

For many years, polyketide natural products have provided the scientific community with a rich source of novel molecular architectures, many of which have become important therapeutics for clinical use.[1] In 1993, the polyketide rhizopodin was isolated from the myxobacterium Myxococcus stipitatus.[2] It was shown to display an interesting array of biological properties, including potent anti-tumor activity against a range of cancer cell lines in the low nanomolar range and the ability to inhibit the polymerization of actin.[2,3] Despite its original structural assignment as the 19-membered monomeric lactone 1a (monorhizopodin), recent studies revealed dimeric structure 2 to be the correct architecture of rhizopodin (Figure 1).[4] These molecules have started to attract attention from the synthetic community, although no total syntheses have been reported to date.[5]
Figure 1.

Structures of monorhizopodin (1a), 16-epi-monorhizopodin (1b) and rhizopodin (2).
We were intrigued as to whether the originally proposed structure (1a) for monorhizopodin might exhibit comparable biological properties to its parent dimer (2), since these molecules contain several common structural elements, including an enamide side-chain, crucial for biological activity. X-ray crystallographic analysis of a rhizopodin-actin complex indicated that the binding of 2 to actin was largely due to favorable van der Waals interactions between the enamide side-chain and certain hydrophobic residues in the binding cleft.[6] Thus, the construction of 1a might shed light on the effect of the macrocycle itself upon the binding affinity of these compounds. Herein, we describe a highly convergent and enantioselective total synthesis of monorhizopodin (1a) and its C16 epimer (1b), as well as preliminary biological evaluation of these compounds.
Scheme 1 depicts, in retrosynthetic format, the outline of the synthetic strategy employed for the construction of monorhizopodin (1a) and 16-epi-monorhizopodin (1b). Thus, rupture of the macrolactone and the C22–C23 and C29-N bonds led, after functional group adjustments, to hydroxy carboxylic acid 3 and ketophosphonate 4 as potential precursors to the target molecules. Disassembly of 3 at the indicated bond (C15–C16) through a SmI2-mediated Barbier reaction revealed iodoester 5 and aldehyde 6 as the required building blocks for its construction. Finally, disconnection of the diene system of 5 through a Stille coupling traced the origins of this compound to vinyl iodide 7 and oxazole-containing vinyl stannane 8. The choice for the SmI2-mediated Barbier coupling over the more conventional Grignard reaction was based on the remarkably mild nature of the former reagent,[7] considered to be essential for the survival of the resulting secondary alcohol, whose facile elimination toward the oxazole moiety was considered to be an issue.
Scheme 1.

Retrosynthetic analysis of monorhizopodin (1). TBS = tert-butyldimethylsilyl, TBDPS = tert-butyldiphenylsilyl, TES = triethylsilyl.
The synthesis of monorhizopodin (1a) and its 16-epi-diastereoisomer (1b) commenced with the construction of the requisite building blocks in their enantiomerically pure forms, starting with ketophosphonate 4. Thus, as shown in Scheme 2, enantioselective crotylation of aldehyde 9[8] with (−)-(Ipc)2-trans-crotyl borane following Brown’s protocol afforded alcohol 10 in 91% yield as a >10:1 mixture of diastereomers and >95% ee. Methylation (NaH, MeI, 76% yield) and ozonolytic cleavage of the terminal olefin (O3; Ph3P, 84% yield) of the latter compound led to aldehyde 12 through intermediate 11. Reaction of aldehyde 12 with the lithium species derived from dimethyl methylphosphonate and nBuLi, followed by DMP oxidation of the resulting mixture of diastereomeric alcohols, furnished ketophosphonate 4 in 98% overall yield.
Scheme 2.

Construction of ketophosphonate 4. Reagents and conditions: a) (−)-(Ipc)2-trans-crotyl borane (1.2 equiv), BF3·OEt2 (1.2 equiv), THF, −78 °C, 1 h; 3 N NaOH (aq.), H2O2, Et2O, 25 °C, 10 h, 91%, d.r. >10:1; b) NaH (1.5 equiv), MeI (1.5 equiv), THF, 0 to 25 °C, 1 h, 76%; c) O3, CH2Cl2, −78 °C; then PPh3 (1.5 equiv), 25 °C, 1 h, 84%; d) MeP(O)(OMe)2 (5.5 equiv), nBuLi (5.5 equiv), THF, −78 °C, 1 h; 12 (1.0 equiv), −78 °C, 2.5 h; DMP (1.4 equiv), CH2Cl2, 25 °C, 10 min, 98% for 2 steps. Ipc = isopinocampheyl, aq. = aqueous, THF = tetrahydrofuran, DMP = Dess–Martin periodinane.
Aldehyde 6 was synthesized from known diol 13[9] as summarized in Scheme 3. Thus, acetonide formation from 13 [Me2C(OMe)2, (±)-CSA (cat.), 96% yield] followed by ozonolysis of the resulting olefin (14, O3; Ph3P, 83% yield) led to aldehyde 15, which was subjected to reagent-controlled crotylation under the Brown conditions [(+)-(Ipc)2-cis-crotyl borane] to afford secondary alcohol 16 in 75% yield as a single diastereomer. Methylation of 16 (NaH, MeI) was followed by ozonolysis (O3; Ph3P) and NaBH4 reduction to give primary alcohol 18, whose silylation [TBDPSCl, imid., DMAP (cat.)] led to acetonide TBDPS ether 19, via intermediates 17 and 18, in 75% overall yield for the four steps. Liberation of the 1,3-diol system of 19 required mild acid conditions (PPTS) and recycling (3×) in order to avoid significant degradation of the TBDPS ether and resulted in 56% yield (plus 33% recovered starting material) of diol 20. Selective oxidation of the primary hydroxyl group of the latter [PhI(OAc)2, TEMPO (cat.)], followed by TES protection (TESOTf, 2,6-lut.), furnished, through intermediate 21, the required aldehyde 6 (75% overall yield for the two steps).
Scheme 3.

Construction of aldehyde 6. Reagents and conditions: a) (±)-CSA (0.010 equiv), Me2C(OMe)2, DMF, 25 °C, 2 h, 96%; b) O3, CH2Cl2, −78 °C; then PPh3 (2.0 equiv), 25 °C, 1 h, 83%; c) (+)-(Ipc)2-cis-crotyl borane (1.2 equiv), BF3·OEt2 (1.2 equiv), THF, −78 °C, 1 h; 3N NaOH (aq.), H2O2, Et2O, 25 °C, 10 h, 75%; d) NaH (1.3 equiv), MeI (2.0 equiv), THF, 0 to 25 °C, 1 h, 89%; e) O3, CH2Cl2, −78 °C; then PPh3 (2.0 equiv), 25 °C, 1 h; NaBH4 (1.1 equiv), MeOH, 0 °C, 30 min, 86% for 2 steps; f) TBDPSCl (1.1 equiv), imidazole (1.3 equiv), DMAP (0.10 equiv), CH2Cl2, 0 to 25 °C, 1 h, 98%; g) PPTS (1.0 equiv), CH2Cl2, MeOH, 25 °C, 16 h, 56% (33% recovered 19); h) TEMPO (0.30 equiv), PhI(OAc)2 (3.0 equiv), CH2Cl2, 25 °C, 12 h, 85%; i) TESOTf (1.2 equiv), 2,6-lutidine (2.4 equiv), CH2Cl2, −78 °C, 30 min, 88%. DMAP = 4-dimethylaminopyridine, PPTS = pyridinium para-toluenesulfonate, TEMPO = 2,2,6,6-tetramethyl-1-piperidinyloxy.
Scheme 4 summarizes the construction of vinyl iodide 7. Following a procedure developed by Denmark and co-workers, α,β-unsaturated aldehyde 22[10] was treated with silyl ketene acetal 23[11] in the presence of catalyst 24 and SiCl4[12] to afford secondary alcohol 25 in 69% yield and >90% ee. Exposure of 25 to KHMDS and benzaldehyde afforded benzylidene acetal 26 (71% yield), whose stereochemical configuration was confirmed by nOe studies. Subsequent reaction of 26 with (±)-CSA in MeOH revealed dihydroxy methyl ester 27 through acetal cleavage and transesterification (54% yield plus 44% recovered starting material). Selective protection of the hydroxyl group proximal to the ester moiety was achieved through δ-lactone formation (K2CO3; pTsOH, 96% overall yield) within 27 and silylation (TBSCl, DMAP, 84% yield), leading to TBS ether 29 via hydroxy compound 28. Subsequent opening of the δ-lactone moiety of 29 (NaOMe, 86% yield), followed by methylation (Me3OBF4, 2,6-di-t-butyl-4-methyl pyridine, 80% yield) and iodination (NIS, 96% yield), furnished vinyl iodide 7 via intermediates 30 and 31.
Scheme 4.

Construction of vinyl iodide 7. Reagents and conditions: a) 23 (1.2 equiv), cat. (R,R)-24 (0.015 equiv), SiCl4 (1.1 equiv), iPr2NEt (0.20 equiv), CH2Cl2, −78 °C, 2 h, 69%; b) PhCHO (3.3 equiv), KHMDS (0.30 equiv), THF, 0 °C, 1 h, 71%; c) (±)-CSA (0.30 equiv), MeOH, 25 °C, 20 h, 54% (44% recovered 26); d) K2CO3 (2.0 equiv), MeOH, H2O, 25 °C, 15 min; pTsOH (1.0 equiv), THF, 25 °C, 1 h, 96% for 2 steps; e) TBSCl (2.4 equiv), DMAP (3.9 equiv), CH2Cl2, 25 °C, 2 h, 84%; f) NaOMe (2.0 equiv), MeOH, 0 °C, 1 h, 86%; g) Me3OBF4 (4.0 equiv), 2,6-di-t-butyl-4-methyl pyridine (5.0 equiv), CH2Cl2, 25 °C, 12 h, 80%; h) NIS (3.0 equiv), MeCN, 60 °C, 10 h, 96%. KHMDS = potassium hexamethyldisilazide, CSA = camphorsulfonic acid, pTsOH = para-toluenesulfonic acid, NIS = N-iodosuccinimide.
Vinyl stannane 8 was constructed from known oxazole aldehyde 32 [13] and converted to advanced iodide 5 as summarized in Scheme 5. Thus, treatment of 32 with allenyl-tri-n-butylstannane in the presence of Ti(OiPr)4 and (S)-BINOL afforded alcohol 33 in 60% yield and >95% ee.[14] Methylation of 33 (NaH, MeI, 93% yield) followed by desilylation (aq. HF, 86% yield) led to primary alcohol 35 via 34. Regio- and stereoselective addition of tri-n-butyltin hydride to the terminal acetylene of 35 was achieved through palladium catalysis [Pd2(dba)3, Cy3P·HBF4, iPr2NEt, (nBu)3SnH, 67% yield] to afford vinyl stannane 8. Coupling of this stannane with vinyl iodide 7 in the presence of CuTC[15] led to alcohol 36 (75% yield), which was converted to the desired advanced iodide 5 by sodium iodide through its mesylate (MsCl, Et3N; NaI, 96% overall yield).
Scheme 5.

Synthesis of vinyl stannane 8 and advanced iodide 5. Reagents and conditions: a) Ti(OiPr)4 (1.0 equiv), (S)-BINOL (1.0 equiv), 4 Å MS, CH2Cl2, 40 °C, 1 h; 32 (1.0 equiv), allenyl-tri-n-butylstannane (1.2 equiv), −24 °C, 72 h, 60% (24% recovered 32); b) NaH (2.0 equiv), MeI (2.0 equiv), THF, 0 to 25 °C, 1 h, 93%; c) 48% aq. HF (2.0 equiv), MeCN, 25 °C, 3 h, 86%; d) Pd2(dba)3 (0.0050 equiv), Cy3P HBF4 (0.020 equiv), iPr2NEt (0.040 equiv), (nBu)3SnH (1.2 equiv), CH2Cl2, 25 °C, 10 min, 67%; e) 7 (1.0 equiv), 8 (1.0 equiv), CuTC (10.0 equiv), NMP, 0 °C, 30 min, 75%; f) MsCl (1.2 equiv), Et3N (1.5 equiv), CH2Cl2, −78 °C, 30 min; NaI (5.0 equiv), acetone, 25 °C, 1 h, 96% for 2 steps. BINOL = 1,1′-bi(2-naphthol), dba = dibenzylidine acetone, CuTC = copper(I) thiophene-2-carboxylate, NMP = N-methyl-2-pyrrolidinone.
With fragments 4–6 now available, the stage was set for their coupling and elaboration to the targeted molecules monorhizopodin (1a) and its 16-epi-diastereoisomer (1b). As shown in Scheme 6, a mixture of 5 (1.0 equiv) and 6 (1.5 equiv) was exposed to the action of SmI2 in THF to afford a 1:1 diastereomeric mixture of alcohols 37 at C16 (56% yield). Given the complexity of these substrates, the performance of SmI2 in this coupling reaction is remarkable and provides further testament for the power of this reagent in organic synthesis.[7] Having achieved coupling of the key fragments, the resulting mixture of alcohols (37) was oxidized through the action of DMP to afford ketone 38 in 95% yield. Saponification of the methyl ester of 38 (LiOH, 60 °C) followed by TES removal (PPTS) then led to hydroxy acid 3 (62% overall yield), which was now primed for macrolactonization. It was after screening of several conditions that we found that optimal results could be obtained by employing the Shiina protocol.[16] Thus, slow addition of hydroxy acid 3 to a toluene solution of MNBA and DMAP at 60 °C afforded keto macrolactone 39 in good yield. Due to its tailing TLC properties, this ketone was difficult to purify and, therefore, was reduced with NaBH4 in the presence of CeCl3 in MeOH at −20 °C to give hydroxy compounds 40a and 40b as a chromatographically separable mixture of diastereomers (ca. 2:1 in favor of 40a; under the high dilution conditions employed in the macrolactonization process, no dimeric material was observed). Interestingly, reduction of ketone 39 with NaBH4 alone delivered alcohol 40b exclusively. Other reducing agents led to unsatisfactory results. The stereochemical configurations of diastereomers 40a and 40b were determined by the Rychnovsky 13C NMR method,[17] employing the acetonides obtained from 40a and 40b upon reductive macrolactone opening, selective primary alcohol TBDPS protection, and acetonide formation [40a gave anti-acetonide 40a′ (13C NMR, δ = 101.61 ppm), whilst 40b gave syn-acetonide 40b′ (13C NMR, δ = 99.04 ppm); see Supporting Information). In preparation for the side-chain attachment, the silyl protecting groups of diastereomer 40a (corresponding to the C16 rhizopodin configuration) were removed (TBAF, AcOH) to afford triol 41a in 89% yield. The latter compound was then converted to its tris-TES derivative 42a (TESOTf, 2,6-lut., 66% yield) which underwent selective mono-desilylation (PPTS, 89% yield) and oxidation (SO3·py, 81% yield) to afford aldehyde 44a through primary alcohol 43a. This aldehyde was then reacted with ketophosphonate 4 in the presence of Ba(OH)2 to afford the corresponding α,β-unsaturated ketone (65%), which was reduced with Stryker’s reagent, leading to its saturated counterpart (45a) in 87% yield. At this juncture, all that remained to complete the synthesis of monorhizopodin (1a) was global deprotection and installation of the enamide moiety. To this end, tris-silylated ketone 45a was first exposed to TASF (to produce triol 46a, 70% yield) and thence to PhI(OAc)2/TEMPO (cat.) to furnish, through selective oxidation of the primary alcohol, dihydroxy aldehyde 47a (74% yield). The latter was condensed with N-methyl formamide (PPTS, 4 Å MS, 80 °C) to afford monorhizopodin (1a) in 78% yield as a mixture of E:Z geometrical isomers (ca. 2:1 in favor of the E isomer). Following the same sequence, 16-epi-monorhizopodin (1b) was also synthesized from diastereoisomer 40b (see Supporting Information). The 1H and 13C NMR and IR spectroscopic data of 1a and 1b were similar to those reported for rhizopodin (2).[4] The monomeric structures of these compounds were confirmed by their mass spectral data.
Scheme 6.

Fragment coupling and completion of the total synthesis of monorhizopodin (1a). Reagents and conditions: a) 5 (1.0 equiv), 6 (1.5 equiv), SmI2 (3.0 equiv), THF, 25 °C, 5 min, 56%; b) DMP (3.0 equiv), CH2Cl2, 25 °C, 15 min, 95%; c) LiOH·H2O (6.0 equiv), (CH3)2CHOH:THF:H2O (4:1:1), 60 °C, 1 h; PPTS (1.0 equiv), CH2Cl2, MeOH, 25 °C, 12 h, 62% for 2 steps; d) 3 (1.0 equiv in THF-toluene 1:1, syringe-pump slow addition), MNBA (2.0 equiv), DMAP (6.0 equiv), 4 Å MS, toluene, 60 °C, 20 h; NaBH4 (3.0 equiv), CeCl3·7H2O (10.0 equiv), MeOH, −20 °C, 30 min, 70% for 2 steps, d.r. 2:1; e) TBAF (10.0 equiv), AcOH (10.0 equiv), DMF, 25 °C, 12 h, 89%; f) TESOTf (10.0 equiv), 2,6-lutidine (20 equiv), CH2Cl2, −78 °C, 30 min, 66%; g) PPTS (0.10 equiv), CH2Cl2, MeOH, 25 °C, 30 min, 89%; h) SO3·py (2.0 equiv), iPr2NEt (6.0 equiv), CH2Cl2, DMSO, 25 °C, 30 min, 81%; i) 4 (2.0 equiv), Ba(OH)2 (0.5 equiv), THF, H2O, 25 °C, 2 h, 65%; j) [CuH(PPh3)]6 (1.0 equiv), benzene, 25 °C, 8 h, 87%; k) TASF (5.0 equiv), DMF, 25 °C, 8 h, 70%; l) TEMPO (0.10 equiv), PhI(OAc)2 (2.0 equiv), CH2Cl2, 25 °C, 4 h, 74%; m) HN(Me)CHO (20 equiv), PPTS (0.14 equiv), 4 Å MS, C6H6, 80 °C, 8 h, 78%. MNBA = 2-methyl-6-nitrobenzoic anhydride, TBAF = tetra-n-butylammonium fluoride, py = pyridine, DMSO = dimethylsulfoxide, TASF = tris(dimethylamino)sulfonium difluorotrimethylsilicate.
With synthetic samples of monorhizopodin (1a) and 16-epi-monorhizopodin (1b) available to us, we were in a position to evaluate their biological properties in actin polymerization and cytotoxicity assays. As shown in Figure 2, monorhizopodin (1a) exhibited potent inhibitory activity of actin polymerization, as expected from its enamide side-chain structural motif. This activity, which is mimicked by monorhizopodin’s 16-epi-isomer (1b), albeit with somewhat lower potency, is comparable to that of latrunculin A (LatA, see figure 2), which was used as a standard in this assay. However, neither monorhizopodin (1a) nor 16-epi-monorhizopodin (1b) exhibited cytotoxicity against MDA-MB-231 breast cancer cells (up to 100 μM concentrations), presenting an interesting dichotomy and a puzzle regarding their divergence from rhizopodin (2). Although further investigations are needed to explain this phenomenon, we hypothesize that either these compounds are unable to displace G-actin binding proteins, such as profilin,[18] within cells, or that they fail to penetrate the cell membrane to reach their target.
Figure 2.
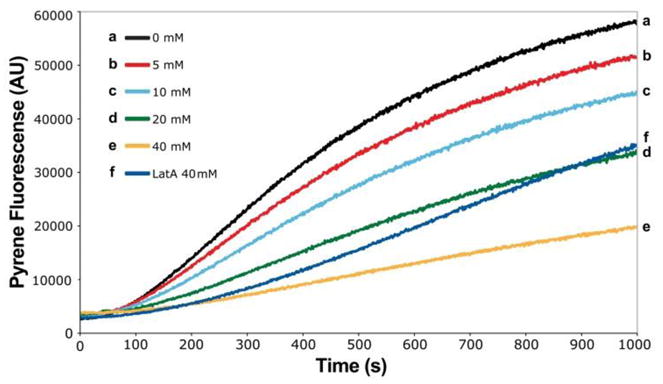
Inhibition of actin polymerization by monorhizopodin (1a). The concentration of actin was 5 μM, that of monorhizopodin (1a) as indicated. For the corresponding graphs obtained with 16-epi-monorhizopodin (1b) and further details of the assay, see Supplementary Information. LatA = latrunculin A.
In conclusion, a highly convergent total synthesis of monorhizopodin (1a) and 16-epi-monorhizopodin (1b) has been developed, rendering these monomeric homologues of the powerful antitumor agent rhizopodin (2) available for biological investigations. Preliminary studies showed these compounds to be endowed with actin-binding properties but devoid of any associated cytotoxicity, posing interesting questions regarding the role of the dimeric nature of rhizopodin (2) in its mode of action. Further studies directed toward the elucidation of the mechanism of action and the differences of rhizopodin (2) and its monomeric homologues, (1a) and (1b), as well as the total synthesis of the former are in progress.
Supplementary Material
Footnotes
Financial support for this work was provided by grants from the National Institute of Health (USA) to K.C.N (CA100101) and to V.M.F (HL083464), a fellowship from Institut de Chimie des Substances Naturelles (ICSN) to A.C., and by funds from The Skaggs Institute for Research. We are indebted to Prof. Scott Denmark for a generous gift of his catalyst (24).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Prof. Dr. K. C. Nicolaou, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037 (USA) and Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA), Fax: (+1) 858-784-2469.
Dr. Xuefeng Jiang, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037 (USA) and Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA), Fax: (+1) 858-784-2469
Dr. Peter J. Lindsay-Scott, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037 (USA) and Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA), Fax: (+1) 858-784-2469
Dr. Andrei Corbu, Department of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037 (USA) and Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA), Fax: (+1) 858-784-2469
Dr. Sawako Yamashiro, Department of Cell Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037 (USA), Fax: (+1) 858-784-8753
Dr. Andrea Bacconi, Department of Cell Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037 (USA), Fax: (+1) 858-784-8753
Prof. Dr. Velia M. Fowler, Email: velia@scripps.edu, Department of Cell Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037 (USA), Fax: (+1) 858-784-8753
References
- 1.a) Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]; b) Newman DJ, Grothaus PG. Chem Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 2.Sasse F, Steinmetz H, Höfle G, Reichenbach H. J Antibiot. 1993;46:741–748. doi: 10.7164/antibiotics.46.741. [DOI] [PubMed] [Google Scholar]
- 3.Gronewold TM, Sasse F, Lünsdorf H, Reichenbach H. Cell Tissue Res. 1999;295:121–129. doi: 10.1007/s004410051218. [DOI] [PubMed] [Google Scholar]
- 4.Jansen R, Steinmetz H, Sasse F, Schubert WD, Hagelüken G, Albrecht SC, Müller R. Tetrahedron Lett. 2008;49:5796–5799. [Google Scholar]
- 5.a) Cheng Z, Song L, Xu Z, Ye T. Org Lett. 2010;12:2036–2039. doi: 10.1021/ol100515m. [DOI] [PubMed] [Google Scholar]; b) Chakraborty TK, Pulukuri KK, Sreekanth M. Tetrahedron Lett. 2010;51:6444–6446. [Google Scholar]; c) Chakraborty TK, Sreekanth M, Pulukuri KK. Tetrahedron Lett. 2010 doi: 10.1016/j.tetlet.2010.10.142. [DOI] [Google Scholar]
- 6.Hagelüken G, Albrecht SC, Steinmetz H, Jansen R, Heinz DW, Kalesse M, Schubert WD. Angew Chem. 2009;121:603–606. doi: 10.1002/anie.200802915. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:595–598. doi: 10.1002/anie.200802915. [DOI] [PubMed] [Google Scholar]
- 7.For selected reviews on the use of SmI2 in organic synthesis, see: Nicolaou KC, Ellery SP, Chen JS. Angew Chem. 2009;121:7276–7301. doi: 10.1002/anie.200902151.Angew Chem Int Ed. 2009;48:7140–7165. doi: 10.1002/anie.200902151.Gopalaiah K, Kagan HB. New J Chem. 2008;32:607–637.Jung DY, Kim YH. Synlett. 2005:3019–3032.Edmonds DJ, Johnston D, Procter DJ. Chem Rev. 2004;104:3371–3403. doi: 10.1021/cr030017a.For specific examples relevant to the present application, see: Wu J, Panek JS. Angew Chem. 2010;122:6301–6304.Angew Chem Int Ed. 2010;49:6165–6168. doi: 10.1002/anie.201002220.Williams DR, Berliner MA, Stroup BW, Nag PP, Clark MP. Org Lett. 2005;7:4099–4102. doi: 10.1021/ol051345v.Williams DR, Kiryanov AA, Emde U, Clark MP, Berliner MA, Reeves JT. Proc Natl Acad Sci. 2004;101:12058–12063. doi: 10.1073/pnas.0402477101.
- 8.Luo J, Li H, Wu J, Xing X, Dai WM. Tetrahedron. 2009;65:6828–6833. [Google Scholar]
- 9.Bode J, Gauthier DR, Carreira EM. Chem Commun. 2001:2560–2561. [Google Scholar]
- 10.Su Q, Dakin LA, Panek JS. J Org Chem. 2007;72:2–24. doi: 10.1021/jo0610412. [DOI] [PubMed] [Google Scholar]
- 11.Denmark SE, Beutner GL, Wynn T, Eastgate MD. J Am Chem Soc. 2005;127:3774–3789. doi: 10.1021/ja047339w. [DOI] [PubMed] [Google Scholar]
- 12.Denmark SE, Fujimori S. J Am Chem Soc. 2005;127:8971–8973. doi: 10.1021/ja052226d. [DOI] [PubMed] [Google Scholar]
- 13.Li DR, Zhang DH, Sun CY, Zhang JW, Yang L, Chen J, Liu B, Su C, Zhou WS, Lin GQ. Chem Eur J. 2006;12:1185–1204. doi: 10.1002/chem.200500892. [DOI] [PubMed] [Google Scholar]
- 14.Keck GE, Krishnamurthy D, Chen X. Tetrahedron Lett. 1994;35:8323–8324. [Google Scholar]
- 15.Allred GD, Liebeskind LS. J Am Chem Soc. 1996;118:2748–2749. [Google Scholar]
- 16.Shiina I, Kubota M, Oshiumi H, Hashizume M. J Org Chem. 2004;69:1822–1830. doi: 10.1021/jo030367x. [DOI] [PubMed] [Google Scholar]
- 17.Rychnovsky SD, Rogers BN, Richardson TI. Acc Chem Res. 1998;31:9–17. [Google Scholar]
- 18.Carlsson L, Nyström LE, Sundkvist I, Markey F, Lindberg U. J Mol Biol. 1977;115:465–483. doi: 10.1016/0022-2836(77)90166-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.