

Abstract
DNA footprinting and nuclease protection studies of PcrA helicase complexed with a 3′-tailed DNA duplex reveal a contact region that covers a significant region of the substrate both in the presence and absence of a non-hydrolysable analogue of ATP, ADPNP. However, details of the interactions of the enzyme with the duplex region are altered upon binding of nucleotide. By combining this information with that obtained from crystal structures of PcrA complexed with a similar DNA substrate, we have designed mutant proteins that are defective in helicase activity but that leave the ATPase and single-stranded DNA translocation activities intact. These mutants are all located in domains 1B and 2B, which interact with the duplex portion of the DNA substrate. Taken together with the crystal structures, these data support an ‘active’ mechanism for PcrA that involves two distinct ATP-dependent processes: destabilization of the duplex DNA ahead of the enzyme that is coupled to DNA translocation along the single strand product.
Keywords: DNA–protein interactions/DNA translocation/helicase/mutagenesis
Introduction
DNA helicases are motor proteins that translocate along DNA and separate it into its component single strands (for reviews see Lohman and Bjornson, 1996; Soultanas and Wigley, 2000). This reaction is an essential precursor to a wide variety of DNA transactions including DNA replication, repair and recombination. Consequently, these enzymes are ubiquitous and organisms encode multiple DNA helicases that each perform specific roles in vivo (Matson et al., 1994). Since the helicase reaction is energetically unfavourable, all DNA helicases couple this reaction to the hydrolysis of nucleoside triphosphates (NTPs). The absolute requirement for this energy source means that, strictly speaking, the helicase reaction is always an active process. However, models for helicase activity have been defined as ‘active’ if the free energy of NTP hydrolysis is used directly to destabilize the DNA duplex (Lohman and Bjornson, 1996). Experiments on the Rep helicase of Escherichia coli have suggested that it unwinds DNA by an active mechanism (Amaratunga and Lohman, 1993). In contrast, a ‘passive’ helicase uses the free energy of ATP hydrolysis for single-stranded DNA (ssDNA) translocation and achieves unwinding by trapping the ssDNA that becomes available due to ‘thermal fraying’ of the junction. Porter et al. (1998) have suggested that the HCV helicase operates by such a mechanism on the basis of stopped flow studies with fluorescently labelled DNA. The question of whether ‘active’ duplex separation is applicable in a general helicase mechanism remains unclear.
The PcrA helicase from Bacillus stearothermophilus is a monomeric 3′–5′ DNA helicase that is involved in DNA repair and rolling circle plasmid replication (Bird et al., 1998; Petit et al., 1998; Soultanas et al., 1999a). As a member of helicase superfamily I, it is closely related to several well studied enzymes from E.coli, including the Rep and UvrD helicases as well as the B subunit of the RecBCD helicase/nuclease (Gorbalenya and Koonin, 1993). We have recently reported two structures of PcrA helicase in complex with a partial duplex DNA substrate (Velankar et al., 1999). In one structure, the enzyme was also in complex with ADPNP, a non-hydrolysable analogue of ATP. This structure is equivalent to a substrate complex and represents a snapshot of the enzyme before ATP hydrolysis takes place. The second structure is a ternary complex of PcrA, partial duplex DNA and a sulfate ion. Since the sulfate ion occupies the expected position of the phosphate ion after ATP hydrolysis, we refer to this structure as a ‘product’ complex. The two structures revealed the location of the ssDNA and duplex DNA binding sites on the PcrA monomer. Importantly, differences in these binding sites between the substrate and product complexes were used to devise a detailed molecular model for the helicase mechanism of PcrA. We argued that helicase activity was the result of the combination of two complementary but structurally distinct functions of the protein. First, the protein contains an ssDNA tracking motor capable of making single base steps that are each coupled to the hydrolysis of one ATP molecule. Secondly, in the ATP-bound state, the protein binds the duplex DNA ahead of the fork in such a way as to cause distortion from regular B-form DNA, thereby destabilizing the duplex DNA ahead of the enzyme and facilitating the helicase activity. This ATP-dependent duplex destabilization would imply an active mechanism for the enzyme.
The first aspect of our model, unidirectional ssDNA tracking, has been demonstrated biochemically using a novel assay (Dillingham et al., 2000). These data also confirmed that one ATP was hydrolysed for each base that is tracked, as predicted by the model. However, it remains unclear to what extent, if any, the force generated by the ssDNA motor could be transmitted mechanically to the duplex to facilitate strand separation. Such an activity may itself be considered ‘active’, and might be sufficient to achieve helicase activity. Indeed, recent work with the bacteriophage helicases gp41 and Dda has demonstrated that they are capable of generating sufficient force to displace streptavidin from biotin-labelled ssDNA (Morris and Raney, 1999). In our present study, we test the second aspect of our model, namely the ATP-dependent duplex destabilization activity. Our structural studies have identified several residues that might be involved in this process. We reasoned that if duplex destabilization occurred via these contacts, then their loss should result in a reduction in helicase activity whose magnitude reflected the contribution of duplex destabilization to unwinding of DNA. However, since the ssDNA tracking motor should remain intact, then essentially all PcrA–ssDNA transactions should be unaffected by the mutations, i.e. we should be able to uncouple ssDNA translocation from helicase activity.
In this study we have employed a combination of footprinting and mutagenesis techniques in order to investigate the interactions of PcrA with duplex DNA, as well as the manner in which these interactions are modulated by ATP binding. Mutation of residues that contact the DNA duplex produced proteins that were deficient in helicase activity, but that had ssDNA-dependent ATPase and translocation activities that were the same as those of the wild-type enzyme. Further analysis demonstrated that the loss in helicase activity was due to a deficiency in affinity of the enzymes for duplex DNA, an activity that is ATP dependent in the wild-type protein. Together with the structural information, our findings show that PcrA actively engages and distorts the duplex DNA in an ATP-dependent process. In addition to providing direct evidence for an ‘active’ mechanism, our results map critical duplex contacts to several amino acid residues in domains 1B and 2B of PcrA and analyse the contributions made by these residues towards duplex binding and destabilization.
Results
Protection from exonuclease III digestion by the wild-type enzyme
Exonuclease III is a double-strand-specific, non-processive 3′–5′ exonuclease that will trim back from a blunt or 3′-recessed DNA duplex end. Duplex ends with 3′ overhangs of greater than four bases in length are protected from exonuclease III digestion. In order to examine the interactions between the protein and DNA substrates, we looked at protection of a 3′-tailed duplex DNA substrate from digestion by exonuclease III, which was afforded by binding of PcrA in the presence and absence of ADPNP (Figure 1A). In the presence of bound nucleotide, the DNA footprint showed protection of the duplex portion of the substrate out to 13 bp from the junction. In the crystal structure of the substrate complex (Velankar et al., 1999), we observed that although our tailed duplex in that structure was only 10 bp in length, a symmetry-related DNA molecule was packed such that the two molecules formed a pseudo-continuous duplex region of 19 bp within the crystal that made additional contacts with the protein (unusual base-pairing at the duplex ends results in the loss of a base pair). Examination of these contacts revealed that the footprint matched perfectly with the first region of the DNA that is contacted by protein in the crystal structure (Figure 2), as approached from the direction of digestion by exonuclease III.

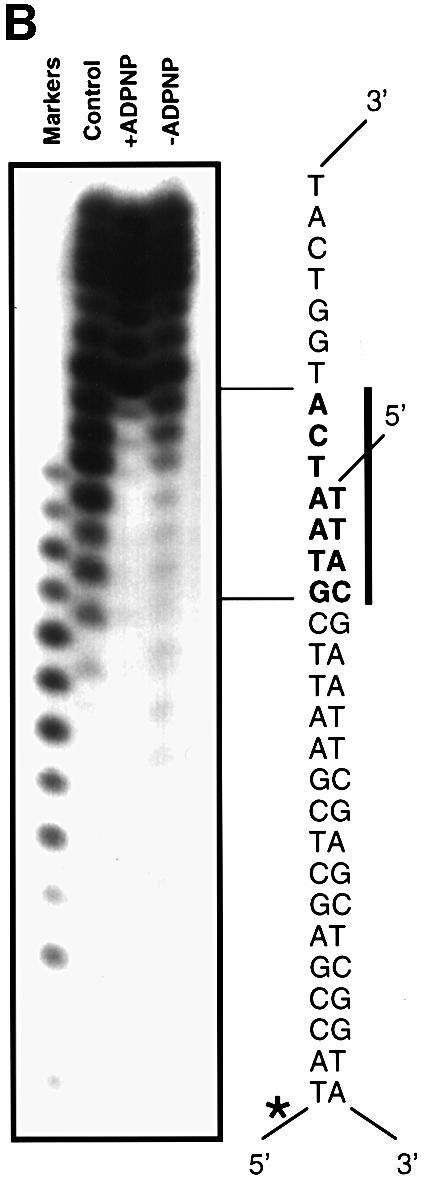
Fig. 1. (A) Exonuclease III protection studies of the 3′-tailed DNA duplex in the presence and absence (control) of PcrA, and the presence and absence of ADPNP as indicated. The amount of exonuclease III in the reaction mixture is 4, 100 or 500 U (lanes 1–3, respectively). The points of protection from 3′-digestion of the labelled strand of the duplex (marked with an asterisk) are shown in bold and marked by a bar and arrows to the sequence of the DNA substrate. Markers are from 20 to five bases in all figures. (B) Hydroxyl radical footprinting of wild-type enzyme bound to a 3′-tailed DNA duplex in the presence and absence of ADPNP, as indicated. The sequence of the protected region is shown in bold alongside the gel and is marked by a bar. The labelled strand is marked with an asterisk.

Fig. 2. (A) Cartoon summarizing the nuclease protection and hydroxyl radical footprinting data with protected areas of the DNA substrate highlighted in magenta. The data are in good agreement with the areas of contact shown by crystal structures (solid bars). The bars are colour coded green, red, yellow and blue to represent the 1A, 2A, 1B and 2B subdomains of PcrA, respectively. (B) Crystal structure of the PcrA ‘substrate’ complex showing the locations of residues that contact the duplex region of the substrate. The four subdomains of PcrA are colour coded as in (A). The tailed duplex DNA substrate and part of its symmetry-equivalent molecule are shown in white. Amino acid residues of interest are shown in magenta. Although not obvious from this orientation, due to limitations of the 2D projection, K137 and K138 from domain 1B (yellow) are close to the longer (tailed) strand near to the junction, while K419, T426 and K456 from domain 2B (blue) contact the same strand one turn higher up the duplex. This figure was created using RIBBONS (Carson, 1991).
An unexpected finding was that protection of the duplex DNA was also seen in the absence of ADPNP, although the main protection was observed from base pair 12 rather than 13 (Figure 1A). The protection by PcrA was overcome at increased levels of exonuclease III, but only in the absence of ADPNP, suggesting that the interaction between the protein and the duplex DNA is tighter when nucleotide is bound to the enzyme. In the absence of ADPNP, it was possible to begin to overcome the protection by PcrA around base pair 12. However, the limited length of the duplex that remains at this stage does not allow complete digestion by exonuclease III even in the absence of protein (see Figure 1A), which precludes detailed analysis of other potential contact regions further down the duplex. This is because of the poor stability of such a short duplex, which dissociates into its component strands before the exonuclease III, which requires a duplex DNA substrate, can complete the digestion. This technique, therefore, was only able to give us useful information about the contacts between the protein and the duplex that are remote from the junction, i.e. the 2B contact region (Figure 2). In order to investigate interactions that are closer to the junction we had to use a different technique.
Cu-phenanthrolate hydroxyl radical footprinting with the wild-type enzyme
For this technique, we had the opposite problem to that of the exonuclease III protection experiments, namely that the short size of the DNA substrate precludes the ability to detect the small fragments that would result from cleavage within the 5′-labelled short strand due to their poor efficiency of precipitation by ethanol. However, data for the longer 5′-labelled strand revealed an apparent region of protection extending across the junction by ∼4 bp into the duplex region and three bases along the ssDNA tail (Figure 1B). This contact region beginning at the first base pair of the duplex and crossing into the beginning of the ssDNA tail is consistent with the crystal structure of the substrate complex (Figure 2). However, the extended protection of the duplex region does not correlate with protein contacts. The specificity of Cu-phenanthrolate attack relies on binding to the minor groove of the DNA (Veal and Rill, 1988), although ssDNA is also cleaved readily (Thederahn et al., 1990). We observed efficient cleavage of the ssDNA tail beyond the third base, and of the duplex beyond the fourth base pair, but protection of the duplex DNA around the junction. Examination of the DNA in the crystal structure of the substrate complex reveals that the phosphates of the single-stranded tail are accessible to the Cu-phenanthrolate beyond the third base and those of the duplex are accessible beyond the second base pair. However, the structure also reveals that the minor groove of 4–5 bp of the duplex is distorted as the DNA begins to separate at the junction (Velankar et al., 1999). One consequence of this distortion is that two bases twist into the minor groove (the third and fourth base pairs of the duplex), thereby blocking the binding of the phenanthrolate (Figure 3). This observation explains the apparent ‘footprint’ because the Cu-phenanthrolate is sterically blocked from binding in the minor groove and hence is unable to promote cleavage. The limited protection seen even in the absence of ADPNP suggests that there is some distortion of the duplex simply as a consequence of binding to the protein (note that the first 4–5 bp of the duplex are cleaved efficiently in the control reaction without protein; Figure 1B). In the presence of ADPNP, however, the footprint becomes more distinct, suggesting a greater distortion and hence a greater protection from binding of phenanthrolate. This is direct evidence that binding of ADPNP promotes a distortion in the DNA duplex that assists duplex unwinding, consistent with the crystal structure of the substrate complex (Velankar et al., 1999).

Fig. 3. Distortion of the DNA duplex on binding to PcrA. Space filling representations of a view along the minor groove of (A) the junction of the DNA substrate and (B) regular B-form DNA for comparison. The bases that protrude into the minor groove in the substrate are coloured magenta, the phosphate groups are coloured orange and other atoms are in cyan. This figure was created using RIBBONS (Carson, 1991).
Importantly, the data from nuclease protection and hydroxyl radical footprinting reveal information about two different contact regions with the duplex DNA. On the tailed duplex strand, there is a contact extending across the junction. There is also protection of a region located ∼12–13 bp from the junction. These two sites of interaction match with the contacts between the protein and DNA substrate observed in the crystal structure of the substrate complex (Velankar et al., 1999) and are summarized in Figure 2. The contacts close to the junction are made by the 1B domain and will be referred to hereafter as the 1B contact region, while those at base pairs 12–13 are with the 2B domain and will be referred to as the 2B contact region. In both cases, the contacts are made with the same strand but displaced by one helical turn. Thus, the contacts made between the protein and the DNA substrate are almost exclusively with the strand along which the protein is translocating. Although interactions between a small region of the duplex DNA and the enzyme were also seen in the crystal structure of the product complex, the high degree of disorder of the remainder of the duplex led us previously to deduce that the contacts might be due to crystal packing. The biochemical data now reveal these contacts to be genuine, but the strength and extent of the interactions are determined by binding of ATP.
Examination of the crystal structures indicated several residues that were close to the two contact regions of the DNA duplex (Figure 2). We selected a number of these residues for analysis by site-specific mutagenesis to test the effects of reducing the ability of the enzyme to bind to duplex DNA. Residues K419, T426 and K456 in domain 2B, which contact the 12–13 bp region, and residues K137 and K138 from domain 1B, which are closer to the junction, were replaced with alanines.
Helicase activity of the mutant enzymes
All of the mutant enzymes were deficient in helicase activity (Figure 4). Two of the single point mutants in domain 2B that showed large defects in helicase activity (K419A and K456A) were then introduced into the same protein to produce a mutant enzyme with the worst helicase activity of all (Figure 4). The data were fitted to estimate the time taken to displace 50% of the oligonucleotide and then compared with the wild type (Table I).

Fig. 4. DNA helicase activity of the mutant enzymes. The standard assay conditions are described in Materials and methods.
Table I. DNA helicase assays.
| Protein | Relative rate (%) |
|---|---|
| Wild type | 100 |
| K137A | 7 |
| K138A | 5 |
| K419A | 4 |
| T426A | 12 |
| K456A | 7 |
| K419A/K456A | 3 |
In order to dissect out the reasons for the reduced helicase activity we carried out a number of different assays to investigate the various biochemical properties that together constitute helicase activity.
ssDNA-dependent ATPase activity
All of the mutants were analysed for their ssDNA-dependent ATPase activity (Table II). In all cases, the kcat and KM values for the ATPase activity were well within 2-fold of that of the wild-type enzyme. We also determined whether the KDNA values (defined in Materials and methods) were affected by the mutations. Again these were generally comparable to that of the wild-type enzyme, although the worst affected was the K419A/K456A double mutant, for which KDNA was increased between 4- and 5-fold (Table II). However, this is much less significant than the 30-fold reduction in helicase activity in this mutant (Table I).
Table II. Steady state kinetic parameters for ATP hydrolysis.
| Protein | kcat (s–1) | KM (µM) | KDNA (nM) |
|---|---|---|---|
| Wild type | 35 | 110 | 200 |
| K137A | 26 | 110 | 370 |
| K138A | 30 | 160 | 730 |
| K419A | 31 | 110 | 450 |
| T426A | 31 | 120 | 420 |
| K456A | 30 | 160 | 340 |
| K419A/K456A | 34 | 130 | 900 |
ssDNA binding affinity
The affinity of the mutant proteins for ssDNA was tested using an ssDNA gel mobility shift assay (Figure 5). The results confirmed those of the ATPase assays with all of the mutant proteins having a similar affinity for ssDNA, most are within 2-fold of that of the wild-type enzyme. The worst affected were the K138A and K419A/K456A mutants (up to 4-fold affected), which also showed the worst KDNA in the ATPase assay (Table II).

Fig. 5. ssDNA gel mobility shift assay. In each case, the first lane (left) is a control containing no protein. The protein concentration increases from left to right across the gel, doubling each time from 27 nM to 1.75 µM. The sequence of the oligonucleotide used was 5′-ACTCTAGAGGATCCCCGGGTACGTTATTGCATGAAAGCCCGGCTG-3′.
ssDNA translocation
We have recently developed an assay to monitor ssDNA translocation in PcrA helicase (Dillingham et al., 2000). Using a fluorescent sensor for inorganic phosphate ions (Pi; Brune et al., 1994), the kinetics of ATP hydrolysis catalysed by PcrA–ssDNA complexes can be followed on the millisecond timescale. The steady state is preceded by a rapid phase of Pi release, the amplitude and duration of which are linearly dependent on ssDNA length. This rapid phase of ATP hydrolysis is interpreted as being associated with processive 3′–5′ tracking along the ssDNA. The slower steady state rate appears to be associated with uncoupled ATP hydrolysis at the 5′ end of the ssDNA and rebinding of PcrA to ssDNA. This fortuitous kinetic phenomenon provides a convenient means by which to measure the step size (bases/ATP hydrolysed) and tracking speed (bases/second) of PcrA, simply by measuring the amplitude and duration of the rapid phase for complexes of PcrA with ssDNA of various lengths. On the basis of these experiments we have proposed a kinetic scheme for ATP hydrolysis during processive DNA tracking by PcrA (Dillingham et al., 2000).
To compare the ability of wild-type and mutant proteins to track along ssDNA we measured the kinetics of Pi release from PcrA–dT8 and PcrA–dT16 complexes. All of the mutant proteins showed qualitatively similar kinetics to the wild type, suggesting that there had been no major change to the rate limiting steps for the rapid and steady state phases. The kinetic parameters for the wild-type and mutant complexes with dT8 and dT16 are shown in Table IIIa and b, respectively. The ssDNA tracking model (Dillingham et al., 2000) predicts a Pi release amplitude of 2.5 Pi/PcrA and a duration of 90 ms for processive DNA tracking on the dT8 oligonucleotide. The equivalent values for dT16 tracking are 6.5 Pi/PcrA and 170 ms. With the exception of the K138A mutant, all of the proteins displayed ssDNA tracking parameters that were similar to the model values. Therefore, these mutants retain the ability to track along ssDNA both processively and at a normal speed. The reduction in Pi release amplitude in the K138A mutant may reflect an increased dissociation of the enzyme before reaching the 5′ end of DNA (i.e. a reduced processivity). This would be consistent with the observation that this was the only mutant protein to require higher DNA concentrations for complete formation of the PcrA–ssDNA complex (see Materials and methods).
Table III. dT8 and dT16 tracking parameters in wild-type and mutant PcrA proteins.
| (a) dT8 tracking parameters in wild-type and mutant PcrA proteins | ||||
|---|---|---|---|---|
| Protein | Rapid phase rate (s–1) | Steady state rate (s–1) | Rapid phase duration (ms) | Rapid phase magnitude (Pi/PcrA) |
| Wild type | 34 | 6.1 | 94 | 3.1 |
| K137A | 23 | 4.8 | 125 | 2.9 |
| K138A | 13 | 5.2 | 125 | 1.7 |
| K419A | 25 | 6.4 | 114 | 2.8 |
| T426A | 29 | 5.8 | 135 | 3.7 |
| K456A | 29 | 5.4 | 101 | 2.9 |
| K419A/K456A |
27 |
5.2 |
106 |
2.9 |
| (b) dT16 tracking parameters in wild-type and mutant PcrA proteins | ||||
| Protein |
Rapid phase rate (s–1) |
Steady state rate (s–1) |
Rapid phase duration (ms) |
Rapid phase magnitude (Pi/PcrA) |
| Wild type | 43 | 6.3 | 160 | 6.9 |
| K137A | 27 | 5.5 | 219 | 5.9 |
| K138A | 24 | 5.7 | 117 | 2.9 |
| K419A | 34 | 6.8 | 184 | 6.2 |
| T426A | 34 | 5.9 | 200 | 6.7 |
| K456A | 40 | 5.5 | 151 | 6.0 |
| K419A/K456A | 35 | 5.7 | 165 | 5.7 |
These data show that the processivity of the mutant enzymes (with the exception of K138A) has not been impaired, at least in terms of translocation along ssDNA. We therefore conclude that we have uncoupled helicase activity from ssDNA translocation activity in these mutants because all of the biochemical properties that relate to ssDNA translocation activity are unaffected in the mutant proteins. Since the lysine mutations showed the greatest degree of uncoupling, we focused on these mutations in subsequent experiments.
Duplex DNA binding affinity
The affinity of the mutant proteins for duplex DNA was compared using a gel mobility shift assay (Figure 6). In contrast to properties involving the ssDNA-binding site, all of the mutants showed defects in their binding to duplex DNA, of at least one order of magnitude.

Fig. 6. Duplex DNA gel mobility shift assay. Protein concentrations (µM) increase from left to right as indicated. The sequence of the duplex DNA used was 5′-ACTCTAGAGGATCCCCGGGTAC-3′ and the complementary strand.
Protection from exonuclease III digestion by the mutant proteins
Since we had uncoupled ssDNA translocation from helicase activity in the mutant enzymes, and we had shown that the DNA duplex binding affinity of the mutants had been affected, we wished to investigate the defect in more detail. We used exonuclease III footprinting to analyse the effects of the most drastic mutations on the interactions in the base pair 12–13 region of the duplex (Figure 7). In the absence of ADPNP, mutations in the 1B domain at positions that contact the duplex close to the junction (K137A, K138A) showed protection around base pairs 12–13 that was similar to that shown by the wild-type protein. In contrast, the K419A and K456A mutants (data not shown) and the K419A/K456A double mutant (Figure 7) showed greatly reduced protection, showing that specific removal of these contacts in the mutant protein results in a loss of protection of the duplex from exonuclease digestion.
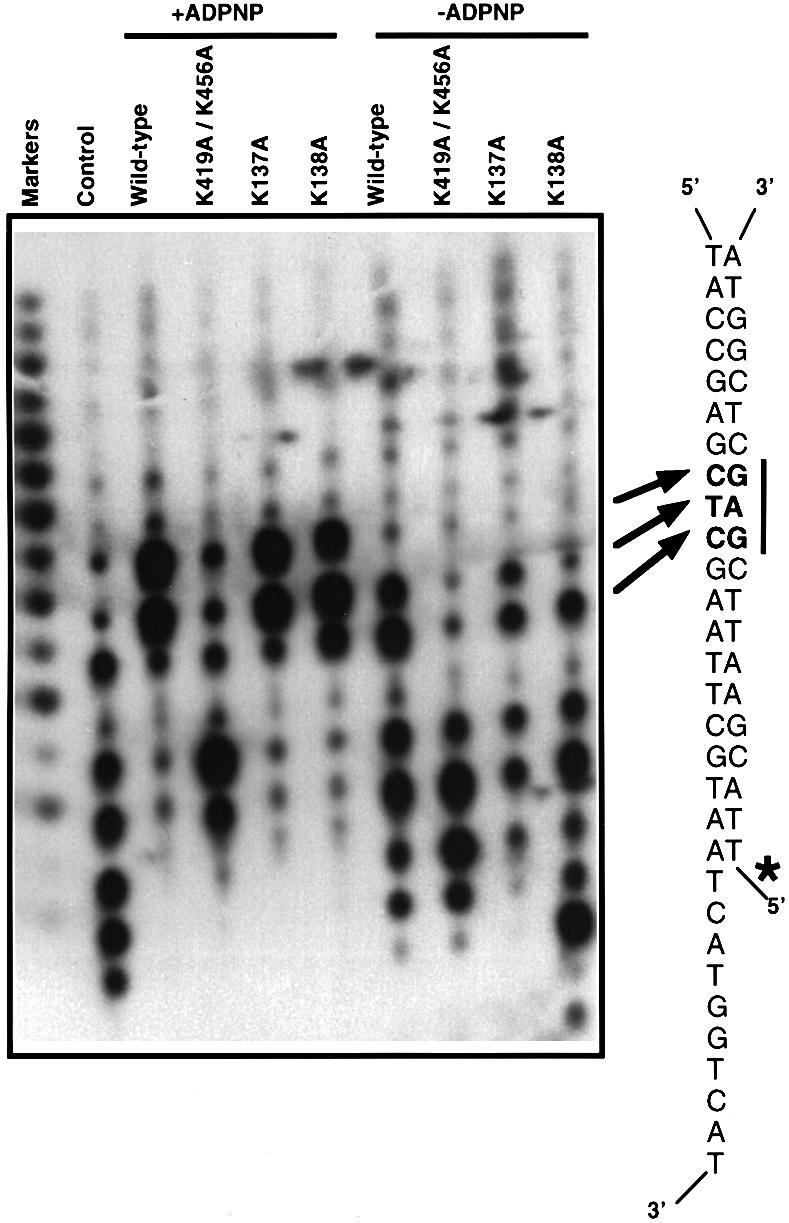
Fig. 7. A comparison of the protection from digestion by exonuclease III with the wild-type and mutant enzymes in the presence and absence of ADPNP, as indicated. Control reactions were carried out in the absence of any protein. The nucleotide sequence of the DNA substrate is shown alongside the gel to indicate the location of the protected region, which is shown in bold and marked by arrows and a bar. The labelled strand is indicated with an asterisk. Markers are from 20 to five bases.
Digestion in the presence of ADPNP also revealed profound differences between the mutant and wild-type enzymes. Mutations in the 1B domain (K137A, K138A) had protection patterns similar to that of the wild-type protein because contacts at the main resistance point (base pair 12–13) are retained and therefore the exonuclease III does not digest beyond this point. For the wild-type, K137A and K138A proteins, protection was greater in the presence of ADPNP than in its absence, showing a tightening of the interaction. In contrast, mutations in the 2B domain (K419A/K456A double mutant), in the contact region with the duplex at base pairs 12–13, showed only a small degree of protection at the main resistance point even in the presence of ADPNP. There were also alterations in the protection closer to the junction, but the limitations of the technique (see above) did not allow us to draw any firm conclusions from these differences. Instead we used Cu-phenanthrolate hydroxyl radical footprinting to examine changes in this region.
Cu-phenanthrolate hydroxyl radical DNA footprinting with the mutant proteins
The results of hydroxyl radical footprinting of the 3′-tailed DNA substrate bound to mutant proteins are shown in Figure 8. For the wild-type protein, there is a small degree of protection around the junction in the absence of ADPNP, which was similar in the K137A and K138A mutant proteins. However, for the K419A/K456A mutant protein there does not seem to be any protection at the junction compared with the control reaction in the absence of protein. Since the domain 2B contact region is distant from the junction in the crystal structure, this is further evidence that the protection at the junction in the absence of ADPNP is not due to protein contacts, but to the distortion observed in the DNA substrate when bound to the protein, which prevents attack by Cu-phenanthrolate as discussed above.


Fig. 8. A comparison of the hydroxyl radical footprinting of wild-type and mutant enzymes in the presence and absence of ADPNP, as indicated. Experiments carried out with mutants K137A and K138A are shown in (A) whereas experiments carried out with the mutant K419A/K456A are shown in (B). Control reactions were carried out in the absence of any protein and are included in (A) only but are appropriate for both parts of this figure. The nucleotide sequence of the DNA substrate is indicated alongside the gel to show the location of the protected region, which is marked in bold and with a bar. The labelled strand is indicated with an asterisk. Markers are from 20 to five bases.
In the presence of ADPNP the situation was different. For the wild-type protein, binding of ADPNP results in significantly enhanced protection of the DNA close to the junction (Figure 1). This arises from two sources, direct contacts between residues in domain 1B and the DNA, and from distortion of the DNA duplex (see above). For the K419A/K456A protein, protection at the junction appears to be the same as that observed for the wild-type enzyme. In contrast, this enhancement was not evident in the K137A and K138A mutants, which showed a similar degree of protection in both the presence and absence of ADPNP. Therefore, the enhanced distortion of the DNA duplex at the junction that is induced by binding of ADPNP does not occur in the K137A and K138A mutant proteins, but is retained in the K419A/K456A double mutant. This suggests that duplex destabilization and protein–DNA contacts at the junction (K137 and K138) are both enhanced by binding of ADPNP, evidence in favour of an active mechanism.
The combined results of the exonuclease III protection, hydroxyl radical footprinting and crystallography reveal that there are two different contact points between the protein and the DNA substrate that involve different domains of the protein. Weakening of the interactions at either of these contact points results in a loss of helicase activity as a consequence of the loss of interactions with the duplex that are enhanced by, although not completely dependent upon, binding of ATP. The data also indicate that the duplex DNA at the junction is distorted upon binding to the enzyme, an effect that is enhanced upon binding of ATP apparently in the main part by interactions with domain 1B (residues K137 and K138).
Discussion
Previously, we have reported crystal structures of PcrA complexed with a 3′-tailed DNA duplex trapped as ‘substrate’ and ‘product’ complexes (Velankar et al., 1999). Based on these crystal structures we proposed an inchworm mechanism for helicase activity that comprised two components: ssDNA translocation and DNA duplex destabilization. The implications for the mechanism that we proposed were manifold. One of these implications is that the DNA translocation activity would have a step size of one base for each ATP that was hydrolysed. This has since been demonstrated using a novel biochemical assay to measure uni-directional translocation along ssDNA substrates (Dillingham et al., 2000). A second consequence of this mechanism is that helicase activity would be an ‘active’ process in that the free energy of ATP hydrolysis would be harnessed by the enzyme to promote duplex destabilization. This suggestion implies that interactions of the enzyme with duplex DNA during the unwinding reaction should be altered by the binding and hydrolysis of ATP.
In order to investigate this aspect of our proposed mechanism, we examined the ATP-dependent changes in DNA duplex binding of the wild-type protein using exonuclease III protection studies and hydroxyl radical footprinting. These experiments revealed several important findings. First, we discovered that the footprint covers the duplex portion of the DNA substrate even in the absence of bound nucleotide. We demonstrated two regions of contact, each being contacted by either domain 1B or 2B. These contacts were in good agreement with those observed in the crystal structure of the substrate complex (Figure 2). However, in the crystal structure of the product complex a large portion of the DNA substrate is disordered so we concluded previously that the contacts we observed between the duplex portion of the substrate and domain 2B might simply be due to crystal packing. In fact, it now appears that these contacts, which are similar in both substrate and product complexes, are present at all stages of the reaction cycle. In other words, during processive helicase activity, the DNA duplex ahead of the fork is not released. However, further experiments revealed that PcrA shows an altered footprint in the presence and absence of a non-hydrolysable ATP analogue, ADPNP. Furthermore, our experiments show that this alteration of the contacts alters the affinity of the protein for duplex DNA, being weakest in the absence of ADPNP. These data suggest that the grip on the DNA is relaxed, allowing the duplex portion of the DNA to slide over the surface of the protein when the conformation of the protein changes after ATP hydrolysis.
Another important implication of our mechanism is that it should be possible to uncouple helicase activity from ssDNA translocation. In order to accomplish this aim, we examined the crystal structures to determine which residues might contribute to binding of the duplex. Several candidates were identified in domains 1B and 2B, and we constructed mutant enzymes in which these residues were replaced with alanines. In all cases, the mutant proteins were deficient in helicase activity. However, these proteins showed wild-type activities in regard to kinetic parameters for ssDNA-dependent ATPase activity, affinity for ssDNA and even in ssDNA translocation. Thus, we had created enzymes in which helicase activity was compromised without affecting ssDNA translocation, confirming that there are at least two independent processes that are coupled by the enzyme to create helicase activity. Electrophoretic gel mobility shift experiments using duplex DNA confirmed that all of the mutants also had a marked defect in their affinity for duplex DNA. In order to investigate the defects in DNA binding in more detail we utilized nuclease protection and hydroxyl radical footprinting.
Nuclease protection studies with the wild-type protein revealed that there is a major region of contact between the protein and the duplex in the region around 12–13 bp from the junction. Although this contact is retained in both the presence and absence of ADPNP, binding of nucleotide results in changes in the conformation of the complex that have a small effect on the digestion pattern. The contacts with this region are exclusively with domain 2B of PcrA, which undergoes substantial rigid body movement when nucleotide binds to the protein–DNA complex (Velankar et al., 1999). Mutation of the residues in domain 2B that contact the duplex DNA (K419, T426, K456) results in proteins that are deficient in helicase activity but that are still able to translocate along ssDNA. These mutations therefore show that contacts between domain 2B of the protein and the DNA duplex are essential for helicase activity but not for translocation along ssDNA or for ssDNA-dependent ATPase activity.
In order to examine the interactions of the protein with the DNA that is closer to the junction, we employed hydroxyl radical footprinting in the presence and absence of ADPNP. For the wild-type protein, these studies revealed several important aspects of the interaction between the protein and the DNA substrate. First, there is a major point of contact with the tailed strand of the duplex in the region adjacent to the junction, even in the absence of bound nucleotide. The crystal structures suggested that this contact region involves two lysine residues (K137 and K138) from domain 1B. Mutations at these positions have a drastic effect on the helicase activity of the protein. For the K137A protein, the ATPase, ssDNA binding and ssDNA translocation activities remained intact, but for the K138A these properties were affected slightly, suggesting that this residue contributes to helicase activity in a more complex manner. However, both mutations show that contacts between domain 1B and the DNA substrate are essential for helicase activity. The binding of ADPNP to the wild-type complex shows that there is increased protection of the DNA at this contact region, suggesting a tighter interaction. However, another important finding was that the Cu-phenanthrolate footprint appeared to cover a region that was not contacted by protein in the crystal structure. This apparent contradiction is resolved upon inspection of the crystal structure, which reveals the reduced cleavage to be due to distortion of the DNA, which sterically blocks binding of phenanthrolate into the minor groove of the DNA substrate. Although present even in the absence of ADPNP, this distortion is increased dramatically when ADPNP binds to the complex. Thus, some of the energy associated with the binding of ATP is utilized by the enzyme to distort the DNA duplex and promote strand separation, i.e. an active mechanism.
Previously, we proposed that helicases might consist of structural modules with defined biochemical functions (Subramanya et al., 1996; Bird et al., 1998). We suggested that domains 1A and 2A, which contain all of the conserved helicase ‘signature’ motifs (Gorbalenya and Koonin, 1993), would comprise the helicase motor, while the less conserved 1B and 2B domains might contribute specificity for different nucleic acid substrates. Our crystal structures of substrate and product complexes of PcrA (Velankar et al., 1999) extended this idea further and suggested a mechanism for how PcrA unwinds DNA that involved two distinct, but coupled processes: ssDNA translocation (provided by domains 1A and 2A) and duplex destabilization (domains 1B and 2B). We have since proved that PcrA can translocate along ssDNA, utilizing one ATP molecule each time it moves along by one base (Dillingham et al., 2000). We now confirm the essential role of two different sets of contacts with the duplex portion of the substrate and, together with the structural evidence, show how ATP binding and hydrolysis are used to destabilize the duplex region of DNA ahead of the junction, producing ssDNA along which the protein is able to translocate, thereby unwinding the duplex. The two sets of contacts with the duplex DNA probably serve different roles. The crystal structure shows that the DNA remote from the junction (the 2B contact region) is in approximate B conformation, while that closer to the junction (the first 4–5 bp) is badly distorted with disrupted base pairing (Velankar et al., 1999). As the ATP-binding site cleft closes around the nucleotide, associated movements of the 1B and 2B domains pull the DNA substrate junction onto the surface of the protein. The domain 2B contact region acts as a lever on the duplex without distorting the DNA locally but causing a distortion of the duplex at the junction. This distortion is enhanced by the conformational changes that take place when ATP binds to the enzyme. The role of the contacts between domain 1B and the DNA closer to the junction is to help to stabilize the distorted DNA duplex, allowing the separated ssDNA strand to be fed through the ssDNA translocation site and thereby facilitate the helicase activity.
The experiments that we present here provide direct evidence in support of an active mechanism for PcrA helicase, with helicase activity resulting from a coupling of two separate ATP-dependent processes provided by different regions of the protein. An important corollary of this idea is that not all proteins that contain the conserved ‘helicase’ motifs need be helicases. In fact, since the motifs are all located in domains 1A and 2A, and are required for the ATPase and associated ssDNA translocation activity, they are more likely to be a characteristic of DNA translocases. This suggestion might explain why a number of ‘helicases’, which have been identified by sequence analysis, do not appear to have helicase activity. Although the role of domains 1B and 2B is duplex destabilization in PcrA, these domains could serve diverse functions in other proteins. Remaining questions about the mechanism of PcrA include the factors that affect the processivity of the enzyme, and the potential biological role of PcrA within multiprotein complexes. Our future efforts will seek to address these aspects of protein function.
Materials and methods
Protein preparations
Mutant protein genes were prepared by PCR as described previously (Soultanas et al., 1999b) and were resequenced to ensure only the required mutation was present. Wild-type and mutant PcrA proteins were purified as described previously (Soultanas et al., 1999b), with the exception of the K419A/K456A double mutant protein. This mutant displayed a poor affinity for the heparin column, and so the flowthrough from this column (containing the mutant protein) was collected and the purification continued as usual from that stage. All proteins were >99% pure as estimated from Coomassie Blue-stained SDS–PAGE gels. Protein concentration was determined spectrophotometrically using a calculated extinction coefficient of 0.76 OD280/mg/ml/cm.
MDCC-modified phosphate binding protein was prepared as described previously (Brune et al., 1998).
Preparation of radioactively labelled probes for phenanthroline-copper footprinting, exonuclease III protection assays and electrophoretic mobility shift assays
HPLC-purified synthetic oligonucleotides were purchased from Oswel. The probe used for the exonuclease III protection assays was prepared by radioactively labelling 10 pmol of a 20mer oligonucleotide (5′-TTACGAATTCGAGCTCGGTA-3′) at the 5′ end with polynucleotide kinase (PNK), as described elsewhere (Sambrook et al., 1989). At the end of the reaction the mixture was incubated at 94°C for 10 min to inactivate the PNK. Subsequently, 1 nmol of a 30mer complementary oligonucleotide (5′-TACCGAGCTCGAATTCGTAATCATGGTCAT-3′) was added to the reaction mixture and the two oligonucleotides were allowed to anneal to each other by incubating the mixture, sequentially, at 94°C for 5 min, 94–80°C in 10 min, 80–60°C in 1 h, 60–45°C in 1 h and 45–20°C in 1 h. The mixture was then centrifuged through a microspin™ S-400 HR column (Pharmacia-Biotech) to purify the labelled probe from unincorporated radioactive [γ-32P]ATP. The final volume was adjusted to 50 µl and the concentration of the probe was considered to be 0.2 pmol/µl, assuming 100% recovery. Radioactively labelled markers were prepared by similar labelling of a series of HPLC-purified synthetic polydeoxythymidylate oligonucleotides ranging from 20 to six bases in length. The probe used in the phenanthroline-copper footprinting experiments was prepared in a similar manner but this time the long 30mer oligonucleotide was radioactively labelled at the 5′ end before being annealed onto the shorter complementary 20mer oligonucleotide. An ssDNA probe for electrophoretic mobility shift assays was prepared by labelling 5 pmol of a single-stranded 45mer (5′-ACTCTAGAGGATCCCCGGGTACGTTATTGCATGAAAGCCCGGCTG-3′) at the 5′ end. A dsDNA probe was prepared in a similar manner by labelling a purified 22 bp duplex DNA (5′-ACTCTAGAGGATCCCCGGGTAC-3′ and complementary strand).
Exonuclease III protection assays
Exonuclease III protection assays were carried out using exonuclease III purchased from Promega. Initially, PcrA protein (8.5 µM) was incubated with radioactively labelled probe (34 nM) at room temperature for 15 min, in a buffer containing 50 mM Tris–HCl pH 7.5, 1 mM EDTA, 200 mM NaCl, 4 mM MgCl2, 2 mM ADPNP, 10% glycerol in a total volume of 20 µl. At the end of the incubation period an equal volume (20 µl) of 1× ExoIII buffer (66 mM Tris–HCl pH 8, 0.66 mM MgCl2) was added and the mixture incubated for a further 5 min at room temperature. At the end of the second incubation period 21 µl of 1× ExoIII buffer, supplemented with 100 mM NaCl, was added. Exonuclease III digestion was initiated by addition of 400 U (2 µl) of exonuclease III unless stated otherwise. Digestion was allowed to proceed for 45 min at room temperature. Reactions were terminated by removing a 5 µl sample and adding it to a microfuge tube on ice, containing 0.5 µl of 0.5 M EDTA and 1 µl of sequencing stop buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol FF). The terminated samples were heated at 94°C for 4 min and loaded onto a 20% polyacrylamide–urea denaturing gel.
1,10-phenanthroline-copper ion nuclease footprinting of PcrA–DNA complexes in situ
Phenanthroline-copper ion nuclease footprinting in situ was carried out as described elsewhere (Papavassiliou, 1994). Wild-type or mutant PcrA (2 µM) was incubated with radioactively labelled probe (67 nM) in the presence or absence of 2.5 mM ADPNP in a total volume of 20 µl, in a buffer containing 20 mM Tris pH 7.5, 50 mM NaCl, 3 mM MgCl2, 2.5 mM ATP, 4 mM dithiothreitol (DTT) and 10% glycerol, at room temperature for 20 min. The radioactive probe used in these experiments was the same as that used in the exonuclease III protection experiments with the only difference being that the 30mer oligonucleotide, instead of the short 20mer oligonucleotide, was radioactively labelled at the 5′ end. After incubation, the samples were loaded onto a 10% non-denaturing polyacrylamide gel and electrophoresis was performed at 120 V. After electrophoresis, the gel was immersed in 200 ml of 10 mM Tris–HCl pH 8 and 5 mM MgCl2, in the presence or absence of 0.75 mM ADPNP, and was gently shaken for 15 min. Solutions of 58 mM 3-mercaptopropionic acid (MPA), 40 mM 1,10-phenanthroline (OP) and 9 mM CuSO4 (Cu2+) were prepared. The [OP]2-Cu2+ chelate was prepared by mixing 1 ml of each of the freshly prepared OP and Cu2+ solutions and waiting for 1 min, with continued mixing. Finally, 18 ml of twice distilled sterile water were added to the mixture to make up the final [OP]2-Cu2+ solution. This solution (20 ml) was added to the gel equilibrating in the 200 ml buffer and the gel was shaken gently for 2 min. The chemical nuclease reaction was initiated by addition of the MPA solution (20 ml) and the gel was again shaken gently for 2 min. The gel was then incubated for a further 13 min without shaking at room temperature. During the last 5 min of the incubation period, 28 mM [OP]2-Cu2+-stop solution (2,9-dimethyl-1,10-phenanthroline) was prepared. The reaction was quenched by adding the [OP]2-Cu2+-stop solution (20 ml) and gently mixing for 2 min. The gel was exposed (3–4 h) to X-ray film in order to locate the appropriate bands on the gel. Gel slices were excised and crushed into sterile 0.5 ml microfuge tubes, with small holes punched through their bottoms with a sterile needle. The small microfuge tubes were then placed into larger tubes and centrifuged at maximum speed (13 000 r.p.m.) for 15 min. The gel slices were crushed through the hole into the lower tube forming a gel paste. A 0.5 ml aliquot of extraction solution (0.5 M ammonium acetate pH 7.5, 1 mM EDTA, 0.1% w/v SDS and 10 mM MgCl2) was added to each gel paste. The tubes were incubated at 37°C overnight with vigorous shaking in order to extract the DNA. The gel paste was pelleted by centrifugation at 13 000 r.p.m. for 15 min. The supernatant containing the DNA was collected and centrifuged again, as described above, in order to remove traces of gel paste. Finally, the DNA was precipitated by adding 1/10th the volume of 3 M potassium acetate pH 7 and 2 vols of cold ethanol, followed by incubation at –20°C for 2 h. The pellet was washed twice with 0.5 ml of 70% ethanol to remove traces of salt. The DNA samples were then dissolved in 5 µl of bidistilled sterile water and, after addition of 0.5 µl of sequencing stop buffer, the samples were electrophoresed through a 20% polyacrylamide–urea sequencing gel.
Helicase assays
3′-tailed substrates were prepared by annealing a radioactively labelled 45mer synthetic oligonucleotide to a slight excess of ssM13mp18 DNA as described previously (Matson and Richardson, 1983; Crute et al., 1988). This creates a substrate with a 22 bp duplex region and a 23 base 3′-terminated ssDNA tail. Helicase time course reactions were carried out at 37°C in a reaction buffer containing 20 mM Tris pH 7.5, 50 mM NaCl, 3 mM MgCl2, 2.5 mM ATP, 4 mM DTT and 10% glycerol, using 0.2 µM protein and 1 nM DNA substrate. After a 2 min pre-incubation the reactions were started with PcrA and later stopped by adding stop buffer (0.4% w/v SDS, 40 mM EDTA, 8% v/v glycerol, 0.1% w/v bromophenol blue). Displaced oligonucleotide was separated from annealed oligonucleotide by electrophoresis through a 10% non-denaturing polyacrylamide gel at constant voltage (150 V). Gels were then dried and quantitative analysis performed using a PhosphorImager and ImageQuant software (Molecular Dynamics).
ATP hydrolysis assays
Steady state ATPase activity was measured by linking ATP hydrolysis to the oxidation of NADH as described previously (Pullman et al., 1960; Tamura and Gellert, 1990). Reactions were carried out with 24 nM protein in a buffer containing 20 mM Tris–HCl pH 7.5, 50 mM NaCl, 3 mM MgCl2 and 4 mM DTT. Values for KDNA, the concentration of dT16 required for half maximal stimulation of ATP hydrolysis, were determined at saturating (2 mM) ATP concentrations. Values of kcat and Km were determined at saturating dT16 concentrations (10× KDNA value). Substrate concentrations were varied at least 5-fold above and below the measured Km and KDNA values. All data were fitted directly to a single site binding equation using the non-linear least squares fitting algorithm of the program GraphPad Prism v2.0 (GraphPad software, San Diego, CA).
Electrophoretic mobility shift assays
Protein at various concentrations was incubated with 2.5 nM ssDNA or dsDNA probe (see above) in buffer (20 mM Tris pH 7.5, 50 mM NaCl, 3 mM MgCl2, 4 mM DTT) at room temperature for 20 min. A small volume of loading buffer (0.25% bromophenol blue, 40% w/v sucrose for ssDNA gel shifts and 25% Ficoll, 0.05% bromophenol blue for duplex DNA shifts) was added to each sample immediately before they were run on a native polyacrylamide gel (9% for ssDNA gel shifts and 6% for duplex DNA gel shifts) to separate the protein–DNA complex from the free DNA. Gels were dried and exposed to X-ray film.
ssDNA translocation assays
Assays for ssDNA translocation were performed essentially as described previously (Dillingham et al., 2000). Stopped flow measurements were made with a HiTech SF61MX apparatus, with excitation at 436 nm through 4 nm slits. Fluorescence emission was measured after a 455 nm cut-off filter. All concentrations quoted are the final ones after mixing and were generally 0.1 µM PcrA, 8 µM (saturating) dT8 or dT16, 500 µM (saturating) ATP and 3 µM MDCC-PBP in a buffer containing 50 mM Tris–HCl pH 7.5, 150 mM NaCl and 3 mM MgCl2. The only exception was for the K138A mutant protein, which required 16 µM DNA for saturation. All experiments were performed at 20°C. A phosphate mop was used throughout to ensure that MDCC-PBP was phosphate free immediately prior to experiments (Dillingham et al., 2000). The fluorescence signal of MDCC-PBP was calibrated using known concentrations of phosphate standard (Merck) as described previously (Brune et al., 1994).
Acknowledgments
Acknowledgements
We thank Jennifer Byrne for technical assistance, Alice Taylor for DNA sequencing and Jackie Hunter for preparing MDCC-PBP. This project was supported by the Wellcome Trust (D.B.W.) and the Medical Research Council (M.R.W.).
References
- Amaratunga M. and Lohman,T.M. (1993) Escherichia coli Rep helicase unwinds DNA by an active mechanism. Biochemistry, 32, 6815–6820. [DOI] [PubMed] [Google Scholar]
- Bird L.E., Subramanya,H.S. and Wigley,D.B. (1998) Helicases, a unifying structural theme? Curr. Opin. Struct. Biol., 8, 14–18. [DOI] [PubMed] [Google Scholar]
- Brune M., Hunter,J.L., Corrie,J.E.T. and Webb,M.R. (1994) Direct real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry, 33, 8262–8271. [DOI] [PubMed] [Google Scholar]
- Brune M., Hunter,J.L., Howell,S.A., Martin,S.R., Hazlett,T.L., Corrie,J.E. and Webb,M.R. (1998) Mechanism of inorganic phosphate interaction with phosphate binding protein from Escherichia coli. Biochemistry, 37, 10370–10380. [DOI] [PubMed] [Google Scholar]
- Carson M. (1991) Ribbons 2.0. J. Appl. Crystallogr., 24, 958–961. [Google Scholar]
- Crute J.J., Mocarski,E.S. and Lehman,I.R. (1988) A DNA helicase induced by herpes simplex virus type I. Nucleic Acids Res., 16, 6585–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillingham M.S., Wigley,D.B. and Webb,M.R. (2000) Demonstration of unidirectional single stranded DNA translocation by PcrA helicase: measurement of step size and translocation speed. Biochemistry, 39, 205–212. [DOI] [PubMed] [Google Scholar]
- Gorbalenya A.E. and Koonin,E.V. (1993) Helicases: amino acid sequence comparisons and structure–function relationships. Curr. Opin. Struct. Biol., 3, 419–429. [Google Scholar]
- Lohman T.M. and Bjornson,K.P. (1996) Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem., 65, 169–214. [DOI] [PubMed] [Google Scholar]
- Matson S.W. and Richardson,C.C. (1983) DNA-dependent nucleoside 5′-triphosphatase activity of the gene 4 protein of bacteriophage T7. J. Biol. Chem., 258, 14009–14016. [PubMed] [Google Scholar]
- Matson S.W., Bean,D.W. and George,J.W. (1994) DNA helicases: enzymes with essential roles in all aspects of DNA metabolism. BioEssays, 16, 13–22. [DOI] [PubMed] [Google Scholar]
- Morris P.D. and Raney,K.D. (1999) DNA helicases displace streptavidin from biotin-labelled oligonucleotides. Biochemistry, 38, 5164–5171. [DOI] [PubMed] [Google Scholar]
- Papavassiliou A.G. (1994) 1,10-phenanthroline-copper ion nuclease footprinting of DNA–protein complexes in situ following mobility-shift electrophoresis assays. Methods Mol. Biol., 30, 43–77. [DOI] [PubMed] [Google Scholar]
- Petit M.A., Dervyn,E., Rose,M., Entian,K.D., McGovern,S., Ehrich,S.D. and Bruand,C. (1998) PcrA is an essential DNA helicase of Bacillus subtilis fulfilling functions in repair and rolling-circle replication. Mol. Microbiol., 29, 261–273. [DOI] [PubMed] [Google Scholar]
- Porter D.J.T., Short,S.A., Hanlon,M.H., Preugschat,F., Wilson,J.E., Willard,D.H. and Consler,T.G. (1998) Product release is the major contributor to kcat for the hepatitis C virus helicase-catalysed strand separation of short duplex DNA. J. Biol. Chem., 273, 18906–18914. [DOI] [PubMed] [Google Scholar]
- Pullman M.E., Penefsky,H.S., Datta,A. and Racker,E. (1960) Partial resolution of the enzymes catalysing oxidative phosphorylation. J. Biol. Chem., 235, 3322–3329. [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular cloning. A laboratory manual. 2nd edn, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Soultanas P. and Wigley,D.B. (2000) DNA helicases: ‘inching forward’. Curr. Opin. Struct. Biol., 10, 124–128. [DOI] [PubMed] [Google Scholar]
- Soultanas P., Dillingham,M.S., Papadopoulos,F., Phillips,S.E.V., Thomas,C.D. and Wigley,D.B. (1999a) Plasmid replication initiator protein RepD increases the processivity of PcrA DNA helicase. Nucleic Acids Res., 27, 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soultanas P., Dillingham,M.S., Velankar,S.S. and Wigley,D.B. (1999b) DNA binding mediates conformational changes and metal ion coordination in the active site of PcrA helicase. J. Mol. Biol., 290, 137–148. [DOI] [PubMed] [Google Scholar]
- Subramanya H.S., Bird,L.E., Brannigan,J.A. and Wigley,D.B. (1996) Crystal structure of a DExx box helicase. Nature, 384, 379–383. [DOI] [PubMed] [Google Scholar]
- Tamura J.K. and Gellert,M. (1990) Characterisation of the ATP binding site on Escherichia coli DNA gyrase. Affinity labelling of Lys-103 and Lys-110 of the B subunit by pyridoxal 5′-diphospho-5′-adenosine. J. Biol. Chem., 265, 21342–21349. [PubMed] [Google Scholar]
- Thederahn T., Spassky,A., Kuwabara,M.D. and Sigman,D.S. (1990) Chemical nuclease activity of 5-phenyl-1,10-phenanthroline-copper ion detects intermediates in transcription initiation by E.coli RNA polymerase. Biochem. Biophys. Res. Commun., 168, 756–762. [DOI] [PubMed] [Google Scholar]
- Veal J.M. and Rill,R.L. (1988) Sequence specificity of DNA cleavage by Bis(1,10-phenanthroline)copper(I). Biochemistry, 27, 1822–1827. [DOI] [PubMed] [Google Scholar]
- Velankar S.S., Soultanas,S., Dillingham,M.S., Subramanya,H.S. and Wigley,D.B. (1999) Crystal structures of complexes of PcrA helicase with a DNA substrate indicate an inchworm mechanism. Cell, 97, 75–84. [DOI] [PubMed] [Google Scholar]