

Abstract
The global regulator Mlc controls several genes implicated in sugar utilization systems, notably the phosphotransferase system (PTS) genes, ptsG, manXYZ and ptsHI, as well as the malT activator. No specific low molecular weight inducer has been identified that can inactivate Mlc, but its activity appeared to be modulated by transport of glucose via Enzyme IICBGlc (PtsG). Here we demonstrate that inactivation of Mlc is achieved by sequestration of Mlc to membranes containing dephosphorylated Enzyme IICBGlc. We show that Mlc binds specifically to membrane fractions which carry PtsG and that excess Mlc can inhibit Enzyme IICBGlc phosphorylation by the general PTS proteins and also Enzyme IICBGlc-mediated phosphorylation of α-methylglucoside. Binding of Mlc to Enzyme IICBGlc in vitro required the IIB domain and the IIC–B junction region. Moreover, we show that these same regions are sufficient for Mlc regulation in vivo, via cross-dephosphorylation of IIBGlc during transport of other PTS sugars. The control of Mlc activity by sequestration to a transport protein represents a novel form of signal transduction in gene regulation.
Keywords: glucose transport/induction/PTS/repressor sequestration
Introduction
Our thinking of bacterial gene regulation has been dominated by the seminal work of Jacob and Monod (1961), which defined the repressor as a protein that binds to an operator site thus preventing transcription. In such a scheme, regulation (induction) is brought about by the inducer, which, by binding to the repressor, changes the repressor’s conformation so that it no longer allows binding to the operator, thus lifting repression. Consequently, to understand regulation the discovery of a bacterial repressor in any new system is usually followed by a search for its cognate inducer. The Mlc (makes large colonies) protein has recently been described as a global regulator that responds to glucose in the growth medium but whose cognate inducer remained elusive. In this paper we present evidence for repressor sequestration by the glucose-specific permease of the phosphotransferase system (PTS), Enzyme IICBGlc (EIICBGlc), as a means of induction.
The Mlc protein was discovered by its ability, when overproduced, to reduce the rate of consumption of glucose in complex media (Hosono et al., 1995). Independently, during a search for mutations that increased mal gene expression, null mutants in mlc were identified (Decker et al., 1998). Mlc was shown to function as a repressor for the malT gene, which codes for the central activator of the mal regulon. Similarly, it was shown that mlc null mutations resulted in an increase of manXYZ expression and that the mlc gene probably corresponds to the previously described dgsA locus (Plumbridge, 1998a). The manXYZ operon encodes a PTS transporter capable of transporting several sugars, including glucose. Subsequent studies revealed that Mlc controlled the expression of ptsG encoding EIICBGlc, the glucose-specific permease of the PTS (Kimata et al., 1998; Plumbridge, 1998b; Oh et al., 1999). In addition, the ptsHI genes encoding the general cytoplasmic components of the PTS were found to be controlled by Mlc (Kim et al., 1999; Plumbridge, 1999; Tanaka et al., 1999). Thus, Mlc was identified as a negative regulator of several genes involved in sugar degradation systems and in particular for PTS genes. Binding sites for Mlc were identifed in all these operons and it was proposed that Mlc represents a new global regulator of carbohydrate metabolism. Mlc belongs to the so-called ROK family (repressors, ORFs, kinases), which contains two identified classes of proteins (Titgemeyer et al., 1994). One are transcriptional regulators, the other glucose/fructose kinases. The latter are missing the first ∼100 amino acids, comprising the DNA binding domain in the transcriptional activators.
From the start it was clear that the regulation of Mlc activity as a repressor was connected to glucose utilization. Yet all efforts to identify an inducer (glucose or glucose-derived metabolites) that would inactivate Mlc binding to its operator failed. In contrast, transport of glucose via EIICBGlc led to derepression of Mlc controlled genes (Plumbridge, 1999; Zeppenfeld et al., 2000). Therefore, transport of glucose via EIICBGlc (PtsG) appeared to be the beginning of a signal transduction chain resulting in the inactivation of Mlc. Support for such a notion came from the analysis of mutations in ptsG that affect not only substrate specificity of PstG but also regulation of ptsG expression (Notley-McRobb and Ferenci, 2000; Plumbridge, 2000; Zeppenfeld et al., 2000). The most straightforward explanation for the control of Mlc activity was that Mlc could be sequestered from the cytoplasm by binding to EIICBGlc but only under conditions of active glucose transport. Here we provide biochemical evidence for the binding of Mlc to EIICBGlc. Binding of Mlc did not require any additional cytoplasmic factor but was controlled by phosphorylation of the IIB domain. Using PtsG variants lacking large portions of EIICBGlc we found that the interaction of Mlc with EIICBGlc requires the IIB domain plus the end of the IIC domain. The IIC domain alone does not take part in Mlc binding and the IIB domain alone is not sufficient.
Results
Mlc binds to PtsG-containing membranes
In order to test the ability of PtsG (EIICB) to bind Mlc we prepared purified membrane fractions of strains harbouring plasmid-encoded PtsG. After removing the cytoplasmic constituents the membranes were resuspended in buffer and purified Mlc (N-terminal His-tag version) was added. Then, membranes were spun down at 132 000 g in the airfuge. Equivalent amounts of membranes and supernatant were analysed by SDS–PAGE, and Mlc was detected by western blotting with anti-His-tag antibodies. Figure 2A shows that Mlc was detectable in the membrane fraction but only when membranes were prepared from cells expressing PtsG (EIICB). Also, from the setup of the experiment using just isolated membranes, it was clear that cytoplasmic components were not needed for Mlc binding. Mlc binding was saturable. Increasing the amount of Mlc resulted in the appearance of Mlc in the supernatant (data not shown). Using a plasmid expressing only the membrane-bound EIIC domain (pACH, amino acids 1–386 with a C-terminal His-tag) (Figure 1), no binding of Mlc could be observed even though the C domain was membrane bound, as seen by its reaction with His-tag antibodies. When the soluble EIIB domain was expressed from pJBH (amino acids 391–477 fused to the first four amino acids and with a C-terminal His-tag) no binding of either EIIB or Mlc to the membrane fraction was observed.



Fig. 2. Mlc binding to PtsG-containing membrane fractions. (A) Inverted membrane vesicles from strains expressing plasmid-encoded PtsG (EIICB) or PtsG deleted variants (EIIC, EIIB, PtsGΔ5–320, PtsGΔ80–320) were incubated with purified MlcH6 (2 µg) and separated by centrifugation (132 000 g) as described in the first procedure in Materials and methods. Equal amounts of supernatant (SN) and pellet (P), the membrane-bound fraction, were separated by SDS–PAGE, and Mlc was detected by western blotting using anti-His-tag antibodies. Lane 14 contains purified MlcH6 run as a control. Lane 1 shows a 50 kDa prestained molecular weight marker (Bio-Rad). (B) The assay was performed according to the second procedure in Materials and methods. The crude extracts, expressing PtsG or deleted variants, were incubated with Glc, PEP or nothing prior to isolation of membranes and incubation with MlcH6. Odd-numbered lanes represent the supernatant fraction (SN), even-numbered lanes represent the pellet fraction (P). Lane 21 has purified MlcH6 as control. (C) Membranes from PtsG (EIICB) expressing cells were treated with MlcH6 (2 µg) in the presence or absence of soluble IIBH6 (10 µg) (His-tag version) as indicated. The lower panel shows the crossreactivity of the His-tag antibodies with EIIBH6. Lane 1 shows a 47.5 kDa prestained molecular weight marker (Bio-Rad).

Fig. 1. Structure of the ptsG region and derived plasmids. The ptsG gene (hatched box) is expressed from two promoters, p1 and p2, and controlled by two Mlc-binding sites shown by shaded boxes. PtsG is divided into a hydrophobic, membrane-bound N-terminal IIC domain (amino acids 1–380) and a soluble cytoplasmic IIB domain (amino acids 389–477) joined by a so-called 8 aa ‘linker’ (Buhr and Erni, 1993). Cys421 is the site of phosphorylation. Relevant restriction sites are indicated. The EcoRI site is derived from the vector. Oligonucleotides are indicated with a star at their 5′ extremity. The extents of DNA present in the various PtsG plasmids are indicated. H6 indicates the presence of a His-tag.
Two other PtsG variants with in-frame deletions within the IIC domain, removing amino acids 80–320 and 5–320 (Figure 1), were tested for their ability to retain Mlc in the membrane fraction. According to the two-dimensional topological model of PtsG by Buhr and Erni (1993), PtsG(Δ80–320) still carries the first two membrane-spanning helices of the IIC domain so this form was expected to be membrane bound. However, a deletion from amino acids 5 to 320 is predicted to remove all eight membrane-spanning segments and PtsG(Δ5–320) was therefore not expected to be membrane bound. To our surprise, when we tested membranes made from cultures expressing PtsG(Δ5–320) and PtsG(Δ80–320) both were still able to retain Mlc in the membrane fraction (Figure 2A). This shows that the entire PtsG protein is not required for Mlc binding but that the IIB domain plus part of the C-terminal region of IIC and the IIC–B linker are sufficient to bind Mlc to membranes.
Mlc binding to PtsG is sensitive to the phosphorylation state
A different experimental setup was used to test the influence of the state of PtsG phosphorylation on Mlc binding. After formation of the crude extract the cytoplasmic constituents were not removed. The crude extract was incubated either with glucose, to dephosphorylate, or phospho-enol-pyruvate (PEP), to phosphorylate, PtsG via the soluble PTS components, EI, HPr and EIIAGlc, present in the crude extract. Following the incubation, membranes were isolated, Mlc added and its binding to membranes tested by centrifugation. Figure 2B shows that the presence of glucose increased binding of Mlc whereas the presence of PEP consistently decreased Mlc binding. Figure 2B also shows the result of this analysis with PtsG variants. The internally deleted PtsG protein, encoded by plasmid pTZ(PtsGΔ80–320), also binds Mlc in a phosphorylation-dependent manner. Dephosphorylation by glucose still appears to stimulate binding of Mlc whereas the presence of PEP has hardly any effect. Possibly, phosphorylation of PtsGΔ80–320 by EIIAGlc is less effective than full-length PtsG. The plasmid expressing just the C domain alone does not elicit Mlc binding and is not affected by incubation with glucose or PEP.
The soluble IIB domain does not interfere with binding of Mlc to PtsG
To test the possibility that the IIB domain alone is sufficient for Mlc binding we added purified IIB domain (amino acids 391–477 fused to the first four amino acids and with a C-terminal His-tag) to the Mlc binding assay. If binding of Mlc occurs exclusively via the IIB domain the addition of soluble IIB should prevent Mlc binding to PtsG membranes. As shown in Figure 2C this is not the case. We measured the capacity of the PtsG-containing membranes to bind Mlc and found that up to 4 µg could be bound in the pellet. In the experiment of Figure 2C (with 2 µg Mlc), purified IIB was added in 2.5-fold excess over the functional Mlc binding capacity of the membranes and did not completely inhibit binding of Mlc to EIICB-containing membranes. However, ∼50% of the Mlc is retained in the supernatant by IIB. This indicates that the soluble IIB domain has a lower binding affinity for Mlc than the membrane-bound PtsG. Taken together these results demonstrate that the IIB domain, harbouring the phosphorylation site, together with part of the IIC–IIB junction region (see below) is necessary for strong Mlc binding.
PtsG prevents Mlc binding to ptsG operator DNA
Binding of Mlc to its DNA operators, upstream of ptsG, can be detected in a band shift assay. Including purified PtsG-containing membranes with Mlc prevents Mlc binding to DNA, while control membranes have no effect (Figure 3).
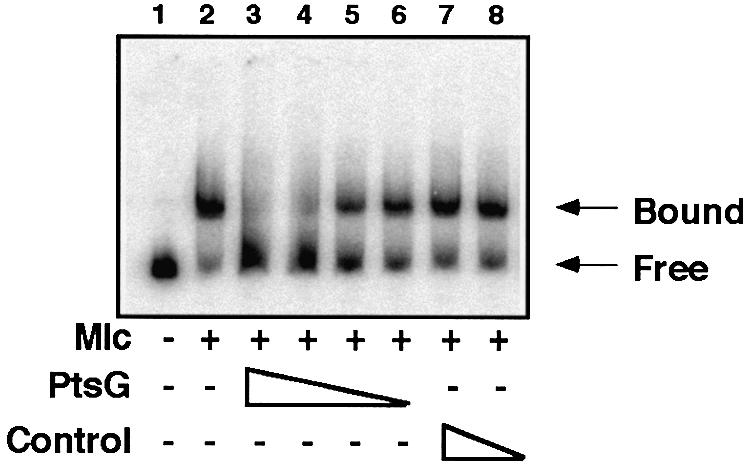
Fig. 3. Effect of PtsG-containing membranes on Mlc binding to its target DNA. The DNA fragment, Glc1–4 (Figure 1) labelled at Glc4, was mixed with Mlc (lanes 2–8) (extract from an overproducing strain, ∼30 µg/ml), and PtsG-containing membranes (lanes 3–6) or control membranes (lanes 7–8) were added (lanes 3 and 7, 100 µg/ml; lanes 4 and 8, 50 µg/ml; lane 5, 25 µg/ml; lane 6, 12.5 µg/ml). Complexes were analysed on small native 5% polyacrylamide gels. The positions of migration of free DNA and DNA complexed with Mlc (Bound) are indicated.
Mlc inhibits PtsG phosphorylation by the general components of the PTS
Addition of [γ-32P]ATP to total sonicated extracts of bacteria results in its conversion to [32P]PEP and the labelling of several PTS proteins (e.g. EI and EIIAGlc) plus certain other major proteins involved in energy production (e.g. succinate thiokinase, SucCD) (Figure 4). The PTS-derived labellings are eliminated in a ΔptsHIcrr strain (Figure 4, lane 9). PtsG overexpressed from a plasmid is also labelled in the extracts (Figure 4, lanes 1–2). Moreover, the plasmids encoding the three internally deleted PtsG proteins (Δ80–320, Δ5–320, Δ5–390), which retain the Cys421 phosphorylation site in the IIB domain, also produce the corresponding labelled proteins (Figure 4, lanes 3–8). Addition of purified Mlc, prior to the [γ-32P]ATP, specifically reduces the intensity of the labelling of the PtsG band by 2- to 4-fold (Figure 4, compare lane 2 with 1). Moreover, Mlc also reduces the phosphorylation of the deleted PtsG molecules Δ80–320 and Δ5–320 (Figure 4, lanes 3–6) but does not affect the phosphorylation of Δ5–390 (lanes 7–8). The simplest interpretation of these results is that Mlc is capable of interacting with unphosphorylated PtsG and thus inhibits its phosphorylation by the PTS. The lack of an effect of Mlc on PtsGΔ5–390 implies that a region necessary for interaction with Mlc is present between amino acids 320 and 390. This region is more or less equivalent to the StuI–BstXI interval on the gene (Figure 1). These 70 amino acids, covering the C-terminus of the IIC domain plus the conserved 8 aa IIC–IIB ‘linker’ sequence defined by Buhr and Erni (1993) will be referred to here as the IIC–IIB ‘junction’ region to distinguish it from the much shorter, previously defined linker.

Fig. 4. Phosphorylation of PtsG in cellular extracts. Extracts of bacteria (JM-G96, ΔptsG, ptsM8) carrying plasmids overexpressing PtsG (lanes 1–2) or deleted variants (lanes 3–8, 10–11) (∼60 µg total protein) were incubated with [γ-32P]ATP as described in Materials and methods. Prior to the addition of [γ-32P]ATP, Mlc (3.5 µg) was added to lanes 2, 4, 6, 8 and 11. Proteins were separated by SDS–PAGE and labelled proteins detected by phosphoimaging after blotting to a Hybond-C membrane. Prestained molecular weight markers (Biolabs) are indicated on the right. The labelled proteins are identified by comparison with Waygood et al. (1984); PpsA, phospho-enol-pyruvate synthase; SucCD, succinate thiokinase α subunit. Lanes 1 and 2, pTZ(PtsG); lanes 3 and 4, pTZ(PtsGΔ80–320); lanes 5 and 6, pTZ(PtsGΔ5–320); lanes 7 and 8, pTZ(PtsGΔ5–390); and lanes 10 and 11, pTZ(PtsGΔ5–320,Δ399–477), which is missing Cys421. Lane 9 has an extract from JM-G77 pTZ(PtsG) (ΔptsHIcrr). The PTS proteins EI and EIIAGlc are not present in this strain and no PTS proteins are labelled.
Mlc inhibits in vitro phosphorylation of α–methylglucoside by PtsG
If Mlc binds to and prevents the phosphorylation of PtsG then PtsG-mediated glucose phosphorylation in crude extracts should also be inhibited by Mlc. We used the non-metabolizable glucose analogue α–methylglucoside (αMG) as PtsG substrate and measured PEP-dependent αMG phosphorylation in crude extracts of strain IT1168 harbouring pTSG11 (encoding wild-type PtsG). Formation of [14C]αMG phosphate was time dependent, it was abolished in the presence of 0.1 mM glucose and, under the chosen experimental conditions, reached a plateau after 3–5 min. At this point ∼15% of the substrate was used up (Figure 5). The addition of Mlc decreased the initial rate of αMG phosphorylation in a concentration-dependent manner (Figure 5). This indicates that not only does PtsG phosphorylation control binding of Mlc but also Mlc binding affects the function of PtsG as an enzyme to phosphorylate its substrate. However, it should be noted that the Mlc added to the in vitro assays, for both PtsG and αMG phosphorylation, is in vast excess compared with PTS proteins present in the extracts. Thus, it is not certain that this inhibition has any physiological relevance but it is further evidence that there is a specific interaction between Mlc and PtsG.

Fig. 5. Mlc inhibits the phosphorylation of the PtsG substrate αMG. Crude cellular extracts (26 µg of protein in 105 µl volume) of strain IT1168 harbouring a PtsG-encoding plasmid were used to phosphorylate [14C]αMG (4 µM) by 20 mM PEP in the presence of no added Mlc (diamonds), 2 µg of Mlc (squares) or 5 µg of Mlc (circles). After the time intervals indicated, samples were removed and processed as described in Materials and methods. The total amount of αMG in the assay was 420 pmol.
The IIB domain with the C-terminal part of IIC (‘junction’ region) is sufficient for Mlc regulation in vivo
Previously we showed that mutations which prevent phosphate transfer from PEP to PtsG (mutations in ptsHIcrr) result in constitutive high expression in a ptsG+ background. In this strain EIICBGlc (PtsG) cannot be phosphorylated and this is accompanied by a derepression of Mlc controlled genes, presumably because Mlc cannot bind to its operators (Plumbridge, 1999). Here we have tested the internally deleted PtsG plasmids for their ability to induce ptsG-lacZ expression in the ptsHIcrr strain (Table I). The two plasmids that carry the IIB domain plus junction region starting at amino acid 320 give almost full complementation of the ptsG defect in the ΔptsHIcrr strain. On the other hand, the plasmids carrying just the soluble IIB domain (with or without the His-tag), but missing amino acids 320–390, are inactive in this test. The IIB domain carrying the phosphorylation site Cys421 is, however, necessary for the induction because the plasmid carrying just the central amino acids 321–398 is inactive.
Table I. Effect of plasmids carrying parts of PtsG on expression of ptsG-lacZ in a ΔptsHIcrr mlc+ strain producing chromosomal levels of Mlc.
| Plasmid | ptsG-lacZ (units) |
|---|---|
| pTZ(PtsG) | 100 |
| pTZ(PtsGΔ80–320) | 75 |
| pTZ(PtsGΔ5–320) | 90 |
| pTZ(PtsGΔ5–320,Δ399–477) | 9 |
| pTZ(PtsGΔ5–390) | 6 |
| pJBH (soluble IIBGlcHis6) | 8 |
| pTZ19 (vector; –IPTG) | 9 |
JM-G77 (ΔptsHIcrr ΔptsG mlc+) was transformed with the plasmids indicated and β-galactosidase activities measured during growth on minimal MOPS medium with 0.4% glycerol, 0.5 mg/ml ampicillin, 1 mM cAMP, 10 µM IPTG. Similar results were obtained with 0.1 mM IPTG.
We previously showed that growth on other PTS sugars besides glucose was also capable of inducing ptsG-lacZ expression (Plumbridge, 1999). The plasmid carrying PtsG amino acids 320–477 (i.e. IIB plus the junction region between IIC and IIB domains) is sufficient to allow induction by these other sugars. Table II shows that the pTZ(PtsGΔ5–320) plasmid produces an induction of ptsG-lacZ expression equal to that produced by the full size PtsG protein during growth on glucose and the other PTS sugars tested: N-acetylglucosamine (GlcNAc), mannitol (Mtl) and mannose (Man). This strain is ptsM+(manXYZ+), which allows growth on Glc and Man. On the other hand pJBH, expressing amino acids PtsG391–477, does not produce the same induction. There is no effect on ptsG-lacZ expression during growth on non-PTS sugars like glycerol or glucose-6-P, with any of the plasmids. This shows that the 320–477 aa region of EIICBGlc is sufficient to signal the activation of the other PTS transporters to Mlc, presumably via a change in its phosphorylation state. Cross phosphorylation between the IIB and IIA domains of PtsG and NagE has been reported previously (Vogler et al., 1988). The present results are further evidence for interdomain complementation and phosphate transfer among the PTS proteins.
Table II. Effect of growth on different sugars on expression of ptsG-lacZ fusion in the presence of plasmids carrying PtsG deletions.
| Plasmid |
ptsG-lacZ |
|||||
|---|---|---|---|---|---|---|
| Gly | Glc-6-P | Glc | GlcNAc | Mtl | Man | |
| pTZ(PtsG) | 10 | 9 | 41 | 57 | 56 | 70 |
| pTZ(PtsGΔ5–320) | 11 | 11 | 70 | 58 | 57 | 70 |
| pJBH (soluble IIBGlcHis6) | 8 | 7 | 8.5 | 10 | 7 | 10 |
JM-G62 (ΔptsG) was transformed with the plasmids indicated and β-galactosidase activities measured during growth in minimal medium with the carbon sources indicated (0.2%, except Gly, glycerol = 0.4%), 0.5 mg/ml ampicillin, 0.1 mM IPTG. GlcNAc, N-acetylglucosamine; Mtl, mannitol; Man, mannose. Results are the mean of two independent cultures.
Buhr et al. (1994) showed that the two PtsG domains, IIB and IIC, carried on separate compatible plasmids, pJBH and pACH, allowed growth on glucose. Although this pair of plasmids is sufficient for growth on glucose in a ptsG, ptsM strain, they do not produce an increase in ptsG-lacZ expression. However, the pACH plasmid in the presence of pTZ(PtsGΔ5–320) produces an increase in ptsG-lacZ expression during growth on glucose (Table III). These results confirm that the junction region between the IIB and IIC domains is necessary for regulation of ptsG expression but not for phosphate transfer to glucose. [Contrary to all the above results, Zeppenfeld et al. (2000) report that a multicopy plasmid carrying the IIC domain results in derepression of a ptsG-lacZ fusion. A trivial explanation of this anomaly could be that their plasmid, which carries an Mlc operator, is producing derepression by titrating Mlc away from the ptsG-lacZ fusion.]
Table III. Effect of plasmids carrying IICGlc and IIBGlc domains on expression of ptsG-lacZ during growth on glucose.
| Plasmid | ptsG-lacZ | |
|---|---|---|
| Gly | Glc | |
| pTZ(PtsG) | 9.5 | 39 |
| pACH (IICGlcHis6) and pTZ(PtsGΔ5–320) | 12.5 | 33 |
| pACH (IICGlcHis6) and pJBH (IIBGlcHis6) | 9.5 | 11 |
| pACH (IICGlcHis6) | 15 | – |
JM-G96 (ΔptsG, ptsM8) was transformed with the plasmids indicated and β-galactosidase activities measured during growth in minimal medium with glycerol (0.4%) or glucose (0.2%), 0.1 mM IPTG, 0.5 mg/ml ampicillin for cultures with pTZ(PtsG), pTZ(PtsGΔ5–320) or pJBH and 40 µg/ml kanamycin for cultures with pACH. Results are the mean of three independent cultures.
Discussion
Previous in vivo experiments showed that PtsG was implicated in the regulation of the expression of its own gene and other genes controlled by Mlc. The induction of ptsG expression was visible during the activation of the PTS system by growth on glucose or other PTS sugars and was dependent upon a functional PtsG. The activity of certain EIIs of the PTS in the regulation of the expression of their cognate operons has been well documented. The EIIs are capable of phosphorylating a particular domain (PRD) within the transcriptional regulator and hence modifying its activity (reviewed in Stülke et al., 1998). This domain is not present in Mlc. An alternative explanation for the involvement of PtsG in regulation of mlc controlled genes was that Mlc senses the activation state of the PTS system by monitoring the phosphorylation state of PtsG (EIICBGlc), and a model was proposed whereby Mlc could be sequestered to the membrane by a physical interaction with the activated PtsG transporter (Figure 6; Plumbridge, 1999; Zeppenfeld et al., 2000). In this work we show that such an interaction between PtsG and Mlc exists. Examples of inhibition of transcription factor activity by interaction with other proteins are well studied in eukaryotic systems, e.g. the binding of IκB to NF-κB transcription factors prevents nuclear localization (for review see Verma et al., 1995). An interesting example in bacteria is the PutA protein, a transcriptional repressor of the put operon when it is present in the cytoplasm but a flavin-linked proline dehydrogenase when associated with the membrane (Ostrovsky de Spicer and Maloy, 1993). Another recent example concerns the MalT activator, whose action is antagonized by binding to MalK, a subunit of the maltose transporter (Panagiotidis et al., 1998).

Fig. 6. A schematic model for the regulation of Mlc controlled genes by PtsG. PtsG is shown with eight transmembrane helices in the IIC domain and the soluble IIB domain in the cytoplasm. (A) In the absence of glucose, Mlc controlled genes are repressed and IICBGlc is predominantly in its phosphorylated form. (B) The transport of glucose results in the dephosphorylation of IICBGlc and derepression of Mlc-controlled genes. The experiments presented in this paper show that PtsG can bind Mlc to membranes in a phosphorylation-dependent manner, supporting the hypothesis that induction of Mlc controlled genes involves sequestration of Mlc to membrane-bound PtsG. The binding of Mlc to PtsG requires the IIB domain and the IIC–B junction region.
An interaction between PtsG and Mlc has been demonstrated in four distinct assays in vitro. Mlc was shown to be bound to PtsG-containing membranes and the binding was enhanced when glucose was added to the vesicles to simulate the transport of glucose. Mlc inhibited both the phosphorylation of PtsG itself and the PEP-dependent phosphorylation of αMG by the PTS. Thus, Mlc can inhibit the PTS functions of PtsG. Moreover, PtsG inhibits Mlc’s function as a repressor, since PtsG was capable of preventing Mlc binding to its target operators on DNA. It should be recalled that the crude extracts used for these experiments contain membrane fragments. Thus, we stress that although the ensemble of experiments presented show biochemically that there is an interaction between Mlc and PtsG, we cannot exclude that some other components of the membrane fractions are necessary for the interaction.
Two in-phase deletion derivatives of the PtsG protein, missing amino acids 80–320 or 5–320, were constructed and shown to be active in fixing Mlc to membranes, showing that the whole IIC domain is not required but only the IIC–IIB junction region and the IIB domain. According to the model of Buhr and Erni (1993) PtsG is composed of eight transmembrane helices (amino acids 17–323) with a soluble cytoplasmic region (amino acids 324–380) in the IIC domain, a conserved ‘linker’ (amino acids 381–388) and a soluble IIB domain (amino acids 389–477) (Figure 1). In this model the Δ80–320 construction should retain the first two transmembrane helices and should be membrane bound, while the Δ5–320 variant should be missing the transmembrane segment. This construct however retained the full capability to bind Mlc to membranes unlike the Δ5–390 (EIIB) form (Figure 2A). An alternative model for the membrane helices of EIICBGlc has been proposed by Lengeler et al. (1994), in which the eighth transmembrane helix is formed by amino acids 354–372, which are still present in PtsG(Δ5–320). The results of this paper support their model. We do not know what conformation the PtsG variants with internal deletions adopt in the membrane and cannot exclude that they form aggregates, but it is clear that these membrane-associated forms are still capable of binding Mlc.
It was important to test whether or not the IIB domain alone would be able to bind Mlc. Since the IIB domain is a soluble protein, binding of Mlc to the membranes could not be tested. Instead we used the possible interference of the soluble IIB domain in the binding of Mlc to intact PtsG. With an excess of the IIB domain, binding of Mlc to membrane-bound PtsG was not strongly inhibited, although somewhat more Mlc was retained in the supernatant than in the absence of EIIB (Figure 2C). This indicates that there is some weak affinity of the isolated IIB domain for Mlc but that the interdomain IIC–B junction region together with the IIB domain, carrying the Cys421 site of phosphorylation, is needed for effective Mlc binding and regulation of Mlc-controlled gene expression.
This result is confirmed by the in vitro protein phosphorylation experiment and the ptsG-lacZ regulation data. Mlc reduces the phosphorylation of the PtsGΔ80–320 and Δ5–320 deleted forms but not that of the PtsGΔ5–390 form, missing the IIC–B junction region (Figure 4). Similarly, both PtsGΔ80–320 and PtsGΔ5–320 deleted forms (but not the PtsGΔ5–390 form) allow Mlc induction of ptsG-lacZ expression in the ΔptsHIcrr strain. Note that neither pTZ(PtsGΔ5–390) nor the IIB His-tag version expressed from pJBH is capable of inducing ptsG-lacZ expression in the ΔptsHIcrr strain (Table I), showing that the His-tag on the B domain encoded by pJBH is not responsible for the lack of activation but it is rather the lack of amino acids in the region 320–390. However, the plasmid expressing just amino acids 320–398 from the PtsG translational start site is not sufficient for regulation, implying that two signals are necessary and that they are supplied by two parts of the PtsG protein: a portion of the protein in the 320–390 amino acid sequence plus the IIB phosphorylation domain. Current experiments are designed to define these contact sites more precisely. At the moment we cannot distinguish between a direct contact between Mlc and the IIC–B junction region (amino acids 320–390) or just a requirement for a membrane localization. It should be emphasized that there is complete correlation between the ability of the deleted PtsG variants to bind Mlc to membranes and their ability to allow regulation of Mlc-controlled genes.
There is a surprising reciprocity in the regulation of Mlc’s function by PtsG. PtsG binds Mlc and prevents it repressing its target sugar utilization operons. At the same time, at least in the in vitro cellular extracts, Mlc inhibits the PTS functions of PtsG, namely its phosphorylation by the general PTS components and the rate of subsequent phosphorylation of αMG. This is an apparent paradox since the conditions that lead to derepression of Mlc controlled genes, i.e. uptake of glucose by the PTS, are those requiring maximum PtsG activity. Thus, Mlc could serve to dampen excessive PtsG transport activity. It is also possible that the inhibition conditions are only achieved in the artificial in vitro extracts, with added excess, purified Mlc, and not under normal in vivo conditions. PtsG, a major sugar transporter, whose level is increased 8-fold during growth on glucose (Erni and Zanolari, 1986; Plumbridge, 1998b), should be in large excess compared with the transcriptional regulator, Mlc, so that only a small fraction of the total PtsG will be involved in Mlc binding. The experiments showing an inhibition of αMG phosphorylation by PtsG in vitro provide additional evidence for the physical interaction between PtsG and Mlc. Our view is summarized in Figure 6. We conclude that the inactivation of the repressor function of the global regulator, Mlc, occurs by sequestration to the membrane-bound PtsG transport protein and that the interaction between the two proteins is regulated by PtsG’s transport activity.
Materials and methods
Bacteriological methods
The Escherichia coli strains (derivatives of JM101) carrying a ptsG-lacZ fusion on a lambda lysogen have been described previously (Plumbridge, 1999). JM-G96 carries ΔptsG::cat and ptsM8 (within the manXYZ operon) alleles eliminating uptake of glucose by the PTS. JM-G62 carries ΔptsG::cat and JM-G77 carries ΔptsG::cat and ΔptsHIcrr::kan. β-galacto sidase activities were measured as described previously in minimal MOPS medium (Miller, 1972). Strain IT1168 (ptsG::kan) (Takahashi et al., 1998) was used for Mlc-binding assays.
Plasmids
A plasmid expressing Mlc with an N-terminal His-tag under isopropyl-β-d-thiogalactopyranoside (IPTG) inducible control was constructed. pKDmlc (Decker et al., 1998) was used as a template for PCR. The synthetic oligonucleotides for N-terminal His6-tagged Mlc were: BamHI-mlc (5′-CGGATCCGATGGTTGCTGAAAACCAGCC-3′) and mlc-PstI (5′-TTTCTGCAGATTAACCCTGCAACAGACGAATC-3′). The amplified PCR product was digested with BamHI and PstI and ligated into pQE31 (Qiagen) harbouring lacIq (described as pGDR11 in Peist et al., 1997) digested with the same enzymes. The MlcH6-encoding plasmid was named pSL104. Plasmids pTSG11, pACH and pJBH have been described (Buhr et al., 1994). pTSG11 expresses wild-type PtsG from both the inducible tac promoter and the ptsG p1 promoter. pACH expresses the IIC domain (amino acids 1–386) with a C-terminal His-tag and pJBH expresses the IIB domain amino acids, 391–477, fused to the first four amino acids and with a C-terminal His-tag (Figure 1). pTZ(PtsG) expresses full size PtsG from the lac promoter; the MluI–EcoRI fragment from pMaG (Buhr et al., 1992) was inserted into pTZ19R. Three plasmids carrying internal deletions within PtsG were constructed (Figure 1). pTZ(PtsGΔ5–320) (the numbers indicate the amino acids missing) was made by deleting between NsiI (made blunt with T4 DNA polymerase) and StuI. pTZ(PtsGΔ80–320) is deleted between EaeI and StuI, and was made by ligating the PstI to EaeI fragment (2.5 kb) of pMaG (EaeI was made blunt by treatment with S1, PstI is within bla) to the StuI–PstI fragment prepared independently. The correct deletions were confirmed by sequencing. These two constructions were made on pMaG and subsequently recloned into pTZ19R. pTZ(PtsGΔ5–390) was made from a PCR fragment using Glc13Nsi (5′-CCAATGCATCTGAAGATGCAA AAGCGACA-3′) and Glc11E (5′-GCGAATTCGTTAACATGGCAA AATCGCGTG-3′). Glc13Nsi carries an extension with a NsiI site adjacent to DNA corresponding to amino acids 391–396. Glc11E carries an extension with an EcoRI site adjacent to DNA hybridizing to DNA ∼300 bp downstream of the ptsG ORF, overlapping the HpaI site (Figure 1). The PCR-generated Glc13–11 fragment was digested with NsiI and EcoRI and inserted into pTZ(PtsG) digested with the same enzymes to replace the full size ptsG gene. This plasmid essentially reconstructs the deletion of pJBH (Buhr et al., 1994) without the His-tag. pTZ(PtsGΔ5–320,Δ399–477) was constructed from pTZ(PtsGΔ5–320) by removing the C-terminal 78 amino acids by deleting between BstXI (made blunt with T4 DNA polymerase) and the EcoRI site at the end of the polylinker. All pTZ(PtsG) plasmids start at the MluI site and are missing the ptsG promoter and Mlc operator (Figure 1). The internally deleted PtsG alleles are all expressed from the wild-type PtsG translational initiation site.
Purification of MlcH6
pSL104 encoding N-terminal His-tagged Mlc was transformed into E.coli DHB4. Cells were grown at 37°C in 1 l of Luria Bertani (LB) broth containing 100 µg/ml ampicillin to an optical density A578 = 0.5, prior to the addition of 0.1 mM IPTG. Further growth was for 5 h. The cells were harvested by centrifugation, washed by lysis buffer (50 mM sodium phosphate pH 8.0, 300 mM NaCl), harvested by centrifugation and kept frozen until further use. They were resuspended in lysis buffer and passed three times through a French pressure cell at 16 000 p.s.i. Unbroken cells were removed by centrifugation (15 min at 25 000 g at 4°C). The supernatant was loaded onto a Ni-NTA Superflow column (Qiagen) and washed with 2 vols of lysis buffer containing 20 mM imidazole. Mlc was eluted with lysis buffer containing 250 mM imidazole. It was dialyzed over night in equilibration buffer (50 mM Tris–HCl pH 7.5 containing 20 mM NaCl and 10 mM β-mercaptoethanol) and loaded onto an anion exchange column (MonoQ from Bio-Rad) equilibrated with the same buffer. After loading the column with the Mlc-containing fraction, it was washed with the same buffer before an NaCl gradient (up to 1 M NaCl) was applied. Mlc eluted as a homogeneous protein (as judged by SDS–PAGE) and was dialyzed against equilibration buffer. It was kept frozen at 1 mg/ml (in 50% glycerol). The yield from 1 l of LB culture was 8 mg.
Membrane preparations and Mlc-binding assays
The method used is similar to that of Nelson et al. (1983) to show an interaction between the lactose permease and EIIA. IT1168 (ptsG::kan) harbouring plasmids encoding PtsG or constructs with parts of PtsG deleted were grown overnight in 500 ml of minimal A medium (Miller, 1972) with 0.2% glycerol as carbon source and ampicillin (0.1 mg/ml). The cells were harvested by centrifugation, washed with 0.9% NaCl and resuspended in 5 ml of HEPES (adjusted to pH 7.0 with 0.5 M NaOH). Cells were broken in a French pressure cell at 16 000 p.s.i. Unbroken cell fragments were removed by centrifugation (at 25 000 g for 15 min at 4°C) and the supernatant was kept on ice (crude cellular extract). Protein concentration was adjusted to 10 mg/ml with HEPES pH 7.0. Membranes were shown to be free of soluble components; residual αMG phosphorylation activity was <1% of the value seen when supernatant was added. To measure binding of Mlc two protocols were used. In the first (Figure 2A), 2 ml of crude cellular extract (20 mg protein) were centrifuged at 100 000 g for 1 h. The supernatant was discarded. The pellet (membrane fraction) was resuspended in 200 µl of HEPES buffer pH 7.0, and 20 µl of purified Mlc (total amount 2 µg) were added and incubated at room temperature for 10 min. The membrane suspension was centrifuged for 1.5 min at 132 000 g in an airfuge. The supernatant was removed and the pellet was resuspended in the same volume of HEPES buffer. Equal samples of supernatant and resuspended membranes were subjected to SDS–PAGE analysis followed by western blotting with anti-His-tag antibodies. In the second protocol (Figure 2B and C), to 200 µl of cellular extract (2 mg protein), 1 mM glucose or 1 mM phosphoenol pyruvate (PEP) or water was added and incubated at 37°C for 10 min. The samples were centrifuged at 132 000 g for 1.5 min and the supernatant discarded. The pellet was resuspended in 100 µl of HEPES buffer containing 2 µg of purified Mlc and incubated for 10 min at room temperature. The samples were again centrifuged for 1.5 min at 132 000 g in the airfuge. The supernatant was removed and the pellet was resuspended in the same volume of HEPES buffer. Equal samples of supernatant and resuspended membranes were subjected to SDS–PAGE analysis followed by western blotting with anti-His-tag antibodies. The antibodies were obtained from Qiagen. Western blotting was done according to the supplier’s protocol. Detection was with alkaline phosphatase.
In vitro PtsG phosphorylation assay
JM-G96 (ΔptsG::cat) transformed with pTZ19R-derived plasmids encoding full size or internally deleted PtsG were grown in MOPS media with 0.4% glycerol, 0.5% casamino acids, 0.5 mg/ml ampicillin and 0.05 mM IPTG to induce expression of PtsG. Bacteria (12 ml) were harvested at A650 = 0.8, washed with TE and resuspended in 0.2 ml of sonication buffer (40 mM Tris–HCl pH 8.0, 1 mM EDTA, 10 mM β-mercaptoethanol and 10% glycerol). Cells were broken by 2× 10 s pulses of sonication with cooling on ice and used immediately for phosphorylation tests. The procedure was based on that described by Waygood et al. (1984). Extracts (6–10 mg/ml; 7 µl) were mixed with 1 µl of purified Mlc (3.5 µg in 50% glycerol) or 1 µl of 50% glycerol on ice and then 5 µCi of [γ-32P]ATP were added in buffer to give (final concentrations) 10 mM HEPES pH 8.0, 5 mM MgCl2 and 0.2 mM ATP, and incubated for 10 min at 30°C. The reaction was terminated with the addition of an equal volume of 2× SDS gel sample buffer. Samples were warmed briefly to 50°C and immediately analysed by SDS–PAGE. The proteins were then transferred by electrophoretic blotting to a Hybond-C membrane (Amersham) in 100 mM Tris, 50 mM boric acid buffer for 4 h at 400 mA. Proteins on the membrane were revealed by Ponceau S staining and radioactively labelled proteins by exposure to a phosphoimager screen.
In vitro αMG phosphorylation
Strain IT1168 harbouring pTSG11 was grown overnight and an extract made using the French pressure cell, as described above for the membrane preparation. The phosphorylation assay (105 µl total) contained 20 µl of buffer A (0.5 M potassium phosphate pH 7.5, 25 mM dithiothreitol, 125 mM potassium fluoride), 20 µl of buffer B (100 mM PEP pH 7.5, 50 mM MgCl2), 40 µl of crude extract (26 µg protein) and 5 µl of water, or buffer containing 2 or 5 µg of purified Mlc. The reaction was started by the addition of 20 µl of [14C]αMG (295 mCi/mmol; Amersham) containing 0.625 µCi radioactivity, resulting in a final concentration of 4 µM substrate. Samples (20 µl) were removed after different time intervals and added to 2.5 µl of 100 mM unlabelled αMG to stop further phosphorylation of [14C]αMG. The total volume was placed on a DE81 ion exchange filter (Whatman), washed extensively with water and counted in a scintillation counter. Control assays were done with strains not containing PtsG, or in the absence of PEP or the presence of 10 mM glucose. No signals were obtained under these conditions. It should be noted that strain IT1168 does contain chromosomally encoded Mlc and that the addition of purified Mlc to the assay represents a vast excess over physiological conditions.
Mlc binding to the ptsG operator
The interaction between Mlc and the ptsG regulatory region DNA was detected by a band shift assay as described previously (Plumbridge, 1998b). The probe used was the Glc1–Glc4 PCR fragment (Figure 1) labelled at Glc4 (2000 c.p.m./lane = ∼1 nM). It was mixed with Mlc (crude extract of an overproducing strain, 30 µg/ml total protein) in binding buffer (25 mM HEPES, 100 mM sodium glutamate pH 8.0 containing 0.5 mg/ml bovine serum albumin), in the presence of varying dilutions of PtsG-containing membranes or control membranes. [MlcH6 bound to DNA in this test but was not displaced by PtsG-containing membranes, unlike wild-type Mlc. Possibly the presence of the N-terminal basic His-tag adjacent to the DNA binding helix–turn–helix motif increased DNA binding non-specifically (c.f. Kolkhof et al., 1995). The footprint of MlcH6 and Mlc on PtsG operator DNA was identical.]
Acknowledgments
Acknowledgements
We are very grateful to Sandra Carolé for constructing the PtsGΔ5–320 and PtsGΔ80–320 deletions. We are indebted to Pieter Postma who suggested using membrane vesicles for measuring binding of Mlc and who supplied the protocol for αMG phosphorylation. We thank Bernard Erni for comments on the manuscript, and Bernard Erni and Hiroji Aiba for the generous gifts of plasmids and strains. Financial support for work in Konstanz was from the Deutsche Forschungsgemeinschaft and Fond der Chemischen Industrie, and in France from the CNRS (UPR9073 and UMR5100) and the Ministère de l’Education Nationale de la Recherche et de la Technologie.
References
- Buhr A. and Erni,B. (1993) Membrane topology of the glucose transporter of Escherichia coli. J. Biol. Chem., 268, 11599–11603. [PubMed] [Google Scholar]
- Buhr A., Daniels,G.A. and Erni,B. (1992) The glucose transporter of Escherichia coli. Mutants with impaired translocation activity that retain phosphorylation activity. J. Biol. Chem., 267, 3847–3851. [PubMed] [Google Scholar]
- Buhr A., Flükiger,K. and Erni,B. (1994) The glucose transporter of Escherichia coli. Overexpression, purification and characterization of functional domains. J. Biol. Chem., 269, 23437–23443. [PubMed] [Google Scholar]
- Decker K., Plumbridge,J. and Boos,W. (1998) Negative transcriptional regulation of a positive regulator: the expression of malT, encoding the transcriptional activator of the maltose regulon of Escherichia coli, is negatively controlled by Mlc. Mol. Microbiol., 27, 381–390. [DOI] [PubMed] [Google Scholar]
- Erni B. and Zanolari,B. (1986) Glucose-permease of the bacterial phosphotransferase system. Gene cloning, overproduction and amino acid sequence of Enzyme IIGlc. J. Biol. Chem., 261, 16398–16403. [PubMed] [Google Scholar]
- Hosono K., Kakuda,H. and Ichihara,S. (1995) Decreasing accumulation of acetate in rich medium by Escherichia coli on introduction of genes on a multicopy plasmid. Biosci. Biotechnol. Biochem., 59, 256–261. [DOI] [PubMed] [Google Scholar]
- Jacob F. and Monod,J. (1961) Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol., 3, 318–356. [DOI] [PubMed] [Google Scholar]
- Kim S.-Y., Nam,T.-W., Shin,D., Koo,B.-M., Seok,Y.-J. and Ryu,S. (1999) Purification of Mlc and analysis of its effects on the pts expression in Escherichia coli.J. Biol. Chem., 274, 25398–25402. [DOI] [PubMed] [Google Scholar]
- Kimata K., Inada,T., Tagami,H. and Aiba,H. (1998) A global repressor (Mlc) is involved in glucose induction of the ptsG gene encoding major glucose transporter in Escherichia coli.Mol. Microbiol., 29, 1509–1519. [DOI] [PubMed] [Google Scholar]
- Kolkhof P., Oehler,S., Alex,R. and Müller-Hill,B. (1995) A basic tail increases repression by dimeric lac repressor. J. Mol. Biol., 247, 396–403. [DOI] [PubMed] [Google Scholar]
- Lengeler J.W., Jahreis,K. and Wehmeier,U.F. (1994) Enzymes II of the phosphoenolpyruvate-dependent phosphotransferase systems: their structure and function in carbohydrate transport. Biochim. Biophys. Acta, 1188, 1–28. [DOI] [PubMed] [Google Scholar]
- Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Nelson S.O., Wright,J.K. and Postma,P.W. (1983) The mechanism of inducer exclusion. Direct interaction between purified IIIGlc of the phosphoenolpyruvate:sugar phosphotransferase system and the lactose carrier of Escherichia coli.EMBO J., 2, 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notley-McRobb L. and Ferenci,T. (2000) Substrate specificity and signal transduction pathways in the glucose-specific enzyme II (EIIGlc) component of Escherichia coli phosphotransferase system. J. Bacteriol., 182, 4437–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H., Park,Y. and Park,C. (1999) A mutated PtsG, the glucose transporter, allows uptake of d-ribose. J. Biol. Chem., 274, 14006–14011. [DOI] [PubMed] [Google Scholar]
- Ostrovsky de Spicer P. and Maloy,S. (1993) PutA protein, a membrane-associated flavin dehydrogenase, acts as redox-dependent transcriptional regulator. Proc. Natl Acad. Sci. USA, 90, 4295–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagiotidis C.H., Boos,W. and Shuman,H.A. (1998) The ATP-binding cassette subunit of the maltose transporter MalK antagonizes MalT, the activator of the Escherichia coli mal regulon. Mol. Microbiol., 30, 535–546. [DOI] [PubMed] [Google Scholar]
- Peist R., Koch,A., Bolek,P., Sewitz,S., Kolbus,T. and Boos,W. (1997) Characterization of the aes gene of Escherichia coli encoding an enzyme with esterase activity. J. Bacteriol., 179, 7679–7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumbridge J. (1998a) Control of the expression of the manXYZ operon in Escherichia coli: Mlc is a negative regulator of the mannose PTS. Mol. Microbiol., 27, 369–381. [DOI] [PubMed] [Google Scholar]
- Plumbridge J. (1998b) Expression of ptsG, the gene for the major glucose PTS transporter in Escherichia coli, is repressed by Mlc and induced by growth on glucose. Mol. Microbiol., 29, 1053–1063. [DOI] [PubMed] [Google Scholar]
- Plumbridge J. (1999) Expression of the phosphotransferase system (PTS) both mediates and is mediated by Mlc regulation in Escherichia coli.Mol. Microbiol., 33, 260–273. [DOI] [PubMed] [Google Scholar]
- Plumbridge J. (2000) A mutation which affects both the specificity of PtsG sugar transport and the regulation of ptsG expression by Mlc in Escherichia coli.Microbiology, 146, 2655–2663. [DOI] [PubMed] [Google Scholar]
- Stülke J., Arnaud,M., Rapoport,G. and Martin-Verstraete,I. (1998). PRD—a protein domain involved in PTS-dependent induction and carbon catabolite repression of catabolic operons in bacteria. Mol. Microbiol., 28, 865–874. [DOI] [PubMed] [Google Scholar]
- Takahashi H., Inada,T., Postma,P. and Aiba,H. (1998) CRP down-regulates adenylate cyclase activity by reducing the level of phosphorylated IIAGlc, the glucose specific phosphotransferase protein in Escherichia coli.Mol. Gen. Genet., 259, 317–326. [DOI] [PubMed] [Google Scholar]
- Tanaka Y., Kimata,K., Inada,T., Tagami,H. and Aiba,H. (1999) Negative regulation of the pts operon by Mlc: mechanism underlying glucose induction in Escherichia coli.Genes Cells, 4, 391–399. [DOI] [PubMed] [Google Scholar]
- Titgemeyer F., Reizer,J., Reizer,A. and Saier,M.H. (1994) Evolutionary relationships between sugar kinases and transcriptional repressors in bacteria. Microbiology, 140, 2349–2354. [DOI] [PubMed] [Google Scholar]
- Verma I.M., Stevenson,J.K., Schwarz,E.M., Van Antwerp,D. and Miyamoto,S. (1995) Rel/NF-κB/IκB family: intimate tales of association and dissociation. Genes Dev., 9, 2723–2735. [DOI] [PubMed] [Google Scholar]
- Vogler A.P., Broekhuizen,C.P., Schuitema,A., Lengeler,J.W. and Postma,P.W. (1988) Suppression of IIIGlc-defects by Enzymes IINag and IIBgl of the PEP::carbohydrate phosphotransferase system. Mol. Microbiol., 2, 719–726. [DOI] [PubMed] [Google Scholar]
- Waygood E.B., Mattoo,R.L. and Peri,K. (1984) Phosphoproteins and the phosphoenolpyruvate:sugar phosphotransferase system in Salmonella typhimurium and Escherichia coli: evidence for IIIMannose, IIIFructose, IIIGlucitol and the phosphorylation of EnzymeIIMannitol and EIIN–acetylglucosamine. J. Cell. Biochem., 25, 139–184. [DOI] [PubMed] [Google Scholar]
- Zeppenfeld T., Larisch,C., Lengeler,J.W. and Jahreis,K. (2000) Glucose transporter mutants of Escherichia coli K-12 with changes in substrate recognition of the IICBGlc and induction behavior of the ptsG gene. J. Bacteriol., 182, 4443–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]