

Abstract
This mini-review summarizes studies my associates and I carried out that are relevant to the topic of the present volume [i.e. glutamate dehydrogenase (GDH)] using radioactive 13N (t½ 9.96 min) as a biological tracer. These studies revealed the previously unrecognized rapidity with which nitrogen is exchanged among certain metabolites in vivo. For example, our work demonstrated that a) the t½ for conversion of portal vein ammonia to urea in the rat liver is ~10–11 sec, despite the need for five enzyme-catalyzed steps and two mitochondrial transport steps, b) the residence time for ammonia in the blood of anesthetized rats is ≤7–8 sec, c) the t½ for incorporation of blood-borne ammonia into glutamine in the normal rat brain is <3 sec, and d) equilibration between glutamate and aspartate nitrogen in rat liver is extremely rapid (seconds), a reflection of the fact that the components of the hepatic aspartate aminotransferase reaction are in thermodynamic equilibrium. Our work emphasizes the importance of the GDH reaction in rat liver as a conduit for dissimilating or assimilating ammonia as needed. In contrast, our work shows that the GDH reaction in rat brain appears to operate mostly in the direction of ammonia production (dissimilation). The importance of the GDH reaction as an endogenous source of ammonia in the brain and the relation of GDH to the brain glutamine cycle is discussed. Finally, our work integrates with the increasing use of positron emission tomography (PET) and nuclear magnetic resonance (NMR) to study brain ammonia uptake and brain glutamine, respectively, in normal individuals and in patients with liver disease or other diseases associated with hyperammonemia.
Keywords: [13N]Ammonia, 13N-labeled amino acids, alanine aminotransferase, aspartate aminotransferase, branched chain amino acid aminotransferase, glutamate dehydrogenase, glutamine cycle, nitrogen flux, transreamination, transdeamination, urea cycle
1. Historical
13N is a positron emitting isotope. In positron (or β+) emission, the particle ejected from the nucleus of the decaying nuclide has the same mass, but opposite charge, to that of the electron. As with the more familiar β− (electron) emission, the ejected positrons are emitted with a spectrum of kinetic energies with characteristic Eaverage and Emax values. In the case of 13N, the Emax value is 1.2 MeV. By comparison, the Emax values of the more familiar β− emissions of 14C and 32P are 0.16 and 1.7 MeV, respectively. After rapidly losing most of its kinetic energy, the ejected positron combines with a nearby electron to form a positronium atom (one electron, one positron) somewhat analogous to a hydrogen atom (one electron, one proton). After a brief interlude of about 140 ns, the two particles annihilate, resulting in the production of pure energy according to Einstein’s famous equation (e = mc2) in the form of two coincident 511 KeV γ-rays ejected at an angle of 180° or almost 180°.
Historically, the first demonstration of induction of radioactivity in an otherwise stable element was accomplished in 1934 by Curie and Joliot in the first description of a radiochemical experiment, and involved the production of [13N]ammonia. In this remarkable experiment, the authors irradiated boron nitride with α particles emitted from a polonium source, thereby generating 13N by the 10B(α,n)13N reaction. After heating the target with caustic soda, [13N]ammonia was detected (Curie and Joliot, 1934). Assisted by the invention of the cyclotron by Ernest O. Lawrence in 1931, by the end of the decade a large number of radionuclides had been produced, including positron-emitting isotopes now generated routinely in biomedical cyclotrons worldwide [e.g. 15O, t½ 2.04 min; 13N, t½ 9.96 min; 11C, t½ 20.38 min; 18F, t½ 109.8 min]. Perhaps the first biochemical study using 13N as a tracer was used to study N2 fixation in plants (Ruben et al., 1940). For a historical survey of the development of the discovery of positron-emitting isotopes and the use of 13N as a tracer see the review by Cooper et al. (1985a).
In the studies described in the present review, 13N was produced by a medical cyclotron in collaboration with my colleague Dr. Alan Gelbard and staff of the Biophysics Laboratory, Memorial Sloan Kettering Cancer Center, New York. Although there are several possibilities for generating 13N in a cyclotron, we routinely used the 16O(p,α)13N reaction. In this reaction, a water target is irradiated with high-energy protons; emission of an α particle generates 13N. The radiochemical species generated is mostly [13N]nitrate with smaller amounts of [13N]nitrite. At the end of the bombardment the water target is allowed to drip into a flask containing NaOH pellets and Devarda’s alloy. [13N]Nitrate/nitrite is reduced to [13N]ammonia in an exothermic reaction generated by dissolution of NaOH, and the evolved [13N]ammonia is collected into a small volume of saline or appropriate buffer (Gelbard et al., 1975). Yields of [13N]ammonia can be as high as a few hundred mCi/ml. The [13N]ammonia can then be used directly in animal studies or in PET studies. Alternatively, the [13N]ammonia may be incorporated into a number of L-amino acids and other nitrogenous compounds by use of appropriate enzymes and substrates. For example, we have enzymatically prepared 13N-labeled glutamine, asparagine, glutamate, alanine, methionine, branched-chain amino acids, phenylalanine, citrulline, carbamyl 1-phosphate and N-carbamyl aspartate (Cooper and Gelbard, 1981; Gelbard et al., 1980, 1985, 1990; Cooper et al., 1985a). A particularly useful procedure involves the use of GDH immobilized onto an inert support for the synthesis of L-[13N]glutamate and some other 13N-labeled amino acids. The L-[13N]amino acid is separated from unreacted [13N]ammonia on a Dowex 50[H+] column.
2. Advantages and disadvantages of using 13N as a biological tracer
The most obvious disadvantages of using 13N as a tracer are that it is expensive to produce and its production is limited to facilities that can generate high energy particle beams. Another disadvantage is the short half life, which means that experiments must be completed within an hour or so. On the other hand, because 13N is radioactive it is relatively easy to quantitate. Moreover, because of its short half life disposal of radioactive waste is not a problem. Another advantage is that exceedingly high specific activities are theoretically possible. The maximum specific activity of a radioisotope is in its carrier-free form and is inversely proportional to its half life. For example, the maximum specific activity of 13N (t½ 9.96 min) is 4.2 × 108 greater than that of 14C (t½ 5,730 years). In practice, however, it is almost impossible to generate carrier-free [13N]ammonia because unlabeled ammonia is a ubiquitous contaminant. Nevertheless, very high specific activities are possible (>mCi/nmol). This is an advantage because isotope can be administered to experimental animals/human volunteers/patients without disturbing the steady state of N pools. Consider the much more commonly used nitrogen tracer, namely 15N. Because 15N is a stable isotope there are no time constraints on production of 15N-labeled compound of interest or in measuring its biological disposition in animal and cell models. There is, however, one major drawback. Most nitrogen in the biosphere is in the form of 14N, but a significant amount (~0.37%) is in the form of 15N. This background 15N means that investigators have oftentimes used unphysiological doses of 15N-labeled compounds as tracers. No such problems are encountered with 13N, especially as turnover of nitrogenous substances are often very rapid, as documented in later sections.
3. The concept of transreamination and transdeamination for assimilating and dissimilating ammonia
The terms transreamination and transdeamination were introduced by Alexander Braunstein (1957), a brilliant Russian biochemist and linguist who discovered transamination (“umaminierung”) while working in Moscow under what must have been very difficult conditions in the 1930s (Braunstein and Kritzmann, 1937). Unfortunately, perhaps, these terms are not used much nowadays, but nevertheless embody useful concepts. [For an appreciation of Braunstein and a historical account of the discovery of transamination and the biological roles of transamination see Cooper and Meister (1989).] Braunstein (1957) pointed out that coupling of an α-ketoglutarate-linked transaminase (aminotransferase) (Eq. 1) to the GDH reaction (Eq. 2) can be used to incorporate ammonia nitrogen into an amino acid (transreamination) or to remove it (transdeamination). The net reaction is shown in Eq. 3.
| (1) |
| (2) |
| (3) |
The forward direction of Eq. 3 is transreamination. The back reaction is transdeamination. These reactions are especially important in the liver as discussed in the following two sections.
4. Importance of the GDH and linked aminotransferase reactions in providing nitrogen for urea synthesis in the liver
In the liver, the major mechanism for removing excess nitrogen derived from the breakdown of nitrogenous substances is through the urea cycle. Aspartate and ammonia are the two points of entry of nitrogen into the urea cycle. In effect, however, the nitrogen atoms of aspartate and ammonia are interchangeable. Consider the aspartate aminotransferase reaction (Eq. 4) coupled to the GDH reaction (Eq. 1). The net reaction is shown in Eq. 5. The forward direction is transdeamination of aspartate yielding oxaloacetate and ammonia; the reverse direction is transreamination generating aspartate from oxaloacetate and ammonia. Equation 5 shows that, in a sense the question of how nitrogen enters the urea cycle is moot, especially because as discussed below nitrogen is exchanged rapidly between ammonia and aspartate.
| (4) |
| (1) |
| (5) |
Another potential mechanism for converting aspartate to ammonia is the purine nucleotide cycle (Lowenstein, 1972). The net reaction is shown in Eq. 6. This pathway is an important source of ammonia in the muscle (Lowenstein and Goodman, 1978) and possibly the brain (Schultz and Lowenstein, 1978), but appears to be of little importance in the liver (Cooper et al., 1987).
| (6) |
By coupling an α-ketoglutarate linked aminotransferase (Eq. 2) to aspartate aminotransferase (Eq. 4) it is possible for amino acid nitrogen to be incorporated into aspartate (Eq. 7). Examples of amino acids that may provide aspartate nitrogen in this manner include alanine, tyrosine and the branched-chain amino acids (leucine, isoleucine and valine). The aspartate can then act as a source of nitrogen for urea synthesis either directly through formation of the urea cycle intermediate argininosuccinate or indirectly through transdeamination to ammonia.
| (2) |
| (4) |
| (7) |
The considerations discussed in this section are important in understanding the metabolic fate of [13N]ammonia in rat tissues and the central role of GDH, as described in the following sections.
5. Rapid metabolism of [13N]ammonia in rat liver
Fig. 1 shows the HPLC elution profile of 13N-metabolites derived from [13N]ammonia in rat liver. In this experiment, a bolus of [13N]ammonia was injected into the portal vein of an anesthetized rat. Fifteen seconds later a piece of the liver was freeze clamped and the animal euthanized. The frozen liver was homogenized and deproteinized, and a sample of the deproteinized solution was analyzed by HPLC (by means of a cation exchange column) with radiochemical detection (Cooper et al., 1987). Remarkably, Fig. 1 shows that despite the need for five enzyme steps and two mitochondrial transport steps more than 50% of the label derived from [13N]ammonia was in a metabolized form. Label in all components of the urea cycle, except carbamyl phosphate (i.e. citrulline, argininosuccinate, arginine, urea) could be detected. [It is conceivable that the mitochondrial pool of carbamyl phosphate is very small and turns over very rapidly.] From these studies we estimated that the t½ for incorporation of portal vein-derived ammonia into urea in the rat liver is about 10–11 sec (Cooper et al., 1987). Because of this rapid conversion of ammonia to urea in the liver, we hypothesized that some substrate channeling may have occurred. Later work by others is consistent with substrate channeling among components of the urea cycle (Cheung et al., 1989; Watford 1991; Maher et al., 2003).
Fig. 1.

Decay-corrected HPLC elution profile of 13N-metabolites obtained from deproteinized liver 16 s after a bolus injection of tracer quantities of [13N]ammonia into the portal vein of an anesthetized adult male rat. Modified from Cooper et al. (1987).
From radioisotope balance studies, we calculated a single-pass extraction of [13N]ammonia by the rat liver of about 93%, in good agreement with the ~90% portal-hepatic vein difference for ammonia (Cooper et al., 1987). These results are consistent with the sinusoidal zonal distribution of enzymes involved in nitrogen metabolism in the liver. As reviewed by Poyck et al. (2008), the liver lobule is classically divided into 3 zones: 1) an upstream periportal zone receiving blood from the terminal branches of the portal vein and the hepatic artery, 2) an intermediate zone, and 3) a downstream pericentral zone surrounding the efferent central vein.
Many techniques have been used to establish the presence of a remarkable gradient (zonation) along the liver sinusoid of different enzyme activities related to carbon and nitrogen metabolism. These techniques include 1) analysis of the biochemical properties of isolated periportal and perivenous hepatocytes, 2) histochemistry, 3) immunohistochemistry, 4) in situ hybridization, 5) determination of gene expression patterns in different populations of hepatocytes, and 6) determination of labeled metabolites following in vivo antegrade or retrograde perfusion of labeled compounds via the hepatic artery, For example, these techniques have revealed that the urea cycle and glutaminase activities are concentrated in the periportal hepatocytes, whereas glutamine synthetase activity is located exclusively in perivenous (pericentral) hepatocytes (e.g. Häussinger, 1990, 1998; Watford and Smith, 1990; Jungermann and Keitzmann, 1996; Moorman et al., 1994; Braeuning et al., 2006; Poyck et al., 2008).
It has been suggested that the upstream urea cycle acts as a high capacity, but relatively low affinity system for removing most of the portal vein ammonia, whereas the downstream glutamine synthetase reaction acts as a backup, higher affinity system for removing most of the remaining portal vein ammonia (Jungerman and Kietzmann, 1996; Häussinger, 1998). Under the conditions of our experiment, about 7% of the administered portal vein [13N]ammonia was incorporated into glutamine relative to that incorporated into urea (Fig. 1).
There are several points of additional interest regarding Fig. 1. A considerable amount of label was present in glutamate, aspartate and alanine, dramatic proof of the central importance of a) GDH in nitrogen metabolism in the liver and b) the linkage of nitrogen flow from an amino acid (in this case alanine) to aspartate via coupled aminotransferase reactions (alanine and aspartate aminotransferases) (scheme 1).
Scheme 1.

Flow of nitrogen through coupled aminotransferases and the GDH reaction in the liver. 1, Alanine aminotransferase; 2, aspartate aminotransferase; 3, GDH; 4, urea cycle enzymes.
Clearly, the alanine aminotransferase, aspartate aminotransferase and GDH reactions are fully reversible in the liver and thus the enzymes may act as a conduit for synthesis (transreamination) or catabolism (transdeamination) of alanine and aspartate as needed. However, for the essential amino acids such as the branched-chain amino acids and tyrosine, the net direction of the GDH reaction coupled to an aminotransferase reaction on balance in the whole body must be toward transdeamination.
Another point of interest is the extreme rapidity with which [13N]ammonia, once it has been incorporated into glutamate by the GDH reaction, is incorporated into aspartate and alanine, attesting to the high in vivo activity of aspartate and alanine aminotransferases in rat liver. From experiments in which rat liver samples were freeze clamped at different intervals after injection of [13N]ammonia via the portal vein, we estimated that once glutamate is labeled by the action of GDH, equilibration of label between glutamate and aspartate occurs very rapidly (maximally in seconds) (Cooper et al.,1987). However, we also noted that it took a few minutes for the specific activity of glutamate and aspartate to reach equilibrium in the liver. An explanation for these seemingly contradictory findings resides in the fact that, in the liver, the GDH reaction is exclusively mitochondrial, whereas the aspartate aminotransferase reaction occurs in both cytosolic and mitochondrial compartments. Thus, initially glutamate and aspartate will be labeled very rapidly only in the mitochondrial compartment and not in the cytosolic compartment. As a result of very different metabolic demands, the equilibrium concentrations of glutamate and aspartate maintained by the aspartate aminotransferase isozymes are different in the mitochondrial and cytosolic compartments of the rat liver. Hepatocytes contain a calcium-activated glutamate/aspartate carrier (also known as citrin) (del Arco et al., 2002). Thus, once glutamate is labeled in the liver mitochondria, this carrier will begin to transport labeled glutamate from the mitochondria into the cytosol, wherein the label in the glutamate will rapidly exchange with cytosolic aspartate. The rate of exchange of glutamate and aspartate between the two compartments by the glutamate/aspartate carrier is evidently slower than the rate of exchange of nitrogen between glutamate and aspartate in the two compartments. Thus, although label exchange between glutamate and aspartate is very rapid in both cytosolic and mitochondrial compartments, the different concentrations of unlabeled glutamate/aspartate in cytosol and mitochondria and the relatively slow transport step, provides an explanation for the somewhat slow rate at which the specific activities of total (i.e. cytosolic plus mitochondrial) glutamate and aspartate reach equilibrium in the whole liver cells.
In other experiments, we injected tracer quantities of L-[13N]glutamate or L[13N]alanine into the portal vein of anesthetized rats and determined the metabolic profile of the label in the liver. In both cases label rapidly (seconds) was found among components of the GDH, aspartate aminotransferase and alanine aminotransferase reactions (Cooper et al., 1988). Components of the aspartate aminotransferase reaction and GDH reactions in the rat liver are near thermodynamic equilibrium (Williamson et al., 1967; Treberg et al., 2010). Our data indicate just how rapidly the nitrogen is shared among the equilibrium components – ammonia, glutamate and aspartate.
Comparison of the metabolic fate of 13N after administration of [13N]glutamate or [13N]alanine provided direct in vivo evidence for metabolic zonation along the sinusoid, consistent with the urea cycle and glutaminase activities predominating in periportal hepatocytes and glutamine synthetase activity predominating in perivenous hepatocytes, respectively. Thus, L-[13N]glutamate derived from portal vein L-[13N]alanine in rat liver was a much better source of urea nitrogen (occurs in periportal hepatocytes) than was portal vein L-[13N]glutamate (taken up predominantly in perivenous hepatocytes). On the other hand, portal vein administered L-[13N]glutamate (metabolized predominantly in perivenous hepatocytes) was a much better source of L-[amide-13N]glutamine than was portal vein administered L-[13N]alanine (metabolized predominantly in the perivenous hepatocytes) (Cooper et al., 1988). In other experiments it was shown that metabolic turnover of L-[amide-13N]glutamine in the liver was slower than that of L-[13N]alanine and L-[13N]glutamate, presumably as a result of the much larger hepatic pool of endogenous glutamine. Despite the relatively slow turnover, we were able to detect [13N]urea as a metabolite. These results strongly suggest, despite occasional reports to the contrary, that the glutaminase action on glutamine in the periportal hepatocytes provides a source of ammonia for urea synthesis (Cooper et al., 1988) (see also Treberg et al., 2010).
6. Rapid clearance of [13N]ammonia from the blood
The rapidity with which selected nitrogen pools turn over can also be appreciated from Fig. 2. In the experiment depicted, we measured the clearance of label from the blood of three anesthetized rats after bolus injection of [13N]ammonia via a femoral vein (Cooper and Freed, 2005). From this experiment we calculated the t½ for removal of tracer quantities of [13N]ammonia from the blood of anesthetized rats is ≤7–8 sec. This calculation is based on the disappearance of label, but labeled metabolites could be detected in the blood by 6 sec, resulting in a possible underestimation of rate of [13N]ammonia clearance from the blood. Evidently, blood-derived ammonia is efficiently trapped as glutamine in extrahepatic tissues and as urea/glutamine in the liver. Similar results have been reported for the clearance of [13N]ammonia in human volunteers except that the rate of clearance from the blood is somewhat slower than that in the rat (Rosenspire et al., 1990).
Fig. 2.
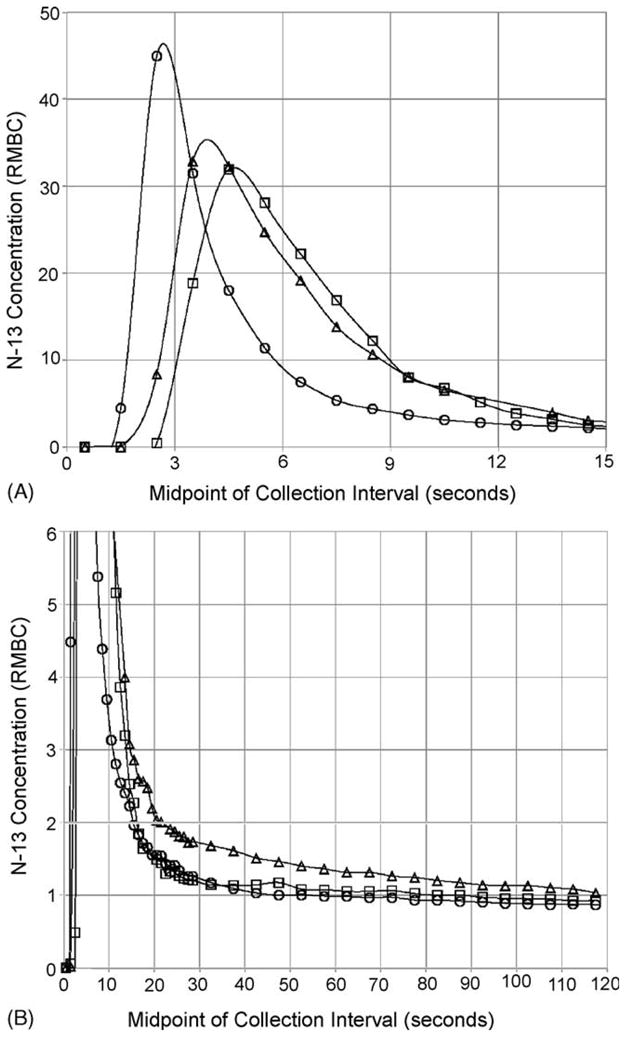
Concentration (RMBC) of 13N activity in arterial rat blood versus time after injection. A bolus (0.3–0.4 ml) of [13N]ammonia solution was injected into the femoral vein of each of three anesthetized 250 g rats and blood samples were collected from a cannula in the tail artery every second for 30 s and every 5 s thereafter for the next 90 s. Using a distinct symbol for each rat, RMBC measurements are plotted against time (the midpoint of each collection interval). (A) and (B) show the measurements for the first 15 s and the first 2 min, respectively. Smoothed curves have been fitted through the data points for each rat. RMBC is “the ratio to mean body concentration” and is defined as the decay-corrected fraction of injected tracer recovered in a specimen divided by the fraction of body weight contained in that specimen. From Freed and Cooper (2005) with permission.
7. Rapid incorporation of [13N]ammonia into glutamine in the rat brain
In our first series of experiments using 13N as a tracer in rat brain, we determined the brain uptake index (BUI; brain uptake relative to a marker that completely enters the brain) of [13N]ammonia in awake, but restrained rats (Cooper et al., 1979). The BUI of [13N]ammonia was measured five seconds after a bolus administration of [13N]ammonia –enough time for the bolus to clear the brain, but at a time when return of label through the circulation was small. The BUI was found to be about 25% and to increase with increasing pH of the bolus (Cooper et al., 1979). Similar results were obtained by Lockwood et al. (1980), who measured the BUI for [13N]ammonia in rats over a pH range of 6.58 to 7.73. These authors stated that “the brain-blood pH gradient plays a major role in determining the forward flux of ammonia from the blood into the brain in the physiological pH range.” The pKa of ammonia (NH3 + H+ ⇆ NH4+) is 9.2 under physiological conditions and temperature. Thus, only ~1% of ammonia will be in the form of the free base (NH3) and ~99% will be in the form of ammonium ion (NH4+) under normal physiological conditions (~pH 7.2). Our data and those of Lockwood et al. (1980) suggest that uptake of ammonia is mostly by diffusion of the free base. This idea was taken a step further by Raichle and Larson (1981) who showed that most blood-derived [13N]ammonia enters the brain by diffusion of 13NH3 but a smaller portion enters as [13N]ammonium ions.
Next, we determined the cerebral metabolic fate of [13N]ammonia infused into immobilized rats via an indwelling cannula in the carotid artery (Cooper et al., 1979). In one experiment, analysis of labeled metabolites in rapidly frozen brain was carried out after a ten-minute continuous infusion (Table 1). There are several points of interest in this table. First, the relative specific activity of L-[amine-13N]glutamine was greater than that of L-[13N]glutamate. The label in L-[13N]glutamate must have arisen by reductive [13N]amination of α-ketoglutarate in a reaction catalyzed by GDH. The only known route for net glutamine synthesis involves the glutamine synthetase reaction, which is very active in brain (Pamiljans et al., 1962). Thus, the L-[amine-13N]glutamine in the brain must have been synthesized from L-[13N]glutamate. If the brain behaves as a simple, single compartment then a classical product-precursor relationship dictates that the specific activity of L-[amine-13N]glutamine cannot be greater than that of L[13N]glutamate. Thus, the data argue strongly in favor of glutamate turning over rapidly to glutamine in a small compartment that is metabolically and kinetically distinct from a much larger pool of more slowly turning over glutamate.
Table 1.
Relative specific activities of 13N-metabolites in rat brain after a 10 min-intracarotid artery infusion of [13N]ammonia
| Metabolite | Controls (n = 4) | MSO-treated (n =4) | P |
|---|---|---|---|
| Glutamine (amide) | [100 ± 29] | 8.0 ± 2.3 | <0.03 |
| Glutamine (amine) | 1.2 ± 0.4 | 0.6 ± 0.2 | NS |
| Glutamate | 0.26 ± 0.07 | 1.4 ± 0.2 | <0.03 |
| Aspartate | 0.56 ± 0.14 | 1.4 ± 0.6 | NS |
Tracer quantities of [13N]ammonia were administered to immobilized rats for 10 min by continuous infusion via a carotid artery. At the end of the infusion, brains were rapidly frozen, deproteinized and analyzed for 13N-labeled metabolites. Endogenous ammonia levels in the brains of control and MSO-treated rats were 0.181 ± 0.14 (n = 12) and 0.818 ± 0.098 (n – 12) μM, respectively. The specific activities of cerebral GDH in the control and MSO-treated rats were 196 ± 6 and 185 ± 4 μmol/h/g wet weight, respectively; the specific activities of cerebral glutamine synthetase in control and MSO-treated rats were 54.2 ± 2.5 and 7.5 ± 2.6 μmol/h/g wet weight, respectively (n = 12–24). Modified from Cooper et al. (1979). The data reported in this table and tables 2 and 3 are the mean ± SEM.
In fact, metabolic compartmentation in brain was first suggested by Berl and colleagues in the early 1960s. These authors found that, after 20 min of infusion of [15N]ammonia via the carotid artery of paralyzed and artificially ventilated cats, the relative enrichment of label in brain metabolites was of the order: glutamate < glutamine (amine) < glutamine (amide). Since glutamate is the only known precursor of glutamine, the findings prompted Berl et al. (1962a) to put forward the hypothesis that cerebral glutamine is synthesized (from blood-derived ammonia) in a compartment of glutamate (small pool) that does not exchange readily with the remainder of brain glutamate (large pool). However, these experiments were conducted with relatively enormous, non-physiological amounts of ammonia such that the animals were comatose, and brain glutamine levels were twice normal. Nevertheless, our experiments supported the Berl hypothesis of brain compartmentation, but under conditions in which the nitrogen steady state was not altered. Another point of interest in Table 1 is that despite the relatively large amount of GDH in rat brain, the overwhelming fate of blood-derived ammonia on entering the brain is incorporation into the amide group of glutamine.
In a second experiment, when [13N]ammonia was infused via a cannula inserted into a cerebral ventricle, similar results were obtained; namely, compartmentation of cerebral ammonia metabolism occurred and the major route for metabolism was incorporation into glutamine (amide). Thus, ammonia entering the brain from the cerebrospinal fluid (CSF), as well as from the blood, is incorporated into glutamine in the small compartment (Cooper et al., 1979).
The anatomical basis for compartmentation and the robust labeling of glutamine (amide) in our experiments can be understood by reference to important work carried out by Norenberg and associates. These workers found that glutamine synthetase in the brain is located predominantly in astrocytes (Martinez-Hernandez et al., 1977; Norenberg and Martinez-Hernandez, 1979). In the brain, astrocyte end feet surround the blood capillaries and underlie the ependyma. Thus, the small compartment is represented by astrocytes, and ammonia entering the brain either from the blood or CSF must first encounter a compartment rich in glutamine synthetase. The large compartment is represented by neurons.
In a third experiment, tracer quantities of [13N]ammonia were administered to rats pretreated with L-methionine S,R-sulfoximine (MSO) – a potent inhibitor of glutamine synthetase (Rowe and Meister, 1970). After a 10-min infusion via an indwelling carotid artery cannula the brains were rapidly frozen and label in various metabolites was determined in a portion of the frozen brain sample (Table 1). The specific activity of GDH and glutamine synthetase in the MSO-treated rat brain was also determined in a separate portion of the frozen brain sample. Treatment with MSO resulted in no significant change in the specific activity of cerebral GDH, whereas the specific activity of glutamine synthetase was reduced by about 86% (Table 1). It is of considerable interest that the relative specific activity of cerebral L-[13N]glutamate in the MSO-treated rats was greater than that of L-[amine-13N]glutamine. This finding shows that compartmentation of ammonia metabolism was no longer intact and that [13N]ammonia had diffused across the small compartment (astrocyte end feet) to the large compartment (neurons) to be acted upon therein in part by GDH. Nevertheless, the amount of label in L-[amide-13N]glutamine was still considerably greater than that in glutamate (plus glutamine amine). From Table 1 it can be seen that when the glutamine synthetase reaction is inhibited there is no significant alternative means to trap ammonia. In this regard, after 10 minutes of infusion only about 20% of label was metabolically trapped in the brains of MSO-treated rats compared to control rats (Cooper et al., 1979). In summary, despite high inherent activity of the GDH reaction in brain (Table 1), this enzyme is not able to compensate for decreased glutamine synthetase activity and the level of ammonia in the brain increased by about 4.5 fold in the MSO-treated rats.
Table 1 shows that after administration of [13N]ammonia, relative labeling of the glutamate pool in the rat brain is much less impressive than the labeling of the glutamate pool in the liver (cf. Fig. 1). Nevertheless, once a portion of the glutamate pool in the brain is labeled, the aspartate pool is also labeled. As with the liver, the activity of aspartate aminotransferase activity is very high in brain and components of the cerebral aspartate aminotransferase reaction are thought to be in thermodynamic equilibrium (Howse and Duffy, 1975).
In a fourth experiment, we measured the short-term metabolic fate of [13N]ammonia in rat brain. After an injection via the carotid artery of a bolus of [13N]ammonia, rats were sacrificed 5 sec later and the forebrain rapidly frozen. Even after a period as short as 5 sec most of the label (>50%) was in a metabolized form, of which ~97% was in the amide group of glutamine. Only small amounts of label were present in glutamate (plus aspartate) and in the amine position of glutamine (Cooper et al., 1979). We estimated that the t½ for incorporation of blood-derived ammonia upon entry into the astrocyte compartment is ≤ 3 sec.
A few years after the studies of normal and MSO-treated rats were published (Cooper et al., 1979) we studied cerebral [13N]ammonia metabolism in chronically hyperammonemic [14-week portacaval-shunted (PCS)] rats (Cooper et al., 1985b). We verified our previous findings (Cooper et al., 1979) that the overwhelming fate of blood-derived [13N]ammonia is incorporation into glutamine in the brains of control rats (Table 2). Furthermore, we showed that this trapping of ammonia as glutamine is also true for the chronically hyperammonemic PCS rats (Table 2). However, the rate of turnover of ammonia to glutamine in the small (astrocyte) compartment was somewhat slower in the PCS rats compared to that of the control rats, possibly as a result in part of a larger ammonia pool and moderately decreased inherent glutamine synthetase activity (Cooper et al., 1985b; Desjardins et al., 1999).
Table 2.
13N-Metabolite profile in rat brain after intracarotid artery injection of a bolus of [13N]ammonia into portacaval-shunted (PCS) rats
| Percent of Label in Various Compounds | |||||
|---|---|---|---|---|---|
| Treatment | Time after Injection (sec) | n | Ammonia | Glutamine | Glutamate |
| None | 5 | 5 | 46.8 ± 0.9 | 52.2 ± 0.8 | 1.0 ± 0.2 |
| None | 8 | 3 | 32.3 ± 1.6 | 66.3 ± 1.8 | 1.4 ± 0.1 |
| 14-wk PCS | 5 | 3 | 61.5 ± 0.9 | 38.0 ± 0.3 | 0.6 ± 0.3 |
| 14-wk PCS | 20 | 3 | 22.9 ± 2.5 | 72.7 ± 2.8 | 1.1 ± 0.2 |
Ammonia concentrations (μM) in control rat blood, PCS rat blood, control brain and PCS brain were 59 ± 8 (n = 19), 158 ± 31 (n = 24), 176 ± 25 (n=14), 274 ± 26 (n = 14), respectively. Modified from Cooper et al., 1985b.
In a second experiment we subjected both normal and 8-week PCS rats to urease treatment (Table 3). Urease treatment is an alternative method to injection of ammonium salts for induction of acute hyperammonemia (Gibson et al., 1974). As expected, this treatment dramatically raised blood and brain ammonia in the control animals (Table 3). As in the chronically hyperammonemic animals (14-week PCS rats; Table 2) the overwhelming metabolic fate of intracarotid administered [13N]ammonia in the acutely hyperammonemic rats was incorporation into L-glutamine (Table 3). However, as noted for the 14-week PCS rats the rate of incorporation of [13N]ammonia into L-glutamine was slowed. In another experiment, 8-week PCS rats were subjected to urease treatment. In both the 8-week PCS rats and the 8-week PCS rats subjected to urease treatment the overwhelming metabolic fate of [13N]ammonia upon entry into the brain was incorporation into L-glutamine (Table 3). To emphasize the point again – relatively little label was found in glutamate.
Table 3.
13N-Metabolite profile in brain after intracarotid artery injection of a bolus of [13N]ammonia into urease-treated rats
| Percent of Label in Various Compounds | |||||
|---|---|---|---|---|---|
| Treatment | Time after Injection (sec) | n | Ammonia | Glutamine | Glutamate |
| None | 5 | 5 | 44.6 ± 1.5 | 54.2 ± 1.8 | 1.3 ± 0.4 |
| 8-wk PCS | 5 | 5 | 60.0 ± 2.4 | 38.5 ± 2.2 | 1.3 ± 0.2 |
| Urease | 5 | 3 | 66.3 ± 6.2 | 32.8 ± 6.0 | 1.0 ± 0.1 |
| 8-wk PCS + urease | 5 | 2 | 60.7, 69.0 | 38.2, 29.6 | 1.1, 1.4 |
Ammonia concentrations (μM) in control rat blood, blood from urease-treated rats, control brain and brains from urease-treated rats were 59 ± 8 (n = 19), 322 ± 99 (n = 7), 176 ± 25 (n=14), 424 ± 49 (n = 14), respectively. Ammonia levels in blood and brain from urease treated PCS rats were not measured. Modified from Cooper et al., 1985b.
8. Integration of the GDH reaction with the glutamine cycle in the brain
The glutamine cycle in brain (Benjamin and Quastel, 1975) is depicted in Fig. 3 (red arrows). Glutamate released into the extracellular space from neurons during neurotransmission is taken up in part by astrocytes, therein to be converted to glutamine by the glutamine synthetase reaction. Glutamine is released from the astrocytes to the neurons, wherein it is converted back to glutamate by the action of glutaminase(s) (Fig. 3). However, the return route for glutamate is unlikely to be stoichiometric with glutamate release as a result in part of loss of glutamine to the circulation. This loss of glutamine is particularly notable during hyperammonemia (reviewed by Cooper and Plum (1987)). Moreover, during hyperammonemia there is a very large increase in brain glutamine, but only small changes in α-ketoglutarate and glutamate (reviewed by Brusilow et al. (2010)). There is very little evidence for net glutamine uptake by the brain. Taken together, the findings indicate that there is a large endogenously generated increase in 5-carbon units (mostly in the form of glutamine) in brain during hyperammonemia. What is the mechanism for the increase in these 5-carbon units? Clearly, the carbon skeleton of glutamine must ultimately be derived from α-ketoglutarate, suggesting that anaplerotic mechanisms are important. Berl and colleagues (1962b) noted that the brain has an unusually robust ability to fix CO2 and that this process is stimulated during hyperammonemia. Three enzymes are known to be present in brain that are capable of fixing CO2, namely pyruvate carboxylase, phosphoenolpyruvate carboxykinase and malic enzyme (Cooper and Plum, 1987). Of these enzymes, astrocytic pyruvate carboxylase (Fig. 3) is probably the most important (Zwingmann, 2007).
Fig. 3.

Cerebral ammonia metabolism. Under normal conditions, ammonia enters the brain mostly by diffusion of the free base (NH3). This ammonia, and that derived from endogenous reactions, is metabolized primarily via incorporation into the amide position of L-glutamine in a reaction catalyzed by astrocytic glutamine synthetase (reaction 1). Although the GDH reaction is freely reversible, the evidence from our 13N-tracer studies suggests that this enzyme is a source of ammonia (reaction 2) rather than a sink for ammonia removal. The glutamate required for glutamine synthesis is derived in part from glutamate released from neurons during neurotransmission. Some of this glutamine may be released to the circulation to maintain nitrogen homeostasis. Another portion may be returned to the neurons, wherein it is converted back to glutamate by the action of glutaminase(s) (reaction 4). The sequence GLU (neurons) → GLU (astrocytes) → GLN (astrocytes) → GLN (neurons) → GLU (neurons) is known as the brain glutamine cycle. Anaplerotic reactions occur in the brain and may be used to replenish 5-C units. Such anaplerotic reactions include CO2 fixation by pyruvate carboxylase (→, reaction 6) and metabolism of branched-chain amino acids (→, reaction 5). BCAAs, branched-chain amino acids; ECS, extracellular space; TCACA, TCA cycle in the astrocytic compartment; TCANA, TCA cycle in the neuronal compartment. From Brusilow et al. (2010) with permission.
The increase in 5-carbon units in brain glutamine during hyperammonemia must also result in increased nitrogen in both the amide and α-amino groups. The amide nitrogen is obviously derived from endogenously produced ammonia and ammonia entering the brain from the blood. But what is the origin of the α-amino nitrogen? Our 13N studies indicate that only a small fraction of the pool of [13N]ammonia in the brain is incorporated into glutamate, even in the hyperammonemic state. The low level of incorporation of label into glutamate relative to that in the amide position of glutamine presumably reflects in part the low Km exhibited by brain glutamine synthetase for ammonia of ~0.18 mM (Pamiljans et al., 1962) compared to the much higher Km value exhibited by brain GDH of ~10 mM (Chee et al., 1979). Because the GDH reaction is fully reversible, the labeling of brain glutamate with 13N does not necessarily imply a small net synthesis of glutamate from ammonia. On the contrary, our data suggest that the GDH reaction is not responsible for a net synthesis of glutamate, but rather is responsible for contributing to the endogenous ammonia pool in the brain (Fig. 3). This role of GDH as an enzyme contributing to the catabolism of glutamate in the brain is now generally accepted (Treberg et al., 2010). Therefore, most of the α-amino nitrogen of glutamate/glutamine in brain must be derived from a source other than the GDH reaction.
The α-amino group of glutamate (and glutamine) may be supplied by linked aminotransferases (Brusilow et al., 2010). There is a net uptake of amino acids from the circulation by the human brain, particularly the branched-chain amino acids (Felig et al., 1973). The only known route for metabolism of the branched-chain amino acids requires an initial transamination step with α-ketoglutarate, thus providing α-amino nitrogen for glutamate synthesis. Many studies suggest that leucine is a particularly important source of glutamate α-amine nitrogen in the brain [reviewed by Cooper and Plum (1987); but see García-Espinosa et al (2007) for a more recent study]. Since leucine is purely ketogenic and its metabolism provides only 2-carbon units for the tricarboxylic acid cycle it cannot be a net source of α-ketoglutarate/glutamate carbon. However, isoleucine can contribute anaplerotic carbon to the TCA cycle, thereby contributing (in addition to CO2 fixation) to the increased net synthesis of α-ketoglutarate/glutamate carbon during hyperammonemia (Johansen et al., 2007).
In summary, the available evidence suggests that although the brain contains considerable amounts of GDH, there is little evidence that the reaction is involved in the net synthesis of glutamate. Rather the consensus is that the GDH contributes to the ammonia pool, which in turn acts as a precursor for the amide group of glutamine. The GDH reaction is linked to aminotransferase reactions, directing the flow of nitrogen toward ammonia (transdeamination). In this regard, the branched chain aminotransferases are especially important. The brain has high levels of both mitochondrial and cytosolic forms of branched-chain aminotransferase (García-Espinosa et al., 2007). In fact, there is recent evidence that the mitochondrial branched-chain aminotransferase forms a metabolon with GDH (Islam et al., 2010). This metabolon would be especially effective in channeling branched-chain amino acid nitrogen toward ammonia (transdeamination), and at the same time providing some carbon for energy metabolism and anaplerosis.
9. Relevance to studies of human brain in vivo
13N has been used as tracer for studying the dynamics of ammonia uptake into human brain for more than 30 years (Lockwood et al., 1979). For example, the permeability-surface area (PS) product of the blood-brain barrier (BBB) for [13N]ammonia has been reported to be increased in liver disease patients (Lockwood et al., 1991). However, this finding has been disputed (Sørensen and Keiding, 2007 Sørensen et al., 2009), but Ahl et al. (2004) previously detected significant changes of the PS product for [13N]ammonia in thalamus, but not in other brain regions, in hepatic encephalopathy patients. Hyperammonemia is a characteristic feature of liver disease patients and is a major contributing factor to the encephalopathy (for a recent review see Brusilow et al., 2010). Moreover, respiratory alkalosis is a feature of hyperammonemic syndromes (Brusilow et al., 2010). Thus, ammonia uptake into brain in hyperammonemic patients may be increased even when blood ammonia levels are near normal by an increase in blood pH and possibly by changes in the BBB PS product.
Recently it has become possible to measure glutamine (or glutamine plus glutamate; Glx) in human brain in vivo by means of nuclear magnetic resonance spectroscopy (MRS) techniques. As expected from previous studies of autopsied brain material (Lavoie et al., 1987), MRS techniques have shown greatly increased levels of brain glutamine (or Glx) in hyperammonemic/liver disease patients (e.g. Córdoba et al., 2002; Rovira et al. 2008; see also Brusilow et al., 2010).
10. Conclusions
Our work with 13N has demonstrated the importance of GDH as a key enzyme in maintaining nitrogen homeostasis in the liver. The GDH reaction is fully reversible and thus can act both as a source of ammonia and as a sink for ammonia removal in the liver as dictated by the needs of that organ (see also Treberg et al., 2010). Although the GDH reaction is presumably also reversible in the brain, the accumulated data suggest that the GDH reaction acts mostly in the direction of ammonia production. The GDH reaction in brain is linked to aminotransferase reactions in channeling catabolized amino acid nitrogen (especially that derived from the branched-chain amino acids) into the metabolic brain ammonia pool (transdeamination). This ammonia then can act as a source of glutamine in the astrocytic compartment. The flow nitrogen may be depicted as follows: Branched chain amino acids → glutamate → ammonia → glutamine. This glutamine can then be exported from the brain as a means of maintaining nitrogen homeostasis and/or act as a precursor for neuronal glutamate (glutamine cycle).
Although our studies with 13N were initiated 30 years ago they are perhaps even more relevant today than they were in the 1980s. Recently, there has been a resurgent interest in the use of PET to study [13N]ammonia uptake and metabolism in patients with hyperammonemic diseases (Sørensen and Keiding, 2007; Sørensen et al., 2009, Goldbecker et al., 2010). Moreover, as noted above MRS techniques to measure glutamine (or Glx) in human brain have been developed (Rovira et al., 2008). It is gratifying to know that our results with [13N]ammonia in rats have stood the test of time and are being quoted in the development of models of nitrogen metabolism in normal and diseased human brain. Any such models must take into account the combined importance of glutamine synthetase, linked aminotransferases and GDH.
Research Highlights.
In the liver GDH and aminotransferases incorporate or remove ammonia from amino acids
In the brain GDH is a source of ammonia for glutamine synthesis
Incorporation of ammonia into glutamine amide in the rat brain is rapid (t1/2 ≤ 3 sec)
Exchange of nitrogen among ammonia, glutamate and aspartate in liver and brain is rapid
Acknowledgments
I thank members of the Cyclotron Facility at the Memorial Sloan Kettering Cancer Center (MSKCC), New York. Special thanks go to Dr. Alan S. Gelbard a retired member of the MSKCC team for his help and encouragement and for providing me with useful criticisms of the present review. The investigations reported in this review were funded in part by United States Public Health Grant AM-16739, NINCDCS grant NS 15665, and Department of Energy Contract EE-77-5-4268.
Abbreviations
- BBB
blood-brain barrier
- BUI
brain uptake index
- CSF
cerebrospinal fluid
- GDH
glutamate dehydrogenase
- HPLC
high performance liquid chromatography
- MSO
L-methionine-S,R-sulfoximine
- PCS
portacaval shunt
- PET
positron emission tomography
- PS
permeability-surface area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahl B, Weissenborn K, van den Hoff J, Fischer-Wasels D, Köstler H, Hecker H, Burchert W. Regional differences in cerebral blood flow and cerebral ammonia metabolism in patients with cirrhosis. Hepatology. 2004;40:73–79. doi: 10.1002/hep.20290. [DOI] [PubMed] [Google Scholar]
- Benjamin AM, Quastel JH. Metabolism of amino acids and ammonia in rat brain cortex slices in vitro: a possible role of ammonia in brain function. J Neurochem. 1975;25:197–206. doi: 10.1111/j.1471-4159.1975.tb06953.x. [DOI] [PubMed] [Google Scholar]
- Berl S, Takagaki G, Clarke DD, Waelsch H. Metabolic compartments in vivo. Ammonia and glutamic acid metabolism in brain and liver. J Biol Chem. 1962a;237:2562–2569. [PubMed] [Google Scholar]
- Berl S, Takagaki G, Clarke DD, Waelsch H. Carbon dioxide fixation in the brain. J Biol Chem. 1962b;237:2570–2573. [PubMed] [Google Scholar]
- Braeuning A, Ittrich C, Köhle C, Hailfinger S, Bonin M, Buchmann A, Schwarz M. Differential gene expression in periportal and perivenous mouse hepatocytes. Faseb J. 2006;273:5051–5061. doi: 10.1111/j.1742-4658.2006.05503.x. [DOI] [PubMed] [Google Scholar]
- Braunstein AE. Principal ways of assimilation & dissimilation of nitrogen in animals. Adv Enzymol Relat Subj Biochem. 1957;19:335–389. (French) [PubMed] [Google Scholar]
- Braunstein AE, Kritzmann MG. Formation of amino-acids by inter-molecular transfer of the amino group. Nature. 1937;140:503–504. [Google Scholar]
- Brusilow SW, Koehler RC, Traystman RJ, Cooper AJL. Astrocyte glutamine synthetase: Importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics. 2010 doi: 10.1016/j.nurt.2010.05.015. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee PY, Dahl JL, Fahien LA. The purification and properties of rat brain glutamate dehydrogenase. J Neurochem. 1979;33:53–60. doi: 10.1111/j.1471-4159.1979.tb11705.x. [DOI] [PubMed] [Google Scholar]
- Cheung C, Cohen NS, Raijman L. Channeling of urea cycle intermediates in situ in permeabilized hepatocytes. J Biol Chem. 1989;264:4038–4044. [PubMed] [Google Scholar]
- Cooper AJL, Gelbard AS. The use of immobilized glutamate dehydrogenase to synthesize 13N-labeled L-amino acids. Anal Biochem. 1981;111:42–48. doi: 10.1016/0003-2697(81)90225-6. [DOI] [PubMed] [Google Scholar]
- Cooper AJL, Plum F. Biochemistry and physiology of brain ammonia. Physiol Rev. 1987;67:440–519. doi: 10.1152/physrev.1987.67.2.440. [DOI] [PubMed] [Google Scholar]
- Cooper AJL, Meister A. An appreciation of Professor Alexander E. Braunstein. The discovery and scope of enzymatic transamination. Biochimie. 1989;71:387–404. doi: 10.1016/0300-9084(89)90169-7. [DOI] [PubMed] [Google Scholar]
- Cooper AJL, Freed BR. Metabolism of [13N]ammonia in rat lung. Neurochem Int. 2005;47:103–118. doi: 10.1016/j.neuint.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Cooper AJL, McDonald JM, Gelbard AS, Gledhill RF, Duffy TE. The metabolic fate of 13N-labeled ammonia in rat brain. J Biol Chem. 1979;254:4982–4992. [PubMed] [Google Scholar]
- Cooper AJL, Nieves E, Coleman AE, Filc-DeRicco S, Gelbard AS. Short-term metabolic fate of [13N]ammonia in rat liver in vivo. J Biol Chem. 1987;262:1073–1080. [PubMed] [Google Scholar]
- Cooper AJL, Nieves E, Rosenspire KC, Filc-DeRicco S, Gelbard AS, Brusilow SW. Short-term metabolic fate of 13N-labeled glutamate, alanine and glutamine (amide) in rat liver. J Biol Chem. 1988;263:12268–12273. [PubMed] [Google Scholar]
- Cooper AJL, Gelbard AS, Freed BR. Nitrogen-13 as a biochemical tracer. Adv Enzymol Relat Areas Mol Biol. 1985a;57:251–356. doi: 10.1002/9780470123034.ch4. [DOI] [PubMed] [Google Scholar]
- Cooper AJL, Mora SN, Cruz NF, Gelbard AS. Cerebral ammonia metabolism in hyperammonemic rats. J Neurochem. 1985b;44:1716–1723. doi: 10.1111/j.1471-4159.1985.tb07159.x. [DOI] [PubMed] [Google Scholar]
- Córdoba J, Sanpedro F, Alonso J, Rovira A. 1H magnetic resonance in the study of hepatic encephalopathy in humans. Metab Brain Dis. 2002;17:415–429. doi: 10.1023/a:1021926405944. [DOI] [PubMed] [Google Scholar]
- Curie I, Joliot F. Artificial production of a new kind of radio-element. Nature. 1934;133:201–202. [Google Scholar]
- del Arco A, Morcillo J, Martínez-Morales JR, Galián C, Martos V, Bovolenta P, Satrústegui J. Expression of the aspartate/glutamate mitochondrial carriers aralar1 and citrin during development and in adult rat tissues. Eur J Biochem. 2002;269:3313–3320. doi: 10.1046/j.1432-1033.2002.03018.x. [DOI] [PubMed] [Google Scholar]
- Desjardins P, Rao KV, Michalak A, Rose C, Butterworth RF. Effect of portacaval anastomosis on glutamine synthetase protein and gene expression in brain, liver and skeletal muscle. Metab Brain Dis. 1999;14:273–280. doi: 10.1023/a:1020741226752. [DOI] [PubMed] [Google Scholar]
- Felig P, Wahren J, Ahlborg G. Uptake of individual amino acids by the human brain. Proc Soc Exp Biol Med. 1973;142:230–231. doi: 10.3181/00379727-142-36994. [DOI] [PubMed] [Google Scholar]
- García-Espinosa MA, Wallin R, Hutson SM, Sweatt AJ. Widespread neuronal expression of branched-chain aminotransferase in the CNS: implications for leucine/glutamate metabolism and for signaling by amino acids. J Neurochem. 2007;100:1458–1468. doi: 10.1111/j.1471-4159.2006.04332.x. [DOI] [PubMed] [Google Scholar]
- Gelbard AS, Clarke LP, McDonald JM, Monahan WG, Tilbury RS, Kuo TYT, Laughlin JS. Enzymatic synthesis and organ distribution studies with 13N-labeled L-glutamine and L-glutamic acid. Radiology. 1975;116:127–132. [Google Scholar]
- Gelbard AS, Benua RS, Reiman RE, McDonald JM, Vomero JJ, Laughlin JS. Imaging of the human heart after administration of L-(N-13)glutamate. J Nucl Med. 1980;21:988–991. [PubMed] [Google Scholar]
- Gelbard AS, Kaseman DS, Rosenspire KC, Meister A. Enzymatic syntheses of carbamyl phosphate, L-citrulline, and N-carbamyl L-aspartate labeled with either 13N or 11C. Int. J Nucl Med Biol. 1985;12:235–242. doi: 10.1016/0047-0740(85)90031-2. [DOI] [PubMed] [Google Scholar]
- Gelbard AS, Cooper AJL, Asano Y, Nieves E, Filc-DeRicco S, Rosenspire KC. Methods for the enzymatic synthesis of tyrosine and phenylalanine labeled with nitrogen-13. Appl Radiat Isot Int J Radiat Appl Instrum Part A. 1990;41:229–233. doi: 10.1016/0883-2889(90)90114-v. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Zimber A, Krook L, Richardson EP, Visek WJ. Brain histology and behavior of mice injected with urease. J Neuropath Exp Neurol. 1974;33:201–221. doi: 10.1097/00005072-197404000-00001. [DOI] [PubMed] [Google Scholar]
- Goldbecker A, Buchert R, Berding G, Bokemeyer M, Lichtinghagen R, Wilke F, Ahl B, Weissenborn K. Blood-brain barrier permeability for ammonia in patients with different grades of liver fibrosis is not different from healthy controls. J Cereb Blood Flow Metab. 2010;30:1384–1393. doi: 10.1038/jcbfm.2010.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häussinger D. Nitrogen metabolism in liver: structural and functional organization and physiological relevance. Biochem J. 1990;267:281–290. doi: 10.1042/bj2670281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häussinger D. Hepatic glutamine transport and metabolism. Adv Enzymol Relat Areas Mol Biol. 1998;72:43–86. doi: 10.1002/9780470123188.ch3. [DOI] [PubMed] [Google Scholar]
- Howse DC, Duffy TE. Control of the redox state of the pyridine nucleotides in the rat cerebral cortex. Effect of electroshock-induced seizures. J Neurochem. 1975;24:935–940. doi: 10.1111/j.1471-4159.1975.tb03658.x. [DOI] [PubMed] [Google Scholar]
- Islam MM, Nautiyal M, Wynn RM, Mobley JA, Chuang DT, Hutson SM. Branched-chain amino acid metabolon: interaction of glutamate dehydrogenase with the mitochondrial branched-chain aminotransferase (BCATm) J Biol Chem. 2010;285:265–276. doi: 10.1074/jbc.M109.048777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen ML, Bak LK, Schousboe A, Iversen P, Sørensen M, Keiding S, Vilstrup H, Gjedde A, Ott P, Waagepetersen HS. The metabolic role of isoleucine in detoxification of ammonia in cultured mouse neurons and astrocytes. Neurochem Int. 2007;50:1042–1051. doi: 10.1016/j.neuint.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Jungermann K, Kietzmann T. Zonation of parenchymal and nonparenchymal metabolism in liver. Annu Rev Nutr. 1996;16:179–203. doi: 10.1146/annurev.nu.16.070196.001143. [DOI] [PubMed] [Google Scholar]
- Lavoie J, Giguère JF, Layrargues GP, Butterworth RF. Amino acid changes in autopsied brain tissue from cirrhotic patients with hepatic encephalopathy. J Neurochem. 1987;49:692–697. doi: 10.1111/j.1471-4159.1987.tb00949.x. [DOI] [PubMed] [Google Scholar]
- Lockwood AH, McDonald JM, Reiman RE, Gelbard AS, Laughlin JS, Duffy TE, Plum F. The dynamics of ammonia metabolism in man. Effects of liver disease and hyperammonemia. J Clin Invest. 1979;63:449–60. doi: 10.1172/JCI109322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood AH, Finn RD, Campbell JA, Richman TB. Factors that affect the uptake of ammonia by the brain: the blood-brain pH gradient. Brain Res. 1980;181:259–266. doi: 10.1016/0006-8993(80)90611-3. [DOI] [PubMed] [Google Scholar]
- Lockwood AH, Yap EW, Wong WH. Cerebral ammonia metabolism in patients with severe liver disease and minimal hepatic encephalopathy. J Cereb Blood Flow Metab. 1991;11:337–341. doi: 10.1038/jcbfm.1991.67. [DOI] [PubMed] [Google Scholar]
- Lowenstein JM. Ammonia production in muscle and other tissues. Physiol Rev. 1972;52:382–414. doi: 10.1152/physrev.1972.52.2.382. [DOI] [PubMed] [Google Scholar]
- Lowenstein JM, Goodman MN. The purine nucleotide cycle in skeletal muscle. Fed Proc. 1978;37:2308–2312. [PubMed] [Google Scholar]
- Maher AD, Kuchel PW, Ortega F, de Atauri P, Centelles J, Cascante M. Mathematical modelling of the urea cycle. A numerical investigation into substrate channelling. Eur J Biochem. 2003;270:3953–3961. doi: 10.1046/j.1432-1033.2003.03783.x. [DOI] [PubMed] [Google Scholar]
- Martinez-Hernandez A, Bell KP, Norenberg MD. Glutamine synthetase: glial localization in brain. Science. 1977;195:1356–1358. doi: 10.1126/science.14400. [DOI] [PubMed] [Google Scholar]
- Moorman AFM, de Boer PA, Watford M, Dingemanse MA, Lamers WH. Hepatic glutaminase mRNA is confined to part of the urea cycle domain in the adult rodent liver lobule. FEBS Lett. 1994;356:76–80. doi: 10.1016/0014-5793(94)01230-x. [DOI] [PubMed] [Google Scholar]
- Norenberg MD, Martinez-Hernandez A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979;161:303–310. doi: 10.1016/0006-8993(79)90071-4. [DOI] [PubMed] [Google Scholar]
- Pamiljans V, Krishnaswamy PR, Dumville G, Meister A. Studies on the mechanism of glutamine synthesis; Isolation and properties of the enzyme from sheep brain. Biochemistry. 1962;1:153–158. doi: 10.1021/bi00907a023. [DOI] [PubMed] [Google Scholar]
- Poyck PP, Hoekstra R, Vermeulen JL, van Wijk AC, Chamuleau RA, Hakvoort TB, van Gulik TM, Lamers WH. Expression of glutamine synthetase and carbamoylphosphate synthetase I in a bioartificial liver: markers for the development of zonation in vitro. Cells Tissues Organs. 2008;188:259–269. doi: 10.1159/000121609. [DOI] [PubMed] [Google Scholar]
- Raichle ME, Larson KB. The significance of the NH3-NH4+ equilibrium on the passage of 13N-ammonia from blood to brain. A new regional residue detection model. Circ Res. 1981;48:913–937. doi: 10.1161/01.res.48.6.913. [DOI] [PubMed] [Google Scholar]
- Rosenspire KC, Schwaiger M, Mangner TJ, Hutchins GD, Sutorik A, Kuhl DE. Metabolic fate of [13N]ammonia in human and canine blood. J Nucl Med. 1990;31:163–167. [PubMed] [Google Scholar]
- Rovira A, Alonso J, Córdoba J. MR imaging findings in hepatic encephalopathy. AJNR Am J Neuroradiol. 2008;29:1612–1621. doi: 10.3174/ajnr.A1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe WB, Meister A. Identification of L-methionine-S-sulfoximine as the convulsant isomer of methionine sulfoximine. Proc Natl Acad Sci USA. 1970;66:500–506. doi: 10.1073/pnas.66.2.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben S, Hassid WZ, Kamen MD. Radioactive nitrogen in the study of N2 fixation by non-leguminous plants. Science. 1940;91:578–579. doi: 10.1126/science.91.2372.578. [DOI] [PubMed] [Google Scholar]
- Schultz V, Lowenstein JM. The purine nucleotide cycle. Studies of ammonia production and interconversions of adenine and hypoxanthine nucleotides and nucleosides by rat brain in situ. J Biol Chem. 1978;253:1938–1943. [PubMed] [Google Scholar]
- Sørensen M, Keiding S. New findings on cerebral ammonia uptake in HE using functional 13N-ammonia PET. Metab Brain Dis. 2007;22:277–284. doi: 10.1007/s11011-007-9066-1. [DOI] [PubMed] [Google Scholar]
- Sørensen M, Munk OL, Keiding S. Backflux of ammonia from brain to blood in human subjects with and without hepatic encephalopathy. Metab Brain Dis. 2009;24:237–242. doi: 10.1007/s11011-008-9126-1. [DOI] [PubMed] [Google Scholar]
- Treberg JR, Brosnan ME, Watford M, Brosnan JT. On the reversibility of glutamate dehydrogenase and the source of hyperammonemia in the hyperinsulinism/hyperammonemia syndrome. Adv Enzyme Reg. 2010;50:34–43. doi: 10.1016/j.advenzreg.2009.10.029. [DOI] [PubMed] [Google Scholar]
- Watford M. The urea cycle: a two-compartment system. Essays Biochem. 1991;26:49–58. [PubMed] [Google Scholar]
- Watford M, Smith EM. Distribution of hepatic glutaminase activity and mRNA in perivenous and periportal rat hepatocytes. Biochem J. 1990;267:265–267. doi: 10.1042/bj2670265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J. 1967;103:514–527. doi: 10.1042/bj1030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwingmann C. The anaplerotic flux and ammonia detoxification in hepatic encephalopathy. Metab Brain Dis. 2007;22:235–249. doi: 10.1007/s11011-007-9069-y. [DOI] [PubMed] [Google Scholar]