

Abstract
Three families of prolyl isomerases have been identified: cyclophilins, FK506-binding proteins (FKBPs) and parvulins. All 12 cyclophilins and FKBPs are dispensable for growth in yeast, whereas the one parvulin homolog, Ess1, is essential. We report here that cyclophilin A becomes essential when Ess1 function is compromised. We also show that overexpression of cyclophilin A suppresses ess1 conditional and null mutations, and that cyclophilin A enzymatic activity is required for suppression. These results indicate that cyclophilin A and Ess1 function in parallel pathways and act on common targets by a mechanism that requires prolyl isomerization. Using genetic and biochemical approaches, we found that one of these targets is the Sin3–Rpd3 histone deacetylase complex, and that cyclophilin A increases and Ess1 decreases disruption of gene silencing by this complex. We show that conditions that favor acetylation over deacetylation suppress ess1 mutations. Our findings support a model in which Ess1 and cyclophilin A modulate the activity of the Sin3–Rpd3 complex, and excess histone deacetylation causes mitotic arrest in ess1 mutants.
Keywords: cyclosporin A/HDAC/parvulin/Pin1/prolyl isomerases
Introduction
Protein primary sequences suffice to direct folding in vitro (Anfinsen, 1973), but the situation in vivo is probably more complex. In vivo, chaperones sequester protein folding intermediates and promote spontaneous folding (Gething and Sambrook, 1992). In addition, two classes of enzyme catalyze protein folding: prolyl isomerases and protein disulfide isomerases (Fischer, 1994; Dolinski and Heitman, 1997).
The peptide bond has partial double bond character and can exist as both cis and trans isomers. For all amino acids except proline, the trans isomer is the preferred conformer because of steric clashes in the cis form. In contrast, the peptide bonds preceding proline residues are equally stable as both cis and trans isomers. In proteins with known structures, ∼10% of peptidyl-prolyl bonds are in the cis isomer (Stewart et al., 1990). The ribosome is thought to synthesize the peptide bond in the trans isomer, and spontaneous or catalyzed isomerization yields the cis form (Fischer and Schmid, 1990).
Three classes of enzymes have been discovered that catalyze prolyl isomerization: cyclophilins, FK506-binding proteins (FKBPs) and parvulins (Handschumacher et al., 1984; Harding et al., 1989; Takahashi et al., 1989; Rahfeld et al., 1994). The cyclophilins and FKBPs are notable because the cyclophilin A and FKBP12 members of these families mediate the effects of the immunosuppressants cyclosporin A (CsA), FK506 and rapamycin (Heitman et al., 1991a; Liu et al., 1991; Foor et al., 1992). Moreover, the cyclophilins and FKBPs are large families that are ubiquitous, highly expressed and conserved from microorganisms to man. The third family of prolyl isomerases, the parvulins, is also conserved from bacteria to man. Homologs of the yeast Ess1/Ptf1 parvulin (Hanes et al., 1989; Hani et al., 1995, 1999) are present in Drosophila (Dodo) and humans (Pin1) (Lu et al., 1996; Maleszka et al., 1996). The human parvulin Pin1 is thought to regulate mitosis, and has an unusual substrate specificity for phospho-serine–proline peptides (Ranganathan et al., 1997; Yaffe et al., 1997; Shen et al., 1998; Winkler et al., 2000). This finding, and the fact that Pin1 and other parvulin homologs have a unique WW protein–protein interaction domain (Lu et al., 1999), distinguishes these enzymes from the cyclophilins and FKBPs.
The yeast Saccharomyces cerevisiae expresses eight different cyclophilin and four different FKBP enzymes (Dolinski et al., 1997). None of these 12 prolyl isomerases is essential, either alone or in combination (Dolinski et al., 1997). In contrast, the one yeast parvulin, Ess1, is essential (Hanes et al., 1989; Lu et al., 1996). These findings suggest that most of the yeast prolyl isomerases do not play an essential protein folding role, and may instead each have unique functions, in accord with studies in Drosophila and mammals revealing specific roles for the NinaA and RanBP2 cyclophilins in opsin maturation (Baker et al., 1994; Ferreira et al., 1996), and for FKBP12 as a subunit of the ryanodine receptor (Brillantes et al., 1994; Shou et al., 1998).
In the accompanying manuscript, Wu et al. (2000) isolated temperature-sensitive (ts) mutants of the Ess1 enzyme and identified multicopy genetic suppressors. Remarkably, overexpression of cyclophilin A suppresses conditional lethal mutations in Ess1. Here we have explored the molecular basis for this first example of functional overlap between the members of two different prolyl isomerase families. We present genetic and biochemical evidence that identifies one common target as components of the Sin3–Rpd3 histone deacetylase complex (HDAC), which regulates transcriptional repression and gene silencing in yeast and mammalian cells (Vidal and Gaber, 1991; Vidal et al., 1991; Pazin and Kadonaga, 1997; Struhl, 1998). Our studies provide the first evidence that the functions of cyclophilins and parvulins are linked in protein folding, and provide insight into the cellular functions and substrates of this enigmatic class of protein folding enzymes.
Results
Cyclophilin A catalytic activity suppresses ess1 conditional mutations
The yeast cyclophilin A gene CPR1 was identified as a multicopy suppressor of temperature-sensitive conditional mutations in the ESS1 gene, which encodes an essential prolyl isomerase (Figure 1A; see also Wu et al., 2000). This finding provided the first link between the functions of these two classes of prolyl isomerase. Two other cyclophilins, homologs of mammalian cyclophilin 40 encoded by the yeast CPR6 and CPR7 genes, also weakly suppressed ess1ts mutations (Figure 1A). The FPR1 gene encoding FKBP12 did not (Figure 1A). These results suggest that at high concentrations the Cpr1, Cpr6 and Cpr7 cyclophilins can replace the essential function of Ess1.

Fig. 1. Cyclophilins suppress ess1ts mutants. (A) The ess1ts mutant strain H164RW303 was transformed with 2µ plasmids expressing no protein (pRS426 vector), the cyclophilins Cpr1 (pTB3), Cpr6 (pKDw10) or Cpr7 (pKS24), FKBP12 (FPR1; plasmid pYJH23), the cyclophilin A active site mutant cpr1H90Y [pCPR1(H90Y)] or Ess1 as a control (YEpESS1). Growth was for 72 h at 37°C. (B) Suppression of ess1ts mutants by cyclophilin A (CPR1) and cyclophilin 40 homologs is CsA sensitive. ess1ts strain H164RW303 transformed with 2µ plasmids expressing CPR1, CPR6 or CPR7 was grown on synthetic dextrose medium–uracil and then transferred to SD–Ura medium with 0, 10 or 100 µg/ml CsA and incubated for 72 h at 35°C. (C) Suppression of ess1 conditional mutants by cyclophilin A requires prolyl isomerase activity. ess1ts mutant H164RW303 was transformed with multicopy plasmids expressing wild-type or active site mutants of human cyclophilin A and growth was tested at 30 and 37°C. The wild-type cyclophilin A gene is indicated as wt hCypA, and the active site mutations and relative level of in vitro prolyl isomerase activity are indicated.
To test whether the prolyl isomerase activity of cyclophilin A is required for suppression of ess1ts mutations, we used the immunosuppressive drug CsA. At high concentrations (100 µg/ml), CsA does not affect yeast cell growth because the target of the cyclophilin–CsA complex, calcineurin, is not essential. CsA does, however, inhibit the peptidyl-prolyl isomerase (PPIase) activity of cyclophilin A; thus, if suppression of ess1ts mutations by cyclophilin A requires enzyme activity, then CsA should abolish suppression. CsA blocked the ability of cyclophilin A (and also Cpr6 and Cpr7) to suppress, suggesting that prolyl isomerase activity is required (Figure 1B). Furthermore, several ess1ts mutant strains were sensitive to CsA at permissive temperature (not shown), suggesting that cyclophilin A maintains viability of these strains. The FKBP12 and calcineurin inhibitor FK506 did not impair growth of ess1ts mutants or inhibit suppression of ess1ts mutations by cyclophilin A, indicating that the effects of CsA are not attributable to calcineurin inhibition.
Cyclophilin active site mutants were used to confirm that prolyl isomerase activity is required for genetic suppression. The H90Y cyclophilin A mutant that lacks prolyl isomerase activity (Cardenas et al., 1995a) failed to restore growth of ess1ts mutants at 37°C (Figure 1A). A series of human cyclophilin A mutant enzymes with systematically reduced activity was also tested (Zydowsky et al., 1992; Scholz et al., 1999). The wild-type and mutant human cyclophilin A genes were expressed under the control of the yeast CPR1 gene promoter and terminator from a yeast 2µ multicopy plasmid. Expression of wild-type human cyclophilin A restored growth of two ess1ts mutants (H164R and A144T) at 37°C (Figure 1C and data not shown). The H54Q and W121A mutants, which retain 15 and 8.7% activity, respectively, partially restored growth at 37°C (Figure 1C). Cyclophilin A mutants with less activity did not support growth (Figure 1C). Thus, suppression of ess1ts mutations is correlated with the level of cyclophilin A prolyl isomerase activity, suggesting that the function of Ess1 and cyclophilin A requires enzymatic activity.
Cyclophilin A and FKBP12 mutations are synthetically lethal with ess1 mutations
The functional relationship between Ess1 and the 12 yeast cyclophilin and FKBP prolyl isomerases was examined using a synthetic lethal genetic approach. Specifically, 12 mutant strains each lacking one prolyl isomerase were mated to isogenic ess1ts mutant strains. The resulting diploid strains were sporulated and tetrads analyzed to ascertain the phenotype of the double mutants.
This analysis revealed that a deletion of the cyclophilin A gene, CPR1, is synthetically lethal with ess1ts mutations (Figure 2; Table I). As shown in Figure 2, when the ess1ts/ESS1 cpr1Δ/CPR1 diploid strain was sporulated and tetrads analyzed, two tetrads consisted of four viable spores, five tetrads contained three viable:one inviable spore, and four tetrads contained two viable:two inviable spores (2 PD:5 TT:4 NPD). The inviable segregants could be inferred to be ess1ts cpr1 double mutants, based on the segregation pattern of the ess1ts mutation and the cpr1Δ::LEU2 mutation (Leu+, CsA + Li+ resistant). No ts (ess1ts) and Leu+ (cpr1Δ::LEU2) viable spores were obtained. A plasmid expressing the cyclophilin A gene restored viability of ess1ts cpr1 double mutants (not shown). Thus, a mutation in the cyclophilin A gene, CPR1, is synthetically lethal with the ess1ts mutation under conditions permissive for the ess1ts single mutant.

Fig. 2. Cyclophilin A and ess1 mutations are synthetically lethal. The ess1ts/ESS1 cpr1Δ::LEU2/CPR1 diploid strain MAY3 × MH250-2c was sporulated, and tetrads were dissected and incubated on YPD medium at 26°C for 4 days. Viable meiotic segregants were replica-plated to YPD medium at 26 and 37°C to score the ess1ts mutation, and to synthetic medium lacking leucine to score the cpr1Δ::LEU2 cyclophilin A mutation.
Table I. Cyclophilin A (cpr1) and FKBP12 (fpr1) mutations are synthetically lethal with ess1ts conditional mutations.
| Cross | Viable:inviable segregants |
||
|---|---|---|---|
| 4:0 | 3:1 | 2:2 | |
| cpr1×ess1ts | 2 | 5 | 4 |
| cpr2× | 10 | 1 | 0 |
| cpr3× | 11 | 0 | 0 |
| cpr4× | 9 | 0 | 0 |
| cpr5× | 8 | 1 | 0 |
| cpr6× | 9 | 0 | 0 |
| cpr7× | 8 | 0 | 0 |
| cpr8× | 9 | 0 | 0 |
| fpr1×ess1ts | 1 | 10 | 1 |
| fpr2× | 7 | 2 | 0 |
| fpr3× | 7 | 1 | 1 |
| fpr4× | 8 | 1 | 0 |
Data presented are for crosses with the H164R ess1ts mutant strain MAY3; similar findings were obtained with an A144T ess1ts mutant (MAY1). Inviable segregants in the cpr1×ess1 cross could be inferred to be cpr1 ess1ts double mutants (predicted to be ts and Leu+). Inviable segregants in the fpr1×ess1ts cross could be inferred to be fpr1 ess1ts double mutants (predicted to be ts and rapamycin resistant). Inviable segregants in all other cases resulted from random spore death or failure to germinate, and cpr ess1ts and fpr ess1ts double mutants were viable in all of these crosses.
A deletion mutation of the FPR1 gene encoding FKBP12 was also synthetically lethal with ess1ts mutations (Table I). Again, sporulation and tetrad analysis of the ess1ts/ESS1 fpr1/FPR1 strain revealed that the majority of tetrads yielded three viable and one inviable spore; the inviable spores could be inferred to be ess1ts fpr1 double mutants based on segregation of the ess1ts and fpr1::ADE2 mutations (rapamycin resistant), and no viable ess1ts fpr1 double mutants (ts, Ade+ and rapamycin resistant) were obtained. Thus, both cyclophilin A and FKBP12 are required for viability of ess1ts mutants.
No other synthetic lethal interactions were observed between ess1ts mutations and any other cyclophilin (cpr2–cpr8) or FKBP (fpr2, fpr3 and fpr4) mutations (see Table I).
Cyclophilin A does not stabilize thermolabile Ess1 mutants
Cyclophilin A might restore viability of ess1ts mutant strains by direct binding to Ess1, stabilizing or refolding the thermolabile enzyme. Ess1 protein levels were determined in strains expressing an Ess1 thermolabile mutant and cyclophilin A from a multicopy plasmid. The levels of the Ess1 H164R and A144T mutant enzymes were reduced at 37°C compared with 26°C (Figure 3A), and the A144T mutant protein was virtually undetectable at 26°C, consistent with the ts phenotype conferred by these mutant enzymes. The levels of the H164R and A144T mutant enzymes were similar in cells overexpressing cyclophilin A compared with cells containing the control plasmid (Figure 3A). Western blot confirmed that cyclophilin A was overexpressed in cells with the CPR1 multicopy plasmid (Figure 3A). Thus, overexpression of cyclophilin A does not stabilize the Ess1 mutant enzymes.

Fig. 3. Cyclophilin A does not stabilize A144T or H164R Ess1 thermolabile mutant proteins, and suppresses an ess1 null mutation. (A) Cells expressing the H164R or A144T Ess1 mutant proteins (strains A144TW303 or H164RW303), and which contained a control plasmid (YEplac195 vector) or a 2µ plasmid (pTB3) overexpressing cyclophilin A (CPR1), were grown at 26°C until log phase, diluted and grown at 26 or 37°C for 12 h. Total protein extracts were analyzed by western blotting with antisera against Ess1 or cyclophilin A (CypA). (B) An ess1Δ::G418/ESS1 diploid strain was transformed with a control 2µ plasmid (vector pRS426), or with 2µ plasmids expressing Ess1 (ESS1) or the cyclophilin A gene CPR1. Strains were sporulated, tetrads dissected and spores germinated on YPD medium at 26°C. Viable segregants were replica-plated to YPD medium containing G418 to score the ess1Δ::G418 mutation, and also to synthetic medium lacking uracil compared with 5-FOA medium (not shown) to detect the plasmid-borne URA3 gene.
Cyclophilin A suppresses an ess1 null mutation
Because cyclophilin A did not stabilize Ess1 ts mutant enzymes, we next considered an alternative hypothesis in which cyclophilin A suppresses ess1 mutations by acting on a common target required for cell viability. We tested this hypothesis by determining whether overexpression of cyclophilin A suppresses an ess1 null mutation.
A diploid strain was constructed in which one copy of the ESS1 gene was deleted with the dominant G418 resistance marker. Next, 2µ multicopy plasmids lacking or expressing the ESS1 or cyclophilin A genes were introduced into the ess1Δ/ESS1 diploid. Overexpression of cyclophilin A restored growth in ess1Δ null mutants. The ess1Δ::G418/ESS1 strain bearing the 2µ CPR1 URA3 plasmid yielded several tetrads in which two spores produced normal colonies, and two spores produced smaller colonies with a slower growth rate, which contained the ess1Δ::G418 mutation (Figure 3B). These viable ess1Δ G418-resistant colonies were all Ura+ and 5-fluoro-orotic acid (5-FOA) sensitive (not shown), indicating that the CPR1 URA3 plasmid expressing cyclophilin A rescues growth of strains lacking Ess1, albeit not to a wild-type level of growth. As expected, a control vector did not rescue inviable ess1Δ segregants whereas an ESS1 plasmid did restore viability (Figure 3B). These results show that cyclophilin A can, in part, bypass the essential function of Ess1, and prompted us to search for a common downstream target of Ess1 and cyclophilin A.
Sap30 requires cyclophilin A to suppress ess1ts mutations
In addition to cyclophilin A (CPR1), Wu et al. (2000) identified five other multicopy suppressors of ess1ts mutations, including YKL005C, Fcp1, Sap30, Cth1 and CaRpb7 (from Candida albicans). Three of these proteins, YKL005C, Cth1 and CaRpb7, suppress the lethal phenotype of an ess1 null mutant (Wu et al., 2000). In contrast, the Fcp1 and Sap30 proteins suppress several ess1ts conditional alleles but not a null mutation (Wu et al., 2000). To establish whether one of these proteins is a common target of Ess1 and cyclophilin A, we tested whether cyclophilin A is required for suppression of ess1 mutations by these genes.
We first tested whether CsA inhibits suppression of the ess1ts mutations. A concentration of 50 µg/ml CsA markedly inhibited suppression by Sap30, modestly inhibited suppression by Cth1 and cyclophilin A, and had no effect on suppression by YKL005C, Fcp1 or CaRpb7 (Figure 4). As a second test, we introduced each of the five multicopy suppressor genes into two different ess1ts/ESS1 cpr1Δ/CPR1 mutant strains (H164R and A144T), and sporulated and dissected tetrads to test whether cyclophilin A is required for suppression of ess1ts mutations. This analysis again revealed that the same two suppressors, Sap30 and Cth1, require cyclophilin A to suppress ess1ts mutations, whereas YKL005C, Fcp1 and CaRpb7 did not (Table II). Sap30 and Cth1 had only very modest effects on cyclophilin A mRNA and protein levels, arguing that suppression is not the result of increased cyclophilin A expression (Wu et al., 2000). This suggests that Sap30 and Cth1 may be direct common targets of Ess1 and cyclophilin A. Little is known about CTH1, so we focused on Sap30, a component of the Sin3A–Rpd3 HDAC (Laherty et al., 1998; Zhang et al., 1998).

Fig. 4. Cyclosporin A blocks suppression of ess1ts mutations by Sap30 and Cth1. An ess1ts mutant strain (H164RW303) containing a control plasmid (vector pRS426) or plasmids expressing ESS1 or the multicopy suppressors YKL005C, FCP1, SAP30, CTH1, CPR1 or CaRPB7 were 5-fold serially diluted, spotted on SD–Ura medium lacking (no CsA) or containing 50 µg/ml CsA, and incubated at 37°C for 3 days.
Table II. Sap30 and Cth1 require cyclophilin A to suppress ess1ts mutations.
| Plasmid | Suppression of ess1 cpr1 double mutanta | CsA sensitivity of ess1ts strain at 37°Cb | |
|---|---|---|---|
| Vector | no | (13%, n = 15) | n.d. |
| YEpESS1 | yes | (43%, n = 23) | resistant |
| pYKL005C | yes | (31%, n = 26) | resistant |
| pFCP1 | yes | (55%, n = 20) | resistant |
| pSAP30 | no | (0%, n = 17) | sensitive |
| pCTH1 | no | (0%, n = 7) | sensitive |
| pCPR1 | yes | (50%, n = 4) | sensitive |
| pCaRPB7 | yes | (33%, n = 12) | resistant |
aDiploid strain ess1A144T/ESS1 cpr1Δ::LEU2/CPR1 was transformed with the multicopy suppressors or control plasmids (pRS426 vector, YEpESS1). Cells were sporulated and at least 20 tetrads were dissected. Similar data were obtained with the ess1H164R allele (not shown). Segregants were replica-plated to medium lacking leucine at 37°C to detect those carrying the cpr1Δ::LEU2 allele, to medium lacking uracil to detect those containing the indicated URA3 plasmids, and to 5-FOA-medium to test the ability to lose the suppressor plasmid. 5-FOA-resistant cells are inferred to contain wild-type ESS1 rather than ess1A144T. The values (% suppression) are given as the percentage of cells that are viable at 37°C and are 5-FOAS among total segregants that are Leu+ (contain the cpr1 disruption) and Ura+ (contain the suppressor plasmid); n = total number of Leu+ Ura+ segregants. Maximum values would be 50% (the ess1ts/ESS1 alleles segregate 2:2).
bResults are for two ess1ts strains (A144T and H164R). Sensitivity was tested on plates containing up to 100 µg/ml CsA. n.d. = not determined, since the ess1ts mutant lacking a suppressor is inviable at 37°C.
HDAC subunits Rpd3 and Sap30 are targets of cyclophilin A and Ess1
We tested the hypothesis that Ess1 and cyclophilin A physically interact with Sap30. For this purpose, His6-tagged versions of cyclophilin A and Ess1 were purified, coupled to Affigel beads and incubated with yeast cell extracts. Bound proteins were eluted and analyzed by western blot. As shown in Figure 5A, both Sap30 and Rpd3 interacted with Ess1 and cyclophilin A, whereas an unrelated protein, aspartokinase (Hom3), did not. In addition, no binding of Rpd3 or Sap30 was detected with control Affigel beads alone (Figure 5A). Silver staining of similar gels revealed a limited number of proteins associated with cyclophilin A or Ess1 (not shown).

Fig. 5. Cyclophilin A and Ess1 physically interact with protein complexes containing Rpd3 and Sap30. (A) Yeast extracts from strain W303-1A transformed with a plasmid expressing a V5 epitope-tagged form of Sap30 (pYMR263W) were incubated with Affigel alone (beads) or coupled to cyclophilin A (CypA) or Ess1. Bound proteins were eluted and analyzed by western blotting with antisera against Rpd3 or the V5 epitope (Sap30). (B) Yeast extracts from wild-type (W303-1A), sin3Δ (MAY6) or rpd3Δ (MAY7) strains transformed with pYMR263W, or from an untransformed sap30Δ strain (MAY8), were incubated with Affigel-coupled cyclophilin A or Ess1, and the bound proteins analyzed as in (A). The lower panel shows western blot analysis of the protein extracts used in these experiments; proteins were resolved and analyzed as in (A) and (B).
To investigate further the interaction of Ess1 and cyclophilin A with the HDAC, similar binding experiments were conducted with protein extracts from yeast strains deleted for the SIN3, RPD3 or SAP30 genes. Deletion of SIN3 disrupted the interaction of both Rpd3 and Sap30 with Ess1. Rpd3 and Sap30 were stably expressed in the sin3Δ mutant (Figure 5B). These findings suggest that Sin3 is a direct target of Ess1. Notably, Sin3 and Rpd3 contain multiple Ser–Pro or Thr–Pro sites that are the preferred substrate for Ess1, whereas Sap30 does not. Binding of Rpd3 to cyclophilin A was somewhat weaker but, in contrast to Ess1, did not require Sin3. This suggests that Rpd3 is a direct target of cyclophilin A, in accord with a previous report which showed that the Cpr6 and Cpr7 yeast cyclophilins bind Rpd3 in vivo and in vitro (Duina et al., 1996). Binding of either Ess1 or cyclophilin A to Sap30 required Rpd3, consistent with previous studies showing that mammalian Sap30 and the Rpd3 homolog HDAC1 directly interact (Zhang et al., 1998), and supporting models in which Rpd3 links Sin3 and Sap30, or Sap30 binds to the Sin3–Rpd3 dimer. Deletion of SAP30 did not prevent interaction between Rpd3 and the prolyl isomerases, indicating that Sap30 occupies a peripheral position and does not mediate the Sin3–Rpd3 interaction. In summary, Ess1 and cyclophilin A specifically interact with the Sin3–Rpd3 HDAC in vitro.
Ess1 and cyclophilin A modulate silencing at the ribosomal DNA loci
Silencing within the rDNA array has been described in yeast as an epigenetic phenomenon involving transcriptional repression of RNA polymerase II-transcribed genes inserted in this region (Bryk et al., 1997; Fritze et al., 1997; Smith and Boeke, 1997). Recently, a general role for the yeast Sin3–Rpd3 HDAC in silencing has been reported, including silencing at the rDNA array; mutations in SIN3, RPD3 or SAP30 genes enhance silencing, implying that the Sin3–Rpd3 complex counteracts silencing (Smith et al., 1999; Sun and Hampsey, 1999).
We investigated the role of Ess1 and cyclophilin A in the activity of the Sin3–Rpd3 complex by testing their effects on expression of a genetic marker inserted in the rDNA array. Yeast strain CFY559 bearing an ADE2–CAN1 double marker integrated in the rDNA array (Fritze et al., 1997) was transformed with 2µ plasmids expressing ESS1 or CPR1 and tested for growth in the presence of the toxic amino acid analog l-canavanine. Sensitivity to canavanine depends on the expression level of the canavanine permease encoded by the CAN1 gene inserted in the rDNA array, and is inversely proportional to the degree of silencing. As shown in Figure 6, overexpression of Ess1 increased canavanine resistance; thus, increased Ess1 enhances silencing at the rDNA array. Substitution of the ESS1 wild-type gene with an ess1ts allele dramatically enhanced sensitivity to canavanine, indicating that silencing at the rDNA array is impaired when Ess1 function is compromised. The wild-type ESS1 gene complemented the ess1ts mutation and restored canavanine resistance (Figure 6). These results reveal a functional role for Ess1 in the modulation of transcriptional silencing and indicate that Ess1 is a negative regulator of the Sin3–Rpd3 HDAC.

Fig. 6. Ess1 and cyclophilin A modulate silencing at the rDNA array. A wild-type (wt) yeast strain (CFY559) containing the ADE2-CAN1 double marker integrated in the rDNA array, or isogenic ESS1 (MAY23) and ess1H164R (MAY22) strains, were transformed with 2µ plasmids encoding ESS1 (YEpESS1) or CPR1 (pTB3), or with an empty vector (YEplac195) as a control. Cells were 5-fold serially diluted, spotted on SD–Ade medium containing l-canavanine, and incubated for 48 h at 30°C.
Paradoxically, cyclophilin A had the opposite effect. When expressed from a multicopy plasmid, cyclophilin A increased sensitivity to canavanine (Figure 6), indicating that overexpression of cyclophilin A reduces silencing in the rDNA array, possibly by positively regulating the Sin3–Rpd3 HDAC. In agreement with this hypothesis, overexpression of cyclophilin A in the ess1ts derivative of CFY559 did not restore resistance to canavanine (Figure 6). Taken together, these results support the hypothesis that the prolyl isomerases Ess1 and cyclophilin A functionally interact with the Sin3–Rpd3 HDAC and modulate the silencing activity of this complex.
Rpd3 histone deacetylase activity mediates mitotic arrest in ess1ts mutants
Our results suggest that expression of genes normally controlled by the Sin3–Rpd3 histone deacetylase may be altered in ess1 mutants. Anomalous expression of one or more of these genes may cause the mitotic arrest exhibited by conditional ess1 mutants at non-permissive temperature. We tested the proposal that the Sin3–Rpd3 complex is a target of negative regulation by Ess1 by testing whether reducing Rpd3 deacetylase activity suppresses ess1ts mutations.
Trichostatin A (TsA), an inhibitor of the Rpd3 HDAC, partially suppressed the ts phenotype of ess1 mutants, supporting the hypothesis that the mitotic arrest of the ess1 mutants is, at least in part, caused by an aberrant increase in the Sin3–Rpd3 HDAC activity. To test this model further, we evaluated the role of Rpd3 in the ess1 phenotype by a genetic approach. The G418 resistance marker was used to delete one copy of RPD3 in two ess1ts/ESS1 strains, which were then sporulated to analyze the phenotype of ess1ts rpd3Δ double mutants. Both diploid strains produced a large proportion of tetrads exhibiting 3:1 or 4:0 segregation of temperature-resistant:temperature-sensitive spores. Interestingly, none of the temperature-sensitive segregants was rpd3Δ::G418, indicating that deletion of RPD3 suppresses the ess1 mutations (not shown). Two temperature-resistant rpd3Δ segregants (Figure 7B) were shown by backcrosses to the wild-type parental strain to be ess1ts rpd3Δ double mutants, confirming that the rpd3Δ mutation suppresses ess1 mutations.

Fig. 7. Rpd3 mediates the mitotic phenotype of ess1 conditional mutants. (A) Isogenic ess1A144T (MAY1), ess1H164R (MAY3) and ESS1 wt (JK9-3dα) strains were 5-fold serially diluted and spotted on YPD with or without 13 µM trichostatin A (TsA) and incubated for 48 h at 30 or 37°C. (B) Yeast strains ess1A144T (MAY12-7d), ess1A144T rpd3Δ (MAY12-7b), ess1H164R (MAY16-5c), ess1H164R rpd3Δ (MAY16-2c), rpd3Δ (MAY12–5b) and wt (JK9-3dα) were 5-fold serially diluted, spotted on YPD and incubated for 48 h at 30 or 37°C.
We further analyzed the function of Rpd3 in ess1 mutants by a complementary genetic approach. Yeast Rpd3 mutants have been characterized that are devoid of catalytic activity and that have dominant-negative activity in RPD3 wild-type strains (Kadosh and Struhl, 1998). Overexpression of two such dominant-negative Rpd3 mutants, rpd3H150A and rpd3H151A (Kadosh and Struhl, 1998), partially suppressed the ts phenotype, indicating that reducing Rpd3 activity rescues ess1 conditional mutants (Figure 8).

Fig. 8. Overexpression of Gcn5 or dominant-negative alleles of RPD3 suppresses an ess1 conditional mutation. Temperature-sensitive ess1 strain H164RW303 (ess1H164R) or wild-type strain W303-1A was transformed with 2µ plasmids expressing RPD3 (YEplac112-RPD3), rpd3H150A (YEplac112-RPD3-H150A), rpd3H151A (YEplac112-RPD3-H151A) or GCN5 (Yep10PGK-exp-ScGCN5), or with an empty plasmid, and tested for growth at 35°C.
Finally, we asked whether overexpression of the yeast histone acetyltransferase Gcn5 could suppress ess1 mutations. Gcn5 and Rpd3 are known to play opposing roles in transcriptional regulation of the HO gene (Perez-Martin and Johnson, 1998). Overexpression of Gcn5 partially restored growth of an ess1ts mutant strain at 37°C (Figure 8).
Taken together, these results support the hypothesis that excess histone deacetylase activity of Rpd3 causes misregulation of mitotic genes, and is responsible, at least in part, for the cell cycle arrest of ess1 mutants.
Discussion
Cyclophilin A mediates inhibition of calcineurin by the immunosuppressive drug CsA, but the cellular functions of cyclophilin A have remained elusive. In the studies reported here, we have found that cyclophilin A is a multicopy suppressor of conditional mutations in the one essential prolyl isomerase in yeast, the parvulin Ess1. The cyclophilin A inhibitor CsA blocks suppression, and CsA also inhibits growth of ess1ts mutant strains under permissive growth conditions. Cyclophilin A active site mutants fail to suppress. Moreover, mutations in the cyclophilin A gene CPR1, or in the FKBP12 gene FPR1, are synthetically lethal with conditional ess1 mutations. Cyclophilin A does not suppress by promoting folding or stability of the thermolabile Ess1 mutant enzymes, and overexpression of cyclophilin A also restores viability in strains in which the ESS1 gene has been deleted, indicating that cyclophilin A can bypass the normal cellular requirement for Ess1. This finding suggests that cyclophilin A and Ess1 can act on common targets required for mitosis, and that prolyl isomerase activity is linked to their essential function.
RNA polymerase II as a target of Ess1
In several recent reports (Albert et al., 1999; Morris et al., 1999) and the accompanying manuscript by Wu et al. (2000) the C-terminal domain (CTD) of RNA polymerase II has been identified as a direct target of Ess1. Wu et al. (2000) failed to find any evidence that cyclophilin A associates with the CTD. High affinity binding of Ess1 to the CTD involves the Ess1 WW domain and not the prolyl isomerase domain (Morris et al., 1999), providing an explanation for why cyclophilin A, which lacks the WW domain, might not interact.
Ess1 and cyclophilin A have common targets
In the accompanying manuscript, Wu et al. (2000) identify multicopy suppressors of ess1 mutations. We show that two suppressors, Cth1 and Sap30, require cyclophilin A to restore viability in ess1 mutants and may represent common targets of cyclophilin A and Ess1.
Sap30 is found in a complex with the Sin3 and Rpd3 proteins, which are components of the yeast HDAC (Laherty et al., 1998; Zhang et al., 1998). While Sap30 lacks serine-prolyl or threonine-prolyl sequences that are the known substrates of Ess1, both Sin3 and Rpd3 have multiple Ser–Pro or Thr–Pro sites. Thus, one plausible model is that Ess1 interacts with Sin3 or Rpd3 to catalyze isomerization events required for protein folding and assembly of the HDAC. In accord with this model, our biochemical studies revealed that interaction of Ess1 with the Sin3–Rpd3 HDAC is mediated by Sin3. Interaction between cyclophilin A and Sap30 was disrupted by deletion of SIN3, although weak binding to Rpd3 was still detected, indicating that interaction between cyclophilin A and Rpd3 can be Sin3 independent. Most interestingly, the yeast cyclophilin 40 homologs Cpr6 and Cpr7 were identified originally in a two-hybrid screen with the Rpd3 subunit of histone deacetylase and shown to interact directly with Rpd3 in vitro and in vivo in the yeast two-hybrid assay (Duina et al., 1996). Our findings suggest that cyclophilin A, as well as Cpr6 and Cpr7, functionally and physically interacts with Rpd3.
Histone deacetylases play a central role in both repression and activation of gene expression. The RPD3 and SIN3 genes positively and negatively regulate transcription of many genes (Vidal and Gaber, 1991; Vidal et al., 1991). The DNA-binding protein Ume6 represses transcription by recruiting the Sin3–Rpd3 complex to target promoters (Kadosh and Struhl, 1997). Recruitment of the Sin3–Rpd3 complex to reporter promoters via LexA hybrids represses transcription (Wang and Stillman, 1993; Kadosh and Struhl, 1997), suggesting a general role for Sin3 and Rpd3 as co-repressors. The Sin3–Rpd3 HDAC fulfills a different role in gene silencing. In yeast, gene silencing prevents expression of the cryptic mating loci (HM) (reviewed in Kingston et al., 1996) and represses transcription of reporter genes inserted near telomeres (Gottschling et al., 1990) or in the rDNA tandem array (Bryk et al., 1997; Fritze et al., 1997; Smith and Boeke, 1997). The Sin3–Rpd3 complex is, paradoxically, required for disruption of silencing at these loci (De Rubertis et al., 1996; Rundlett et al., 1996; Kadosh and Struhl, 1998; Magnaghi-Jaulin et al., 1998; Sun and Hampsey, 1999; Smith et al., 1999).
We studied the influence of Ess1 and cyclophilin A on silencing at the rDNA array. Both prolyl isomerases modulate the expression of an RNA polymerase II-transcribed reporter gene inserted in this region, although with opposite effects; Ess1 enhances silencing, whereas cyclophilin A inhibits silencing. We propose that Ess1 and cyclophilin A influence silencing by physically interacting with the Sin3–Rpd3 complex (Figure 9).
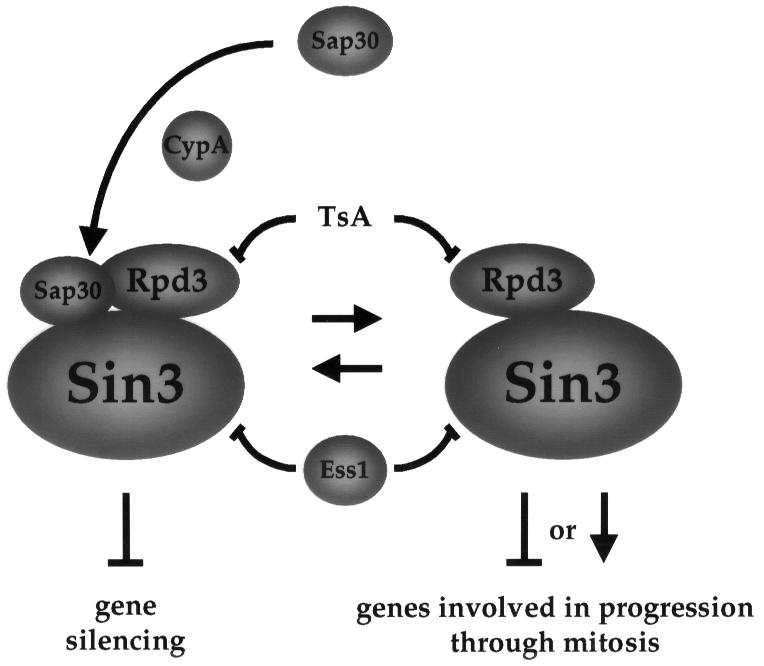
Fig. 9. Model for the mechanism of suppression of ess1 mutations by CypA and Sap30. In this model, Ess1 is a negative modulator of the Sin3–Rpd3 complex, which in turn functions as a global transcriptional regulator. In ess1 mutants, hyperactive Sin3–Rpd3 complexes deregulate expression of genes involved in cell cycle control, causing mitotic arrest. Cyclophilin A (CypA) recruits Sap30 to the complex, reduces activity with mitotic targets and suppresses cell cycle arrest of ess1 mutants.
Rpd3 mediates mitotic arrest of ess1 conditional mutants
We have shown that deletion of RPD3 partially restores viability of ess1ts mutants (Figure 7B). Suppression by RPD3 deletion was not inhibited by CsA and did not increase cyclophilin A levels, indicating that cyclophilin A is not required (not shown). Overexpression of dominant-negative Rpd3 active site mutants, the histone acetyltransferase Gcn5 (Figure 8) or treatment with the HDAC inhibitor TsA (Figure 7A), also suppressed ess1ts mutations. In addition, ess1ts mutant strains show a phenotype consistent with an altered function of the Sin3–Rpd3 complex (decreased silencing at the rDNA locus). Taken together, these findings support a model in which Ess1 inhibits Rpd3 function, and ess1 mutants have increased Rpd3 histone deacetylase activity that results in mitotic arrest.
What is the connection between Rpd3 and the cell cycle arrest exhibited by ess1 mutants? In mammalian cells, the histone deacetylase inhibitors TsA and trapoxin induce G1 and G2 phase cell cycle arrest, indicating an involvement of histone deacetylase activity in cell cycle progression (Yoshida et al., 1995). Deletion of RPD3 increases the life span in yeast, suggesting a correlation between aging and rDNA silencing (Kim et al., 1999). Loss of silencing at the rDNA array observed in the ess1ts mutant is not likely to be the cause of the mitotic arrest, because deletion of the SIN3 or SAP30 genes, both of which are required for disruption of silencing, did not rescue ess1ts mutants (data not shown). Moreover, overexpression of Sap30 suppresses ess1ts mutations.
What is the mechanism by which Sap30 suppresses ess1ts mutations? One possibility is that overexpression of Sap30 diverts Rpd3 from its cellular targets involved in cell cycle arrest. It has been found that yeast Sap30 can repress transcription in a sin3Δ mutant, suggesting that Sap30 can interact with and recruit Rpd3 directly to a promoter in a Sin3-independent manner (Zhang et al., 1998). Taken together, these findings support a model in which overexpression of Sap30 recruits Rpd3 to function in silencing, relieving Rpd3 repression of downstream target genes that are required for mitotic progression (see Figure 9).
Regulation of Sin3–Rpd3 HDAC by Ess1 and cyclophilin A
Sap30 requires cyclophilin A for suppression (Figure 4; Table II). Overexpression of cyclophilin A decreases silencing at the rDNA loci, indicating that cyclophilin A counteracts this function of Ess1. However, cyclophilin A also suppresses ess1 mutations. Our proposal is that both prolyl isomerases catalyze protein conformational changes required for the assembly or activity of the Sin3–Rpd3 complex. Our biochemical studies are consistent with a model in which Ess1 directly interacts with the Sin3 component of HDAC, whereas cyclophilin A interacts directly with Rpd3. In this model, Ess1 functions to down-regulate the histone deacetylase activity of Rpd3; the decreased silencing observed in ess1 mutants then results from deregulated, hyperactive Sin3–Rpd3 complexes. In the model depicted in Figure 9, cyclophilin A catalyzes conformational changes in Rpd3 and/or Sap30 that recruit Sap30 to the Rpd3–Sin3 complex and regulate silencing. Overexpression of cyclophilin A (or of Sap30, in a cyclophilin A-dependent fashion) drives the equilibrium towards the formation of a Sin3–Rpd3–Sap30 complex that is still competent to disrupt silencing (Figure 6), but that has a reduced capacity to interact with downstream targets mediating cell cycle arrest (Figure 9). This model is consistent with that proposed in the accompanying manuscript (Wu et al., 2000) in which mitotic arrest in ess1 mutants is the result of misregulation of genes caused by aberrant RNA polymerase II transcription. Further studies will be required to examine in detail the physical interactions of Ess1 and cyclophilin A with the Sin3–Rpd3 HDAC, and to examine how Ess1 and cyclophilin regulate histone deacetylase activity.
Materials and methods
Yeast strains
Yeast strains used in this work are listed in Table III. Strain MAYX47-4b is a product of a cross of strains JK9-3da and KDY27-7b. Strain MAYX48-6d is from a cross of strains JK9-3da and KDY28. Strain MAY1 was obtained from strain JK9-3dα by introduction of the ess1-A144T mutation using plasmid YIpess1A144T (Wu et al., 2000) via two-step gene replacement. For this purpose, strain JK9-3dα was first transformed with this plasmid linearized with BstXI, excision of the plasmid was selected on 5-FOA at 24°C and ts colonies were selected. Strain MAY3 was obtained in a similar way using plasmid YIpess1H164R. MAY22 was obtained from CFY559 in a similar way; MAY23 is a wild-type ESS1, 5-FOA-resistant strain obtained from the same YIpess1H164R transformant as MAY22. Strain MAY5 was derived from strain JK9-3da/α by replacing the entire open reading frame (ORF) of one ESS1 allele with kanMX (Wach et al., 1994). MAY6 was obtained from W303-1A by replacing the entire SIN3 ORF with kanMX. MAY7 was obtained from W303-1A by replacing the entire RPD3 ORF with kanMX. Strains MAY12 and MAY16 were obtained in a similar fashion from diploids ess1A144T/ESS1 and ess1H164R/ESS1, respectively, by replacing one allele of RPD3 with kanMX. MAY8 was obtained from W303-1A by replacing the entire SAP30 ORF with kanMX. Gene disruptions were all verified by PCR. MAY12-5b, MAY12-7b and MAY12-7d are meiotic products of MAY12. MAY16-2c and MAY16-5c are meiotic products of MAY16.
Table III. Yeast strains.
| Strain | Genotype | Reference |
|---|---|---|
| JK9-3da | MATa his4 HMLa leu2-3, 112 rme1 trp1 ura3-52 | Heitman et al. (1991a) |
| JK9-3dα | MATα (JK9-3da) | Heitman et al. (1991a) |
| MH250-2c | cpr1Δ::LEU2 (JK9-3da) | Cardenas et al. (1994) |
| MAYX47-4b | cpr2Δ::TRP1 (JK9-3da) | this study |
| MAYX48-6d | cpr3::HIS3 (JK9-3da) | this study |
| KDY9 | cpr4Δ::URA3 (JK9-3da) | Dolinski et al. (1997) |
| KDY19 | cpr5Δ::LEU2 (JK9-3da) | Dolinski et al. (1997) |
| KDY46 | cpr6::G418 (JK9-3da) | Dolinski et al. (1997) |
| KDY65 | cpr7Δ::G418 (JK9-3da) | Dolinski et al. (1997) |
| SMY98-1 | cpr8Δ::G418 (JK9-3da) | Dolinski et al. (1997) |
| JHY2-1c | fpr1::ADE2 (JK9-3da) | Cardenas et al. (1994) |
| KDY61 | fpr2Δ::URA3 (JK9-3da) | Dolinski et al. (1997) |
| KDY86-6a | fpr3Δ::URA3 (JK9-3da) | Dolinski et al. (1997) |
| KDY54 | fpr4Δ::G418 (JK9-3da) | Dolinski et al. (1997) |
| KDY27-7b | MATα cpr2Δ::TRP1 cpr4Δ::URA3 (JK9-3da) | this study |
| KDY28 | MATα cpr3::HIS3 cpr4Δ::URA3 cpr5Δ::LEU2 (JK9-3da) | this study |
| W303-1A | MATa ura3-1 leu2-3, 112 trp1-1 can1-100 ade2-1 his3-11,15 [PSI+] | R.Rothstein |
| A144TW303 | ess1-A144T (W303-1A) | S.Hanes |
| H164RW303 | ess1-H164R (W303-1A) | S.Hanes |
| MAY1 | ess1-A144T (JK9-3dα) | this study |
| MAY3 | ess1-H164R (JK9-3dα) | this study |
| MAY5 | CPR1/cpr1Δ::LEU2 ESS1/ess1Δ::G418 (JK9-3da/α) | this study |
| MAY6 | sin3Δ::G418 (W303-1A) | this study |
| MAY7 | rpd3Δ::G418 (W303-1A) | this study |
| MAY8 | sap30Δ::G418 (W303-1A) | this study |
| MAY12 | ESS1/ess1-A144T RPD3/rpd3Δ::G418 (JK9-3da/α) | this study |
| MAY16 | ESS1/ess1-H164R RPD3/rpd3Δ::G418 (JK9-3da/α) | this study |
| MAY12-5b | MATa rpd3Δ::G418 (JK9-3da) | this study |
| MAY12-7b | MATa ess1-A144T rpd3Δ::G418 (JK9-3da) | this study |
| MAY12-7d | MATα ess1-A144T (JK9-3da) | this study |
| MAY16-2c | MATa ess1-H164R rpd3Δ::G418 (JK9-3da) | this study |
| MAY16-5c | MATa ess1-H164R (JK9-3da) | this study |
| CFY559 | MATa ade2Δ::hisG can1Δ::hisG his4 leu2 lys2 pfx1 tyr1-2 ura3-52rDNA::pCAR1 (CAN1-ADE2) | Fritze et al. (1997) |
| MAY22 | ess1-H164R (CFY559) | this study |
| MAY23 | ESS1 (CFY559) | this study |
| MAY22 | ess1-H164R (CFY559) | this study |
Plasmids
YIpess1ts plasmids used for integration of the ess1ts alleles A144T and H164R are described in Wu et al. (2000). Plasmid pTB3 (2µ CPR1 URA3) has been described (Cardenas et al., 1995b). Plasmid pCPR1(H90Y) (2µ CPR1H90Y URA3) is a derivative of pRS426 that carries an H90Y allele of CPR1 that was PCR-amplified from mutant strain TB24-M141 (Cardenas et al., 1995a). Human wild-type and mutant cyclophilin A genes under the control of the 5′- and 3′-untranslated regions of the yeast cyclophilin A CPR1 gene were kindly provided by Jeremy Luban and were subcloned into the YEplac195 vector (2µ URA3), giving rise to plasmids phCypA (wild-type cyclophilin A), phCypA(H54Q), phCypA(R55A), phCypA(F60A), phCypA(F113A), phCypA(W121A) and phCypA(H126Q), used for the overexpression suppression studies described here. Plasmids for CPR6/7 were pKDw10 (2µ CPR6 URA3) and pKS24 (2µ CPR7 URA3) (Dolinski et al., 1998). Plasmid pYJH23 (Heitman et al., 1991b) has a 2 kb EcoRI fragment containing the FPR1 gene cloned in the EcoRI site of pSEY8 (2µ URA3). Plasmid YEpESS1 (2µ ESS1 URA3) has a BamHI fragment containing the ESS1 gene cloned in the BamHI site of YEp24. Plasmids containing ess1ts suppressor genes CTH1, SAP30, YKL005C, FCP1 and CaRPB7 are described in Wu et al. (2000). The 2µ plasmid pYMR263W, expressing a V5 epitope-tagged version of yeast Sap30 under control of the GAL1 promoter, was from Invitrogen. Plasmid YEplac112-RPD3 was described in Kadosh and Struhl (1997). Plasmids YEplac112-RPD3-H150A and YEplac112-RPD3-H151A were described in Kadosh and Struhl (1998). Plasmid Yep10PGK-exp-ScGCN5 (2µ, GCN5, TRP1) was kindly provided by Kevin Struhl. Plasmid pET-ESS1.1 derived from pET28a (Novagen), encodes a His6-tagged version of Ess1.
Binding to Sap30 and Rpd3
His6-FKBP12 and His6-cyclophilin A were purified as described (Cardenas et al., 1994, 1995a). Purification of His6-Ess1p was carried out in the same way, using plasmid pET-ESS1.1. The mutant Escherichia coli strain RY3041 (from Ry Young) lacking the non-essential histidine-rich slyD peptidyl-prolyl isomerase was used to produce His6-tagged proteins (Roof et al., 1997). Yeast strains transformed with plasmid pYMR263W (V5-Sap30) were grown in SD–Ura medium to an OD600 of 0.7, transferred to SGal–ura medium, and incubated for 12 h to induce Sap30 expression. Whole-cell protein extracts were prepared by glass bead disruption in a lysis buffer containing 20 mM HEPES pH 7.9, 150 mM NaCl, 10 mM MgCl2, 10% glycerol, 0.2% Tween-20 and a cocktail of protease inhibitors consisting of 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 1 µg/ml pepstatin, 1 mM benzamidine, 100 µM leupeptin, 1% trasylol and 1 µg/ml TPCK. Affinity chromatography assays were performed with His6-cyclophilin A or His6-Ess1 coupled to Affigel 10 (Bio-Rad) as described (Cardenas et al., 1994) using the lysis buffer described above to wash PPIase-coupled beads after incubation with cell extracts. Bound proteins were resolved by SDS–PAGE in a 12% acrylamide gel, transferred to an Immobilon PVDF membrane (Immun-Blot; Bio-Rad) and analyzed by western blotting. Sap30 and Rpd3 were detected with anti-V5 monoclonal antibody (Invitrogen) or a rabbit polyclonal antiserum specific for Rpd3 (Santa Cruz). Rabbit polyclonal antibodies against cyclophilin A and Ess1 were as described (Cardenas et al., 1995a; Wu et al., 2000).
Acknowledgments
Acknowledgements
We thank Ry Young, Jeremy Luban, Rochelle Esposito, Kevin Struhl, Carl Falco, Kara Dolinski and Rick Gaber for reagents. These studies were supported in part by R01 grant AI39115 (NIAID) to J.H. and M.E.C., K01 award CA77075 (NCI) to M.E.C. and ROI grant GM55108 (NIH) to S.D.H. J.H. is an associate investigator of the Howard Hughes Medical Institute.
References
- Albert A., Lavoie,S. and Vincent,M. (1999) A hyperphosphorylated form of RNA polymerase II is the major interphase antigen of the phosphoprotein antibody MPM-2 and interacts with the peptidyl-prolyl isomerase Pin1. J. Cell Sci., 112, 2493–2500. [DOI] [PubMed] [Google Scholar]
- Anfinsen C.B. (1973) Principles that govern the folding of protein chains. Science, 181, 223–230. [DOI] [PubMed] [Google Scholar]
- Baker E.K., Colley,N.J. and Zuker,C.S. (1994) The cyclophilin homolog NinaA functions as a chaperone, forming a stable complex in vivo with its protein target rhodopsin. EMBO J., 13, 4886–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brillantes A.-M.B. et al. (1994) Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell, 77, 513–523. [DOI] [PubMed] [Google Scholar]
- Bryk M., Banerjee,M., Murphy,M., Knudsen,K.E., Garfinkel,D.J. and Curcio,M.J. (1997) Transcriptional silencing of Ty1 elements in the RDN1 locus of yeast. Genes Dev., 11, 255–269. [DOI] [PubMed] [Google Scholar]
- Cardenas M.E., Hemenway,C., Muir,R.S., Ye,R., Fiorentino,D. and Heitman,J. (1994) Immunophilins interact with calcineurin in the absence of exogenous immunosuppressive ligands. EMBO J., 13, 5944–5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas M.E., Lim,E. and Heitman,J. (1995a) Mutations that perturb cyclophilin A ligand binding pocket confer cyclosporin A resistance in Saccharomyces cerevisiae. J. Biol. Chem., 270, 20997–21002. [DOI] [PubMed] [Google Scholar]
- Cardenas M.E., Muir,R.S., Breuder,T. and Heitman,J. (1995b) Targets of immunophilin–immunosuppressant complexes are distinct highly conserved regions of calcineurin A. EMBO J., 14, 2772–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubertis F., Kadosh,D., Henchoz,S., Pauli,D., Reuter,G., Struhl,K. and Spierer,P. (1996) The histone deacetylase RPD3 counteracts genomic silencing in Drosophila and yeast. Nature, 384, 589–591. [DOI] [PubMed] [Google Scholar]
- Dolinski K. and Heitman,J. (1997) Peptidyl-prolyl isomerases (PPIases). In Tooze,S. (ed.), Guidebook to Molecular Chaperones and Protein Folding Catalysts. Oxford University Press, Oxford, UK, pp. 359–369. [Google Scholar]
- Dolinski K., Muir,R.S., Cardenas,M.E. and Heitman,J. (1997) All cyclophilins and FK506 binding proteins are, individually and collectively, dispensable for viability in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 94, 13093–13098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinski K.J., Cardenas,M.E. and Heitman,J. (1998) CNS1 encodes an essential p60/Stil homolog in Saccharomyces cerevisiae that suppresses cyclophilin 40 mutations and interacts with Hsp90. Mol. Cell. Biol., 18, 7344–7352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duina A.A., Marsh,J.A. and Gaber,R.F. (1996) Identification of two CyP-40-like cyclophilins in Saccharomyces cerevisiae, one of which is required for normal growth. Yeast, 12, 943–952. [DOI] [PubMed] [Google Scholar]
- Ferreira P.A., Nakayama,T.A., Pak,W.L. and Travis,G.H. (1996) Cyclophilin-related protein RanBP2 acts as chaperone for red/green opsin. Nature, 383, 637–640. [DOI] [PubMed] [Google Scholar]
- Fischer G. (1994) Peptidyl-prolyl cis/trans isomerases and their effectors. Angew. Chem. Int. Ed. Engl., 33, 1415–1436. [Google Scholar]
- Fischer G. and Schmid,F.X. (1990) The mechanism of protein folding. Implications of in vitro refolding models for de novo protein folding and translocation in the cell. Biochemistry, 29, 2205–2212. [DOI] [PubMed] [Google Scholar]
- Foor F., Parent,S.A., Morin,N., Dahl,A.M., Ramadan,N., Chrebet,G., Bostian,K.A. and Nielsen,J.B. (1992) Calcineurin mediates inhibition by FK506 and cyclosporin of recovery from α-factor arrest in yeast. Nature, 360, 682–684. [DOI] [PubMed] [Google Scholar]
- Fritze C.E., Verschueren,K., Strich,R. and Esposito,R.E. (1997) Direct evidence for SIR2 modulation of chromatin structure in yeast rDNA. EMBO J., 16, 6495–6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething M.-J. and Sambrook,J. (1992) Protein folding in the cell. Nature, 355, 33–45. [DOI] [PubMed] [Google Scholar]
- Gottschling D.E., Aparicio,O.M., Billington,B.L. and Zakian,V.A. (1990) Position effect at S.cerevisiae telomeres: reversible repression of Pol II transcription. Cell, 63, 751–762. [DOI] [PubMed] [Google Scholar]
- Handschumacher R.E., Harding,M.W., Rice,J. and Drugge,R.J. (1984) Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science, 226, 544–547. [DOI] [PubMed] [Google Scholar]
- Hanes S.D., Shank,P.R. and Bostian,K.A. (1989) Sequence and mutational analysis of ESS1, a gene essential for growth in Saccharomyces cerevisiae. Yeast, 5, 55–72. [DOI] [PubMed] [Google Scholar]
- Hani J., Stumpf,G. and Domdey,H. (1995) PTF1 encodes an essential protein in Saccharomyces cerevisiae, which shows strong homology with a new putative family of PPIases. FEBS Lett., 365, 198–202. [DOI] [PubMed] [Google Scholar]
- Hani J., Schelbert,B., Bernhardt,A., Domdey,H., Fischer,G., Wiebauer,K. and Rahfeld,J. (1999) Mutations in a peptidylprolyl-cis/trans-isomerase gene lead to a defect in 3′-end formation of a pre-mRNA in Saccharomyces cerevisiae. J. Biol. Chem., 274, 108–116. [DOI] [PubMed] [Google Scholar]
- Harding M.W., Galat,A., Uehling,D.E. and Schreiber,S.L. (1989) A receptor for the immunosuppressant FK506 is a cis–trans peptidyl-prolyl isomerase. Nature, 341, 758–760. [DOI] [PubMed] [Google Scholar]
- Heitman J., Movva,N.R. and Hall,M.N. (1991a) Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science, 253, 905–909. [DOI] [PubMed] [Google Scholar]
- Heitman J., Movva,N.R., Hiestand,P.C. and Hall,M.N. (1991b) FK506-binding protein proline rotamase is a target for the immunosuppressive agent FK506 in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 88, 1948–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadosh D. and Struhl,K. (1997) Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell, 89, 365–371. [DOI] [PubMed] [Google Scholar]
- Kadosh D. and Struhl,K. (1998) Histone deacetylase activity of Rpd3 is important for transcriptional repression in vivo. Genes Dev., 12, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Benguria,A., Lai,C.Y. and Jazwinski,S.M. (1999) Modulation of life-span by histone deacetylase genes in Saccharomyces cerevisiae. Mol. Biol. Cell, 10, 3125–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston R.E., Bunker,C.A. and Imbalzano,A.N. (1996) Repression and activation by multiprotein complexes that alter chromatin structure. Genes Dev., 10, 905–920. [DOI] [PubMed] [Google Scholar]
- Laherty C.D. et al. (1998) SAP30, a component of the mSin3 corepressor complex involved in N-CoR-mediated repression by specific transcription factors. Mol. Cell, 2, 33–42. [DOI] [PubMed] [Google Scholar]
- Liu J., Farmer,J.D., Lane,W.S., Friedman,J., Weissman,I. and Schreiber,S.L. (1991) Calcineurin is a common target of cyclophilin–cyclosporin A and FKBP–FK506 complexes. Cell, 66, 807–815. [DOI] [PubMed] [Google Scholar]
- Lu K.P., Hanes,S.D. and Hunter,T. (1996) A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature, 380, 544–547. [DOI] [PubMed] [Google Scholar]
- Lu P.-J., Zhou,X.Z., Shen,M. and Lu,K.P. (1999) Function of WW domains as phosphoserine- or phosphothreonine-binding modules. Science, 283, 1325–1328. [DOI] [PubMed] [Google Scholar]
- Magnaghi-Jaulin L., Groisman,R., Naguibneva,I., Robin,P., Lorain,S., Villian,J.P.L., Troalen,F., Trouche,D. and Harel-Bellan,A. (1998) Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature, 391, 601–605. [DOI] [PubMed] [Google Scholar]
- Maleszka R., Hanes,S.D., Hackett,R.L., Couet,H.G.D. and Miklos,G.L.G. (1996) The Drosophila melanogaster dodo (dod) gene, conserved in humans, is functionally interchangeable with the ESS1 cell division gene of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 93, 447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris D.P., Phatnani,H.P. and Greenleaf,A.L. (1999) Phospho-carboxyl-terminal domain binding and the role of prolyl isomerase in pre-mRNA 3′-end formation. J. Biol. Chem., 274, 31583–31587. [DOI] [PubMed] [Google Scholar]
- Pazin M.J. and Kadonaga,J.T. (1997) What’s up and down with histone deacetylation and transcription? Cell, 89, 325–328. [DOI] [PubMed] [Google Scholar]
- Perez-Martin J. and Johnson,A.D. (1998) Mutations in chromatin components suppress a defect of Gcn5 protein in Saccharomyces cerevisiae. Mol. Cell. Biol., 18, 1049–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahfeld J.-U., Rucknagel,K.P., Schelbert,B., Ludwig,B., Hacker,J., Mann,K. and Fischer,G. (1994) Confirmation of the existence of a third family among peptidyl-prolyl cis/trans isomerase. Amino acid sequence and recombinant production of parvulin. FEBS Lett., 352, 180–184. [DOI] [PubMed] [Google Scholar]
- Ranganathan R., Lu,K.P., Hunter,T. and Noel,J.P. (1997) Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell, 89, 875–886. [DOI] [PubMed] [Google Scholar]
- Roof W.D., Fang,H.Q., Young,K.D., Sun,J. and Young,R. (1997) Mutational analysis of slyD, an Escherichia coli gene encoding a protein of the FKBP immunophilin family. Mol. Microbiol., 25, 1031–1046. [DOI] [PubMed] [Google Scholar]
- Rundlett S.E., Carmen,A.A., Kobayashi,R., Bavykin,S., Turner,B.M. and Grunstein,M. (1996) HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc. Natl Acad. Sci. USA, 93, 14503–14508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz C., Maier,P., Dolinski,K., Heitman,J. and Schmid,F.X. (1999) R73A and H144Q mutants of the yeast mitochondrial cyclophilin Cpr3 exhibit a low prolyl isomerase activity in both peptide and protein-folding assays. FEBS Lett., 443, 367–369. [DOI] [PubMed] [Google Scholar]
- Shen M., Stukenberg,P.T., Kirschner,M.W. and Lu,K.P. (1998) The essential mitotic peptidyl-prolyl isomerase Pin1 binds and regulates mitosis-specific phosphoproteins. Genes Dev., 12, 706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou W. et al. (1998) Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature, 391, 489–492. [DOI] [PubMed] [Google Scholar]
- Smith J.S. and Boeke,J.D. (1997) An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev., 11, 241–254. [DOI] [PubMed] [Google Scholar]
- Smith J.S., Caputo,E. and Boeke,J.D. (1999) A genetic screen for ribosomal DNA silencing defects identifies multiple DNA replication and chromatin-modulating factors. Mol. Cell. Biol., 19, 3184–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart D.E., Sarkar,A. and Wampler,J.E. (1990) Occurrence and role of cis peptide bonds in protein structures. J. Mol. Biol., 214, 253–260. [DOI] [PubMed] [Google Scholar]
- Struhl K. (1998) Histone acetylation and transcriptional regulatory mechanisms. Genes Dev., 12, 599–606. [DOI] [PubMed] [Google Scholar]
- Sun Z.-W. and Hampsey,M. (1999) A general requirement for the Sin3–Rpd3 histone deacetylase complex in regulating silencing in Saccharomyces cerevisiae. Genetics, 152, 921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N., Hayano,T. and Suzuki,M. (1989) Peptidyl-prolyl cis–trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature, 337, 473–475. [DOI] [PubMed] [Google Scholar]
- Vidal M. and Gaber,R.F. (1991) RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in Saccharomyces cerevisiae. Mol. Cell. Biol., 11, 6317–6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M., Strich,R., Esposito,R.E. and Gaber,R.F. (1991) RPD1 (SIN3/UME4) is required for maximal activation and repression of diverse yeast genes. Mol. Cell. Biol., 11, 6306–6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A., Brachat,A., Pohlmann,R. and Philippsen,P. (1994) New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast, 10, 1793–1808. [DOI] [PubMed] [Google Scholar]
- Wang H. and Stillman,D.J. (1993) Transcriptional repression in Saccharomyces cerevisiae by a SIN3–LexA fusion protein. Mol. Cell. Biol., 13, 1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler K.E., Swenson,K.I., Kornbluth,S. and Means,A.R. (2000) Requirement of the prolyl isomerase Pin1 for the replication checkpoint. Science, 287, 1644–1647. [DOI] [PubMed] [Google Scholar]
- Wu X., Wilcox,C.B., Devasahayam,G., Hackett,R.L., Arévalo-Rodríguez,M., Cardenas,M.E., Heitman,J. and Hanes,S.D. (2000) The Ess1 prolyl isomerase is linked to chromatin remodeling complexes and the general transcription machinery. EMBO J., 19, 3727–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe M.B. et al. (1997) Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science, 278, 1957–1960. [DOI] [PubMed] [Google Scholar]
- Yoshida M., Horinouchi,S. and Beppu,T. (1995) Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. BioEssays, 17, 423–430. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Sun,Z.W., Iratni,R., Erdjument-Bromage,H., Tempst,P., Hampsey,M. and Reinberg,D. (1998) SAP30, a novel protein conserved between human and yeast, is a component of a histone deacetylase complex. Mol. Cell, 1, 1021–1031. [DOI] [PubMed] [Google Scholar]
- Zydowsky L.D., Etzkorn,F.A., Chang,H.Y., Ferguson,S.B., Stolz,L.A., Ho,S.I. and Walsh,C.T. (1992) Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci., 1, 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]