

Abstract
Lactococcus lactis degrades exogenous proteins such as β-casein to peptides of 4–30 amino acids, and uses these as nitrogen sources. The binding protein or receptor (OppALl) of the oligopeptide transport system (Opp) of L.lactis has the unique capacity to bind peptides from five up to at least 20 residues. To study the binding mechanism of OppALl, nonameric peptides were used in which the cysteine at position 1, 3, 4, 5, 6, 7 or 9 was selectively labeled with either bulky and non-fluorescent or bulky and fluorescent groups. Also, nonameric peptides with a non-natural residue, azatryptophan, at positions 3 or 7 were used. The fluorescence of azatryptophan reports on the polarity of the environment. The studies indicate that the binding protein encloses the first six amino acids of the peptide, whereas the remaining residues stick out and interact with the surface of the binding protein. The peptide binding mechanism of OppALl is discussed in relation to known three-dimensional structures of members of this class of proteins, and an adaptation of the general binding mechanism is proposed.
Keywords: ABC transporter/binding mechanism/binding protein-dependent transport/oligopeptide
Introduction
The interaction between peptides and proteins plays an important role in biology, e.g. in class I molecules of the major histocompatibility complex (Pamer and Cresswell, 1998), in transporters associated with antigen-processing proteins (Koopmann et al., 1996; Uebel et al., 1997), and in peptide binding proteins of the ATP binding cassette (ABC)-type transporter family (Tame et al., 1994; Dunten and Mowbray, 1995; Nickitenko et al., 1995; Lanfermeijer et al., 1999; Sanz et al., 2000). The interaction between these proteins and their peptide substrates can be amino acid sequence dependent or independent. Sequence-dependent peptide binding is achieved through specific and direct interactions between the side chains of the substrate and those of the protein. Sequence-independent peptide binding is characterized by a major contribution between the peptide backbone of the substrate and the protein. In this case, the side chains are in highly adaptive binding pockets or do not interact with the protein.
Bacterial binding protein-dependent peptide uptake systems have an extracellular receptor protein (Ames, 1986) that is either free floating in the periplasm (Gram-negative bacteria; Tam and Saier, 1993), attached to the cell membrane via a lipid modification of the N-terminal cysteine (Gram-positive bacteria; Tam and Saier, 1993), attached to the cell membrane via a hydrophobic transmembrane segment (Archaea; Albers et al., 1999) or fused with the integral membrane subunit (Gram-positive bacteria; van der Heide and Poolman, 2000). All the ligand binding proteins seem to share a common tertiary fold and a similar binding mechanism, the so-called Venus fly-trap mechanism, as can be deduced from the >14 crystal structures available (Tame et al., 1994, 1995; Dunten and Mowbray, 1995; Nickitenko et al., 1995; Quiocho and Ledvina, 1996; Sleigh et al., 1997).
The crystal structures of the unliganded and liganded forms of binding proteins of the dipeptide transporter (Dpp) of Escherichia coli (Dunten and Mowbray, 1995; Nickitenko et al., 1995) and the oligopeptide transporter (Opp) of Salmonella typhimurium (Tame et al., 1994, 1995, 1996; Sleigh et al., 1997) show that the basic mechanism of peptide binding by these two proteins is similar. Parallel and anti-parallel β-sheet interactions are formed between the peptide backbone and residues of the binding protein, and salt bridges are formed between the N- and C-terminus of the peptide and specific acidic and basic residues of the protein, respectively. The side chains of the peptides are accommodated in voluminous hydrated pockets, in which the number of water molecules present is adjusted according to the size and the nature of the side chain (Tame et al., 1996; Davies et al., 1999; Sleigh et al., 1999). The Opp system of Lactococcus lactis differs from Opp of S.typhimurium and Dpp of E.coli in the sense that much larger peptides are bound and transported (Detmers et al., 1998; Kunji et al., 1998). In fact, the Kd for binding of peptides to the binding protein or receptor (OppA) decreases with increasing peptide length, and significant binding is only observed for peptides longer than five residues (Lanfermeijer et al., 1999). On the basis of the structure of OppA of S.typhimurium and its known binding mechanism, it is difficult to envisage how peptides longer than five residues are bound. Owing to the sheer size of the protein, the number of binding pockets will most likely not reflect the size of the longest peptides bound. It is also unlikely that positively charged residues are positioned in the binding protein to interact with the C-terminus of peptides ranging from five to >20 residues. An important question, therefore, is how does OppA of L.lactis bind these longer peptides?
In this communication, we address the interaction with the OppA protein of L.lactis of a series of nonapeptides, modified at selected positions with either 2-(4′-maleimidylanilino)naphthalene-6-sulfonate (Mal-ANS) or p-chloromercuribenzene sulfonate (pCMBS) (Figure 1A and B). The basic structure of the nonapeptides is SLSQSLSQS, and derivatives for selective labeling were synthesized by introducing a cysteine at position 1, 3, 4, 5, 6, 7 or 9. In addition, peptides with a modified N- or C-terminus and peptides with the non-natural azatryptophan residue were used (Figure 1C). The data are discussed in the light of the known binding mechanism of the homologous DppA and OppA binding proteins of E.coli and S.typhimurium, respectively. The proposed model for the binding of oligopeptides by OppA of L.lactis clearly sets this protein apart from the others.
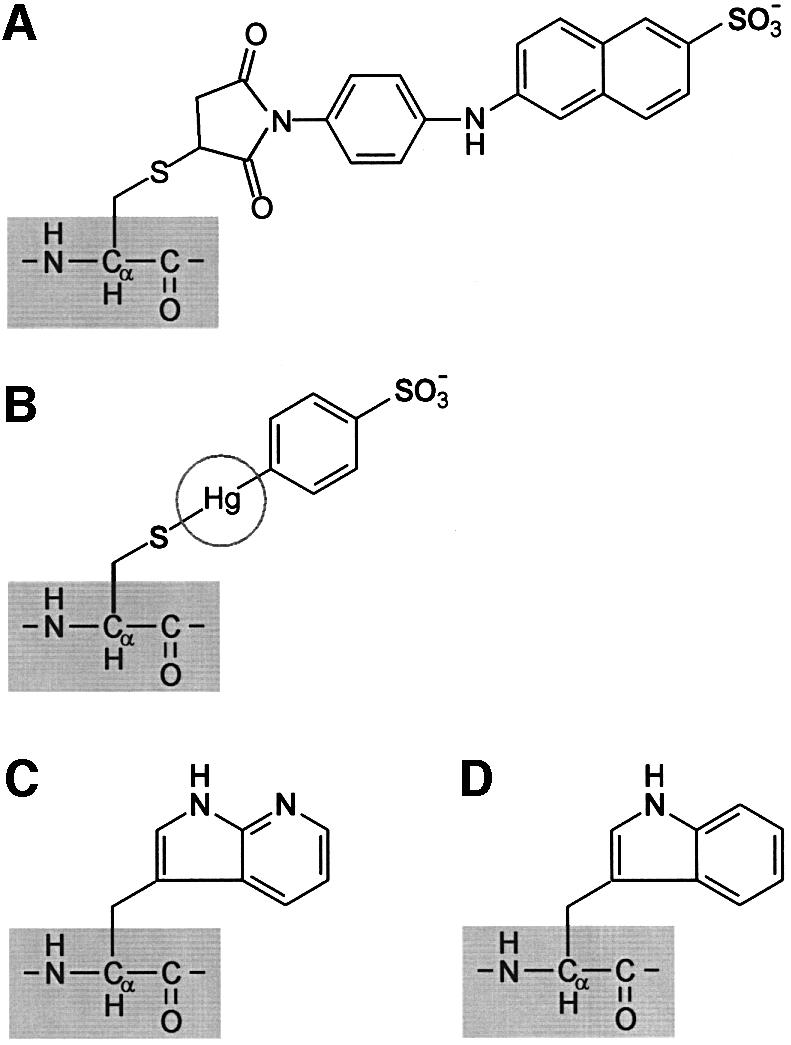
Fig. 1. Structures of azatryptophan and probes used to modify single cysteine-containing peptides. (A) Mal-ANS. (B) pCMBS; the radius of Hg+ is indicated by a circle. (C) Azatryptophan. (D) Tryptophan (shown for comparison with azatryptophan).
Results
Binding of peptides with modified termini
To test whether or not free termini are critical for high affinity binding to OppA* (see Materials and methods), peptides were synthesized that had an acetylated N-terminal amino group (Ac-C0; Table I) or an amidated C-terminal carboxyl group (C0-CON; Table I). After correction for solvent [dimethylsulfoxide (DMSO)] effects, the modified peptides displayed saturable kinetics upon binding with OppA*. The dissociation constants were 2.6 and 4.8 µM for Ac-C0 and C0-CON, respectively (Table I), which are only 3.7- and 6.8-fold larger than the dissociation constant for binding of the unmodified peptide SLSQSLSQS (C0). The maximum fluorescence changes observed with these peptides were 2-fold lower than that observed with the control peptide.
Table I. Binding characteristics of peptides.
| Peptide | Abbreviation | Kd (µM) | ΔFmax (%) |
|---|---|---|---|
| SLSQSLSQS | C0 | 0.71 ± 0.11 | 13.2 ± 0.1 |
| CLSQSLSQS | C1 | 0.85 ± 0.22 | 13.7 ± 0.8 |
| SLCQSLSQS | C3 | 0.28 ± 0.15 | 14.7 ± 0.8 |
| SLSCSLSQS | C4 | 0.27 ± 0.16 | 15.0 ± 0.7 |
| SLSQCLSQS | C5 | 0.11 ± 0.02 | 7.1 ± 0.4 |
| SLSQSCSQS | C6 | 0.62 ± 0.03 | 6.4 ± 0.1 |
| SLSQSLCQS | C7 | 0.29 ± 0.10 | 10.7 ± 0.8 |
| SLSQSLSQC | C9 | 0.66 ± 0.21 | 12.6 ± 1.0 |
| Ac-SLSQSLSQS | Ac-C0a | 2.6 ± 0.5 | 7.0 ± 1.3 |
| SLSQSLSQS-NH2 | C0-CONa | 4.8 ± 0.2 | 5.5 ± 0.3 |
| ΔFmax (AU) | |||
| SLZQSLSQSb | Z3c | 0.63 ± 0.22 | 1.78 ± 0.21 |
| SLSQSLZQSb | Z7c | 1.62 ± 0.15 | 1.19 ± 0.10 |
The concentration dependence of peptide binding was analyzed by the general binding equation as described in Lanfermeijer et al. (1999). Data represent averages of at least three independent experiments ± SD.
aThe concentration dependence of the binding of peptides with modified termini was analyzed as described in Materials and methods. For both Ac-C0 and C0-CON, a linear component with a constant (k) of 0.07 ± 0.03%/µM was observed.
bThe letter Z corresponds to dl-azatryptophan.
cThe concentration dependence of the binding of azatryptophan-containing peptides was analyzed as described in Materials and methods. In both cases, a linear component with a constant (k) of 0.006 ± 0.002 AU/µM was observed.
Binding of cysteine-containing peptides
All cysteine-containing peptides bound to OppA* with simple saturable binding kinetics. Dissociation constants varied from 0.1 to 0.8 µM (Table I), but there was no clear trend between the dissociation constant and the position of the cysteine. In most cases, the addition of saturating amounts of peptides resulted in maximum fluorescence changes in the range of 12–14% (Table I), which is comparable to the maximum changes previously observed with SLSQSKVLP, SLSQSKVLPVPQ, RDMPIQA and RDMPIQAF (Lanfermeijer et al., 1999). However, peptides with the cysteine at position 5 or 6 showed a maximum fluorescence change of only 6–7% (Table I). This reduced fluorescence is in accordance with the thiol of the cysteine being in close proximity to a tryptophan residue(s). Cysteines can quench tryptophan fluorescence via electron capture, which is a diffusion-controlled process when the cysteine and the acetyl tryptophan amide are in solution (Steiner and Kirby, 1969). From the crystal structures of DppA of E.coli and OppA of S.typhimurium (Tame et al., 1994; Dunten and Mowbray, 1995; Nickitenko et al., 1995), it is known that two tryptophans and several tyrosines participate in the formation of the binding pockets, and those residues are well conserved in the group of peptide binding proteins.
Binding of Mal-ANS-labeled peptides
Fluorescence of Mal-ANS-labeled peptides in buffer is characterized by an excitation maximum and an emission maximum at 328 and 445 nm, respectively (Figure 2), which is in accordance with the ANS moiety being in a hydrophilic environment. The presence of OppA* did not affect the fluorescence of C1 and C3 labeled with Mal-ANS (Figure 2A and B; Table II), which could be due to a failure to bind. For the other peptides, two types of response were observed. First, addition of OppA* to the labeled C7 and C9 resulted in a shift of the emission maximum from 450 to 420 nm, an increase in the fluorescence quantum yield, and the appearance of a second excitation maximum at 280 nm (Figure 2C and D; Table II). Secondly, addition of OppA* to the labeled C4, C5 and C6 resulted in a shift of the emission maximum from 450 to 400 nm, an increase in the fluorescence quantum yield and the appearance of a second excitation maximum at 280 nm (Figure 2E and F; Table II). With C4, C5 and C6, however, binding was slow (association rate constants of the order of several minutes), and the same slow equilibration was observed when the labeled peptides were competed out by the high affinity peptide bradykinin (RPPGFSPFR) (data not shown). This kinetic behavior indicates that binding of the Mal-ANS-labeled C4, C5 and C6 is hindered (Table II).



Fig. 2. Fluorescence of Mal-ANS-labeled peptides. (A and B) Fluorescence spectra of Mal-ANS-modified C1, which are representative of both C1 and C3 labeled peptides. (C and D) Fluorescence spectra of the Mal-ANS-modified C9, which are representative of the C7 and C9 labeled peptides. (E and F) Fluorescence spectra of the Mal-ANS-modified C5, which are representative of the C4, C5 and C6 labeled peptides. (A, C and E) Excitation spectra of the labeled peptides (0.5 µM) in the absence (–) and in the presence of 0.5 µM OppA* (filled circles) and in the presence of 0.5 µM OppA* plus 5 µM bradykinin (open circles; bradykinin was added after spectra of the labeled peptides in the presence of OppA* were collected). Emission wavelengths were 450, 400 and 420 nm for the modified C1, C5 and C9, respectively. (B, D and F) Emission spectra of 0.5 µM of the labeled peptides in the absence (–) and in the presence of 0.5 µM OppA* (filled circles) and in the presence of 0.5 µM OppA* plus 5 µM bradykinin (open circles; bradykinin was added after spectra of the labeled peptides in the presence of OppA* were collected). The excitation wavelength was 328 nm for the modified C1, C5 and C9 (see also Table III).
Table II. Fluorescence characteristics of Mal-ANS-labeled peptides in the presence of OppA*.
| Peptide | Emission wavelength maximum (nm) | Excitation wavelength(s) maximum (nm) | Energy transfer | Competition by bradykinin | Equilibration |
|---|---|---|---|---|---|
| ANS-C1a | 450 | 328 | no | n.r.b | n.r. |
| ANS-C3 | 450 | 328 | no | n.r. | n.r. |
| ANS-C4 | 400 | 280 and 328 | yes | yes | slowc |
| ANS-C5 | 400 | 280 and 328 | yes | yes | slowc |
| ANS-C6 | 400 | 280 and 328 | yes | yes | slowc |
| ANS-C7 | 420 | 280 and 328 | yes | yes | fastd |
| ANS-C9 | 420 | 280 and 328 | yes | yes | fastd |
aANS-C1 to ANS-C9 refer to the Mal-ANS-labeled peptides.
bn.r., not relevant because no changes were observed upon mixing the labeled peptide with OppA*.
cAssociation rate constants of binding were of the order of several minutes.
dThe binding process was completed within seconds, i.e. the mixing time of the experiment.
The fluorescence changes observed with C4, C5, C6, C7 and C9 suggest that the ANS moiety of the peptide has moved into an environment more hydrophobic than water. The fluorescence emission peaks of the bound labeled peptides at 420 and 400 nm compare to those of free Mal-ANS-labeled peptide in methanol and ethanol/propanol, respectively. The fluorescence changes observed upon binding of Mal-ANS-labeled C4, C5, C6, C7 and C9 to OppA* were reversed upon addition of bradykinin (Figure 2, open circles) (Table II). The data indicate that Mal-ANS-labeled C4, C5 and C6 seem to bind more strongly than C7 and C9, i.e. a 10-fold excess of bradykinin only partly reverses the signal of bound C5 (Figure 2E and F, open symbols), C4 and C6 (not shown). Determination of the dissociation constants for the labeled peptides was not pursued because of interference of the Mal-ANS fluorescence with the intrinsic fluorescence of the protein.
Binding of azatryptophan-containing peptides
Fluorescence of azatryptophan is characterized by a strong quenching by water (Ross et al., 1997), implying that peptides containing this non-natural amino acid are potentially good indicators of the environment of the ligand. To substantiate the observed differences in binding of Mal-ANS-labeled C1 and C3 versus C7 and C9, SLZQSLSQS and SLSQSLZQS were synthesized, where Z refers to dl-azatryptophan. These peptides exhibited a low fluorescence in aqueous media (MP buffer) (Figure 3A) with an excitation peak at 287 nm and an emission peak at 397 nm with a small shoulder at 372 nm. When these peptides were dissolved in 96% ethanol, the emission at 372 nm became more pronounced, resulting in an emission spectrum with two peaks (Figure 3A). This bimodal type of emission has been associated with the concerted proton transfer from N1 to N7 of the azatryptophan facilitated by one ethanol molecule (Chen et al., 1993a,b).



Fig. 3. Fluorescence of azatryptophan-containing peptides. (A) Excitation and emission spectra of Z3, representative of both Z3 and Z7, in water (open circles) and ethanol (–). All spectra were normalized. (B) Emission spectra of 0.5 µM OppA* (filled circles) and 10 µM Z3 in the absence (–) or presence (open circles) of 0.5 µM OppA*. The spectrum of Z3 bound to OppA*, but corrected for the fluorescence contribution of the protein, is shown as a dotted line. (C) Emission spectra of 0.5 µM OppA* (filled circles) and 10 µM Z7 in the absence (–) or presence (open circles) of 0.5 µM OppA*. The spectrum of Z7 bound to OppA*, but corrected for the fluorescence contribution of the protein, is shown as a dotted line. In (B) and (C), emission spectra were obtained by exciting at 280 ± 4 nm.
Upon binding of the azatryptophan-containing peptides to OppA*, a large increase in fluorescence quantum yield was observed (Figure 3B and C). In addition, for Z7 this was accompanied by the appearance of a second emission maximum at 372 nm next to the peak at 397 nm. Thus, except for the increased quantum yield, the emission maxima of bound Z3 and Z7 displayed characteristics typical of azatryptophan-containing peptide dissolved in aqueous media and ethanol, respectively. Bradykinin competed with the binding of both azatryptophan peptides, as in the presence of this peptide the emission values at 372 and 397 nm were reduced to those of the peptides in buffer (data not shown). These data indicate that the azatryptophan residues in both peptides are shielded from water when bound to OppALl, but the environments in which both azaindole rings reside are clearly different.
Concentration dependence of binding of azatryptophan-containing peptides
The fluorescence quantum yield increase of both peptides allowed us to determine the binding characteristics of the Z3 and Z7 peptides, independent of the intrinsic protein fluorescence (Figure 4). Binding of the Z3 and Z7 peptides could in both cases be described by the sum of a saturable and a linear component, in which the linear component reflected the fluorescence of the unbound peptide. The saturable components could be described by dissociation constants of 0.6 and 1.6 µM for the Z3 and Z7 peptide, respectively (Table I). The observed difference in the maximum fluorescence change (1.8 and 1.2 AU, respectively) between the two peptides is caused by the difference in emission of the bound peptides. In the case of Z3, the absorbed radiation is released at one peak, whereas the absorbed energy of Z7 is emitted at two peaks, of which only the one at 397 nm was measured.

Fig. 4. Concentration dependence of the fluorescence increase induced by Z3 (filled circles) and Z7 (filled squares).The OppA* concentration was 0.5 µM. The excitation wavelength was 280 nm and emission was observed at 400 nm. Data were analyzed according to equation (1) as described in Material and methods. The solid lines through the data points represent the best fits.
Binding of cysteine-containing peptides labeled with a non-fluorescent group
The binding of the cysteine-containing peptides was observed via intrinsic protein fluorescence, using a peptide concentration of three times the dissociation constant to OppA*. After binding of the peptide, a 16-fold excess of pCMBS was added and the changes in fluorescence were monitored. pCMBS reduced binding of C1, C3, C4, C5 and C6 (Table III). For the other two positions, 7 and 9, binding was unaffected by pCMBS. The effect of pCMBS in the case of C1, C3, C4, C5 and C6 could be reversed by adding 100 µM dithiothreitol (DTT). Binding was affected in a similar way when the peptides were labeled with pCMBS before they were added to OppA* (data not shown), which indicates that the different effects of pCMBS are not the result of differences in labeling in the presence of OppA*. Overall, we conclude that OppA* tolerates large bulky groups attached to the cysteine side chains at positions 7 and 9 in the peptides, but not when these groups are attached to the first, third, fourth, fifth and sixth positions.
Table III. Effect of pCMBS on peptide binding.
| Peptide | ΔFmax peptide (AUa) | ΔFmax peptide + pCMBS (AU) | ΔFmax peptide + pCMBS + DTTc (AU) |
|---|---|---|---|
| C0 | 10.0 | 9.7 (97b) | 9.0 (90d) |
| C1 | 10.3 | 2.3 (23) | 8.5 (83) |
| C3 | 9.1 | 1.3 (14) | 8.9 (98) |
| C4 | 13.8 | 1.9 (14) | 13.1 (95) |
| C5 | 4.9 | 0.3 (6) | 4.7 (96) |
| C6 | 4.6 | 0.6 (13) | 4.4 (97) |
| C7 | 7.8 | 7.7 (99) | 6.9 (89) |
| C9 | 10.1 | 8.7 (86) | 9.1 (90) |
| No peptide | 0.0 | –0.3 | –0.2 |
Data represent averages of at least three independent experiments, the standard deviation never exceeded 10%.
aThe fluorescence signal of OppA in the absence of any solute was set at 100 AU.
bFluorescence change elicited by the peptide in the presence of pCMBS as a percentage of the change in the absence of pCMBS.
cThe fluorescence signal was corrected for quenching by DTT (see Materials and methods).
dFluorescence change elicited by the peptide in the presence of pCMBS plus DTT as a percentage of the change in the absence of pCMBS and DTT.
Discussion
To date, all the peptide binding proteins studied that form part of an ABC uptake system share the same binding mechanism, often referred to as the Venus fly-trap mechanism (Quiocho and Ledvina, 1996). An important feature of this mechanism is the total enclosure of the ligand. The different ligands are oriented and bound in the binding site through Coulomb, van der Waals and/or hydrogen interactions with specific amino acid residues of the protein. However, as already noted by Sleigh et al. (1999), the hydrogen bonds and Coulomb interactions formed between the peptide backbone and the binding protein drive binding.
The oligopeptide binding protein of S.typhimurium (OppASt) is able to bind peptides with lengths from two to five amino acid residues. The three-dimensional structure of this binding protein reveals how these peptides are accommodated and shows that di-, tri- and tetrapeptides are totally enclosed by the binding protein. The three-dimensional structure also suggests that pentapeptides may be totally enclosed, whereas longer peptides, if bound at all, would be destined partially to stick out of the binding protein.
In this paper, we define the binding mechanism of the oligopeptide binding protein of L.lactis (OppALl) by using a series of homologous peptides in which either the termini or side chains are selectively modified. We conclude that peptide binding to OppALl has features in common with peptide binding to OppASt and to the dipeptide binding protein of E.coli (DppAEc), but our study also reveals a number of notable differences. The following characteristics of peptide binding by OppALl are worth emphasizing: (i) OppALl binds peptides of up to at least 20 amino acid residues (Detmers et al., 1998; Lanfermeijer et al., 1999; F.J.M.Detmers, F.C.Lanfermeijer, R.Abele, R.Jack, R.Tampé, W.N.Konings and B.Poolman, in preparation); (ii) a free N-terminal amino group is not that critical for binding as in OppASt, OppAEc, DppAEc and DppALl; (iii) the first six N-terminal residues are enclosed in the binding protein; (iv) the remaining C-terminal residues are exposed to the medium but interact with the surface of the binding protein; and (v) peptide binding to OppA* lacks the interaction of the carboxyl group of the peptide with a positively charged residue, a feature observed for the interactions of peptides with OppASt, DppAEc and DppALl.
No strict requirement for free amino and carboxyl groups
Acetylation of the N-terminal amino group or amidation of the C-terminal carboxyl group of the peptide SLSQSLSQS only moderately lowered the affinity for the peptide. Thus, the presence of an unmodified α-amino or α-carboxyl group is less critical for OppALl than for DppAEc, OppAEc and DppALl (Payne and Gilvarg, 1968; Payne, 1971, 1974; Guyer et al., 1986; Sanz et al., 2000). For the α-amino group, this is in agreement with the observation that the asparate residue, which interacts with the positively charged α-amino group of the peptide in OppASt and DppAEc (Asp419 and Asp408, respectively), seems to be absent in OppALl (Picon et al., 2000). The absence of an effect of modification of the α-carboxyl group suggests that the interaction between a positive residue of the binding protein and the negatively charged C-terminus is lacking or not as important as for the binding of small peptides to OppAEc or DppAEc. The lack of this Coulomb interaction may be compensated for by the additional residues in their binding with the surface of the protein. As a consequence, only relatively long peptides, e.g. longer than five or six residues, bind to OppALl with high affinity (Lanfermeijer et al., 1999).
The first six amino acid residues are enclosed by the binding protein
The fluorescence of derivatized Mal-ANS is dependent on its environment. In a hydrophilic environment, the emission maximum of ANS is at 450 nm and the fluorescence quantum yield is low, whereas in a hydrophobic environment the emission maximum can be red-shifted as much as 50 nm and the quantum yield is increased accordingly. Moreover, energy transfer may take place when the ANS moiety is in close proximity to intrinsic protein fluorophores, which results in a second peak of excitation at ∼280 nm. Since the fluorescence of the Mal-ANS-labeled C1 and C3 was the same in the absence and presence of OppA*, it is most probable that these peptides are not bound. We cannot entirely exclude the possibility that the ANS moiety in C1 and C3 is in a hydrophilic pocket, but, if so, the distance between any intrinsic protein fluorophores and the ANS moiety would be large as energy transfer is not observed.
In contrast to Mal-ANS-labeled C1 and C3, the modified amino acid side chains of C7 and C9 are in a relatively hydrophobic environment and in close proximity to protein fluorophores (tryptophans and/or tyrosines), allowing fluorescence resonance energy transfer. Modification of the cysteine in C4, C5 and C6 resulted in a binding process that was slow, but when these peptides were eventually bound, the Mal-ANS fluorescence suggested an environment even more hydrophobic than that observed with labeled C7 or C9. Moreover, energy transfer between the protein fluorophores and Mal-ANS was observed.
The observation that peptides with cysteine at positions 1, 3, 4, 5 and 6 are released from the binding protein when pCMBS is added, whereas those with the cysteine at positions 7 and 9 are not, suggests that the first six binding pockets do not allow modification of the cysteine with pCMBS. We suggest that the pockets for residues 1–6 are too small for or incompatible with the pCMBS modification. Along similar lines of reasoning, one could expect that Mal-ANS modification of residues 1–6 is also incompatible with binding to OppA*. For C1 and C3, this seems to be the case as no fluorescence change is observed upon addition of OppA*, but Mal-ANS-labeled C4, C5 and C6 clearly interact with OppA*, albeit with low association rate constants.
What are the differences in observing the binding of peptides labeled with Mal-ANS or pCMBS? Besides size/structural differences in the probes (Mal-ANS is larger but may adopt a more planar structure than pCMBS; the radius of Hg+ in pCMBS is ∼0.13 nm), there is an important experimental difference in the testing of C4, C5 and C6 modified with Mal-ANS or pCMBS. Ligand binding in the pCMBS experiments is studied via intrinsic protein fluorescence and, therefore, reports on a binding-associated conformational change in the protein (Lanfermeijer et al., 1999). In the Mal-ANS experiments, the fluorescent characteristics of the ANS moiety are observed, which report a change in environment of the probe and not a conformational change in the binding protein. It is, therefore, possible that Mal-ANS-labeled C4, C5 and C6 interact with the binding protein, and may even be positioned in the binding cleft, but these peptides do not necessarily provoke the final conformational change (closure) of the binding protein. On the other hand, the binding pockets for side chains 4, 5 and 6 might be large enough to accommodate the Mal-ANS moieties, whereas the hydrodynamic volume and/or charge of pCMBS prevents binding of the corresponding peptides. The fact that binding is not affected by either pCMBS or Mal-ANS modification of C7 and C9 strongly suggests that the C-terminal residues of nonameric peptides are not restricted by defined pockets in the protein, but rather interact with the surface of the protein.
Labeling of the peptides with Mal-ANS or pCMBS introduces relatively large groups into the side chain moieties of the amino acids. Importantly, the conclusions drawn from these experiments are supported by the observations made with the peptides with the tryptophan mimetic azatryptophan as an environment-sensitive probe. Both Z3 and Z7 showed a dramatic increase in quantum yield in the presence of OppA*, indicating that both azatryptophan residues were shielded from free water by OppA*. When azatryptophan is free in solution, water molecules interact with the N1 and N7 of the azaindole ring structure and thereby quench the fluorescence of the molecule via excited deprotonation (Chen et al., 1993a,b). The fluorescence of the azatryptophan in bound Z3 indicates the absence of both concerted (absence of bimodal emission) and non-concerted excited proton transfer (absence of the quenching by free water) (Chen et al., 1993a,b). Sleigh et al. (1999) described the binding by OppASt as engulfment of peptides together with their solvation water. However, the water molecules in the binding pockets of the liganded binding protein are highly ordered, which might impair both concerted and non-concerted proton transfer in an azaindole moiety. Overall, the fluorescence properties of bound Z3 are consistent with the azatryptophan on the third position being enclosed by the binding protein. The fluorescence of bound Z7 displays characteristics indicative of an environment that potentiates excited concerted double proton transfer between N1 and N7, comparable to the azatryptophan-containing peptide in ethanol (Chen et al., 1993a,b). A residue of the binding protein or a structured water molecule could facilitate the concerted proton transfer necessary for the bimodal emission. The azatryptophan residue on the seventh position is shielded from the aqueous solution and, therefore, must interact with the binding protein, as quenching by free water would otherwise be observed.
Overall, the observations agree very well with a model in which the binding protein encloses the six N-terminal residues, whereas the remaining C-terminal residues of the peptide interact with the binding protein but are in principle accessible from the solvent (see also below). Whatever the structure of this peripheral region, the Mal-ANS and azatryptophan fluorescence studies indicate that the side chains at positions 7 and 9 are in an environment that clearly differs from the aqueous medium. This binding configuration also explains the observation that amidation of the C-terminal carboxyl group of the peptide has only a minimal effect on the binding kinetics.
Mechanistic and structural implications of the binding of long peptides
As discussed above, the formation of Coulomb interactions between the charged N- and C-terminal groups of a peptide and oppositely charged residues in OppALl is not critical for peptide binding. Therefore, alternative interactions between the peptides and the protein must be formed to fulfill the energy requirement for the closure of the binding protein. The observation that the binding affinity of OppALl increases with increasing length of the peptide (optimal peptide length is nine residues; F.J.M.Detmers, F.C.Lanfermeijer, R.Abele, R.Jack, R.Tampé, W.N.Konings and B.Poolman, in preparation) indicates that these interactions are made via the extra C-terminal residues of the peptide. The nature of these interactions could be specific, i.e. a defined region at the surface of the binding protein could accommodate the C-terminal residues. Alternatively, and perhaps more likely, the different C-terminal residues of the peptides may interact in an opportunistic and energetically most favorable way with different surface residues of the protein.
It has been shown that small peptides (four and five residues) can be transported by the Opp system (Detmers et al., 1998). However, binding of these short peptides exhibits a very low affinity. This can now be explained by the fact that binding of these short peptides to OppALl does not involve the additional interactions with the surface of the protein. For example, reducing the length of a peptide from nine residues (SLSQSKVLP) to five residues (SLSQS) results in an increase of the dissociation constant for binding to OppA* by three orders of magnitude, which is comparable to the decrease in binding affinity of OppAEc and DppALl upon methylation of the C-terminus of a dipeptide (Guyer et al., 1986; Sanz et al., 2000).
The fact that the C-terminal part of the peptide is not enclosed by the binding pocket of OppALl implies that this C-terminus is accessible for modifications, which allows several applications. First of all, long peptides could be used as couriers for solutes normally not transported by the Opp system. One could think of antibiotics that do not resemble peptides, but also of peptide drugs whose solubility is increased by addition of hydrophilic residues. Intriguing questions that remain relate to the interactions between liganded OppALl, especially with large ligands, and the membrane complex, the size-exclusion limits for translocation by the membrane component of the Opp system, and the mechanism by which long peptides are translocated through the membrane complex.
Materials and methods
Purification of OppA* and removal of the endogenous ligand
OppA* was purified from L.lactis strain AMP2/pAMP21 and its endogenous ligand was removed as described in Lanfermeijer et al. (1999). OppA* lacks its class II signal peptide and, consequently, the protein is not exported and does not contain the modification of the N-terminal cysteine that anchors the wild-type protein to the external face of the cell membrane. OppA* carries a C-terminal His6 tag (Lanfermeijer et al., 1999).
Peptide labeling
A series of peptides was synthesized with a cysteine residue at the first, third, fourth, fifth, sixth, seventh or ninth position (Table I) of SLSQSLSQS (Isogen, The Netherlands). Peptides were obtained as lyophilized powder and stored at –20°C under nitrogen. Before use, the peptides were dissolved in 25 mM potassium phosphate pH 7.0 supplemented with 10 mM Tris-(2-carboxyethyl)phosphine–HCl at a final concentration of 2.5 mM. The peptides were labeled with Mal-ANS in 50 mM potassium phosphate pH 7.5. Both the peptide and Mal-ANS concentrations were 0.25 mM. After incubation for at least 10 min at room temperature in the dark, the labeled peptides were added to the reaction cuvette (see below).
Binding assays
Fluorescence data were obtained with an Aminco 4800 spectrofluorimeter as described by Lanfermeijer et al. (1999). A quartz cuvette contained 1 ml of a filtered and thoroughly degassed MP buffer (25 mM potassium phosphate, 100 mM KCl, 1 mM K-EDTA, 10% v/v glycerol pH 6.0), which was continuously stirred and kept at 15°C with a circulating flow of water. Concentrations of OppA* varied between 0.25 and 2 µM, depending on the experiment.
Emission spectra of Mal-ANS-labeled peptides were obtained by excitation at 280 ± 4 or 328 ± 4 nm. Excitation spectra were obtained by monitoring emission at a wavelength of 450 ± 4, 420 ± 4 or 400 ± 4 nm.
Binding of unlabeled or pCMBS-labeled peptides to OppA* was assessed by exciting at 280 ± 1 nm, and measuring the emission at 315 ± 4 nm in MP buffer. The buffer was supplemented with 2 mM DTT when the concentration dependence of binding of non-labeled cysteine-containing peptides was studied. DTT was omitted from the buffer when the effect of pCMBS on the binding of the cysteine-containing peptides was assessed. For the reversal of pCMBS labeling of peptides, DTT was added up to 500 µM in steps of 100 µM. The fluorescence signal was corrected for quenching by DTT, which was proportional to the concentration up to at least 500 µM.
Emission spectra of the azatryptophan-containing peptides (Table I; Anaspec, USA) were obtained by exciting at 280 ± 4 nm and observing the emission with a slit width of 8 nm. Excitation spectra were obtained by observing the emission at 400 ± 4 nm and exciting at various wavelengths with a slit width of 8 nm. The kinetics of binding of the azatryptophan-containing peptides to OppA* was assessed by exciting at 280 ± 4 nm and observing emission at 400 ± 4 nm.
The peptides with modified termini were dissolved in 100% DMSO, resulting in the introduction of solvent in the binding assays up to a maximum concentration of 4%. Since the addition of DMSO resulted in an apparent increase of fluorescence of the protein, corrections had to be made for the solvent addition. For this, a linear component was included in the kinetic analysis of binding of terminally modified peptides (see below).
Protein determination
The concentration, purity and stability of the purified OppA* binding protein were assessed by measuring the absorption spectrum of the sample between 240 and 340 nm on a Cary 100 spectrophotometer. The concentration was calculated assuming an extinction coefficient (ε280) of 1.605 (mg/ml)–1cm–1 for OppA* (Pace et al., 1995).
Analysis of the binding data
The binding data were analyzed as described (Lanfermeijer et al., 1999). For the analysis of the binding of the terminally modified peptides and the azatryptophan-containing peptides, a linear component was added to the hyperbolic binding equation [equation (1)] to allow for correction of the DMSO effect or fluorescence of free azatryptophan-containing peptide, respectively.

where ΔF is the observed fluorescence at the free peptide concentration L, ΔFmax is the fluorescence change at infinite peptide concentration, KdL is the equilibrium dissociation constant and (k × L) is the linear component resulting from the increase in fluorescence as a result of the addition of DMSO or the presence of unbound azatryptophan-containing peptide. Using a reiterative fit procedure, the free peptide concentration was estimated using the binding parameters, the protein concentration and a binding stoichiometry of one (Lanfermeijer et al., 1999).
Acknowledgments
Acknowledgements
We acknowledge the support of Professor Dr J.W.Petrich of the Department of Chemistry at the Iowa State University in the interpretation of the azatryptophan fluorescence data. This work is supported by grants from the European Union (Bio-4-CT-960016) and the Gratama Foundation (NL).
References
- Albers S.V., Elferink,M.G., Charlebois,R.L., Sensen,C.W., Driessen,A.J.M. and Konings,W.N. (1999) Glucose transport in the extremely thermoacidophilic Sulfolobus solfataricus involves a high-affinity membrane-integrated binding protein. J. Bacteriol., 181, 4285–4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames G.F. (1986) Bacterial periplasmic transport systems: structure, mechanism and evolution. Annu. Rev. Biochem., 55, 397–425. [DOI] [PubMed] [Google Scholar]
- Chen Y., Gai,F. and Petrich,J.W. (1993a) Solvation of 7-azaindole in alcohols and water: evidence for concerted, excited-state, double-proton transfer on alcohols. J. Am. Chem. Soc., 115, 10158–10166. [Google Scholar]
- Chen Y., Rich,R.L., Gai,F. and Petrich,J.W. (1993b) Fluorescent species of 7-azaindole and 7-azatryptophan in water. J. Phys. Chem., 97, 1770–1780. [Google Scholar]
- Davies T.G., Hubbard,R.E. and Tame,J.R.H. (1999) Relating structure to thermodynamics: the crystal structures and binding affinity of eight OppA–peptide complexes. Protein Sci., 8, 1432–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detmers F.J.M., Kunji,E.R.S., Lanfermeijer,F.C., Poolman,B. and Konings,W.N. (1998) Kinetics and specificity of peptide uptake by the oligopeptide transport system of Lactococcus lactis.Biochemistry, 37, 16671–16679. [DOI] [PubMed] [Google Scholar]
- Dunten P. and Mowbray,S.L. (1995) Crystal structure of the dipeptide binding protein from Escherichia coli involved in active transport and chemotaxis. Protein Sci., 4, 2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyer C.A., Morgan,D.G. and Staros,J.V. (1986) Binding specificity of the periplasmic oligopeptide-binding protein from Escherichia coli. J. Bacteriol., 168, 775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmann J.O., Post,M., Neefjes,J.J., Hämmerling,G.J. and Momburg,F. (1996) Translocation of long peptides by transporters associated with antigen processing (TAP). Eur. J. Immunol., 26, 1720–1728. [DOI] [PubMed] [Google Scholar]
- Kunji E.R., Fang,G., Jeronimus-Stratingh,C.M., Bruins,A.P., Poolman,B. and Konings,W.N. (1998) Reconstruction of the proteolytic pathway for use of β-casein by Lactococcus lactis. Mol. Microbiol., 27, 1107–1118. [DOI] [PubMed] [Google Scholar]
- Lanfermeijer F.C., Picon,A., Konings,W.N. and Poolman,B. (1999) Kinetics and consequences of binding of nona- and dodecapeptides to the oligopeptide binding protein (OppA) of Lactococcus lactis. Biochemistry, 38, 14440–14450. [DOI] [PubMed] [Google Scholar]
- Nickitenko A.V., Trakhanov,S. and Quiocho,F.A. (1995) 2Å resolution structure of DppA, a periplasmic dipeptide transport/chemosensory receptor. Biochemistry, 34, 16585–16595. [DOI] [PubMed] [Google Scholar]
- Pace C.N., Vajdos,F., Fee,L., Grimsley,G. and Gray,T. (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci., 4, 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamer E. and Cresswell,P. (1998) Mechanisms of MHC class I-restricted antigen processing. Annu. Rev. Immunol., 16, 323–358. [DOI] [PubMed] [Google Scholar]
- Payne J.W. (1971) The requirement for the protonated α-amino group for the transport of peptides in Escherichia coli. Biochem. J., 123, 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne J.W. (1974) Peptide transport in Escherichia coli: permease specificity towards terminal amino group substituents. J. Gen. Microbiol., 80, 269–276. [DOI] [PubMed] [Google Scholar]
- Payne J.W. and Gilvarg,C. (1968) The role of the terminal carboxyl group in peptide transport in Escherichia coli. J. Biol. Chem., 243, 335–340. [PubMed] [Google Scholar]
- Picon A., Kunji,E.R.S., Lanfermeijer,F.C., Konings,W.N. and Poolman,B. (2000) Specificity mutants in the binding protein of the oligopeptide transport system of Lactococcus lactis. J. Bacteriol., 182, 1600–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiocho F.A. and Ledvina,P.S. (1996) Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol. Microbiol., 20, 17–25. [DOI] [PubMed] [Google Scholar]
- Ross A.J.B., Szabo,A.G. and Hogue,C.W.V. (1997) Enhancement of protein spectra with tryptophan analogs: fluorescence spectroscopy of protein–protein and protein–nucleic acid interactions. Methods Enzymol., 278, 151–190. [DOI] [PubMed] [Google Scholar]
- Sanz Y., Lanfermeijer,F.C., Konings,W.N. and Poolman,B. (2000) Kinetics and structural requirements for the binding protein of the di-tripeptide transport system of Lactococcus lactis. Biochemistry, 39, 4855–4862. [DOI] [PubMed] [Google Scholar]
- Sleigh S.H., Tame,J.R.H., Dodson,E.J. and Wilkinson,A.J. (1997) Peptide binding in OppA, the crystal structures of the periplasmic oligopeptide binding protein in the unliganded form and in complex with lysyllysine. Biochemistry, 36, 9747–9758. [DOI] [PubMed] [Google Scholar]
- Sleigh S.H., Seavers,P.R., Wilkinson,A.J., Ladbury,J.E. and Tame,J.R.H. (1999) Crystallographic and calorimetric analysis of peptide binding to OppA protein. J. Mol. Biol., 291, 393–415. [DOI] [PubMed] [Google Scholar]
- Steiner R.F and Kirby,E.P. (1969) The interaction of the ground and excited states of indole derivatives with electron scavengers. J. Phys. Chem., 73, 4130–4135. [DOI] [PubMed] [Google Scholar]
- Tam R. and Saier,M.H. (1993) Structural, functional and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev., 57, 320–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tame J.R.H., Murshudov,G.N., Dodson,E.J., Neil,T.K., Dodson,G.G., Higgins,C.F. and Wilkinson,A.J. (1994) The structural basis of sequence-independent peptide binding by OppA protein. Science, 264, 1578–1581. [DOI] [PubMed] [Google Scholar]
- Tame J.R.H., Dodson,E.J., Murshudov,G., Higgins,C.F. and Wilkinson,A.J. (1995) The crystal structures of the oligopeptide-binding protein OppA complexed with tripeptide and tetrapeptide ligands. Structure, 3, 1395–1406. [DOI] [PubMed] [Google Scholar]
- Tame J.R.H., Sleigh,S.H., Wilkinson,A.J. and Ladbury,J.E. (1996) The role of water in sequence-independent ligand binding by an oligopeptide transporter protein. Nature Struct. Biol., 3, 998–1001. [DOI] [PubMed] [Google Scholar]
- Uebel S., Kraas,W., Kienle,S., Wiesmüller,K.-H., Jung,G. and Tampé,R. (1997) Recognition principle of the TAP transporter disclosed by combinatorial peptide libraries. Proc. Natl Acad. Sci. USA, 11, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Heide T. and Poolman,B. (2000) Osmoregulated ABC-transport system of Lactococcus lactis senses water stress via changes in the physical state of the membrane. Proc. Natl Acad. Sci. USA, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]