

Abstract
Heart valve disease is unique in that it affects both the very young and very old, and does not discriminate by financial affluence, social stratus, or global location. Research over the past decade has transformed our understanding of heart valve cell biology, yet still more remains unclear regarding how these cells respond and adapt to their local microenvironment. Recent studies have identified inflammatory signaling at nearly every point in the life cycle of heart valves, yet its role at each stage is unclear. While the vast majority of evidence points to inflammation as mediating pathological valve remodeling and eventual destruction, some studies suggest inflammation may provide key signals guiding transient adaptive remodeling. Though the mechanisms are far from clear, inflammatory signaling may be a previously unrecognized ally in the quest for controlled rapid tissue remodeling, a key requirement for regenerative medicine approaches for heart valve disease. This paper summarizes the current state of knowledge regarding inflammatory mediation of heart valve remodeling and suggests key questions moving forward.
1. Introduction
The heart valves are the sole mediators of unidirectional flow through the cardiovascular system. These valves flex open and close 30 million times per year, subjecting the thin and flexible cusps or leaflets to demanding tissue strains and hemodynamic stresses. The fact that these tissues thrive can only be attributed to the remarkable stamina and remodeling capacity of the indigenous valve endothelial and interstitial cells that populate these valves. Over the past decade, many exciting discoveries have been made regarding the unique phenotypes of these cells, yet it has only framed the beginning of our understanding of valve function and dysfunction. Heart valve disease remains a serious and increasing clinical problem for which no solution exists save prosthetic replacement. These come in the form of mechanical or processed biological tissue valves. While providing over 20 years of function in elderly patients, these technologies perform dismally in children and young adults, with undesirable lifestyle restrictions and significant medical requirements. Tissue engineering has the potential to alleviate these limitations by providing a living valve conduit that can grow and remodel with the patient. Current results in animal trials are very promising, but human trials to date suggest that there is still much more to learn. Foremost among these needs is to understand how valve tissue remodels in the midst of the complex mechanical and biological signaling environment in which it resides. This includes natural tissue remodeling that occurs over embryonic development into adulthood, homeostatic and pathological adaptation over the course of life and disease, and with different living tissue replacement strategies. Being a biological structure that evolves and adapts over the entire lifespan, it seems likely that similar signaling mechanisms would be utilized across this continuum. While literally hundreds of regulatory genes have been identified in valve phenotypes and discussed in several reviews, this review will focus on the role of inflammation. While it is well appreciated that inflammation is a major driver of valve pathology, recent evidence suggests that inflammatory cytokines are present in embryonic development and in remodeling valves, which suggests its presence may not be singularly negative.
2. Inflammation and Wound Healing
The general healing response to tissue injury involves three phases: inflammation, tissue formation, and tissue remodeling [1]. The healing response begins when the tissue is injured, blood comes into contact with collage or other components of the extracellular matrix, and a blood clot forms. The blood clot platelets release chemotactic factors that recruit leukocytes to the injury site and initiate the inflammation phase [2]. These leukocytes then secrete chemokines and inflammatory cytokines to enhance the inflammatory response [2]. Next, neutrophils enter the wound site to remove foreign materials, bacteria, and damaged tissue; macrophages follow to continue the process of phagocytosis [3]. Fibroblasts deposit new extracellular matrix in the tissue formation phase [3]. In the remodeling phase, the newly deposited extracellular matrix is cross-linked and organized [3]. There are many cell signaling events required for this tightly controlled repair process to take place. The cytokine transforming growth factor-β (TGF-β) has been shown to play a part in all three phases of healing [4].
The inflammatory phase is initiated when TGFβ and other growth factors are released from platelets [5]. TGFβ has been shown to be chemotactic and mitogenic for neutrophils, lymphocytes, monocytes, macrophages, and fibroblasts [6]. During the tissue formation phase inflammatory cells migrate to the wound and secrete additional TGFβ, which at higher concentrations may induce the expression of other growth factors and increase cell proliferation at the wound site, stimulate angiogenesis, and promote collagen deposition [6–8]. During tissue remodeling TGFβ continues to promote extracellular matrix production and inhibit its breakdown, which has been implicated the cytokine in scar formation [6, 9]. Scars are fibrous tumors characterized by overabundant collagen deposition [10]. Treatment with TGFβ has been shown to increase endogenous TGFβ production, collagen deposition, and scar formation; while exposure to anti-TGFβ antibody decreases endogenous TGFβ production, collagen deposition, and scarring [9–11].
3. Inflammation and Valve Homeostasis
Each valve is comprised of thin, fibrous leaflets or cusps that are attached to a relatively rigid annulus or root [12]. The atrioventricular valves are further supported by tendinous chords that connect the leaflet free edge to the papillary muscles [12]. The leaflets/cusps are a multilayer composite of collagen, elastin, and glycosaminoglycans that assist in its efficient biomechanical function [12]. The surfaces of these tissues are lined with endothelial cells (VEC) while the underlying matrix is populated with interstitial cells (VIC)—a constellation of subphenotypes with incompletely understood individual roles [13]. In general, the endothelial cells are responsible for sensing and integrating biological and mechanical signals from the blood, and transmitting signals to the interstitial cells [14]. The VIC in turn proliferate and remodel the surrounding matrix [15]. Both cell types are also affected by other microenvironmental cues, such as mechanical strain, tissue stiffness, and the presence of other cell types such as inflammatory macrophages and circulating cells [16]. Mitral valve cell biology is much less understood because the tissue is much more heterogeneous in structure and composition, potentially reflective of its more diverse mechanical environment [17]. The mitral valve is unsurprisingly susceptible to a much wider range of dysfunctional conditions (explained later). Therefore, this section will discuss results from aortic valves.
The inflow (atrialis/ventricularis) surface is exposed to a rapid, pulsatile, unidirectional shear stress with cycle averaged magnitude of 20 dynes/cm2 [18]. The outflow (fibrosa) surface experiences a much lower magnitude, nearly oscillatory shear stress [19]. VEC align perpendicular to the direction of flow in vitro and in vivo whereas vascular EC align parallel [20]. Microarray comparisons between aortic valvular and vascular endothelium in static culture and under fluid flow identify hundreds of significantly different genes, suggesting that VEC are a distinct endothelial phenotype [21]. Steady shear stress was protective against pro-oxidation and proinflammation in both cell types, but VEC were inherently less inflammatory than arterial endothelial cells [21]. Simmons and colleagues probed side-specific differences in aortic VEC gene expression [22]. Aortic side endothelial cells showed significantly less expression of multiple inhibitors of cardiovascular calcification, enhanced antioxidative gene expression, and a lack of differential expression of proinflammatory molecules; suggesting that the aortic side endothelium may be primed to protect against inflammation and lesion initiation in the normal valve. In a follow-on study, Guerraty et al. investigated the side-specific aortic valve endothelial gene expression of hypercholesterolemic pigs [23]. They identified differential expression on the aortic side of caspase 3, peroxisome proliferator-activated receptor-γ, tumor necrosis factor-α (TNF-α), and nuclear factor-κB- (NF-κB-) related pathways that were consistent with a protective endothelial phenotype that persisted at 6 months. In contrast to these results, Sucosky et al. showed that high magnitude pulsatile shear stress applied to fibrosa-side VEC (an “altered” shear stress state) induced upregulation of inflammatory receptors and expression of BMP-4 [24]. Similar to shear stress, cyclic mechanical strain or pressure can induce and modulate an inflammatory phenotype in aortic VEC [25–27]. Cyclic tissue strain at 5% or 20% magnitude increased inflammatory cytokine expression in aortic valve endothelium, but decreased at 10–15% [25, 26].
VIC are a heterogeneous cell population with up to five different cell phenotypes (fibroblasts, smooth muscle cells, myofibroblasts) in adult aortic valves [13, 15, 28–31], but generally exhibiting fibroblastic phenotypic characteristics. Normal aortic valve interstitial cells secrete and turnover proteins and glycosaminoglycans at a dramatically increased rate in comparison to other cell types in vivo, with a significantly higher index of proliferation [32]. This suggests that VIC continually repair mechanically induced tissue microdamage to enable long-term durability. VIC freshly isolated from higher pressure left-sided valves (aortic, mitral) were significantly stiffer and had more collagen biosynthesis than cells isolated from right-sided valve (tricuspid, pulmonary) [33]. Collagen synthesis by valve interstitial cells was shown to be dependent upon the degree and duration of stretch by Ku et al., as there was a significant increase in [3H]-proline incorporation into stretched valve cells at 10%, 14%, and 20% stretch [34]. These results indicate that VIC-mediated matrix remodeling is regulated in part by the magnitude of local mechanical signaling. VIC exposed to 15% “pathological” cyclic strain increased expression of α-smooth muscle actin (α-SMA), bone morphogenic protein (BMP)-2/4, matrix metalloproteases (MMP) and cathepsin activity, apoptosis, and osteoblastic protein expression, but not at 10% [27]. Hypertensive (170 mmHg) cyclic pressure also increased expression of VCAM-1 and downregulated osteopontin [35]. VIC-VEC cocultures within 3D type I collagen scaffolds suggest that VEC help maintain a quiescent VIC fibroblastic phenotype [36, 37]. The presence of endothelial cells stabilized VIC proliferation, promoted a quiescent VIC phenotype, increased VIC protein synthesis, and decreased glycosaminoglycan loss [37]. The addition of steady shear stress to the cocultures further enhanced the effects of the endothelial cells, including a further decrease in myofibroblastic markers and increase in protein synthesis [37].
4. Inflammation and Calcific Aortic Valve Disease
The cause of calcific aortic valve disease (CAVD) is not completely defined, but inflammation plays a lead role in the initiation and progression of CAVD. Adhesion molecules, such as ICAM-1, VCAM-1, PECAM-1, CD34, and E-selectin, promote the participation of endothelial cells (EC) in both physiological and pathological inflammatory responses through the recruitment of leukocytes [38]. EC become activated, which is a phenotypic change characterized by the production of a variety of biologically active products (cytokines, growth factors, proteolytic enzymes, adhesion molecules), an increase in adhesion molecule expression, endothelial-leukocyte interaction, and permeability, in response to stimuli including circulating inflammatory cytokines, lipopolysaccharides, activation of the renin-angiotensin system, hypercholesteremia, CD40/CD40 ligand interactions, ischemia-reperfusion, physical trauma, diabetes, and hemodynamic forces [38–41]. In diseased aortic valves, VEC upregulate ICAM-1, VCAM-1, and E-selectin, and this occurs preferentially on the fibrosa surface of the valve [24, 38, 40]. The fibrosa or aortic side of the valve is also exposed to disturbed, oscillatory flow, high bending stresses, and is where calcific degeneration initiates [19, 42–45]. The hemodynamics on the valve fibrosa may make the cells more susceptible to inflammatory cell infiltration. In vitro, disturbed flow has been shown to cause pro-inflammatory cytokine release (BMP-2/4) and a pro-oxidant phenotype (NADPH, ROS) in VEC [21, 24, 46]. Oxidative, inflammatory, and chondrogenic/osteogenic gene expression profiles are upregulated in vitro in VEC grown under static conditions, which mimic hemodynamic conditions on the fibrosa side, when compared with steady shear stress conditions, which recreate ventricularis side hemodynamics [21, 47]. Evidence of leaflet stress promoting CAVD is the discrepancy in age at the time of presentation with tricuspid and bicuspid valves [48]. Patients with bicuspid valves, which are subjected to higher mechanical stresses, on average present with CAVD two decades younger than those with tricuspid valves [49, 50].
Atherosclerotic risk factors, such as increased low-density lipoprotein cholesterol, increased lipoprotein(a), male gender, cigarette smoking, hypertension, elevated body mass index, and diabetes, increase the incidence of aortic stenosis and likely contribute to valve endothelial dysfunction [44, 51, 52]. The dysfunctional, activated VEC in early valve disease have increased permeability and upregulated adhesion molecule expression. Monocytes attach to adhesion molecules, migrate into the subendothelial space of the valve, and differentiate into macrophages [38, 40, 53]. Macrophages and T-lymphocytes have been shown to be present in aortic valve lesions [54–56]. These invading inflammatory cells, and likely the resident activated endothelial cells, secrete a number of cytokines and/or chronic inflammation effector molecules (e.g., HLA-DR IL-1β/2/6, TNF-α, TGFβ-1, BMP-2/4/7) [43, 56–59]. In addition, circulating low density lipoproteins (LDL) are able to migrate through the permeable endothelial layer, and oxidized forms are capable of deep tissue penetration [60, 61]. Sub-endothelial LDL accumulation can recruit additional inflammatory cells by mechanisms including the induction of macrophage chemoattractants, adhesion molecules, and cytokines [62, 63]. Circulating LDL particles may also deliver angiotensin converting enzyme (ACE) to valve lesions [64]. Angiotensin II, which is generated from angiotensin I by ACE, can compound the inflammatory responses already present by stimulating inflammation and macrophage cholesterol accumulation, increasing oxidant stress, and impairing fibrinolysis [65].
The cytokines and inflammatory effector molecules secreted into the subendothelium and fibrosa by immune cells and activated endothelial cells contribute to a local biochemical environment that promotes VIC differentiation, matrix remodeling, neovascularization, fibrosis, and calcification. TGFβ-1, BMP-2/4/7, IL-1β, TNF-α, lipopolysaccharide, and peptidoglycan have been shown to induce myofibroblastic differentiation, the expression of proinflammatory mediators, or the upregulation of osteogenesis-associated factors in VIC when applied individually in vitro [43, 57, 59, 66–69]. Impaired anti-inflammation mechanisms may also contribute to VIC-mediated pathogenesis of aortic stenosis as interleukin-1 receptor antagonist, which is the antagonist of interleukin-1β, was shown to be abundant in nonstenotic aortic valve leaflets and nearly absent in leaflets from stenotic valves [70]. Myofibroblastic activation is characterized by an increase in myofibroblast markers such as vimentin, α-SMA, and embryonic nonmuscle myosin heavy chain (SMemb); and increased cell migration, proliferation, and contractility [13, 29, 31, 71–73]. Activated VIC and inflammatory cells secrete MMP, and cathepsins that progressively destroy the primarily collagen and elastin valve matrix ultrastructure [71, 74–79]. Enzymatic cleavage of ECM can release bound growth factors such as TGFβ-1, which further promotes VIC myofibroblast differentiation and MMP expression [80–83]. MMP and cathepsin inhibitors, such as TIMPs and cystatin C, are also expressed by activated VIC, but their role in CAVD is not yet known [77, 84]. Following healthy ECM destruction, activated VIC deposit a remodeled, fibrotic matrix characterized by disorganized collagen bundle accumulation, proteoglycan degradation, and fragmentation and stratification of elastin fibers [84–86]. This ECM remodeling results in a stiff aortic valve that is prone to restricted movement, stenosis, and eventual calcification [85].
5. Myxomatous Degeneration of the Mitral Valve
Myxomatous degeneration of the mitral valve or mitral valve prolapse (MVP) is a condition diagnosed with echocardiographically and characterized by abnormally thickened, redundant, floppy leaflets that are displaced into the left atrium during systole [87]. MVP is estimated to affect 1–3% of the US population, and some serious complications of the condition include progressive heart failure, thromboembolism, infective endocarditis, and sudden death [71, 87–90]. The mechanisms for the changes within the valve leaflets are unknown, but myxomatous valvular degeneration is characterized by collagen degradation, proteoglycan accumulation, and elastin fragmentation [85, 87]. This valve matrix remodeling allows stretching of the leaflets, resulting in a floppy valve that is prone to prolapse and regurgitation. It has been hypothesized that the leaflet remodeling may be a response to repeated mechanical stress [91].
There are several connective tissue inherited disorders that are associated with mitral valve dysfunction including Marfan syndrome (fibrillin-1 mutations), Williams syndrome (elastin mutations), osteogenesis imperfecta (collagen 1A1 and 1A2 mutations), Ehlers-Danlos syndrome (mutations in collagen 1A1, 3A1, 1A2, 5A1, and tenascin-x), and Stickler syndrome (collagen 2A1 and collagen 11A1 mutations) [92–100]. MVP is generally sporadic, however, and it is unlikely that more than 1-2% of MVP cases are associated with a connective tissue disorder [87]. A Marfan syndrome mouse model has indicated that increased TGF-β signaling contributes to collagen dysregulation and loss of valve matrix integrity in Marfan syndrome-related and possibly other forms of MVP [101]. Fibrillin-1 interaction with latent TGF-β binding proteins (LTBP) regulates TGF-β activation [102, 103]. LTBP forms a bridge between matrix microfibrils and latency-associated peptide (LAP), which remains noncovalently linked to TGF-β and aids in matrix sequestration. Work by Ng et al. suggests that TGF-β sequestered in the TGF-β, LAP, and LTBP complex is stabilized and/or less prone to activation due to interaction with fibrillin-1 and potentially other components of the extracellular matrix [101]. Pharmacological inhibition of TGF-β signaling with losartan has been shown to reduce Marfan syndrome pathology in mice and humans [104, 105]. Increased TGF-β signaling also supports the interstitial cell activated myofibroblast phenotype and excessive proteolytic activity found in myxomatous valves, as TGF-β has been shown to activate valve interstitial cells and upregulate the expression of ECM-degrading enzymes [71, 82].
6. Rheumatic Heart Valve Disease and Infective Endocarditis
Rheumatic fever (RF) is an inflammatory complication that may develop after a untreated throat infection by the group A β-hemolytic streptococcal bacteria Streptococcus pyogenes in susceptible children and teenagers [106]. Carditis, which is one of the most serious RF complications, occurs about 20 days after the infection in 40–50% of patients and can lead to valvular heart disease, heart failure, or death [107–109]. The streptococcal cell structures include the cell wall, capsule, fimbriae, peptidoglycans, cytoplasmic membrane, group-specific carbohydrates, and the M, T, and R antigenic proteins [110]. The streptococcal M protein and hyaluronic acid capsule have been established as the most important virulence factors in human infections, as both confer antiphagocytic properties upon the streptococcal cell and patients with acute RF have a high level of antibodies to streptococcal M protein [111–115]. The M protein is attached to the bacterial cell wall and membrane and extends from the cell surface as an alpha-helical coiled-coil dimer [116, 117].
RF-related cardiac complications are the result of an autoimmune reaction induced by molecular mimicry of human tissues by streptococcal M proteins [108]. The alpha-helical coiled-coil dimer streptococcal M protein has been shown to be structurally and immunologically similar to cardiac myosin, a known mediator of inflammatory heart disease, and other alpha-helical coiled-coil molecules [118]. The antistreptococcal/antimyosin monoclonal antibody mAb 3B6 from rheumatic carditis has shown that the group A streptococcal M protein N-acetylglucosamine, which is the dominant group A carbohydrate epitope, and cardiac myosin in the myocardium are the cross-reactive antigens involved in antibody deposition on the valve [119]. Cardiac myosin is not part of the valve, but mAB 3B6 was also shown to recognize laminin, a valvular extracellular matrix protein with alpha-helical coiled-coil domains that are highly homologous with streptococcal M proteins and cardiac myosins [118, 119].
In acute rheumatic carditis host M-protein antibodies bind the valve surface endothelium and/or the valve basement membrane structure protein laminin, which upregulates the endothelial expression of inflammatory adhesion molecules such as VCAM-1 [120]. The inflamed valvular endothelium leads to T-cell recruitment and infiltration through the endothelial layer [118]. T-lymphocytes enter the valve interstitium and cause further inflammation, degeneration, and remodeling. The resulting valve pathologies include neovascularization, chronic inflammation, commissural fusion, thickening, calcification, and thickened and shortened chordae in the atrioventricular valves [108].
Infective endocarditis (IE) is inflammation of the endocardial surfaces of the heart, most commonly the heart valve, caused by the presence of bacteria in the bloodstream and bacterial vegetations forming on valve leaflets [121]. The bacterial strains that cause IE include staphylococci, streptococci, and enterococcus, and the complications from this type of infection include severe valvular dysfunction, congestive heart failure, and death [122]. If there is altered blood flow around the valves or the valves have been damaged, from mitral valve regurgitation and thickening due to rheumatic fever, for example, the risk of bacterial attachment increases [121].
The bacterial vegetations that form on the valve surface are composed of platelets, fibrin, microorganisms, and inflammatory cells and are the result of mechanical or inflammatory valve lesions [121, 123]. Mechanically denuded endothelial layer lesions promote microbial adherence to the endothelium when bacteria is present in the bloodstream [123]. Endothelial denudation results in direct contact between blood and proteins within the valve interstitium, which include extracellular matrix proteins, thromboplastin, and tissue factor, and causes blood coagulation [123]. The damaged endothelium is colonized when microorganisms bind to the fibrin and platelet-containing blood clots and initiate monocyte activation and production of cytokines and tissue factor activity (TFA) [124]. The vegetation grows when cytokines and TFA activate coagulation cascades, attract and activate blood platelets, and induce cytokine, integrin, and TFA production from nearby endothelial cells [123]. Inflammatory lesions are the result of endothelial cells responding to local inflammation by expressing β1 integrins, including very late antigen (VLA) [125]. β1 integrins bind fibronectin to the valve endothelial surface [123]. IE-associated pathogens have fibronectin-binding proteins on their surface, therefore β1 integrins provide an adhesive surface for the circulating microorganisms [123]. Following pathogen adhesion, endothelial cells internalize the bacteria; which causes endothelial TFA and cytokines production, blood clotting, the extension of inflammation, and vegetation growth [123, 126]. Internalised bacteria eventually lyse endothelial cells by secreting membrane active proteins such as hemolysins [123]. The vegetation growth and tissue damage caused by mechanical or inflammatory lesions can result in abscess formation and septic emboli may circulate to other organs [127].
7. Serotonin Metabolism-Related Valve Disorders
Clinical studies have identified an increased incidence of fibrotic aortic valve disease in patients using several classes of drugs that are structurally similar to serotonin (5-hydroxytryptamine, 5-HT) and in patients with carcinoid syndrome, which can result in a high serotonin concentration on the right side of the heart [128–131]. External serotonin administration in vitro and in vivo has been shown to increase VIC proliferation and α-SMA, collagen, and TGFβ expression, which are all indicative of myofibroblastic activation and fibrotic matrix remodeling [132–135]. No studies have directly linked aortic valve calcification and elevated serotonin levels; however, serotonin may mediate intermediate stage valve fibrosis. Recent evidence supports serotonin synthesis by valve cells [136]. This may contribute to valve calcification through increased TGFβ signaling by serotonin receptor antagonists or transporter agonists in mitral valves [136]. The effects of serotonin on VEC are less well understood, but endothelial function is required for serotonin-mediated valve relaxation [137, 138]. These results suggest that controlling serotonin metabolism may be a novel means of selectively modulating valve cell phenotype in vivo.
8. Bone Marrow Stem Cell Contribution to Valve Repair and Disease
Bone marrow-derived mesenchymal stem cells (BMSCs) are found in adult circulation at a low concentration and are thought to regulate the immune response in settings such as tissue injury, transplantation, and autoimmunity [139]. Mouse and human valve studies have shown that at least 10% of VIC are BMSCs, but the specific role of BMSCs in valve disease is not well understood [140, 141]. The progenitors appear to be recruited with other inflammatory cells into the valve interstitium, but whether they initiate repair or promote disease is unclear. Tanaka et al. identified bone marrow-derived cells expressing myofibroblast and osteoblast markers near calcific nodules in aged mice, which was supported in humans by Helske et al. [79]. Understanding the recruitment and function of circulating stem cells in valve homeostasis and pathogenesis could have clinical benefits, as autologous BMSCs may be treated to express antifibrotic and anticalcific proteins before their mobilization to valves [142].
9. Inflammation and Valve Tissue Engineering
Tissue engineered heart valves could provide a great clinical benefit to patients with valve disease, especially children who require valve growth and younger patients who cannot tolerate the side effects of nonliving prosthetics [143, 144]. Understanding the role of host inflammatory response is essential to the successful implantation of tissue engineered valves, however. Repopulation and regeneration are the two main heart valve tissue engineering approaches that have been attempted [145]. A repopulated heart valve is created when a patient's cells repopulate an implanted decellularized porcine aortic valve [145]. A heart valve is regenerated by implantation of a resorbable matrix that remodels in vivo, resulting in a functional valve composed of the patient's cells and connective tissue proteins [145]. Neither of these approaches have achieved clinical success, however.
Long-term sheep implantation studies with decellularized heart valves and acellular valves seeded with cells before implantation showed significant tissue overgrowth, infiltration with inflammatory cells, and dilatation [146, 147]. The SynerGraft valve by CryoLife is a decellularized pulmonary valve that has been implanted in humans. Adult clinical trials have been encouraging, but in children, rapid inflammatory reactions caused death in 3 out of 4 patients [148, 149]. All pediatric valves displayed severe inflammation, including fibrosis, encapsulation, perforation, and deterioration of the leaflet tissues [149]. Matrix P plus valves, which are decellularized pulmonary porcine valves, displayed early obstruction following implantation in pediatric patients [150]. A foreign body-type reaction accompanied by severe fibrosis and massive neointima formation around the decellularized porcine valve wall was found. Examination of the explanted valves showed inflammatory infiltrates, composed of T cells, B cells, plasma cells, dendritic cells, macrophages, and mast cells, in the tissue surrounding the porcine matrix. In vitro studies have shown that the inflammatory mechanism of decellularized valves may involve porcine collagen type I, but not porcine elastin [151].
In one of the few long-term in vivo studies with polymeric tissue engineered heart valves, composite biodegradable polymer valves were seeded with mesenchymal stem cells, cultured for 4 weeks, and implanted into a sheep model for up to 20 weeks [152]. In vivo monitoring showed leaflet coaptation, and explanted valves showed near-native trilaminar matrix striation with both endothelial-like and interstitial-like cell phenotypes. Elevated transvalvular pressure gradients, however, suggest that the cusps are still too stiff. Interestingly, these valves exhibited a marked inflammatory activation during their early remodeling period, characterized by the presence of endothelial inflammatory receptors (VCAM-1, ICAM-1), matrix metalloproteases, and interstitial cell myofibroblastic activation. Early matrix structure was virtually absent and disorganized. After 2-week in vitro conditioning, and further enhanced with in vivo implantation, the progression to organized tissue striation correlated inversely with the degree of activated valve cell phenotypes. Collectively, these results demonstrate that the degree of inflammation has a significant role on the fate of engineered tissues, and suggests that progressively reduced inflammation can help develop and maintain quiescent tissue.
10. Inflammatory Regulation of Valve Formation
The morphogenesis of the atrioventricular and semilunar valves is a complex process that occurs along with the changing cardiac morphology and hemodynamics of the growing embryo. The early embryonic heart is a tube of endocardium surrounded by myocardium [153]. Soon after the linear heart tube begins to loop the myocardium secretes cardiac jelly, a hyaluronan and chondroitin sulfate rich gelatinous matrix, into the atrioventricular (AV) junction and outflow tract (OFT) lumen [154–157]. At chick stage HH14 (mouse E9.0, human day 20), valvulogenesis begins when a subset of endocardial cells receive growth factor signals from the myocardium that initiate a cascade of signal pathways resulting in an endocardial to mesenchymal transformation (EMT) [153, 158]. EMT is characterized by endocardial cell activation; three antigens expressed only by activated endocardial cells include ES130, JB3/fibrillin, and TGFβ3 [159–161]. Activated endocardial cells lose cell-cell contacts proteins (PECAM1, NCAM1, DS-CAM), gain cell-matrix adhesions (integrins), gain mesenchymal markers (α-smooth muscle actin, α-SMA), and the acquire cell migratory and invasive capacity (transformation) [153, 157, 158, 162, 163]. The transformed endocardial cells invade the cardiac jelly and form the endocardial cushions that will eventually develop into mature atrioventricular and semilunar valves [157, 162].
Few studies have been performed to determine the role of inflammation on valve development. A recent study showed that leptin, a member of the IL-6 superfamily, mediates embryonic EMT [164]. TNF-α serum levels were found to be high in children with congenital heart disease and TNF-α and IL-6 levels were elevated in the myocardium of infants with tetralogy of Fallot or ventricular septal defects [165, 166]. Neonates with hypoplastic left-heart syndrome (HLHS) have evidence of an activated inflammatory response, as IL-6 levels are significantly elevated when compared with controls [167]. Fetal aortic valve endothelial cells express ICAM-1 and VCAM-1, but whether this is due to inflammation is unknown [41]. VCAM-1 deficient mice survive to E11.5–12.5 and display severe heart abnormalities including a reduction of the compact layer of the ventricular myocardium and intraventricular septum, blood in the pericardial space, and an absent epicardium [168].
Abnormal inflammatory signaling may play a role in the development of congenital heart defects. Children born to mothers' with the connective tissue disease lupus erythematosus (LE) have an increased risk for congenital heart block (CHB), a type of arrhythmia, and valve regurgitation [169, 170]. CHB can result in early death from delayed pacemaker placement or hemodynamic compromise from associated congenital heart disease [171]. The fetal heart defects resulting from maternal LE are thought to be the result of transplacental passage of certain maternal autoantibodies, including immunoglobulin G (IgG) antibodies to SSA/Ro and SSB/La ribonucleoproteins [169, 172]. These autoantibodies damage the fetal AV conduction tissue by inflammation in the early stage and later by fibrosis [173]. Maternal inflammatory insult has also been shown to result in fetal cardiac dysfunction [174]. Administration of intra-amniotic lipopolysaccharide caused an increase in maternal TNF-α and IL-6 serum levels, and the placenta showed severe maternal vascular dilatation and congestion [175]. No inflammatory activation was found in the fetal tissues, and the amniotic fluid revealed no significant increase in cytokines; but ultrasonographic examination of the fetal hearts showed that 65% of the fetuses exhibited atrioventricular valve regurgitation [175]. The maternal inflammatory insult was shown to activate placental labyrinthine macrophages, which then leads to an acute increase in placental vascular resistance and fetal cardiac dysfunction [175].
Abnormal nuclear factor-Kappa B (NF-κB) activation may play a role in congenital heart defects. NF-κB is a transcription factor that coordinates inflammation and cellular proliferation. Upregulated NF-κB activation was found in the myocardium of children with congenital heart disease and in the myocardium of infants with tetralogy of Fallot or ventricular septal defects [165, 166]. The inhibition of NF-κB during chicken heart development led to impaired OFT development and resulted in interventricular communication, double outlet right ventricle, and valvular and great arteries stenosis [176]. Nuclear factor of activated T cells (NF-ATc) is a transcription factors that may be needed for cytokine gene expression in activated lymphocytes. Mice lacking NF-ATc died in utero from congestive heart failure at days 13.5–17.5 of gestation due to the absence pulmonary and aortic valve leaflets [177]. Adults with congenital heart disease were found to have elevated levels of endotoxin and of the inflammatory cytokines TNF-α and IL-6 when compared to controls, which indicates that adults with congenital heart disease could potentially benefit from novel anti-inflammatory therapies [178].
11. Conclusions
These studies highlight our evolving understanding of the complex role of inflammation in heart valve remodeling, and underscore how much more is left to be learned. While it is true that inflammation mediates the ugliest of pathological valve remodeling, the fact that it is also present during normal embryonic development suggests a potential benefit. Indeed, inflammation-mediated tissue formation, for example, during foreign body encapsulation, is among the most rapid processes available in our natural arsenal. Embryonic morphogenesis progresses on a significantly more rapid scale than wound healing, yet instead of fibrosis results in scar-free, mature tissue architectures. It is therefore time to reconsider the myopic view of inflammation as the harbinger of destruction, but rather as a universal agent of tissue remodeling, for good or ill. In this light, many more questions are revealed rather than answered (Figure 1). How does inflammation promote positive remodeling, and what are the triggers for pathological change? Are there signaling paradigms in embryonic valve formation that are reactivated in pathological valve remodeling? If so, how are these controlled by inflammation? The answers will likely be heavily dependent on the microenvironmental context (patient age, gender, underlying inflammatory state, gene mutations, mechanical stress, etc.). However, this transformative notion suggests a new avenue to promote and control tissue remodeling in adaptive and regenerative heart valve applications. It is not yet known whether or how this may be achieved in tissue engineering applications, but strongly suggests that complete cessation of inflammatory signaling may not be the best approach. We hope with future research in this area these answers will lead to new clinically translatable therapies for patients with valve disease.
Figure 1.
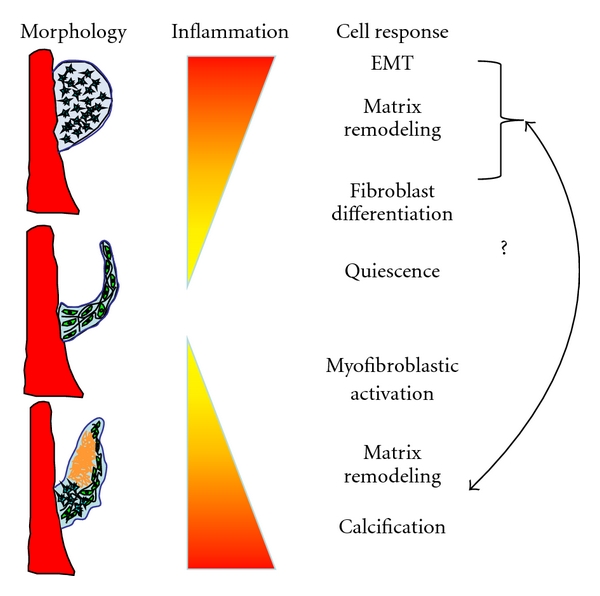
Inflammation and its signaling partners are involved in every stage of the heart valve life cycle, from embryonic morphogenesis (top), quiescent adult valve homeostasis (middle), and in valve pathology (bottom). Many structural and biological features of diseased valves resemble those from embryonic/fetal valves, suggesting similar signaling pathways are reactivated. Identifying and controlling these pathways represents an important and challenging task with potential for large payoff for heart valve disease diagnosis and treatment.
Acknowledgments
This paper was supported by grants from the American Heart Association (J. T. Butcher), The National Science Foundation (J. T. Butcher), and The Hartwell Foundation (J. T. Butcher and G. J. Mahler). The authors have no financial conflicts of interests to disclose.
References
- 1.Singer AJ, Clark RAF. Cutaneous wound healing. The New England Journal of Medicine. 1999;341(10):738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 2.Wang XJ, Han G, Owens P, Siddiqui Y, Li AG. Role of TGFβ-mediated inflammation in cutaneous wound healing. Journal of Investigative Dermatology Symposium Proceedings. 2006;11(1):112–117. doi: 10.1038/sj.jidsymp.5650004. [DOI] [PubMed] [Google Scholar]
- 3.Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Frontiers in Bioscience. 2004;9:283–289. doi: 10.2741/1184. [DOI] [PubMed] [Google Scholar]
- 4.O’Kane S, Ferguson MWJ. Transforming growth factor βs and wound healing. International Journal of Biochemistry & Cell Biology. 1997;29(1):63–78. doi: 10.1016/s1357-2725(96)00120-3. [DOI] [PubMed] [Google Scholar]
- 5.Massague J. The transforming growth factor-β family. Annual Review of Cell Biology. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- 6.Crowe MJ, Doetschman T, Greenhalgh DG. Delayed wound healing in immunodeficient TGF-β1 knockout mice. Journal of Investigative Dermatology. 2000;115(1):3–11. doi: 10.1046/j.1523-1747.2000.00010.x. [DOI] [PubMed] [Google Scholar]
- 7.Roberts AB, Sporn MB, Assoian RK, et al. Transforming growth factor type β: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(12):4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck LS, Deguzman L, Lee WP, Xu Y, McFatridge LA, Amento EP. TGF-β1 accelerates wound healing: reversal of steroid-impaired healing in rats and rabbits. Growth Factors. 1991;5(4):295–304. doi: 10.3109/08977199109000293. [DOI] [PubMed] [Google Scholar]
- 9.Shah M, Foreman DM, Ferguson MWJ. Neutralising antibody to TGF-β1,2 reduces cutaneous scarring in adult rodents. Journal of Cell Science. 1994;107(5):1137–1157. doi: 10.1242/jcs.107.5.1137. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Smith P, Pu LLQ, Kim YJ, Ko F, Robson MC. Exogenous transforming growth factor β2 modulates collagen I and collagen III synthesis in proliferative scar xenografts in nude rats. The Journal of Surgical Research. 1999;87(2):194–200. doi: 10.1006/jsre.1999.5757. [DOI] [PubMed] [Google Scholar]
- 11.Shah M, Foreman DM, Ferguson MWJ. Neutralisation of TGF-β1 and TGF-β2 or exogenous addition of TGF-β3 to cutaneous rat wounds reduces scarring. Journal of Cell Science. 1995;108(3):985–1002. doi: 10.1242/jcs.108.3.985. [DOI] [PubMed] [Google Scholar]
- 12.Christie GW. Anatomy of aortic heart valve leaflets: the influence of glutaraldehyde fixation on function. European Journal of Cardio-Thoracic Surgery. 1992;6(1):S25–S33. [PubMed] [Google Scholar]
- 13.Taylor PM, Batten P, Brand NJ, Thomas PS, Yacoub MH. The cardiac valve interstitial cell. International Journal of Biochemistry & Cell Biology. 2003;35(2):113–118. doi: 10.1016/s1357-2725(02)00100-0. [DOI] [PubMed] [Google Scholar]
- 14.Poggianti E, Venneri L, Chubuchny V, Jambrik Z, Baroncini LA, Picano E. Aortic valve sclerosis is associated with systemic endothelial dysfunction. Journal of the American College of Cardiology. 2003;41(1):136–141. doi: 10.1016/s0735-1097(02)02622-0. [DOI] [PubMed] [Google Scholar]
- 15.Mulholland DL, Gotlieb AI. Cell biology of valvular interstitial cells. Canadian Journal of Cardiology. 1996;12(3):231–236. [PubMed] [Google Scholar]
- 16.Butcher JT, Simmons CA, Warnock JN. Mechanobiology of the aortic heart valve. The Journal of Heart Valve Disease. 2008;17(1):62–73. [PubMed] [Google Scholar]
- 17.Grande-Allen KJ, Liao J. The heterogeneous biomechanics and mechanobiology of the mitral valve: implications for tissue engineering. Current Cardiology Reports. 2011;13(2):113–120. doi: 10.1007/s11886-010-0161-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weston MW, LaBorde DV, Yoganathan AP. Estimation of the shear stress on the surface of an aortic valve leaflet. Annals of Biomedical Engineering. 1999;27(4):572–579. doi: 10.1114/1.199. [DOI] [PubMed] [Google Scholar]
- 19.Kilner PJ, Yang GZ, Wilkest AJ, Mohladdlin RH, Firmin DN, Yacoub MH. Asymmetric redirection of flow through the heart. Nature. 2000;404(6779):759–761. doi: 10.1038/35008075. [DOI] [PubMed] [Google Scholar]
- 20.Butcher JT, Penrod AM, García AJ, Nerem RM. Unique morphology and focal adhesion development of valvular endothelial cells in static and fluid flow environments. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24(8):1429–1434. doi: 10.1161/01.ATV.0000130462.50769.5a. [DOI] [PubMed] [Google Scholar]
- 21.Butcher JT, Tressel S, Johnson T, et al. Transcriptional profiles of valvular and vascular endothelial cells reveal phenotypic differences: influence of shear stress. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(1):69–77. doi: 10.1161/01.ATV.0000196624.70507.0d. [DOI] [PubMed] [Google Scholar]
- 22.Simmons CA, Grant GR, Manduchi E, Davies PF. Spatial heterogeneity of endothelial phenotypes correlates with side-specific vulnerability to calcification in normal porcine aortic valves. Circulation Research. 2005;96(7):792–799. doi: 10.1161/01.RES.0000161998.92009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guerraty MA, Grant GR, Karanian JW, Chiesa OA, Pritchard WF, Davies PF. Hypercholesterolemia induces side-specific phenotypic changes and peroxisome proliferator-activated receptor-γ pathway activation in swine aortic valve endothelium. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(2):225–231. doi: 10.1161/ATVBAHA.109.198549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4- and TGF-β1-dependent pathway. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29(2):254–260. doi: 10.1161/ATVBAHA.108.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Metzler SA, Pregonero CA, Butcher JT, Burgess SC, Warnock JN. Cyclic strain regulates pro-inflammatory protein expression in porcine aortic valve endothelial cells. The Journal of Heart Valve Disease. 2008;17(5):571–578. [PubMed] [Google Scholar]
- 26.Smith KE, Metzler SA, Warnock JN. Cyclic strain inhibits acute pro-inflammatory gene expression in aortic valve interstitial cells. Biomechanics and Modeling in Mechanobiology. 2010;9(1):117–125. doi: 10.1007/s10237-009-0165-2. [DOI] [PubMed] [Google Scholar]
- 27.Balachandran K, Sucosky P, Jo H, Yoganathan AP. Elevated cyclic stretch induces aortic valve calcification in a bone morphogenic protein-dependent manner. American Journal of Pathology. 2010;177(1):49–57. doi: 10.2353/ajpath.2010.090631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yperman J, De Visscher G, Holvoet P, Flameng W. Molecular and functional characterization of ovine cardiac valve-derived interstitial cells in primary isolates and cultures. Tissue Engineering. 2004;10(9-10):1368–1375. doi: 10.1089/ten.2004.10.1368. [DOI] [PubMed] [Google Scholar]
- 29.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. American Journal of Pathology. 2007;171(5):1407–1418. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Messier RH, Bass BL, Aly HM, et al. Dual structural and functional phenotypes of the porcine aortic valve interstitial population: characteristics of the leaflet myofibroblast. The Journal of Surgical Research. 1994;57(1):1–21. doi: 10.1006/jsre.1994.1102. [DOI] [PubMed] [Google Scholar]
- 31.Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. The Journal of Heart Valve Disease. 2004;13(5):841–847. [PubMed] [Google Scholar]
- 32.Schneider PJ, Deck JD. Tissue and cell renewal in the natural aortic valve of rats: an autoradiographic study. Cardiovascular Research. 1981;15(4):181–189. doi: 10.1093/cvr/15.4.181. [DOI] [PubMed] [Google Scholar]
- 33.Merryman WD, Youn I, Lukoff HD, et al. Correlation between heart valve interstitial cell stiffness and transvalvular pressure: implications for collagen biosynthesis. American Journal of Physiology. 2006;290(1):H224–H231. doi: 10.1152/ajpheart.00521.2005. [DOI] [PubMed] [Google Scholar]
- 34.Ku CH, Johnson PH, Batten P, et al. Collagen synthesis by mesenchymal stem cells and aortic valve interstitial cells in response to mechanical stretch. Cardiovascular Research. 2006;71(3):548–556. doi: 10.1016/j.cardiores.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 35.Warnock JN, Burgess SC, Shack A, Yoganathan AP. Differential immediate-early gene responses to elevated pressure in porcine aortic valve interstitial cells. Journal of Heart Valve Disease. 2006;15(1):34–42. [PubMed] [Google Scholar]
- 36.Rothenburger M, Völker W, Vischer P, et al. Tissue engineering of heart valves: formation of a three-dimensional tissue using porcine heart valve cells. ASAIO Journal. 2002;48(6):586–591. doi: 10.1097/00002480-200211000-00003. [DOI] [PubMed] [Google Scholar]
- 37.Butcher JT, Nerem RM. Valvular endothelial cells regulate the phenotype of interstitial cells in co-culture: effects of steady shear stress. Tissue Engineering. 2006;12(4):905–915. doi: 10.1089/ten.2006.12.905. [DOI] [PubMed] [Google Scholar]
- 38.Müller AM, Cronen C, Kupferwasser LI, Oelert H, Müller KM, Kirkpatrick CJ. Expression of endothelial cell adhesion molecules on heart valves: up-regulation in degeneration as well as acute endocarditis. The Journal of Pathology. 2000;191(1):54–60. doi: 10.1002/(SICI)1096-9896(200005)191:1<54::AID-PATH568>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 39.Alom-Ruiz SP, Anilkumar N, Shah AM. Reactive oxygen species and endothelial activation. Antioxidants and Redox Signaling. 2008;10(6):1089–1100. doi: 10.1089/ars.2007.2007. [DOI] [PubMed] [Google Scholar]
- 40.Ghaisas NK, Foley JB, O’Briain DS, Crean P, Kelleher D, Walsh M. Adhesion molecules in nonrheumatic aortic valve disease: endothelial expression, serum levels and effects of valve replacement. Journal of the American College of Cardiology. 2000;36(7):2257–2262. doi: 10.1016/s0735-1097(00)00998-0. [DOI] [PubMed] [Google Scholar]
- 41.Aikawa E, Whittaker P, Farber M, et al. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113(10):1344–1352. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- 42.Mohler ER. Are atherosclerotic processes involved in aortic-valve calcification? The Lancet. 2000;356(9229):524–525. doi: 10.1016/S0140-6736(00)02572-1. [DOI] [PubMed] [Google Scholar]
- 43.Mohler ER, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation. 2001;103(11):1522–1528. doi: 10.1161/01.cir.103.11.1522. [DOI] [PubMed] [Google Scholar]
- 44.Mohler ER. Mechanisms of aortic valve calcification. The American Journal of Cardiology. 2004;94(11):1396–1402. doi: 10.1016/j.amjcard.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 45.Thubrikar MJ, Aouad J, Nolan SP. Patterns of calcific deposits in operatively excised stenotic or purely regurgitant aortic valves and their relation to mechanical stress. The American Journal of Cardiology. 1986;58(3):304–308. doi: 10.1016/0002-9149(86)90067-6. [DOI] [PubMed] [Google Scholar]
- 46.Sorescu GP, Song H, Tressel SL, et al. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a Nox1-based NADPH oxidase. Circulation Research. 2004;95(8):773–779. doi: 10.1161/01.RES.0000145728.22878.45. [DOI] [PubMed] [Google Scholar]
- 47.Yip CYY, Simmons CA. The aortic valve microenvironment and its role in calcific aortic valve disease. Cardiovascular Pathology. 2011;20(3):177–182. doi: 10.1016/j.carpath.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 48.Freeman RV, Otto CM. Spectrum of calcific aortic valve disease: pathogenesis, disease progression, and treatment strategies. Circulation. 2005;111(24):3316–3326. doi: 10.1161/CIRCULATIONAHA.104.486738. [DOI] [PubMed] [Google Scholar]
- 49.Beppu S, Suzuki S, Matsuda H, Ohmori F, Nagata S, Miyatake K. Rapidity of progression of aortic stenosis in patients with congenital bicuspid aortic valves. The American Journal of Cardiology. 1993;71(4):322–327. doi: 10.1016/0002-9149(93)90799-i. [DOI] [PubMed] [Google Scholar]
- 50.Pachulski RT, Chan KL. Progression of aortic valve dysfunction in 51 adult patients with congenital bicuspid aortic valve: assessment and follow up by Doppler echocardiography. British Heart Journal. 1993;69(3):237–240. doi: 10.1136/hrt.69.3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stewart BF, Siscovick D, Lind BK, et al. Clinical factors associated with calcific aortic valve disease. Journal of the American College of Cardiology. 1997;29(3):630–634. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 52.Aronow WS, Ahn C, Kronzon I, Goldman ME. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. The American Journal of Cardiology. 2001;88(6):693–695. doi: 10.1016/s0002-9149(01)01821-5. [DOI] [PubMed] [Google Scholar]
- 53.Shahi CN, Ghaisas NK, Goggins M, et al. Elevated levels of circulating soluble adhesion molecules in patients with nonrheumatic aortic stenosis. The American Journal of Cardiology. 1997;79(7):980–982. doi: 10.1016/s0002-9149(97)00027-1. [DOI] [PubMed] [Google Scholar]
- 54.Olsson M, Dalsgaard CJ, Haegerstrand A, Rosenqvist M, Ryden L, Nilsson J. Accumulation of T lymphocytes and expression of interleukin-2 receptors in nonrheumatic stenotic aortic valves. Journal of the American College of Cardiology. 1994;23(5):1162–1170. doi: 10.1016/0735-1097(94)90606-8. [DOI] [PubMed] [Google Scholar]
- 55.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD. Characterization of the early lesion of ’degenerative’ valvular aortic stenosis: histological and immunohistochemical studies. Circulation. 1994;90(2):844–853. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- 56.Olsson M, Rosenqvist M, Nilsson J. Expression of HLA-DR antigen and smooth muscle cell differentiation markers by valvular fibroblasts in degenerative aortic stenosis. Journal of the American College of Cardiology. 1994;24(7):1664–1671. doi: 10.1016/0735-1097(94)90172-4. [DOI] [PubMed] [Google Scholar]
- 57.Kaden JJ, Dempfle CE, Grobholz R, et al. Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis. 2003;170(2):205–211. doi: 10.1016/s0021-9150(03)00284-3. [DOI] [PubMed] [Google Scholar]
- 58.Kaden JJ, Dempfle C-E, Grobholz R, et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovascular Pathology. 2005;14(2):80–87. doi: 10.1016/j.carpath.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 59.Jian B, Narula N, Li QY, Mohler ER, Levy RJ. Progression of aortic valve stenosis: TGF-β1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Annals of Thoracic Surgery. 2003;75(2):457–465. doi: 10.1016/s0003-4975(02)04312-6. [DOI] [PubMed] [Google Scholar]
- 60.Tompkins RG, Schnitzer JJ, Yarmush ML. Macromolecular transport within heart valves. Circulation Research. 1989;64(6):1213–1223. doi: 10.1161/01.res.64.6.1213. [DOI] [PubMed] [Google Scholar]
- 61.Mohty D, Pibarot P, Després J-P, et al. Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(1):187–193. doi: 10.1161/ATVBAHA.107.154989. [DOI] [PubMed] [Google Scholar]
- 62.Mehrabi MR, Sinzinger H, Ekmekcioglu C, et al. Accumulation of oxidized LDL in human semilunar valves correlates with coronary atherosclerosis. Cardiovascular Research. 2000;45(4):874–882. doi: 10.1016/s0008-6363(99)00389-2. [DOI] [PubMed] [Google Scholar]
- 63.Côté C, Pibarot P, Després JP, et al. Association between circulating oxidised low-density lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis. Heart. 2008;94(9):1175–1180. doi: 10.1136/hrt.2007.125740. [DOI] [PubMed] [Google Scholar]
- 64.O’Brien KD, Shavelle DM, Caulfield MT, et al. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation. 2002;106(17):2224–2230. doi: 10.1161/01.cir.0000035655.45453.d2. [DOI] [PubMed] [Google Scholar]
- 65.O’Brien KD. Pathogenesis of calcific aortic valve disease: a disease process comes of age (and a good deal more) Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(8):1721–1728. doi: 10.1161/01.ATV.0000227513.13697.ac. [DOI] [PubMed] [Google Scholar]
- 66.Osman L, Yacoub MH, Latif N, Amrani M, Chester AH. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation. 2006;114(1):I547–I552. doi: 10.1161/CIRCULATIONAHA.105.001115. [DOI] [PubMed] [Google Scholar]
- 67.Kaden JJ, Kiliç R, Sarikoç A, et al. Tumor necrosis factor alpha promotes an osteoblast-like phenotype in human aortic valve myofibroblasts: a potential regulatory mechanism of valvular calcification. International Journal of Molecular Medicine. 2005;16(5):869–872. [PubMed] [Google Scholar]
- 68.Babu AN, Meng X, Zou N, et al. Lipopolysaccharide stimulation of human aortic valve interstitial cells activates inflammation and osteogenesis. Annals of Thoracic Surgery. 2008;86(1):71–76. doi: 10.1016/j.athoracsur.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 69.Meng X, Ao L, Song Y, et al. Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: potential roles in aortic valve inflammation and stenosis. American Journal of Physiology. 2008;294(1):C29–C35. doi: 10.1152/ajpcell.00137.2007. [DOI] [PubMed] [Google Scholar]
- 70.Lee JH, Meng X, Weyant MJ, Reece TB, Cleveland JC, Jr., Fullerton DA. Stenotic aortic valves have dysfunctional mechanisms of anti-inflammation: implications for aortic stenosis. Journal of Thoracic and Cardiovascular Surgery. 2011;141(2):481–486. doi: 10.1016/j.jtcvs.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 71.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104(21):2525–2532. doi: 10.1161/hc4601.099489. [DOI] [PubMed] [Google Scholar]
- 72.Tamura K, Jones M, Yamada I, Ferrans VJ. Wound healing in the mitral valve. Journal of Heart Valve Disease. 2000;9(1):53–63. [PubMed] [Google Scholar]
- 73.Yip CYY, Chen JH, Zhao R, Simmons CA. Calcification by valve interstitial cells is regulated by the stiffness of the extracellular matrix. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29(6):936–942. doi: 10.1161/ATVBAHA.108.182394. [DOI] [PubMed] [Google Scholar]
- 74.Aikawa E, Nahrendorf M, Sosnovik D, et al. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115(3):377–386. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- 75.Aikawa E, Aikawa M, Libby P, et al. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009;119(13):1785–1794. doi: 10.1161/CIRCULATIONAHA.108.827972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dreger SA, Taylor PM, Allen SP, Yacoub MH. Profile and localization of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) in human heart valves. Journal of Heart Valve Disease. 2002;11(6):875–880. [PubMed] [Google Scholar]
- 77.Wahlgren J, Maisi P, Sorsa T, et al. Expression of MMP2, MMP9, MTI-MMP, TIMP1, and TIMP2 mRNA in valvular lesions of the heart. The Journal of Pathology. 2001;194(2):225–231. doi: 10.1002/path.850. [DOI] [PubMed] [Google Scholar]
- 78.Helske S, Syväranta S, Lindstedt KA, et al. Increased expression of elastolytic cathepsins S, K, and V and their inhibitor cystatin C in stenotic aortic valves. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(8):1791–1798. doi: 10.1161/01.ATV.0000228824.01604.63. [DOI] [PubMed] [Google Scholar]
- 79.Helske S, Syväranta S, Kupari M, et al. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. European Heart Journal. 2006;27(12):1495–1504. doi: 10.1093/eurheartj/ehi706. [DOI] [PubMed] [Google Scholar]
- 80.Merryman WD, Lukoff HD, Long RA, Engelmayr GC, Hopkins RA, Sacks MS. Synergistic effects of cyclic tension and transforming growth factor-β1 on the aortic valve myofibroblast. Cardiovascular Pathology. 2007;16(5):268–276. doi: 10.1016/j.carpath.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nature Reviews Molecular Cell Biology. 2007;8(3):221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-β: implications for pathological extracellular matrix remodeling in heart valve disease. Circulation Research. 2004;95(3):253–260. doi: 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- 83.Clark-Greuel JN, Connolly JM, Sorichillo E, et al. Transforming growth factor-β1 mechanisms in aortic valve calcification: increased alkaline phosphatase and related events. Annals of Thoracic Surgery. 2007;83(3):946–953. doi: 10.1016/j.athoracsur.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 84.Fondard O, Detaint D, Iung B, et al. Extracellular matrix remodelling in human aortic valve disease: the role of matrix metalloproteinases and their tissue inhibitors. European Heart Journal. 2005;26(13):1333–1341. doi: 10.1093/eurheartj/ehi248. [DOI] [PubMed] [Google Scholar]
- 85.Hinton RB, Yutzey KE. Heart valve structure and function in development and disease. Annual Review of Physiology. 2011;73:29–46. doi: 10.1146/annurev-physiol-012110-142145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hinton RB, Lincoln J, Deutsch GH, et al. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circulation Research. 2006;98(11):1431–1438. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- 87.Schoen FJ. Cardiac valves and valvular pathology: update on function, disease, repair, and replacement. Cardiovascular Pathology. 2005;14(4):189–194. doi: 10.1016/j.carpath.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 88.Guthrie RB, Edwards JE. Pathology of the myxomatous mitral value. Nature, secondary changes and complications. Minnesota Medicine. 1976;59(9):637–647. [PubMed] [Google Scholar]
- 89.Davies MJ, Moore BP, Braimbridge MV. The floppy mitral valve. Study of incidence, pathology, and complications in surgical, necropsy and forensic material. British Heart Journal. 1978;40(5):468–481. doi: 10.1136/hrt.40.5.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Freed LA, Levy D, Levine RA, et al. Prevalence and clinical outcome of mitral-valve prolapse. The New England Journal of Medicine. 1999;341(1):1–7. doi: 10.1056/NEJM199907013410101. [DOI] [PubMed] [Google Scholar]
- 91.Durbin AD, Gotlieb AI. Advances towards understanding heart valve response to injury. Cardiovascular Pathology. 2002;11(2):69–77. doi: 10.1016/s1054-8807(01)00109-0. [DOI] [PubMed] [Google Scholar]
- 92.Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 93.Ewart AK, Morris CA, Atkinson D, et al. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nature Genetics. 1993;5(1):11–16. doi: 10.1038/ng0993-11. [DOI] [PubMed] [Google Scholar]
- 94.Pihlajaniemi T, Dickson LA, Pope FM, et al. Osteogenesis imperfecta: cloning of a pro-α2(I) collagen gene with a frameshift mutation. Journal of Biological Chemistry. 1984;259(21):12941–12944. [PubMed] [Google Scholar]
- 95.Wong RS, Follis FM, Shively BK, Wernly JA. Osteogenesis imperfecta and cardiovascular diseases. Annals of Thoracic Surgery. 1995;60(5):1439–1443. doi: 10.1016/0003-4975(95)00706-Q. [DOI] [PubMed] [Google Scholar]
- 96.Burch GH, Gong Y, Liu W, et al. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nature Genetics. 1997;17(1):104–108. doi: 10.1038/ng0997-104. [DOI] [PubMed] [Google Scholar]
- 97.Burrows NP, Nicholls AC, Yates JRW, et al. The gene encoding collagen α1(V) (COL5A1) is linked to mixed Ehlers-Danlos syndrome type I/II. Journal of Investigative Dermatology. 1996;106(6):1273–1276. doi: 10.1111/1523-1747.ep12348978. [DOI] [PubMed] [Google Scholar]
- 98.Malfait F, Symoens S, Coucke P, Nunes L, De Almeida S, De Paepe A. Total absence of the alpha2(I) chain of collagen type I causes a rare form of Ehlers-Danlos syndrome with hypermobility and propensity to cardiac valvular problems. Journal of Medical Genetics. 2006;43(7):p. e36. doi: 10.1136/jmg.2005.038224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Annunen S, Körkkö J, Czarny M, et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. The American Journal of Human Genetics. 1999;65(4):974–983. doi: 10.1086/302585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liberfarb RM, Goldblatt A. Prevalence of mitral valve prolapse in the Stickler syndrome. American Journal of Medical Genetics. 1986;24(3):387–392. doi: 10.1002/ajmg.1320240302. [DOI] [PubMed] [Google Scholar]
- 101.Ng CM, Cheng A, Myers LA, et al. TGF-β-dependent pathogenesis of mitral valve prolapse in a mouse model of Marfan syndrome. The Journal of Clinical Investigation. 2004;114(11):1586–1592. doi: 10.1172/JCI22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dallas SL, Miyazono K, Skerry TM, Mundy GR, Bonewald LF. Dual role for the latent transforming growth factor-β binding protein in storage of latent TGF-β in the extracellular matrix and as a structural matrix protein. Journal of Cell Biology. 1995;131(2):539–549. doi: 10.1083/jcb.131.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Isogai Z, Ono RN, Ushiro S, et al. Latent transforming growth factor β-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. Journal of Biological Chemistry. 2003;278(4):2750–2757. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- 104.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC. Angiotensin II blockade and aortic-root dilation in marfan’s syndrome. The New England Journal of Medicine. 2008;358(26):2787–2795. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hilário MOE, Terreri MTSLRA. Rheumatic fever and post-streptococcal arthritis. Best Practice and Research: Clinical Rheumatology. 2002;16(3):481–494. [PubMed] [Google Scholar]
- 107.Grover A, Dhawan A, Iyengar SD, Anand IS, Wahi PL, Ganguly NK. Epidemiology of rheumatic fever and rheumatic heart disease in a rural community in northern India. Bulletin of the World Health Organization. 1993;71(1):59–66. [PMC free article] [PubMed] [Google Scholar]
- 108.Veinot JP. Pathology of inflammatory native valvular heart disease. Cardiovascular Pathology. 2006;15(5):243–251. doi: 10.1016/j.carpath.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 109.da Silva CH. Rheumatic fever: a multicenter study in the state of São Paulo. Pediatric Committee—São Paulo Pediatric Rheumatology Society. Revista do Hospital das Clínicas. 1999;54(3):85–90. doi: 10.1590/s0041-87811999000300004. [DOI] [PubMed] [Google Scholar]
- 110.Rullan E, Sigal LH. Rheumatic fever. Current Rheumatology Reports. 2001;3(5):445–452. doi: 10.1007/s11926-996-0016-4. [DOI] [PubMed] [Google Scholar]
- 111.Fischetti VA. Streptococcal M protein. Scientific American. 1991;264(6):58–65. doi: 10.1038/scientificamerican0691-58. [DOI] [PubMed] [Google Scholar]
- 112.Wessels MR, Moses AE, Goldberg JB, DiCesare TJ. Hyaluronic acid capsule is a virulence factor for mucoid group A streptococci. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(19):8317–8321. doi: 10.1073/pnas.88.19.8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wessels MR, Goldberg JB, Moses AE, DiCesare TJ. Effects on virulence of mutations in a locus essential for hyaluronic acid capsule expression in group A streptococci. Infection and Immunity. 1994;62(2):433–441. doi: 10.1128/iai.62.2.433-441.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dale JB, Washburn RG, Marques MB, Wessels MR. Hyaluronate capsule and surface M protein in resistance to opsonization of group A streptococci. Infection and Immunity. 1996;64(5):1495–1501. doi: 10.1128/iai.64.5.1495-1501.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bhatnagar A, Grover A, Ganguly NK. Superantigen-induced T cell responses in acute rheumatic fever and chronic: rheumatic heart disease patients. Clinical and Experimental Immunology. 1999;116(1):100–106. doi: 10.1046/j.1365-2249.1999.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pancholi V, Fischetti VA. Isolation and characterization of the cell-associated region of group A streptococcal M6 protein. Journal of Bacteriology. 1988;170(6):2618–2624. doi: 10.1128/jb.170.6.2618-2624.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fischetti VA. Streptococcal M protein: molecular design and biological behavior. Clinical Microbiology Reviews. 1989;2(3):285–314. doi: 10.1128/cmr.2.3.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cunningham MW. T cell mimicry in inflammatory heart disease. Molecular Immunology. 2004;40(14-15):1121–1127. doi: 10.1016/j.molimm.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 119.Galvin JE, Hemric ME, Ward K, Cunningham MW. Cytotoxic mAb from rheumatic carditis recognizes heart valves and laminin. The Journal of Clinical Investigation. 2000;106(2):217–224. doi: 10.1172/JCI7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Roberts S, Kosanke S, Dunn ST, Jankelow D, Duran CMG, Cunningham MW. Pathogenic mechanisms in rheumatic carditis: focus on valvular endothelium. Journal of Infectious Diseases. 2001;183(3):507–511. doi: 10.1086/318076. [DOI] [PubMed] [Google Scholar]
- 121.Mylonakis E, Calderwood SB. Infective endocarditis in adults. The New England Journal of Medicine. 2001;345(18):1318–1330. doi: 10.1056/NEJMra010082. [DOI] [PubMed] [Google Scholar]
- 122.Wilson W, Taubert KA, Gewitz M, et al. Prevention of infective endocarditis: guidelines from the American Heart Association. Circulation. 2007;116(15):1736–1754. doi: 10.1161/CIRCULATIONAHA.106.183095. [DOI] [PubMed] [Google Scholar]
- 123.Moreillon P, Que YA. Infective endocarditis. The Lancet. 2004;363(9403):139–149. doi: 10.1016/S0140-6736(03)15266-X. [DOI] [PubMed] [Google Scholar]
- 124.Veltrop MHAM, Bancsi MJLMF, Bertina RM, Thompson J. Role of monocytes in experimental Staphylococcus aureus endocarditis. Infection and Immunity. 2000;68(8):4818–4821. doi: 10.1128/iai.68.8.4818-4821.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hemler ME, Elices MJ, Parker C, Takada Y. Structure of the integrin VLA-4 and its cell-cell and cell-matrix adhesion functions. Immunological Reviews. 1990;(114):45–65. doi: 10.1111/j.1600-065x.1990.tb00561.x. [DOI] [PubMed] [Google Scholar]
- 126.Sinha B, Francois P, Que YA, et al. Heterologously expressed staphylococcus aureus fibronectin-binding proteins are sufficient for invasion of host cells. Infection and Immunity. 2000;68(12):6871–6878. doi: 10.1128/iai.68.12.6871-6878.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Prendergast BD. The changing face of infective endocarditis. Heart. 2006;92(7):879–885. doi: 10.1136/hrt.2005.067256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roth BL. Focus on research: drugs and valvular heart disease. The New England Journal of Medicine. 2007;356(1):6–9. doi: 10.1056/NEJMp068265. [DOI] [PubMed] [Google Scholar]
- 129.Schade R, Andersohn F, Suissa S, Haverkamp W, Garbe E. Dopamine agonists and the risk of cardiac-valve regurgitation. The New England Journal of Medicine. 2007;356(1):29–38. doi: 10.1056/NEJMoa062222. [DOI] [PubMed] [Google Scholar]
- 130.Setola V, Hufeisen SJ, Grande-Allen KJ, et al. 3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) induces fenfluramine-like proliferative actions on human cardiac valvular interstitial cells in vitro. Molecular Pharmacology. 2003;63(6):1223–1229. doi: 10.1124/mol.63.6.1223. [DOI] [PubMed] [Google Scholar]
- 131.Gustafsson BI, Hauso O, Drozdov I, Kidd M, Modlin IM. Carcinoid heart disease. International Journal of Cardiology. 2008;129(3):318–324. doi: 10.1016/j.ijcard.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 132.Gustafsson BI, Tømmerås K, Nordrum I, et al. Long-term serotonin administration induces heart valve disease in rats. Circulation. 2005;111(12):1517–1522. doi: 10.1161/01.CIR.0000159356.42064.48. [DOI] [PubMed] [Google Scholar]
- 133.Rajamannan NM, Caplice N, Anthikad F, et al. Cell proliferation in carcinoid valve disease: a mechanism for serotonin effects. Journal of Heart Valve Disease. 2001;10(6):827–831. [PubMed] [Google Scholar]
- 134.Jian B, Xu J, Connolly J, et al. Serotonin mechanisms in heart valve disease I: serotonin-induced up-regulation of transforming growth factor-β1 via G-protein signal transduction in aortic valve interstitial cells. American Journal of Pathology. 2002;161(6):2111–2121. doi: 10.1016/s0002-9440(10)64489-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Connolly JM, Bakay MA, Fulmer JT, et al. Fenfluramine disrupts the mitral valve interstitial cell response to serotonin. American Journal of Pathology. 2009;175(3):988–997. doi: 10.2353/ajpath.2009.081101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Disatian S, Orton EC. Autocrine serotonin and transforming growth factor beta 1 signaling mediates spontaneous myxomatous mitral valve disease. The Journal of Heart Valve Disease. 2009;18(1):44–51. [PubMed] [Google Scholar]
- 137.El-Hamamsy I, Balachandran K, Yacoub MH, et al. Endothelium-dependent regulation of the mechanical properties of aortic valve cusps. Journal of the American College of Cardiology. 2009;53(16):1448–1455. doi: 10.1016/j.jacc.2008.11.056. [DOI] [PubMed] [Google Scholar]
- 138.Chester AH, Misfeld M, Sievers HH, Yacoub MH. Influence of 5-hydroxytryptamine on aortic valve competence in vitro. Journal of Heart Valve Disease. 2001;10(6):822–826. [PubMed] [Google Scholar]
- 139.Singer NG, Caplan AI. Mesenchymal stem cells: mechanisms of inflammation. Annual Review of Pathology: Mechanisms of Disease. 2011;6:457–478. doi: 10.1146/annurev-pathol-011110-130230. [DOI] [PubMed] [Google Scholar]
- 140.Deb A, Wang SH, Skelding K, Miller D, Simper D, Caplice N. Bone marrow-derived myofibroblasts are present in adult human heart valves. Journal of Heart Valve Disease. 2005;14(5):674–678. [PubMed] [Google Scholar]
- 141.Visconti RP, Ebihara Y, LaRue AC, et al. An in vivo analysis of hematopoietic stem cell potential: hematopoietic origin of cardiac valve interstitial cells. Circulation Research. 2006;98(5):690–696. doi: 10.1161/01.RES.0000207384.81818.d4. [DOI] [PubMed] [Google Scholar]
- 142.Wu Y, Ip JE, Huang J, et al. Essential role of ICAM-1/CD18 in mediating EPC recruitment, angiogenesis, and repair to the infarcted myocardium. Circulation Research. 2006;99(3):315–322. doi: 10.1161/01.RES.0000235986.35957.a3. [DOI] [PubMed] [Google Scholar]
- 143.Karamlou T, Jang K, Williams WG, et al. Outcomes and associated risk factors for aortic valve replacement in 160 children: a competing-risks analysis. Circulation. 2005;112(22):3462–3469. doi: 10.1161/CIRCULATIONAHA.105.541649. [DOI] [PubMed] [Google Scholar]
- 144.Yacoub MH, Takkenberg JJM. Will heart valve tissue engineering change the world? Nature Clinical Practice Cardiovascular Medicine. 2005;2(2):60–61. doi: 10.1038/ncpcardio0112. [DOI] [PubMed] [Google Scholar]
- 145.Vesely I. Heart valve tissue engineering. Circulation Research. 2005;97(8):743–755. doi: 10.1161/01.RES.0000185326.04010.9f. [DOI] [PubMed] [Google Scholar]
- 146.Hilbert SL, Yanagida R, Souza J, et al. Prototype anionic detergent technique used to decellularize allograft valve conduits evaluated in the right ventricular outflow tract in sheep. The Journal of Heart Valve Disease. 2004;13(5):831–840. [PubMed] [Google Scholar]
- 147.Opitz F, Schenke-Layland K, Cohnert TU, et al. Tissue engineering of aortic tissue: dire consequence of suboptimal elastic fiber synthesis in vivo. Cardiovascular Research. 2004;63(4):719–730. doi: 10.1016/j.cardiores.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 148.Konuma T, Devaney EJ, Bove EL, et al. Performance of CryoValve SG decellularized pulmonary allografts compared with standard cryopreserved allografts. Annals of Thoracic Surgery. 2009;88(3):849–855. doi: 10.1016/j.athoracsur.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 149.Simon P, Kasimir MT, Seebacher G, et al. Early failure of the tissue engineered porcine heart valve SYNERGRAFT™ in pediatric patients. European Journal of Cardio-Thoracic Surgery. 2003;23(6):1002–1006. doi: 10.1016/s1010-7940(03)00094-0. [DOI] [PubMed] [Google Scholar]
- 150.Cicha I, Rüffer A, Cesnjevar R, et al. Early obstruction of decellularized xenogenic valves in pediatric patients: involvement of inflammatory and fibroproliferative processes. Cardiovascular Pathology. 2011;20(4):222–231. doi: 10.1016/j.carpath.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 151.Bayrak A, Tyralla M, Ladhoff J, et al. Human immune responses to porcine xenogeneic matrices and their extracellular matrix constituents in vitro. Biomaterials. 2010;31(14):3793–3803. doi: 10.1016/j.biomaterials.2010.01.120. [DOI] [PubMed] [Google Scholar]
- 152.Sutherland FWH, Perry TE, Yu Y, et al. From stem cells to viable autologous semilunar heart valve. Circulation. 2005;111(21):2783–2791. doi: 10.1161/CIRCULATIONAHA.104.498378. [DOI] [PubMed] [Google Scholar]
- 153.Person AD, Klewer SE, Runyan RB, Kwang WJ. International Review of Cytology 287. New York, NY, USA: Academic Press; 2005. [DOI] [PubMed] [Google Scholar]
- 154.Krug EL, Runyan RB, Markwald RR. Protein extracts from early embryonic hearts initiate cardiac endothelial cytodifferentiation. Developmental Biology. 1985;112(2):414–426. doi: 10.1016/0012-1606(85)90414-2. [DOI] [PubMed] [Google Scholar]
- 155.Manasek FJ. Sulfated extracellular matrix production in the embryonic heart and adjacent tissues. Journal of Experimental Zoology. 1970;174(4):415–439. doi: 10.1002/jez.1401740406. [DOI] [PubMed] [Google Scholar]
- 156.Manasek FJ, Reid M, Vinson W, Seyer J, Johnson R. Gycosaminoglycan synthesis by the early embryonic chick heart. Developmental Biology. 1973;35(2):332–348. doi: 10.1016/0012-1606(73)90028-6. [DOI] [PubMed] [Google Scholar]
- 157.Markwald RR, Fitzharris TP, Manasek FJ. Structural development of endocardial cushions. American Journal of Anatomy. 1977;148(1):85–119. doi: 10.1002/aja.1001480108. [DOI] [PubMed] [Google Scholar]
- 158.Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. 1951. Developmental Dynamics. 1992;195(4):231–272. doi: 10.1002/aja.1001950404. [DOI] [PubMed] [Google Scholar]
- 159.Sinning AR, Hewitt CC. Identification of a 283-kDa protein component of the particulate matrix associated with cardiac mesenchyme formation. Acta Anatomica. 1996;155(4):219–230. doi: 10.1159/000147810. [DOI] [PubMed] [Google Scholar]
- 160.Wunsch AM, Little CD, Markwald RR. Cardiac endothelial heterogeneity defines valvular development as demonstrated by the diverse expression of JB3, an antigen of the endocardial cushion tissue. Developmental Biology. 1994;165(2):585–601. doi: 10.1006/dbio.1994.1278. [DOI] [PubMed] [Google Scholar]
- 161.Ramsdell AF, Markwald RR. Induction of endocardial cushion tissue in the avian heart is regulated, in part, by TGFβ-3-mediated autocrine signaling. Developmental Biology. 1997;188(1):64–74. doi: 10.1006/dbio.1997.8637. [DOI] [PubMed] [Google Scholar]
- 162.Bernanke DH, Markwald RR. Migratory behavior of cardiac cushion tissue cells in a collagen-lattice culture system. Developmental Biology. 1982;91(2):235–245. doi: 10.1016/0012-1606(82)90030-6. [DOI] [PubMed] [Google Scholar]
- 163.Nakajima Y, Mironov V, Yamagishi T, Nakamura H, Markwald RR. Expression of smooth muscle alpha-actin in mesenchymal cells during formation of avian endocardial cushion tissue: a role for transforming growth factor β3. Developmental Dynamics. 1997;209(3):296–309. doi: 10.1002/(SICI)1097-0177(199707)209:3<296::AID-AJA5>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 164.Nath AK, Brown RM, Michaud M, Sierra-Honigmann MR, Snyder M, Madri JA. Leptin affects endocardial cushion formation by modulating EMT and migration via Akt signaling cascades. Journal of Cell Biology. 2008;181(2):367–380. doi: 10.1083/jcb.200708197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mou SS, Haudek SB, Lequier L, et al. Myocardial inflammatory activation in children with congenital heart disease. Critical Care Medicine. 2002;30(4):827–832. doi: 10.1097/00003246-200204000-00018. [DOI] [PubMed] [Google Scholar]
- 166.Qing M, Schumacher K, Heise R, et al. Intramyocardial synthesis of pro- and anti-inflammatory cytokines in infants with congenital cardiac defects. Journal of the American College of Cardiology. 2003;41(12):2266–2274. doi: 10.1016/s0735-1097(03)00477-7. [DOI] [PubMed] [Google Scholar]
- 167.Appachi E, Mossad E, Mee RBB, Bokesch P. Perioperative serum interleukins in neonates with hypoplastic left-heart syndrome and transposition of the great arteries. Journal of Cardiothoracic and Vascular Anesthesia. 2007;21(2):184–190. doi: 10.1053/j.jvca.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 168.Kwee L, Baldwin HS, Shen HM, et al. Defective development of the embryonic and extraembryonic circulatory systems in vascular cell adhesion molecule (VCAM-1) deficient mice. Development. 1995;121(2):489–503. doi: 10.1242/dev.121.2.489. [DOI] [PubMed] [Google Scholar]
- 169.Tseng CE, Buyon JP. Neonatal lupus syndromes. Rheumatic Disease Clinics of North America. 1997;23(1):31–54. doi: 10.1016/s0889-857x(05)70313-6. [DOI] [PubMed] [Google Scholar]
- 170.Krishnan AN, Sable CA, Donofrio MT. Spectrum of fetal echocardiographic findings in fetuses of women with clinical or serologic evidence of systemic lupus erythematosus. Journal of Maternal-Fetal and Neonatal Medicine. 2008;21(11):776–782. doi: 10.1080/14767050802388446. [DOI] [PubMed] [Google Scholar]
- 171.Moak JP, Barron KS, Hougen TJ, et al. Congenital heart block: development of late-onset cardiomyopathy, a previously underappreciated sequela. Journal of the American College of Cardiology. 2001;37(1):238–242. doi: 10.1016/s0735-1097(00)01048-2. [DOI] [PubMed] [Google Scholar]
- 172.Buyon JP, Winchester R. Congenital complete heart block. A human model of passively acquired autoimmune injury. Arthritis and Rheumatism. 1990;33(5):609–614. doi: 10.1002/art.1780330502. [DOI] [PubMed] [Google Scholar]
- 173.Buyon JP, Hiebert R, Copel J, et al. Autoimmune-associated congenital heart block: demographics, mortality, morbidity and recurrence rates obtained from a national neonatal lupus registry. Journal of the American College of Cardiology. 1998;31(7):1658–1666. doi: 10.1016/s0735-1097(98)00161-2. [DOI] [PubMed] [Google Scholar]
- 174.Rounioja S, Räsänen J, Glumoff V, Ojaniemi M, Mäkikallio K, Hallman M. Intra-amniotic lipopolysaccharide leads to fetal cardiac dysfunction: a mouse model for fetal inflammatory response. Cardiovascular Research. 2003;60(1):156–164. doi: 10.1016/s0008-6363(03)00338-9. [DOI] [PubMed] [Google Scholar]
- 175.Rounioja S, Räsänen J, Ojaniemi M, Glumoff V, Autio-Harmainen H, Hallman M. Mechanism of acute fetal cardiovascular depression after maternal inflammatory challenge in mouse. American Journal of Pathology. 2005;166(6):1585–1592. doi: 10.1016/S0002-9440(10)62469-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hernández-Gutierrez S, García-Peláez I, Zentella-Dehesa A, et al. NF-κB signaling blockade by Bay 11-7085 during early cardiac morphogenesis induces alterations of the outflow tract in chicken heart. Apoptosis. 2006;11(7):1101–1109. doi: 10.1007/s10495-006-6984-z. [DOI] [PubMed] [Google Scholar]
- 177.Ranger AM, Grusby MJ, Hodge MR, et al. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392(6672):186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- 178.Sharma R, Bolger AP, Li W, et al. Elevated circulating levels of inflammatory cytokines and bacterial endotoxin in adults with congenital heart disease. The American Journal of Cardiology. 2003;92(2):188–193. doi: 10.1016/s0002-9149(03)00536-8. [DOI] [PubMed] [Google Scholar]