likely the result of decreased body weight driven by increased O2 consumption since food intake was not chronically decreased in the MC4R deficient mice.
Chronic CoPP treatment increased in both O2 consumption and CO2 production in obese loxTB MC4R deficient mice which had significantly lower O2 consumption and CO2 production under basal conditions as compared to lean control mice when corrected for total body mass. This finding is in agreement with previous studies in MC4R deficient mice which also found decreased oxygen consumption (11;12). It is noteworthy that in these studies O2 consumption is normalized to total body mass as well. The underlying decrease in metabolism in MC4R deficient mice is believed to be the initial driving force behind the obesity as hyperphagia in this model is not observed until after 14 weeks of age (12). Our results in the present study are consistent with these findings as we observed a significant increase in food intake in the obese MC4R deficient mice at 18 weeks of age as compared to control lean mice. However, in the present study, we did not detect any differences in basal O2 consumption between obese MC4R deficient and lean mice and observed a greater VCO2 in MC4R deficient as compared to lean mice when no correction for body mass was made. Normalization of metabolic data is an area of great controversy that was recently addressed in an excellent review (16). The body composition between MC4R deficient mice and lean control mice differs in the amount of lean versus fat mass in each model. In order to account for the differences in lean versus fat mass between the models the amounts of each tissue mass would need to be measured using non-invasive methods such as duel-energy X-ray absorptiometry or nuclear magnetic resonance imaging. These measurements were not performed in the current study. Despite the discrepancy between the values for VO2 and VCO2 between the MC4R deficient and lean mice depending on the normalization method used, the effects of chronic CoPP treatment to increase both of these parameters were consistent irrespective of how the data was normalized.
Chronic CoPP treatment resulted in a significant increase in O2 consumption, heat production and locomotor activity in obese loxTB MC4R deficient mice. While the exact mechanism by which chronic induction of HO-1 affects these metabolic parameters is unknown, one potential target of HO-1 induction is the mitochondria. Previous studies have demonstrated that HO-1 localizes to the mitochondria where it can affect the levels of mitochondrial transport proteins (17-19). Further studies of cardiac myocytes have demonstrated that the HO metabolite, carbon monoxide (CO), is an important regulator of mitochondrial biogenesis through enhancement of the peroxisome proliferator-activated receptor gamma-1 (PGC-1) transcriptional co-activator as well as activation of nuclear respiratory factor 2 (NRF2) (20;21). The increase in mitochondrial biogenesis could result in an increase in mitochondrial mass in peripheral tissues such as adipose, liver, or muscle which could in turn increase in O2 consumption. The increase in mitochondrial biogenesis may also result in greater exercise capacity in the obese loxTB mice and help increase locomotor activity. This hypothesis is supported by a recent study in humans which demonstrated increase muscle biogenesis and switching to a more aerobic muscle fiber type following intermittent breathing of CO (22). Further studies specifically examining mitochondrial density and function in obese MC4R deficient mice are needed in order to fully test this hypothesis.
Chronic CoPP treatment significantly attenuated obesity, lowered blood glucose and insulin, and increased plasma adiponectin levels in obese, MC4R deficient, loxTB mice. These findings were confirmed in obese MC4R KO mice as well. The results obtained with chronic induction of CoPP in MC4R deficient mice are similar to those obtained in another mouse model of obesity, the leptin deficient ob/ob mouse. HO induction with CoPP in ob/ob mice had similar effects on weight gain, fat distribution, blood glucose, plasma levels of insulin and adiponectin all of which were prevented by blockade of HO activity with a specific HO inhibitor (23). Similar effects on these parameters were also observed after chronic treatment of ob/ob mice with a different HO inducer, D-4F (2). However, the effects of HO induction on O2 consumption, CO2 and heat production, and activity in obese ob/ob mice were not determined in these studies. Our study demonstrates that chronic induction of HO-1 with CoPP results in increases in O2 consumption, CO2 and heat production, and activity in obese MC4R deficient mice. Furthermore, it should be noted that treatment with metalloporphyrins like CoPP may have effects which are mediated independently of HO-1 activation though such mechanisms as direct activation of soluble guanyl cyclase (24;25). Future experiments in HO-1 deficient models is one approach to determine if any of the observed effects of chronic CoPP administration occur independently of HO-1 induction.
Our results support the important role of the recently established HO-1-adiponectin axis in the lowering of body weight and improvement of insulin sensitivity in obese, MC4R deficient, loxTB mice. Adiponectin levels were significantly decreased in obese, MC4R deficient, loxTB mice and were significantly increased by chronic CoPP treatment. Similar results were also obtained in obese MC4R KO mice. Interestingly, chronic CoPP treatment resulted in a significant increase in adiponectin levels in lean control mice as well. Several studies in obese rat and mouse models have revealed an important link between HO-1, adiponectin and the improvement of insulin-sensitivity and reduction in inflammation in obesity (2;23;26). Chronic HO-1 induction has been reported to transform the phenotype of adipocytes in obesity from large, insulin-resistant, cytokine producing to smaller, insulin-sensitive and adiponectin producing adipocytes (3;27). The mechanism by which induction of HO-1 increases adiponectin levels is not fully understood but may be related to the ability of HO-1 to act as a chaperone protein as it is a member of the heat shock protein family (28). HO-1 may also increase adiponectin levels by its ability to decrease reactive oxygen species through the production of bilirubin or via stimulation of other important cellular antioxidants (29;30).
Consistent with the remodeling of adipocytes by chronic HO-1 induction, we observed significant alterations in important metabolic signaling pathways after chronic CoPP treatment in loxTB mice. Increases in the levels of phosphorylated AKT, AMPK, mTOR were observed in the adipocytes of CoPP treated loxTB mice. These results are in agreement with previous studies in obese leptin deficient, ob/ob mice and in leptin receptor deficient obese Zucker rats (3;27). Consistent with the improvements in plasma glucose and insulin in CoPP treated loxTB MC4R deficient mice, we also observed significant increases in glucose 4 transporter (Glut 4) levels in the adipose tissue. While chronic CoPP treatment resulted in significant enhancement of metabolic pathways in adipose tissue, similar effects were not observed in the liver or muscle. Chronic HO-1 induction in the liver was not associated with changes in pAKT, pAMPK or Glut 4 levels and was associated with a decrease in mTOR levels. Chronic HO-1 induction in the muscle resulted in no changes in the levels of pAMPK and a decrease in the levels of Glut 4. The decrease in the levels of Glut 4 in the muscle in response to chronic HO-1 induction is in contrast with previous studies in the rat which demonstrated an increase in the levels of Glut 4 after chronic HO-1 induction (31;32). The reasons for this discrepancy are not known but could be due to species differences in the metabolic response to chronic HO-1 induction or an effect due to specific loss of MC4R in the muscle of loxTB mice. Our results suggest that the major effect of chronic HO-1 induction to improve glucose utilization and insulin action occur independently of changes to the liver or skeletal muscle in obese loxTB MC4R deficient mice.
Central activation of MC3/4R receptors mediates the effects of hormones such as leptin and serotonin on food intake and energy balance (33-35). Central administration of MC4R agonists results in increases in metabolic rate and decreases in food intake while similar administration of MC4R antagonists has the opposite effect on these parameters (36-38). Chronic HO-1 induction with CoPP resulted in attenuation of weight gain and increases in metabolism in MC4R deficient loxTB mice. These findings would argue against an essential role of central MC4R activation in mediating the effects of HO-1 induction on body weight and metabolism and suggest that changes in these parameters occur via a mechanism independent of central MC4R activation. However, given the important role for MC3R receptors in the regulation of metabolism, it is possible that CoPP may act through these receptors to exert its chronic effects on metabolism (39;40). This possibility needs to be examined in future studies in MC3R deficient mice. While CoPP administration resulted in significant attenuation of obesity in both models of MC4R deficiency, the effect was more pronounced in MC4R KO mice as opposed to loxTB mice. MC4R KO mice exhibited a greater decrease in epididymal fat that did loxTB deficient mice.
The reason for this differential response in reduction of epididymal fat and total body weight between the two MC4R deficient models is unknown but may be due to the different genetic backgrounds of the two strains. MC4R KO mice are on a C57 genetic background while the loxTB mice are on a mixed 129 genetic background. Further studies will be necessary to confirm if epigenetic factors in the 129 background contribute to the lack of effect of CoPP on reduction of epididymal fat and attenuate the anorectic effects of CoPP.
In summary, we report that chronic induction of HO-1 with CoPP significantly increases O2 consumption, heat production and locomotor activity in obese MC4R deficient loxTB mice. The alterations in these metabolic parameters were associated with a decrease in body weight and fat distribution which were also confirmed in obese MC4R KO mice. Chronic CoPP treatment also improved insulin sensitivity, normalized blood glucose levels, and significantly increased plasma adiponectin levels in obese loxTB and MC4R KO mice. Chronic CoPP treatment also resulted in increased pAKT, and pAMPK levels in the adipose tissue of obese loxTB mice. These results demonstrate that chronic CoPP administration lowers body weight and increases metabolism independent of central MC4R activation and that increases in metabolism may mediate the anorectic effect of chronic HO-1 induction in vivo.
Acknowledgments
The authors would like to acknowledge the technical support of Megan V. Storm. We also thank Drs. Roger D. Cone and Bradford B. Lowell for providing initial breeding pairs of the melanocortin-4 receptor deficient mice. This work was supported by a grant from the National Institutes of Health, PO1HL-51971.
Footnotes
Conflict of Interest The authors declare no conflict of interest.
References
- 1.Galbraith RA, Kappas A. Regulation of food intake and body weight by cobalt porphyrins in animals. Proc Natl Acad Sci U S A. 1989;86(19):7653–7657. doi: 10.1073/pnas.86.19.7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peterson SJ, Drummond G, Kim DH, Li M, Kruger AL, Ikehara S, et al. L-4F treatment reduces adiposity, increases adiponectin levels, and improves insulin sensitivity in obese mice. J Lipid Res. 2008;49(8):1658–1669. doi: 10.1194/jlr.M800046-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peterson SJ, Kim DH, Li M, Positano V, Vanella L, Rodella LF, et al. The L-4F mimetic peptide prevents insulin resistance through increased levels of HO-1, pAMPK, and pAKT in obese mice. J Lipid Res. 2009;50(7):1293–1304. doi: 10.1194/jlr.M800610-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galbraith RA, Kappas A. Intracerebroventricular administration of cobalt protoporphyrin elicits prolonged weight reduction in rats. Am J Physiol. 1991;261(6 Pt 2):R1395–R1401. doi: 10.1152/ajpregu.1991.261.6.R1395. [DOI] [PubMed] [Google Scholar]
- 5.Galbraith RA, Kow LM, Pfaff D, Kappas A. Injection of cobalt protoporphyrin into the medial nuclei of the hypothalamus elicits weight loss. Am J Physiol. 1992;263(4 Pt 2):R805–R812. doi: 10.1152/ajpregu.1992.263.4.R805. [DOI] [PubMed] [Google Scholar]
- 6.Galbraith RA, Chua SC, Jr, Kappas A. Hypothalamic mechanism for cobalt protoporphyrin-induced hypophagia and weight loss: inhibition of the feeding response to NPY. Brain Res Mol Brain Res. 1992;15(3-4):298–302. doi: 10.1016/0169-328x(92)90121-q. [DOI] [PubMed] [Google Scholar]
- 7.Turner MB, Corp ES, Galbraith RA. Lack of NPY-induced feeding in cobalt protoporphyrin-treated rats is a postreceptor defect. Physiol Behav. 1994;56(5):1009–1014. doi: 10.1016/0031-9384(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 8.Li M, Vizzard MA, Jaworski DM, Galbraith RA. The weight loss elicited by cobalt protoporphyrin is related to decreased activity of nitric oxide synthase in the hypothalamus. J Appl Physiol. 2006;100(6):1983–1991. doi: 10.1152/japplphysiol.01169.2005. [DOI] [PubMed] [Google Scholar]
- 9.Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, et al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem. 2010;285(23):17271–17276. doi: 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88(1):131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 11.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123(3):493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 12.Ste ML, Miura GI, Marsh DJ, Yagaloff K, Palmiter RD. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc Natl Acad Sci U S A. 2000;97(22):12339–12344. doi: 10.1073/pnas.220409497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20(2):113–114. doi: 10.1038/2407. [DOI] [PubMed] [Google Scholar]
- 14.Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20(2):111–112. doi: 10.1038/2404. [DOI] [PubMed] [Google Scholar]
- 15.Ma L, Tataranni PA, Bogardus C, Baier LJ. Melanocortin 4 receptor gene variation is associated with severe obesity in Pima Indians. Diabetes. 2004;53(10):2696–2699. doi: 10.2337/diabetes.53.10.2696. [DOI] [PubMed] [Google Scholar]
- 16.Butler AA, Kozak LP. A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes. 2010;59(2):323–329. doi: 10.2337/db09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Converso DP, Taille C, Carreras MC, Jaitovich A, Poderoso JJ, Boczkowski J. HO-1 is located in liver mitochondria and modulates mitochondrial heme content and metabolism. FASEB J. 2006;20(8):1236–1238. doi: 10.1096/fj.05-4204fje. [DOI] [PubMed] [Google Scholar]
- 18.Slebos DJ, Ryter SW, van der Toorn M, Liu F, Guo F, Baty CJ, et al. Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am J Respir Cell Mol Biol. 2007;36(4):409–417. doi: 10.1165/rcmb.2006-0214OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Di Noia MA, Van Driesche S, Palmieri F, Yang LM, Quan S, Goodman AI, et al. Heme oxygenase-1 enhances renal mitochondrial transport carriers and cytochrome C oxidase activity in experimental diabetes. J Biol Chem. 2006;281(23):15687–15693. doi: 10.1074/jbc.M510595200. [DOI] [PubMed] [Google Scholar]
- 20.Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty-Wolf KE, Piantadosi CA. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest. 2007;117(12):3730–3741. doi: 10.1172/JCI32967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103(11):1232–1240. doi: 10.1161/01.RES.0000338597.71702.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rhodes MA, Carraway MS, Piantadosi CA, Reynolds CM, Cherry AD, Wester TE, et al. Carbon monoxide, skeletal muscle oxidative stress, and mitochondrial biogenesis in humans. Am J Physiol Heart Circ Physiol. 2009;297(1):H392–H399. doi: 10.1152/ajpheart.00164.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li M, Kim DH, Tsenovoy PL, Peterson SJ, Rezzani R, Rodella LF, et al. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes. 2008;57(6):1526–1535. doi: 10.2337/db07-1764. [DOI] [PubMed] [Google Scholar]
- 24.Ignarro LJ, Degnan JN, Baricos WH, Kadowitz PJ, Wolin MS. Activation of purified guanylate cyclase by nitric oxide requires heme. Comparison of heme-deficient, heme-reconstituted and heme-containing forms of soluble enzyme from bovine lung. Biochim Biophys Acta. 1982;718(1):49–59. doi: 10.1016/0304-4165(82)90008-3. [DOI] [PubMed] [Google Scholar]
- 25.Makino R, Matsuda H, Obayashi E, Shiro Y, Iizuka T, Hori H. EPR characterization of axial bond in metal center of native and cobalt-substituted guanylate cyclase. J Biol Chem. 1999;274(12):7714–7723. doi: 10.1074/jbc.274.12.7714. [DOI] [PubMed] [Google Scholar]
- 26.Ndisang JF, Jadhav A. Heme arginate therapy enhanced adiponectin and atrial natriuretic peptide, but abated endothelin-1 with attenuation of kidney histopathological lesions in mineralocorticoid-induced hypertension. J Pharmacol Exp Ther. 2010;334(1):87–98. doi: 10.1124/jpet.109.164871. [DOI] [PubMed] [Google Scholar]
- 27.Nicolai A, Li M, Kim DH, Peterson SJ, Vanella L, Positano V, et al. Heme oxygenase-1 induction remodels adipose tissue and improves insulin sensitivity in obesity-induced diabetic rats. Hypertension. 2009;53(3):508–515. doi: 10.1161/HYPERTENSIONAHA.108.124701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ewing JF, Haber SN, Maines MD. Normal and heat-induced patterns of expression of heme oxygenase-1 (HSP32) in rat brain: hyperthermia causes rapid induction of mRNA and protein. J Neurochem. 1992;58(3):1140–1149. doi: 10.1111/j.1471-4159.1992.tb09373.x. [DOI] [PubMed] [Google Scholar]
- 29.Rodella L, Lamon BD, Rezzani R, Sangras B, Goodman AI, Falck JR, et al. Carbon monoxide and biliverdin prevent endothelial cell sloughing in rats with type I diabetes. Free Radic Biol Med. 2006;40(12):2198–2205. doi: 10.1016/j.freeradbiomed.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 30.Turkseven S, Kruger A, Mingone CJ, Kaminski P, Inaba M, Rodella LF, et al. Antioxidant mechanism of heme oxygenase-1 involves an increase in superoxide dismutase and catalase in experimental diabetes. Am J Physiol Heart Circ Physiol. 2005;289(2):H701–H707. doi: 10.1152/ajpheart.00024.2005. [DOI] [PubMed] [Google Scholar]
- 31.Ndisang JF, Jadhav A. Up-regulating the hemeoxygenase system enhances insulin sensitivity and improves glucose metabolism in insulin-resistant diabetes in Goto-Kakizaki rats. Endocrinology. 2009;150(6):2627–2636. doi: 10.1210/en.2008-1370. [DOI] [PubMed] [Google Scholar]
- 32.Ndisang JF, Lane N, Jadhav A. The heme oxygenase system abates hyperglycemia in Zucker diabetic fatty rats by potentiating insulin-sensitizing pathways. Endocrinology. 2009;150(5):2098–2108. doi: 10.1210/en.2008-0239. [DOI] [PubMed] [Google Scholar]
- 33.Tallam LS, da Silva AA, Hall JE. Melanocortin-4 receptor mediates chronic cardiovascular and metabolic actions of leptin. Hypertension. 2006;48(1):58–64. doi: 10.1161/01.HYP.0000227966.36744.d9. [DOI] [PubMed] [Google Scholar]
- 34.Zhou L, Williams T, Lachey JL, Kishi T, Cowley MA, Heisler LK. Serotonergic pathways converge upon central melanocortin systems to regulate energy balance. Peptides. 2005;26(10):1728–1732. doi: 10.1016/j.peptides.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 35.Lam DD, Przydzial MJ, Ridley SH, Yeo GS, Rochford JJ, O’Rahilly S, et al. Serotonin 5-HT2C receptor agonist promotes hypophagia via downstream activation of melanocortin 4 receptors. Endocrinology. 2008;149(3):1323–1328. doi: 10.1210/en.2007-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen AS, Metzger JM, Trumbauer ME, Guan XM, Yu H, Frazier EG, et al. Role of the melanocortin-4 receptor in metabolic rate and food intake in mice. Transgenic Res. 2000;9(2):145–154. doi: 10.1023/a:1008983615045. [DOI] [PubMed] [Google Scholar]
- 37.Cepoi D, Phillips T, Cismowski M, Goodfellow VS, Ling N, Cone RD, et al. Assessment of a small molecule melanocortin-4 receptor-specific agonist on energy homeostasis. Brain Res. 2004;1000(1-2):64–71. doi: 10.1016/j.brainres.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 38.Markison S, Foster AC, Chen C, Brookhart GB, Hesse A, Hoare SR, et al. The regulation of feeding and metabolic rate and the prevention of murine cancer cachexia with a small-molecule melanocortin-4 receptor antagonist. Endocrinology. 2005;146(6):2766–2773. doi: 10.1210/en.2005-0142. [DOI] [PubMed] [Google Scholar]
- 39.Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, et al. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141(9):3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 40.Butler AA, Cone RD. Knockout studies defining different roles for melanocortin receptors in energy homeostasis. Ann N Y Acad Sci. 2003;994:240–245. doi: 10.1111/j.1749-6632.2003.tb03186.x. [DOI] [PubMed] [Google Scholar]