

Abstract
Carboxylesterases (CE) are ubiquitous enzymes found in both human and animal tissues and are responsible for the metabolism of xenobiotics. This includes numerous natural products, as well as a many clinically used drugs. Hence the activity of these agents is likely dependent upon the levels and location of CE expression. We have recently identified benzil is a potent inhibitor of mammalian CEs, and in this study, we have assessed the ability of analogues of this compound to inhibit these enzymes. Three different classes of molecules were assayed: One containing different atoms vicinal to the carbonyl carbon atom and the benzene ring [PhXC(O)C(O)XPh, where X = CH2, CHBr, N, S, or O]; a second containing a panel of alkyl 1,2-diones demonstrating increasing alkyl chain length; and a third consisting of a series of 1-phenyl-2-alkyl-1,2-diones. In general, with the former series of molecules, heteroatoms resulted in either loss of inhibitory potency (when X =N), or conversion of the compounds into substrates for the enzymes (when X = S or O). However, the inclusion of a brominated methylene atom resulted in potent CE inhibition. Subsequent analysis with the alkyl diones [RC(O)C(O)R, where R ranged from CH3 to C8H17] and 1-phenyl-2-alkyl-1,2-diones [PhC(O)C(O)R where R ranged from CH3 to C6H13], demonstrated that the potency of enzyme inhibition directly correlated with the hydrophobicity (clogP) of the molecules. We conclude from these studies that that the inhibitory power of these 1,2-dione derivatives depends primarily upon the hydrophobicity of the R group, but also on the electrophilicity of the carbonyl group.
1. Introduction
Carboxylesterases (CE1) are enzymes found in a wide range of organisms, from humans to bacteria [1]. CEs are known to be involved in the hydrolysis of ester-containing xenobiotics [1] using a catalytic serine within a Ser-His-Glu triad to initiate hydrolysis of the molecule. The products that result from this reaction are the respective alcohol and carboxylic acid [2, 3]. Two major CEs exist in humans, human liver CE (hCE1; CES1) and human intestinal CE (hiCE; CES2) [2, 3] with hCE1 being primarily expressed in the liver, while hiCE is found in both the liver and the small intestine. A third human CE, hBr3 (CES3), has been described, but very little is known about the levels of expression and/or the substrate specificity of this enzyme [4]. While the exact role of these proteins in mammals is unclear, and endogenous substrates have not been definitively identified, the patterns of expression are consistent with them playing a protective role. Furthermore, since the carboxylic ester chemotype is present in numerous agents including natural products, pesticides and clinically used drugs, de facto they a substrates for these enzymes [5–10]. Hence drug hydrolysis, which can result in either activation or inactivation of the molecule, is dependent upon the levels of CE expressed in exposed tissues and the substrate specificity of the protein.
One such chemotherapeutic agent that is metabolized by CEs is the anticancer drug irinotecan (CPT-11, 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin [6, 9, 11]). CPT-11 is a carbamate-derived prodrug that is hydrolyzed by hiCE into its active metabolite, SN-38 (7-ethyl-10-hydroxycamptothecin) [12, 13]. The latter is a potent topoisomerase I poison which exerts its toxicity at low nanomolar concentrations. We have previously demonstrated the efficient activation of CPT-11 by a rabbit liver CE (rCE) and used this enzyme to modulate tumor cells sensitivity to this drug [14–16]. The development of clinical approaches using this technology is currently underway. However, the toxicity of CPT-11 (delayed diarrhea) is in part due to high levels of hiCE that are expressed in the intestine [6, 17]. Therefore, identifying specific hiCE inhibitors which could be used in combination with CPT-11 to ameliorate the delayed diarrhea, may have clinical utility [2, 3]. Previously, we determined that small molecules containing the ethane 1,2-dione moiety were potent inhibitors of CEs [18–21]. These compounds demonstrated no activity toward human acetyl- or butyrylcholinesterase, and one class, the benzils, inhibited hiCE intracellularly and modulated cellular response to CPT-11 [22]. Preliminary studies indicated that the planarity of the ethane-1,2-dione group could determine specificity of enzyme inhibition and that that inhibitor potency was increased when phenyl groups were present within the molecule. In this study, we sought to determine the chemical requirements for inhibition of CEs by ethane 1,2-diones and to assess whether nucleophilic attack by the serine Oγ atom within the active site is the mechanism by which enzyme inhibition occurs. This included evaluating the role of the atoms immediately adjacent to the carbonyl groups towards inhibitor potency and to assess the necessity for the inclusion of the phenyl rings. Our results indicate that the atoms bonded to the carbonyl groups in the 1,2-diones play a major role towards inhibitor potency, both by moderating carbonyl electrophilicity, and compound hydrophobicity. Indeed, molecules with clogP > 2.75 were much potent inhibitors of mammalian CEs than more hydrophilic compounds (clogP < 2.75). Furthermore, aromaticity within the molecule is not a requirement for enzyme inhibition.
2. Materials and Methods
2.1 Chemicals and General Chemistry
All solvents and starting materials were purchased from Sigma Aldrich (St. Louis, MO) and were ACS grade or better. Benzil (1), butane-2,3-dione (8), hexane-3,4-dione (9), and 1-phenyl-1,2-propanedione (17) were obtained from Sigma Aldrich. Diphenyl ethanedioate (4), 1,4-diphenylbutane-2,3-dione (5), 1,4-dibromo-1,4-diphenylbutane-2,3-dione (6) and 1-bromo-1,4-diphenylbutane-2,3-dione (7) were purchased from MDPI (Basel, Switzerland). Octane-4,5-dione (10) was obtained from the Developmental Therapeutics Program at NIH and 1,2-dicyclohexylethane-1,2-dione (16) was synthesized as previously described [20, 23]. The structures of the compounds described in this paper are illustrated in Table 1.
Table 1.
Structures, enzyme inhibition constants and kinetic parameters for the aromatic ethane-1,2-diones.
| ID | Name | Structure | clogP | Ki (nM) | hiCE irreversible inhibition (%) | ||||
|---|---|---|---|---|---|---|---|---|---|
| hCE1 | hiCE | rCE | hAChE | hBChE | |||||
| 1 | Benzil |
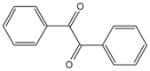
|
3.02 | 15 ± 1.9a | 45 ± 3.2a | 103 ± 19a | >100,000 | >100,000 | <8 |
| 2 | Oxanilide |
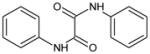
|
2.24 | >100,000 | >100,000 | >100,000 | >100,000 | >100,000 | ND |
| 3 | S1, S2-Diphenyl ethane bis(thioate) |
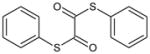
|
3.62 | >100,000 | >100,000 | >100,000 | >100,000 | >100,000 | ND |
| 4 | Diphenyl ethanedioate |
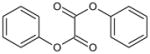
|
3.39 | >100,000 | >100,000 | >100,000 | >100,000 | >100,000 | ND |
| 5 | 1,4-Diphenylbutane-2,3-dione |
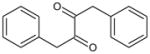
|
3.19 | 3,080 ± 230 | 21,610 ± 2,270 | >100,000 | >100,000 | >100,000 | ND |
| 6 | 1,4-Dibromo-1,4-diphenyl butane-2,3-dione |
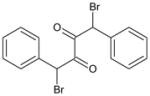
|
4.07 | 107 ± 21 | 334 ± 25 | 20.5 ± 3.3 | >100,000 | >100,000 | <5 |
| 7 | 1-Bromo-1,4-diphenyl butane-2,3-dione |
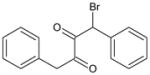
|
3.71 | 2,630 ± 70 | 267 ± 38 | 3,680 ± 320 | >100,000 | >100,000 | ND |
Data taken from Wadkins et al. [21]
For compounds synthesized as part of these studies, melting points were determined using a Mel-temp (Barnstead International, Dubuque, IA) and purity and structures were assessed by TLC, NMR, HRMS and total C, H, N, O, S analysis (as appropriate). Complete physical data is presented as Supplementary Information.
2.2 Enzymes
Pure hCE1, hiCE, and rabbit liver CE (rCE) were obtained by purification from baculovirus infected Spodoptera frugiperda cells as previously described [24]. Human acetylcholinesterase (hAChE) and butyrylcholinesterase (hBChE) were obtained from Sigma Biochemicals (St. Louis, MO).
2.3 Synthesis of oxanilide (N, N′-diphenylethanediamide (2))
Oxanilide was synthesized from aniline and oxalyl chloride as previously described [25]. Physical parameters were consistent with that reported previously. Identity was confirmed by melting point, NMR, and elemental analysis.
2.4 Synthesis of S1, S2-diphenyl ethanebis(thioate) (3)
The named compound was synthesized by condensation of thiophenol with oxalyl chloride in dichloromethane [26]. Identity was confirmed by melting point, NMR, and elemental analysis.
2.5 Synthesis of alkyl diones and phenyl alkyl diones
Alkyl diones (11–15) were synthesized by oxidation of the corresponding alkyne using potassium permanganate in an acetone/water phase transfer system [27]. Routinely, 10.8 mM 1-phenyl-1-alkyne dissolved in 420 ml acetone was added to 240 ml aqueous 32 mM NaHCO3 and 22 mM MgSO4. Potassium permanganate (6.65 g, 42 mmol) was added in one portion and the reaction stirred for 4 hours at rt. Small portions of NaNO2 and 10% H2SO4 were added until the reaction was clear (~5 g and 50 ml, respectively) and solid NaCl was then added to achieve saturation. Samples were worked up using standard approaches and compound identity was confirmed by 1H NMR, elemental analysis (Microlab Inc., Norcross, GA), and HRMS (UIUC School of Chemical Sciences, Urbana, IL). Typically yields were greater than 70% and compound physical parameters were consistent with those previously reported.
2.6 cLogP calculations
cLogP values were calculated using ChemSilico Predict v2.0 software (ChemSilico LLC, Tewksbury, MA).
2.7 Determination of Ki values using o-nitrophenyl acetate (o-NPA) as a substrate
Inhibition of CEs was assessed using a multiwell plate spectrophotometric assay with 3 mM o-NPA as a substrate [21, 28]. Inhibitors (100μM) and substrate were aliquoted into wells of a 96-well plate and enzyme was added. The rate of change in absorbance at 420 nm was recorded at 15 s intervals for 5 min and data was compared to wells without inhibitor. Data was expressed as the yield of o-nitrophenol per minute per milligram of protein. Compounds demonstrating greater than 50% reduction of CE activity were re-screened with dilutions ranging from 1 pM to 100 μM. Data was fitted to the following equation to determine the mode of inhibition [29]:
where i is the fractional inhibition, [I] is the inhibitor concentration, [s] is the substrate concentration, α is the change in affinity of the substrate for enzyme, β is the change in the rate of enzyme substrate complex decomposition, Ks is the dissociation constant for the enzyme substrate complex, and Ki is the inhibitor constant. Examination of the curve fits where α ranged from 0 to ∞ and β ranged from 0 to 1 was performed using GraphPad Prism software and Perl data language [21, 28]. Curves were generated and analyzed using Akaike’s information criteria [30, 31]. Using the equation generated by Prism considered to be the best fit for the datasets, Ki values were computed [21, 28].
2.8 Inhibition of acetylcholinesterase and butyrylcholinesterase
Inhibition of hAChE and hBChE by compounds was determined as previously described using 1 mM of either acetylthiocholine (AcTCh) or butyrylthiocholine (BuTCh), respectively, as substrates [32–35].
2.9 Irreversible enzyme inhibition assays
To assess irreversible enzyme inhibition by selected compounds, inhibitors were added to hiCE at a final concentration of 10 μM in Hepes pH 7.4 and incubated on ice for 40 mins. At this time, samples were diluted 1:500 and enzyme activity was determined using o-NPA as a substrate.
2.10 Enzyme inhibition assays and determination of Ki values using CPT-11 as a substrate
CPT-11 (20 μM) and inhibitor concentrations ranging from 0–100 μM were pre-incubated for 2 min at 37ºC in 50 mM HEPES pH 7.4. Purified hiCE (25 U) was then added and the sample was incubated at 37ºC for 5 min. The reaction was terminated by addition of 100 μl of acid methanol and the amount of SN-38 in the sample was determined using high performance liquid chromatography [14, 36]. Ki values were then determined using Prism software as described earlier.
2.11 Determination of ability of compounds to act as substrates
Compounds 2, 3, or 4 (200 μM) were incubated with enzyme for varying time periods (1, 5, or 20 min) at either rt or 37°C in a total volume of 200 μl of 50 mM Hepes pH 7.4 and reactions were terminated by addition of acetonitrile. Samples were then evaluated by reverse-phase high-performance liquid chromatography and products were identified by UV detection. These results were compared to samples without enzyme and to samples with marker compounds consisting of aniline, thiophenol, or phenol.
2.12 Intracellular CE inhibition assay
Intracellular CE inhibition was assessed using the substrate 4-methylumbelliferone acetate (4-MUA) as previously described [22]. Briefly, 106 cells were suspended in PBS with stirring in a cuvette and were pre-incubated with inhibitor (10 μM) for 1 hour. 4-MUA was then added (100 μM) and fluorescence was determined using a Hitachi F-2000 fluorimeter, with an excitation wavelength of 365 nm and an emission wavelength of 460 nm. Data was collected over 2 minutes and the percentage of inhibition of product formation as compared to cell not treated with inhibitor was determined by assessing the difference in fluorescence intensity at 30 s.
2.13 Growth inhibition assay
Growth inhibition assays using inhibitors and CPT-11 were undertaken as previously described [15, 22, 37]. Briefly, cells were plated in 6-well plates (50,000 cells/well) and allowed to adhere overnight. Following pre-incubation with inhibitor (10 μM) for 1 hour, cells were treated for 2 hours with both inhibitor and varying concentrations of CPT-11 (0–100 μM). Cells were then washed and allowed to grow in drug-free medium for 4 days (~3 cell doublings), prior to harvesting and counting using a Coulter Z2 counter (Beckman Coulter, Fullerton, CA). Routinely, all concentrations were conducted in triplicate and the percentage of surviving cells was determined for each treatment condition and compared to control cells not exposed to inhibitor. Survival curves were generated and IC50 values were calculated using Prism software (GraphPad, San Diego, CA).
2.14 Molecular dynamics simulations
Molecular dynamics simulations to evaluate the dihedral angle of the 1,2-dione bond [(O)CC(O)] were performed as previously described [19]. Briefly, analyses were run using the MM3 module of Alchemy (Tripos Inc., St. Louis, MO) with angle measurements recorded every 10 fs, over a total time frame of 15 ps, using a time step of 1 fs.
2.15 Computational chemistry
All computational analyses were performed using Gaussian 03 software (Gaussian, Wallingford, CT). Molecules were constructed using Gauss-View and geometry optimization was performed using the density functional theory method using the B3LYP hybrid functional [38] with the 6-31G(d, p) basis set [39]. Charge distribution was then visualized using Molden (Centre for Molecular and Biomolecular Informatics, Radboud University Nijmegen Medical Centre, Nijmegen, Netherlands).
3. Results
3.1 Analysis of CE inhibition by 1,4-diphenylbutane-2,3-dione analogues
To assess the role of the atom vicinal to the carbonyl carbon atoms in the molecule towards enzyme inhibition, we synthesized a series of 1,4-diphenylbutane-2,3-dione analogues containing a variety of different moieties in the α-position. In general, symmetrical compounds were used to ensure that that any inhibitory effects would not be as a result of competitive inhibition by two different sites of nucleophilic attack by the protein.
3.1.1 Oxanilide, S1, S2-diphenyl ethanebis(thioate) and diphenyl ethanedioate
Oxanilide (2) was not an inhibitor of hCE1, hiCE or rCE (Table 1) and also lacked activity towards the cholinesterases. Similarly, analogues containing either sulfur (3; S1, S2-diphenyl ethanebis(thioate)) or oxygen (4; diphenyl ethanedioate) yielded molecules that were ineffective at enzyme inhibition (Table 1). However, unlike oxanilide, the latter two compounds underwent spontaneous hydrolysis in aqueous solution liberating either thiophenol or phenol, respectively (data not shown). Furthermore, this reaction was not enhanced by the addition of CE. While detailed kinetics were not determined, greater than 40% of each compound was hydrolyzed within 5 mins of addition to buffered aqueous (pH 7.4) solution. While this was expected for 3 (as it is an activated thioester), it was unclear whether compound 4 would be subject to hydrolysis. Our results confirm that the ester is also labile under these conditions.
3.1.2 1,4-Diphenylbutane-2,3-dione, 1-bromo-1,4-diphenylbutane-2,3-dione and 1,4-dibromo-1,4-diphenylbutane-2,3-dione
Insertion of a carbon atom in the α-position (5; 1,4-diphenylbutane-2,3-dione) resulted in a molecule that was effective at CE inhibition, but the potency was ~200- to 480-fold weaker than that seen with benzil (Table 1). The mode of enzyme inhibition was deemed to be partially competitive (i.e., indicating that while the small molecule structurally resembled a CE substrate, they were unable to reduce substrate hydrolysis to 0%, even at infinite inhibitor concentrations). The brominated derivatives, 1,4-dibromo-1,4-diphenylbutane-2,3-dione (compound 6) and 1-bromo-1,4-diphenylbutane-2,3-dione (compound 7), were considerably more active, with the dibromo analogue ~30- to 5000-fold more potent inhibitor than 5. These results suggest that the activation of the carbonyl group was a major effector in improving inhibitor function. While compound 7 was generally more potent than 5, dramatic improvements in the inhibition constants for this molecule were only observed with hiCE (Table 1).
It is possible the bromine analogues may change the mechanism of enzyme inhibition by allowing nucleophilic attack on the methylene atom rather than the carbonyl carbon. This would then result in irreversible inhibition of the protein. Therefore, we evaluated whether compound 6 could inactivate hiCE following incubation with the protein. As indicated in Table 1 however, no irreversible inhibition of the enzyme was observed with this analogue.
3.1.3 Flexibility of the (O)C-C(O) bond
In previous studies, we have seen a correlation between the flexibility of rotation around the 1,2-dione bond and the inhibitory potency of the molecules. Typically, compounds that adopted a more rigid structure tended to be more potent CE inhibitors [19]. Therefore using molecular dynamics approaches, we evaluated the rotation of this bond for benzil and compound 5. As indicated in Figure 1, the 1,2-dione in benzil rapidly adopted a stable conformation with a dihedral angle of ~ 230°. In contrast, with 1,4-diphenylbutane-2,3-dione, rotation around the dione continued to oscillate over the entire course of the dynamics run. Since compound 5 is a poorer inhibitor than benzil, these results are consistent with those seen previously for a panel of heteroaromatic ethane-1,2-diones, where increased oscillation was observed with weaker molecule potency [19].
Figure 1.

Plot of the dihedral angle for the 1,2-dione group [(O)CC(O)] versus time for benzil (black line) and 1,4-diphenylbutane-2,3-dione (5; blue line) for the molecular dynamics simulations.
3.2 Synthesis and enzyme inhibition by alkyl-1,2-diones
Since the active sites of the mammalian CEs are highly hydrophobic [2], we hypothesized that small molecules with larger clogP values would be more potent enzyme inhibitors. Therefore, we obtained, or generated, a series of alkyl-1,2-diones ranging from butane-2,3-dione to octadecane-9,10-dione. Synthesis was accomplished by oxidation of the corresponding alkyne using potassium permanganate in an acetone/water phase transfer system. Yields were routinely greater than 70% and most compounds were pale yellow crystalline solids. Chemical and physical properties for these compounds are presented as supplementary information.
CE inhibition by the alkyl-1,2-diones was assessed for both hCE1 and hiCE, as well as rCE, using o-NPA as a substrate (Table 2). As can be seen, as the compounds become larger, the molecules become more potent inhibitors. Since this is likely due to the increase in the hydrophobicity of the compounds, we calculated the clogP of the alkyl diones using commercially available software. As indicated in Table 2, both inhibitor potency and the clogP values increased with increasing chain length. As an example, compound 15 containing eight carbon atoms in the alkyl group, was a very potent inhibitor of the mammalian CEs (0.84 nM and 5.7 nM for hCE1 and hiCE, respectively) and demonstrated a clogP value of 6.03. As before, the mode of enzyme inhibition by these compounds was found to be partially competitive.
Table 2.
Structure, logP and Ki values for enzyme inhibition for alkyl 1,2-diones.
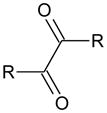 | ||||||||
|---|---|---|---|---|---|---|---|---|
| ID | Name | R | clogP | Ki (nM) | ||||
| hCE1 | hiCE | rCE | hAChE | hBChE | ||||
| 8 | Butane-2,3-dione | CH3 | −0.47 | >100,000 | >100,000 | >100,000 | >100,000 | >100,000 |
| 9 | Hexane 3,4-dione | C2H5 | 0.75 | >100,000 | >100,000 | >100,000 | >100,000 | >100,000 |
| 10 | Octane-4,5-dione | C3H7 | 1.89 | 8,060 ± 330 | 4,670 ± 260 | 17,780 ± 1,290 | >100,000 | >100,000 |
| 11 | Decane-5,6-dione | C4H9 | 2.92 | 46.6 ± 9.3 | 189 ± 15 | 944 ± 75 | >100,000 | >100,000 |
| 12 | Dodecane-6,7-dione | C5H11 | 3.94 | 3.0 ± 0.4 | 57.7 ± 2.2 | 38.6 ± 1.6 | >100,000 | >100,000 |
| 13 | Tetradecane-7,8-dione | C6H13 | 4.89 | 4.8 ± 0.3 | 76.4 ± 9.4 | 15.1 ± 0.9 | >100,000 | >100,000 |
| 14 | Hexadecane-8,9-dione | C7H15 | 5.68 | 7.6 ± 1.6 | 25.6 ± 2.3 | 6.5 ± 1.9 | >100,000 | >100,000 |
| 15 | Octadecane-9,10-dione | C8H17 | 6.03 | 0.84 ± 0.28 | 5.7 ± 1.4 | 9.0 ± 3.8 | >100,000 | >100,000 |
| 16 | 1,2-Dicyclohexylethane-1,2-dione | Cyc. C6H11 | 3.93a | 5.0 ± 0.7a | 72 ± 7.0a | 27 ± 7.7 | >100,000 | >100,000 |
Data taken from Hyatt et al [20].
3.3 Carboxylesterase inhibition by 1-phenyl-2-alkyl-1,2-diones
Having observed potent inhibition by the alkyl-1,2-diones, we next evaluated a series of phenyl alkyl derivatives. These were chosen since our original data demonstrated that benzil (1) was an excellent inhibitor and that potency, and potentially selectivity, might be afforded by the alkyl chain. Therefore, we synthesized a series of 1-phenyl-1,2-alkyldiones using similar methodology to that described for the alkyl derivatives. Similar yields (~70%) were obtained and all physical parameters were consistent with their structure (see Supporting Information).
As indicated in Table 3, potent enzyme inhibition was observed when the alkyl chain exceeded 4 carbon atoms. For example, 1-phenyl-1,2-octanedione (22), the most potent compound synthesized, yielded Ki values of 2.2 nM, 28.7 nM and 28.1 nM for hCE1, hiCE and rCE, respectively. In general, these molecules did not generate inhibition constants as low as that seen for the alkyl-1,2-diones, however these molecules were clearly effective CE inhibitors. Again, the mode of enzyme inhibition was determined to be partially competitive, and inhibitor potency appeared to correlate with the clogP of the compounds.
Table 3.
Structure, logP and Ki values for enzyme inhibition for 1-phenyl-alkyl-1,2-diones.
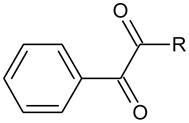 | ||||||||
|---|---|---|---|---|---|---|---|---|
| ID | Name | R | clogP | Ki (nM) | ||||
| hCE1 | hiCE | rCE | hAChE | hBChE | ||||
| 17 | Phenyl-1,2-propanedione | CH3 | 1.07 | 5,270 ± 1,730 | 1,840 ± 260 | 4,930 ± 1,320 | >100,000 | >100,000 |
| 18 | Phenyl-1,2-butanedione | C2H5 | 1.66 | 1,550 ± 380 | 2,420 ± 150 | 12,500 ± 1,980 | >100,000 | >100,000 |
| 19 | Phenyl-1,2-pentanedione | C3H7 | 2.25 | 102 ± 12 | 401 ± 21 | 2,710 ± 170 | >100,000 | >100,000 |
| 20 | Phenyl-1,2-hexanedione | C4H9 | 2.83 | 26.1 ± 2.2 | 72.7 ± 5.1 | 337 ± 22 | >100,000 | >100,000 |
| 21 | Phenyl-1,2-heptanedione | C5H11 | 3.42 | 6.65 ± 0.22 | 45.5 ± 6.1 | 110 ± 10 | >100,000 | >100,000 |
| 22 | Phenyl-1,2-octanedione | C6H13 | 4.01 | 2.19 ± 0.08 | 28.7 ± 2.3 | 28.1 ± 1.8 | >100,000 | >100,000 |
3.4 Correlation between clogP and inhibitory potency for the 1,2-diones
Since we saw a trend towards increased inhibitory potency with higher clogP values for both the alkyl-1,2-diones, and 1-phenyl-1,2-alkyldiones, we evaluated whether correlations could be determined for these parameters. As indicated in Figure 2, linear correlations could be described for both individual datasets (panel A – alkyl-1,2-diones; panel B - 1-phenyl-2-alkyl-1,2-diones), as well as the combined values for all compounds (panel C). The linear regression analyses yielded r2 values ranging from 0.72 to 0.99, depending upon both the enzyme and the class of compounds. Overall, these data indicate that the clogP and Ki values for all of the CEs and inhibitors that were evaluated appear to be related, with increasing clogP resulting in more potent enzyme inhibition.
Figure 2.

Graphs demonstrating the correlation between clogP and Ki values for the alkyl 1,2-diones (A), the 1-phenyl-1,2-alkyldiones (B) and composite results for both classes of compounds (C). Data are shown for the three different enzymes hiCE (blue), hiCE (red) and rCE (black) and the corresponding linear correlation coefficient is indicated on the graph.
3.5 Modulation of CPT-11 metabolism and intracellular carboxylesterase inhibition
Having identified several inhibitors that could potently inhibit hiCE, we sought to determine the Ki values for this enzyme when using CPT-11 as a substrate. This was undertaken only with compounds that demonstrated reasonable efficacy at inhibiting o-NPA hydrolysis (compounds 11–15 and 18–22). As indicated in Table 4, the majority of these molecules were effective at inhibiting hiCE-mediated CPT-11 metabolism, with compound 15 (octadecane-9,10-dione) yielding the lowest Ki value (~24 nM). Having established that these inhibitors could modulate CPT-11 hydrolysis in vitro, we assessed the ability of these compounds to inhibit hiCE intracellularly. This was determined using 4-MUA as a substrate in a fluorometric assay and would provide a measure of the cell permeability of the molecules [22]. As shown in Table 4, all of the inhibitors were effective at inhibiting hiCE within U373MG cells, with the most potent inhibition observed with compound 22 (1-phenyl-2-octane-1,2-dione).
Table 4.
Ki values for the inhibition of CPT-11 hydrolysis by mammalian CEs with alkyl- and phenyl alkyl-1,2-diones and intracellular enzyme inhibition using 4-MUA as a substrate.
| ID | Alkyl chain length (carbon atoms) | hiCE (CPT-11) (nM) | Intracellular hiCE inhibition (%)a |
|---|---|---|---|
| 11 | 4 | 140 ± 18.0 | 85.0 |
| 12 | 5 | 193 ± 26.8 | 90.0 |
| 13 | 6 | 271 ± 42.7 | 91.2 |
| 14 | 7 | 66.8 ± 9.75 | 92.9 |
| 15 | 8 | 24.5 ± 2.35 | 80.4 |
| 18 | 2 | 2,010 ± 189 | 70.3 |
| 19 | 3 | 495 ± 39 | 86.1 |
| 20 | 4 | 168 ± 16.6 | 93.0 |
| 21 | 5 | 98.1 ± 20.6 | 93.6 |
| 22 | 6 | 130 ± 11.0 | 95.1 |
| Benzil (1) | NA | 175 ± 29b | 95.1c |
To assess whether intracellular inhibition could modulate CPT-11 cytotoxicity, we undertook drug survival assays in cells expressing hiCE, following exposure to selected inhibitors. As indicated in Table 5, both benzil and 1-phenyl-2-octane-1,2-dione reduced the toxicity of CPT-11 to U373MGhiCE cells by ~6- and 3-fold respectively. Interestingly, compound 13 (tetradecane-7,8-dione) was ineffective in this assay, despite demonstrating good in vitro biochemical properties (Tables 2 and 4), and inhibiting hiCE by ~91% in the fluorometric assay.
Table 5.
Modulation of IC50 values for CPT-11 in U373MG cells expressing hiCE by selected 1,2-dione inhibitors
| Compound | IC50 (μM)a |
|---|---|
| CPT-11 | 3.6 |
| Benzil (1) | >100 |
| CPT-11 + Benzil (1) | 22.9 |
| 13 | >100 |
| CPT-11 + 13 | 4.0 |
| 22 | >100 |
| CPT-11 + 22 | 10.2 |
Data are representative values from several experiments.
4. Discussion
Past publications have demonstrated the importance of the 1,2-dione chemotype within molecules for the selective inhibition of CE [19–21]. Specifically, benzil was found to be an excellent inhibitor of hiCE, hCE1 and rCE. We previously hypothesized that the mechanism of enzyme inhibition by these compounds was due to abortive nucleophilic attack on one of the carbonyl carbon atoms of the molecule by the catalytic serine [21]. Since the tetrahedral intermediate that would be formed from this reaction would contain a C-C bond (rather than the C-O bond in the ester), we postulated that the enzyme would be unable to cleave this bond. Hence, an equilibrium would be established between the free inhibitor and the enzyme-inhibitor complex, resulting in reversible inhibition. We reasoned that the simplest way to test this hypothesis would be to generate compounds containing either deactivated (e.g. oxanilide) or activated (e.g., diphenyl ethanedioate, 1,4-dibromo-1,4-diphenylbutane-2,3-dione) carbonyl groups, and then assess their activity towards the CEs. Therefore the studies presented here detail the interaction of the mammalian CEs with small molecules containing either highly electrophilic, or essentially inactivated, carbonyl carbon atoms.
For oxanilide (2), where the α-atom with respect to the carbonyl carbon is nitrogen, no enzyme inhibition was observed (Table 1). This is likely due to the decreased electrophilicity of the carbonyl. This renders the molecule less susceptible to nucleophilic attack by the enzyme and as a consequence, oxanilide is unable to inhibit the mammalian CEs. This would suggest that activated electrophiles, such as the carbonyl groups present in compounds 3 and 4, would be effective CE inhibitors. However, both analogues were both subject to spontaneous hydrolysis by water and this reaction was not enhanced by the addition of enzyme (data not shown). Since oxygen and sulfur are both electron withdrawing in nature, and each carbonyl group is further activated by the adjacent one, the 1,2-dione in these compounds is highly reactive, to the extent that even poor nucleophiles such as water, can readily hydrolyze these molecules.
1,4-Diphenylbutane-2,3-dione (5) was a much poorer inhibitor of the CEs than benzil (Table 1) indicating that the proximity of the benzene ring is important towards biological activity. In benzil, the phenyl group is directly attached to this moiety and it would be anticipated that due to resonance effects, this would reduce the electrophilicity of the carbonyl group. Furthermore with compound 5, such an effect would be negated by the methylene atom, and inductive effects from the phenyl ring would actually activate this group. However, the biological data (Table 1) clearly demonstrate that this is not the case. This suggests that other parameters play a role in inhibitor potency, rather than just the electrophilicity of the carbonyl carbon atom.
We have previously demonstrated the flexibility of the 1,2-dione bond is an important parameter with respect to compound potency [19]. For a series of heteroaromatic ethane-1,2-diones, we observed that the ability of the dione bond to adopt a relatively stable conformation correlated with biological activity. In addition, if the final dihedral angle that the molecule adopted was to close to 230°, it could be predicted that the compound would be a potent CE inhibitor. As indicated in Figure 1, the final dihedral angle for benzil was ~230°, whereas for compound 5, the 1,2-dione was still oscillating and never achieved a stable conformation at the end of the dynamics simulations. The exact reason why this parameter correlates with inhibitor potency is not clear, but may be due to the alignment of the small molecule in a configuration such that nucleophilic attack by the catalytic serine is facile.
Furthermore, as indicated in Figure 3, the stability of the conformation adopted by benzil is presumably due to conjugation of the π electrons between the phenyl ring and the adjacent carbonyl group. This accounts for the high frequency 1660cm−1 carbonyl IR stretch seen with this compound [20]. However, no conjugation occurs between the carbonyl group themselves. With molecule 5, no conjugation is observed between the phenyl group and the carbonyl moiety (the IR carbonyl wave number is 1709cm−1 [40]) and hence the rotation around the carbon – carbon bonds within the butane moiety is less restricted. These observations are consistent with the molecular dynamics simulations presented in Figure 1. Also, as can be seen in Figure 3, the electron distribution on the aromatic rings, coincident with the side of attack by a nucleophile in benzil, is reduced as compared to compound 5. Hence, this may make the carbonyl carbon atom in 1,4-diphenylbutane-2,3-dione less prone to interaction with the serine Oγ atom due to increased repulsion from the charge localized on the benzene rings. Alternatively, interaction of benzil with the enzyme may result in a conformational change in the former that results in increased affinity for the protein. Since both the molecular dynamics simulations and the computational approaches (Figures 1 and 3, respectively), indicate that benzil and compound 5 adopt different poses, it is possible that reaction of these molecules with the CEs, would result in a significant change in the energetics of the system. Since the biological data indicates that the interaction of benzil with these proteins is clearly favored, as compared to 5, we are currently undertaking very detailed computational analyses of the proposed enzyme-inhibitor complexes to assess whether significant changes in small molecule conformation can be used to determine and/or predict inhibitor potency.
Figure 3.

Charge distribution patterns in 1,4-diphenylbutane-2,3-dione (5; A) and benzil (1; B). Images are oriented with the oxygen atoms projecting either towards (left panel) or away (right panel) from the viewer. Red indicates relative negative charge and blue relative positive charge.
Activation of the carbonyl carbon by the addition of a bromine atom on the vicinal carbon atom (compounds 6 and 7) resulted in decreased Ki values for enzyme inhibition. Due to the electron withdrawing properties of the halogen, and the fact that bromine is a relatively good leaving group, it is possible that nucleophilic attack by the Oγ within the catalytic serine could occur either at the carbonyl, or the methylene, carbon atom. The latter would result in covalent bond formation and irreversible inhibition of the enzyme. However, as indicated in Table 1, no loss of enzyme activity was observed following dilution of the inhibitor from the reaction. These results imply that interaction of the protein with the inhibitor occurred at the carbonyl carbon and not at the vicinal carbon atom. Additionally, since the kinetic analyses indicated that the mode of enzyme inhibition was partially competitive, this infers that these inhibitors bind to the same site within the CE, and that they structurally resemble the substrate. This would argue that a similar mechanism of interaction likely occurs between the enzyme and the inhibitor/substrate. Again, these data support the hypothesis that the catalytic serine undergoes nucleophilic attack of the carbonyl carbon atom. Furthermore, the halogens also increased the hydrophobicity of the molecules (Table 1), likely allowing for a further increase inhibitor potency. Since the active site gorges of CE are very hydrophobic [2, 21], compounds demonstrating greater clogP values would preferentially localize within this domain. Evidence to this effect has been noticed with a series of isatin analogues [18]. Overall, these results suggest that the electrophilicity of the carbonyl carbon atom plays a role in determining the potency of inhibition towards CEs.
To further examine the effect of small molecule hydrophobicity on enzyme inhibition, we synthesized a series of alkyl-1,2-diones and evaluated their inhibitory potency against the CEs. We reasoned that by simply increasing the size of the alkyl group, we could effect an increase in the clogP of the compounds, without significantly altering the electrostatic configuration of the molecule. As indicated in Table 2, increasing the length of the alkyl side chains resulted in dramatically reduced Ki values for the inhibitors with the CEs. This correlated well with the increasing clogP values of the compounds (Figure 2). This phenomenon was observed for all three mammalian CEs, suggesting that the environment within the active site was similar for each protein. As indicated above, since the active sites of these enzymes are highly hydrophobic, this increase in potency is likely due to preferred localization of the longer chain compounds within this domain. Indeed, since good correlation coefficients were observed between the Ki values for enzyme inhibition and the clogP values of these molecules (Figure 2), this suggests that this parameter is an important factor in determining inhibitor potency. Based upon our observations with the 1,4-diphenylbutane-2,3-dione analogues and the alkyl-1,2-diones, we hypothesized that compounds containing a phenyl group and alkyl chain on either side of the dione moiety should yield very potent CE inhibitors. As displayed in Table 3, we saw selective enzyme inhibition by such molecules, with inhibitory potency correlating with the hydrophobicity of the molecule (Figure 2). These results are in agreement with those described for the alkyl-1,2-diones and further reinforce the importance of hydrophobicity towards enzyme inhibition.
Having identified the necessary properties for these molecules to effect CE inhibition in vitro, we evaluated the ability of selected compounds to modulate enzyme activity in cells and to determine whether these inhibitors could alter CPT-11 cytotoxicity in cells expressing hiCE. We confirmed that the majority of the alkyl and phenylalkyl derivatives were cell permeable and were effective at inhibition hydrolysis of CPT-11 by hiCE. Furthermore, in cell viability studies, benzil and compound 22 were effective at modulating the toxicity of this drug. Surprisingly, tetradecane-7,8-dione (13) was ineffective at reducing CPT-11 toxicity in cells expressing hiCE. Attempts to identify a mechanism for this lack of activity (e.g. the use of serum-free culture media to eliminate protein binding, the use of glass culture dishes to minimize interaction with the plasticware, etc) have so far been unsuccessful. Potentially, these compounds maybe subjected to metabolic processes that may inactivate them, but as yet we have not identified a specific mechanism for the lack of activity of this molecule. However it is clear that compounds of this type can be used to modulate intracellular levels of CE activity and can be used as biochemical tools to evaluate enzyme function.
5. Conclusions
Overall, our studies have delineated the roles of selected atoms within the 1,2-dione CE-specific inhibitors and demonstrated that the electrophilicity of the carbonyl group, the conformation that the molecules adopt, and the hydrophobicity of these compounds play major roles in determining inhibitor potency. Our data further support the hypothesis that nucleophilic attack by the protein on the carbonyl carbon atom represents the most likely mechanism of enzyme inhibition. This has yielded several series of small molecules that demonstrate potency and selectivity towards CE, and elucidated the chemical requirements for the design of further compounds. These studies are currently underway.
Supplementary Material
Acknowledgments
The authors wish to thank Drs. Thomas Webb and Kip Guy for helpful discussion and critical reading of the manuscript. This work was supported in part by NIH Grants CA108775, an NIH Cancer Center Core Grant CA21765, and by the American Lebanese Syrian Associated Charities and St Jude Children’s Research Hospital (SJCRH).
Footnotes
Abbreviations: CE - carboxylesterase CPT-11 - Irinotecan, 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin; hAChE - human acetylcholinesterase; hBChE - human butyrylcholinesterase; hBr3 - human brain carboxylesterase; hCE1 - human carboxylesterase 1; hiCE - hCE2, human intestinal carboxylesterase; HPLC - high performance liquid chromatography; 4-MUA - 4-methylumbelliferone acetate; o-NPA - nitrophenyl acetate; rCE - rabbit liver carboxylesterase; SN-38 - 7-ethyl-10-hydroxycamptothecin.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cashman J, Perroti B, Berkman C, Lin J. Environ Health Perspect. 1996;104:23–40. doi: 10.1289/ehp.96104s123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Potter PM, Wadkins RM. Curr Med Chem. 2006;13:1045–1054. doi: 10.2174/092986706776360969. [DOI] [PubMed] [Google Scholar]
- 3.Redinbo MR, Potter PM. Drug Discov Today. 2005;10:313–325. doi: 10.1016/S1359-6446(05)03383-0. [DOI] [PubMed] [Google Scholar]
- 4.Mori M, Hosokawa M, Ogasawara Y, Tsukada E, Chiba K. FEBS Lett. 1999;458:17–22. doi: 10.1016/s0014-5793(99)01111-4. [DOI] [PubMed] [Google Scholar]
- 5.Fleming CD, Bencharit S, Edwards CC, Hyatt JL, Tsurkan L, Bai F, Fraga C, Morton CL, Howard-Williams EL, Potter PM, Redinbo MR. J Mol Biol. 2005;352:165–177. doi: 10.1016/j.jmb.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 6.Khanna R, Morton CL, Danks MK, Potter PM. Cancer Res. 2000;60:4725–4728. [PubMed] [Google Scholar]
- 7.Ross MK, Borazjani A, Edwards CC, Potter PM. Biochem Pharmacol. 2006;71:657–669. doi: 10.1016/j.bcp.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 8.Ross MK, Potter PM, Borazjani A. Tox Sci. 2005;84:S1, A1569. [Google Scholar]
- 9.Satoh T, Hosokawa M, Atsumi R, Suzuki W, Hakusui H, Nagai E. Biol Pharm Bull. 1994;17:662–664. doi: 10.1248/bpb.17.662. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Burnell JC, Dumaual N, Bosron WF. J Pharmacol Exp Ther. 1999;290:314–318. [PubMed] [Google Scholar]
- 11.Humerickhouse R, Lohrbach K, Li L, Bosron W, Dolan M. Cancer Res. 2000;60:1189–1192. [PubMed] [Google Scholar]
- 12.Hsiang YH, Liu LF. Cancer Res. 1988;48:1722–1726. [PubMed] [Google Scholar]
- 13.Tanizawa A, Fujimori A, Fujimori Y, Pommier Y. J Natl Cancer Inst. 1994;86:836–842. doi: 10.1093/jnci/86.11.836. [DOI] [PubMed] [Google Scholar]
- 14.Danks MK, Morton CL, Krull EJ, Cheshire PJ, Richmond LB, Naeve CW, Pawlik CA, Houghton PJ, Potter PM. Clin Cancer Res. 1999;5:917–924. [PubMed] [Google Scholar]
- 15.Potter PM, Pawlik CA, Morton CL, Naeve CW, Danks MK. Cancer Res. 1998;52:2646–2651. [PubMed] [Google Scholar]
- 16.Wierdl M, Morton CL, Weeks JK, Danks MK, Harris LC, Potter PM. Cancer Res. 2001;61:5078–5082. [PubMed] [Google Scholar]
- 17.Hatfield MJ, Tsurkan L, Garrett M, Shaver T, Edwards CC, Hyatt JL, Hicks LD, Potter PM. Biochem Pharmacol. 2011;81:24–31. doi: 10.1016/j.bcp.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyatt JL, Moak T, Hatfield JM, Tsurkan L, Edwards CC, Wierdl M, Danks MK, Wadkins RM, Potter PM. J Med Chem. 2007;50:1876–1885. doi: 10.1021/jm061471k. [DOI] [PubMed] [Google Scholar]
- 19.Hyatt JL, Stacy V, Wadkins RM, Yoon KJ, Wierdl M, Edwards CC, Zeller M, Hunter AD, Danks MK, Crundwell G, Potter PM. J Med Chem. 2005;48:5543–5550. doi: 10.1021/jm0504196. [DOI] [PubMed] [Google Scholar]
- 20.Hyatt JL, Wadkins RM, Tsurkan L, Hicks LD, Hatfield MJ, Edwards CC, Ii CR, Cantalupo SA, Crundwell G, Danks MK, Guy RK, Potter PM. J Med Chem. 2007;50:5727–5734. doi: 10.1021/jm0706867. [DOI] [PubMed] [Google Scholar]
- 21.Wadkins RM, Hyatt JL, Wei X, Yoon KJ, Wierdl M, Edwards CC, Morton CL, Obenauer JC, Damodaran K, Beroza P, Danks MK, Potter PM. J Med Chem. 2005;48:2905–2915. doi: 10.1021/jm049011j. [DOI] [PubMed] [Google Scholar]
- 22.Hyatt JL, Tsurkan L, Wierdl M, Edwards CC, Danks MK, Potter PM. Mol Cancer Therap. 2006;5:2281–2288. doi: 10.1158/1535-7163.MCT-06-0160. [DOI] [PubMed] [Google Scholar]
- 23.Babudri F, Fiandanese V, Marchese G, Punzi A. Tetrahedron Lett. 1995;36:7305–7308. [Google Scholar]
- 24.Morton CL, Potter PM. Mol Biotechnol. 2000;16:193–202. doi: 10.1385/MB:16:3:193. [DOI] [PubMed] [Google Scholar]
- 25.Santana MD, Garcia G, Julve M, Lloret F, Perez J, Liu M, Sanz F, Cano J, Lopez G. Inorg Chem. 2004;43:2132–40. doi: 10.1021/ic030050f. [DOI] [PubMed] [Google Scholar]
- 26.Jones HO, Tasker HS. J Chem Soc. 1909;95:1904–1909. [Google Scholar]
- 27.Srinivasan NS, Lee DG. J Org Chem. 1979;44:1574–1574. [Google Scholar]
- 28.Wadkins RM, Hyatt JL, Yoon KJ, Morton CL, Lee RE, Damodaran K, Beroza P, Danks MK, Potter PM. Mol Pharmacol. 2004;65:1336–1343. doi: 10.1124/mol.65.6.1336. [DOI] [PubMed] [Google Scholar]
- 29.Webb JL. Enzyme and Metabolic Inhibitors. Volume 1. General Principles of Inhibition. Academic Press Inc; New York: 1963. [Google Scholar]
- 30.Akaike H. Information theory and an extension of the maximum likelihood principle. In: Petrov BN, Csaki F, editors. Second International Symposium on Information Theory; Budapest. Akademiai Kiado; 1973. pp. 267–281. [Google Scholar]
- 31.Akaike H. IEEE Trans Automatic Control. 1974;AC–19:716–723. [Google Scholar]
- 32.Doctor BP, Toker L, Roth E, Silman I. Anal Biochem. 1987;166:399–403. doi: 10.1016/0003-2697(87)90590-2. [DOI] [PubMed] [Google Scholar]
- 33.Ellman GL, Courtney KD, Anders V, Featherstone RM. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 34.Morton CL, Wadkins RM, Danks MK, Potter PM. Cancer Res. 1999;59:1458–1463. [PubMed] [Google Scholar]
- 35.Wadkins RM, Potter PM, Vladu B, Marty J, Mangold G, Weitman S, Manikumar G, Wani MC, Wall ME, Von_Hoff DD. Cancer Res. 1999;59:3424–3428. [PubMed] [Google Scholar]
- 36.Guichard S, Morton CL, Krull EJ, Stewart CF, Danks MK, Potter PM. Clin Cancer Res. 1998;4:3089–3094. [PubMed] [Google Scholar]
- 37.Danks MK, Morton CL, Pawlik CA, Potter PM. Cancer Res. 1998;58:20–22. [PubMed] [Google Scholar]
- 38.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 39.Petersson GA, Bennett A, Tensfeldt TG, Al-Laham MA, Shirley WA, Mantzaris J. J Chem Phys. 1988;89:2193–2218. [Google Scholar]
- 40.Stetter H, Raemsch RY. Synthesis. 1981;6:477–478. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.