

Abstract
The bacteriophage φ29 replication protein p1 (85 amino acids) is membrane associated in Bacillus subtilis-infected cells. The C-terminal 52 amino acid residues of p1 are sufficient for assembly into protofilament sheet structures. Using chemical cross-linking experiments, we demonstrate here that p1ΔC43, a C-terminally truncated p1 protein that neither associates with membranes in vivo nor self-interacts in vitro, can interact with the primer terminal protein (TP) in vitro. Like protein p1, plasmid-encoded protein p1ΔC43 reduces the rate of φ29 DNA replication in vivo in a dosage-dependent manner. We also show that truncated p1 proteins that retain the N-terminal 42 amino acids, when present in excess, interfere with the in vitro formation of the TP⋅dAMP initiation complex in a reaction that depends on the efficient formation of a primer TP–φ29 DNA polymerase heterodimer. This interference is suppressed by increasing the concentration of either primer TP or φ29 DNA polymerase. We propose a model for initiation of in vivo φ29 DNA replication in which the viral replisome attaches to a membrane-associated p1-based structure.
Keywords: compartmentalization/protein–protein interactions/viral DNA replication
Introduction
Studies with eukaryotic cells have shown that DNA replication occurs at numerous fixed locations within the nucleus. Each site constitutes a replication factory containing many polymerizing machines working on different templates (Newport and Yan, 1996). Replication of adenovirus, a linear DNA genome with a terminal protein (TP) bound to the ends, also takes place at distinct subnuclear sites (Pombo et al., 1994). In this case, the nuclear matrix is thought to function as a scaffold to which the different replication proteins are recruited (for review see de Jong and van der Vliet, 1999). All eukaryotic positive-stranded RNA viruses appear to utilize intracellular membranes as a site of replication. Membranes appear to function not just as a way of compartmentalizing virus RNA replication, but also appear to have a central role in the organization and function of the replication complex (Buck, 1996; Ahola et al., 1999).
In the bacterium Bacillus subtilis, the machinery for replicating DNA, as well as the transcriptional and translational machinery, have specific subcellular localizations (Lemon and Grossman, 1998; Lewis et al., 2000). Direct visualization of the replicative DNA polymerase in living B.subtilis cells has revealed that it is localized at discrete intracellular positions, predominantly at or near midcell, rather than being randomly distributed along the nucleoid mass (Lemon and Grossman, 1998). This finding supports the idea that in bacteria, as in eukaryotes, the replisome remains static by becoming attached to larger structures, and DNA is pulled through (Cook, 1999).
The genome of the B.subtilis bacteriophage φ29 consists of a linear double-stranded DNA molecule with a TP covalently linked to each 5′ end (TP-DNA). Recently, it has been reported that phage φ29 DNA replication occurs at sites different from the host cell replication factories (Meijer et al., 2000). At the onset of replication, φ29 DNA nearly always localizes as a single focus towards one end of the host cell nucleoid. Later on, phage replication is redistributed to multiple sites around the nucleoid periphery. The early viral protein p16.7 is thought to be involved in the spatial redistribution of phage DNA replication sites (Meijer et al., 2000). Furthermore, cell fractionation studies have shown that parental viral DNA, newly synthesized φ29 DNA molecules and some viral replication proteins are membrane associated in infected cells, suggesting an essential role of the bacterial membrane for φ29 DNA replication (Ivarie and Pène, 1973; Bravo and Salas, 1997; W.J.J.Meijer, A.Serna-Rico and M.Salas, submitted). Despite these observations, little is known of the molecular basis for the compartmentalization of φ29 DNA replication. Experiments performed in the 1970s (Ivarie and Pène, 1973) indicated that association of parental φ29 DNA with the bacterial membrane is mediated by early viral-encoded proteins. Subsequently, we found that the viral protein p1, which is expressed early after infection, behaves as a membrane-associated protein during φ29 DNA replication. Membrane association of protein p1 also occurs in the absence of other viral components, suggesting that protein p1 contacts the bacterial membrane directly (Bravo and Salas, 1997). This small protein (85 amino acids) affects the rate of φ29 DNA replication in vivo in a temperature-dependent manner (Bravo and Salas, 1998). These features, together with its capacity to self-interact in vitro, generating highly ordered structures, led us to propose that protein p1 could assemble in vivo to form a viral structure in close association with the bacterial membrane (Bravo and Salas, 1998). This membrane-associated p1-based structure might provide a site for recruitment of viral replication proteins before the initiation of φ29 DNA synthesis by a protein-priming mechanism (for review see Salas et al., 1996). In fact, when initiation of in vivo viral DNA replication is blocked due to the lack of the viral protein p6 (histone-like protein), large amounts of φ29 DNA polymerase are recovered in membrane fractions together with protein p1 and free TP, which acts as a primer (Bravo and Salas, 1997). Nevertheless, full understanding of the function of protein p1 requires a detailed knowledge of the molecular interactions between protein p1 and other viral replication components.
In this report, we demonstrate that the N-terminal 42 amino acid residues of protein p1 are sufficient for binding to the primer TP in vitro. In addition, we show that an excess of protein p1 at the beginning of the infective process reduces the rate of φ29 DNA synthesis. The function of protein p1 involved in this in vivo interference has also been located within the N-terminal 42 amino acid region. Truncated p1 proteins that retain the N-terminal 42 amino acids, when present in excess, interfere with the in vitro formation of the TP⋅dAMP initiation complex in a reaction that depends on the efficient formation of a primer TP–φ29 DNA polymerase heterodimer. This in vitro interference can be explained as competition between the truncated p1 protein and the φ29 DNA polymerase for binding to the primer TP. A model for the initiation of in vivo φ29 DNA replication in which the viral replisome attaches to a membrane-associated p1-based structure is discussed.
Results
Plasmid-encoded protein p1 specifically decreases the rate of φ29 DNA synthesis
The gene encoding protein p1 (gene 1) is located at the left end of the φ29 genome, adjacent to gene 2, which encodes the viral DNA polymerase (Figure 1A). Gene 1 is mainly transcribed from two strong early promoters, PA2b and PA2c, in a polycistronic RNA, which contains other replication genes (for review see Salas and Rojo, 1993). In addition, in vivo S1 mapping experiments revealed the existence of a weak and early transcription initiation site within the DNA polymerase coding region (Barthelemy et al., 1986). This transcription initiation site is close to the reported Escherichia coli RNA polymerase binding site, A1IV (Sogo et al., 1984). These findings suggest that gene 1 could also be transcribed from the weak promoter PA1IV (Figure 1A). To construct a protein p1-producing B.subtilis strain, plasmid pw21, which carries gene 1 and the PA1IV promoter, was introduced into B.subtilis 110NA cells (Figure 1B). To find out whether such a strain synthesizes protein p1, whole-cell extracts were pre pared and total proteins were analyzed by immunoblot techniques using polyclonal antibodies against protein p1. As a control, extracts from cells containing plasmid pow3, which lacks gene 1, were analyzed. As shown in Figure 1B, protein p1 was only detected in plasmid pw21-containing cells.

Fig. 1. (A) Genetic and transcriptional map of the left end of the phage φ29 genome. Only relevant features are shown. The position and approximate length of each gene are indicated. Genes 6 (encoding protein p6), 5 (single-stranded DNA-binding protein), 3 (terminal protein), 2 (DNA polymerase) and 1 (protein p1) are involved in viral DNA replication. Gene 4 encodes protein p4, which activates late transcription. Transcripts from the different promoters are shown. A significant number of transcripts starting from the main early promoters PA2b and PA2c terminate at the transcription terminator TA1 (reviewed by Salas and Rojo, 1993). (B) Left: partial genetic map of plasmids carrying different φ29 DNA sequences. Maps are not drawn to scale. φ29 genes 3 (encoding TP), 2 (DNA polymerase) and 1 (protein p1) are indicated. Gene Δ1 has a deletion of one nucleotide, which leads to a truncated p1 protein of 45 amino acids, conserving the 42 N-terminal residues (protein p1ΔC43) (Bravo and Salas, 1997). The position of the promoters PA1IV and PR is shown. Open boxes indicate pUB110 DNA (McKenzie et al., 1986). The hatched box represents a DNA fragment containing the transcriptional termination sites (TT) of the E.coli rrnB rRNA operon (Brosius et al., 1981). Right: immunoblot analysis of whole-cell extracts from plasmid-containing B.subtilis 110NA cells using anti-p1 serum. Equivalent amounts of the cell extracts were loaded on to the gel. Purified protein p1 was run in the same gel. The molecular weight of pre-stained proteins used as markers is indicated in kilodaltons.
Preliminary experiments showed that the plating efficiency of phage φ29 on B.subtilis cells containing plasmid pw21 (protein p1-producing cells) was much less than that obtained on plasmid pow3-carrying cells. Phage φ29 formed very tiny plaques on protein p1-producing cells (not shown). These observations suggest that plasmid-encoded protein p1 interfered with phage φ29 growth. To get a more accurate measurement of this interference, phage development experiments under one-step growth conditions were performed (Figure 2A). To this end, cells producing or not producing protein p1 were infected with phage φ29 or the related phage Nf. When protein p1-producing cells (plasmid pw21) were infected with phage φ29, a viral yield of ∼40 phages/cell was obtained. This production was 45-fold lower than that obtained in cells not producing protein p1 (plasmid pow3). Interestingly, no reduction in viral production was observed when protein p1-producing cells (plasmid pw21) were infected with phage Nf, indicating that the amount of protein p1 provided by plasmid pw21 specifically interfered with phage φ29 growth.

Fig. 2. In vivo interference mediated by protein p1. (A) Development of φ29 and Nf phages in B.subtilis 110NA cells carrying plasmid pow3 (open circles) or pw21 (filled circles). The production of infective particles (measured as p.f.u./ml) was determined by plating on 110NA cells without plasmid. (B) Time course of the accumulation of φ29 DNA in the indicated strains under one-step phage growth conditions.
The interference phenotype was further analyzed by measuring synthesis of viral DNA under one-step growth conditions (Figure 2B). In this experiment, cells producing or not producing protein p1 were infected with phage φ29 and, at various times after infection, total intracellular DNA was isolated and analyzed by agarose gel electrophoresis. In protein p1-producing cells (plasmid pw21), phage DNA replication was markedly delayed and the amount of viral DNA accumulated in 55 min was much lower than that accumulated in cells not producing protein p1 (plasmid pow3).
Collectively, the above results suggest that intracellular accumulation of protein p1 before φ29 infection specifically decreases the rate of viral DNA synthesis and, as a consequence, viral production is reduced.
The N-terminal 42 amino acid residues of protein p1 are sufficient for in vivo interference
In contrast to protein p1, a truncated p1 protein that lacks the C-terminal 43 amino acids (p1ΔC43) does not behave like a membrane-associated protein in vivo (Bravo and Salas, 1997). It cannot self-interact in solution, as determined by sedimentation through glycerol gradients (see below). To investigate whether protein p1ΔC43 was still able to interfere with phage φ29 growth, we constructed plasmids pA1IVΔ1 and pPR53Δ1 (Figure 1B). In these plasmids, the gene encoding p1ΔC43 (gene Δ1) would be transcribed from the weak promoter PA1IV or from the strong promoter PR, respectively. As a control, plasmid pPR53oΔ1, which carries gene Δ1 inserted in the opposite orientation to the PR promoter, was used. Extracts from B.subtilis cells carrying the different plasmids were prepared, and total proteins were analyzed by immunoblot techniques using anti-p1 polyclonal antibodies. In cells containing plasmid pPR53Δ1, a polypeptide migrating more quickly than protein p1 (plasmid pw21) and with the mobility expected for protein p1ΔC43 (5.1 kDa) was detected (Figure 1B). This product was not detected in cells carrying pA1IVΔ1 or pPR53oΔ1, even when more extract or different blotting conditions were used. However, phage development experiments performed with the different strains (see below) indicated that cells carrying plasmid pA1IVΔ1 synthesize protein p1ΔC43, although less of it than plasmid pPR53Δ1-containing cells. Since gene Δ1 in plasmid pA1IVΔ1 is transcribed from the PA1IV promoter, as is the case for gene 1 in plasmid pw21 (see Figure 1B), we assume that these two genes are similarly transcribed. Therefore, the lack of detection of protein p1ΔC43 in cells carrying plasmid pA1IVΔ1, but not of protein p1 in pw21-containing cells, suggests that the stability of the truncated p1 protein could be lower than that of the intact protein and/or that the anti-p1 antibodies could react less efficiently with the truncated p1 protein.
To study the effect of plasmid-encoded protein p1ΔC43 on φ29 development, cells carrying plasmid pA1IVΔ1 or plasmid pPR53Δ1 were infected with phage φ29 under one-step growth conditions (Figure 3A). In cells containing pA1IVΔ1, the latent and rise periods were slightly extended, and phage production was ∼25-fold lower than that in cells carrying the control plasmid (pPR53oΔ1). Therefore, the amount of protein p1ΔC43 accumulated in cells carrying plasmid pA1IVΔ1, although not detected by immunoblot analysis (Figure 1B), was sufficient to decrease viral production. The effect of protein p1ΔC43 on φ29 development was much more severe in cells carrying plasmid pPR53Δ1 (which have higher levels of p1ΔC43). In this case, phage growth was totally blocked (Figure 3A). These results demonstrate that plasmid-encoded protein p1ΔC43 interferes with phage φ29 development in a dosage-dependent manner.

Fig. 3. In vivo interference mediated by protein p1ΔC43. (A) Phage φ29 development under one-step conditions in B.subtilis 110NA cells carrying plasmid pPR53oΔ1 (open circles), pA1IVΔ1 (filled circles) or pPR53Δ1 (filled squares). (B) Phage Nf development under one-step conditions in the same strains.
We also analyzed the growth of the φ29-related phage Nf in cells producing different levels of protein p1ΔC43 (Figure 3B). In contrast to phage φ29, low levels of protein p1ΔC43 (encoded by plasmid pA1IVΔ1) did not significantly affect Nf viral production. However, like phage φ29, Nf development was totally blocked in cells producing high levels of p1ΔC43 (encoded by plasmid pPR53Δ1).
All these observations led us to conclude that the N-terminal region of protein p1 is involved in its interference function. Therefore, such interference cannot be due to the capacity of protein p1 to associate with the bacterial membrane nor to its ability to self-associate; rather, it is probably due to its capacity to interact with other viral replication protein(s). In addition, it appears that protein p1 from phage φ29 is able to interact with its corresponding Nf target, although with less affinity.
p1ΔC11 and p1ΔC43 form a complex with primer TP in vitro
In contrast to protein p1, which self-associates into highly ordered structures (Bravo and Salas, 1998), protein p1ΔC11 (8.7 kDa), which lacks the C-terminal 11 amino acids, cannot self-interact in solution, as determined by sedimentation assays through glycerol gradients. In such experiments, only one population of p1ΔC11 molecules, sedimenting at a position corresponding to monomers, was observed (Figure 4). A similar result was obtained with the smaller protein p1ΔC43 (not shown).

Fig. 4. Oligomeric state of protein p1ΔC11 in solution. The oligomeric state of p1ΔC11 was studied by sedimentation through glycerol gradients. A MalE-p1ΔC11 protein preparation (21 µM) was completely digested with protease factor Xa at a w/w ratio of 2% the amount of fusion protein. The reaction mixture was incubated at 4°C. This protease cleaves after the sequence IEGR, which separates the MalE (42.4 kDa) and p1ΔC11 (8.7 kDa) domains. Two hundred microliters of the totally digested preparation were loaded on to a glycerol gradient (15–30% in 20 mM Tris–HCl pH 7.5, 200 mM NaCl, 10 mM β-mercaptoethanol) and subjected to centrifugation at 62 000 r.p.m. and 4°C in a Beckman SW65 rotor for 22 h. As a marker, φ29 single-stranded DNA-binding protein (SSB; 13.3 kDa), which behaves as a monomer in solution (Soengas et al., 1994), was loaded in the same gradient. After fractionation of the gradient, aliquots from each fraction were analyzed by SDS–Tricine–PAGE (Schagger and Jagow, 1987). The gel was stained with Coomassie Blue. The fraction at which the maximal amount of each protein appears is indicated. The molecular weight of the proteins is indicated in kilodaltons.
To search for viral replication proteins that interact directly with protein p1, in vitro chemical cross-linking experiments, using dithiobis(sulfosuccinimidylpropionate) (DTSSP) as a cross-linking agent, were performed. To this end, we used both C-terminal truncated p1 proteins, p1ΔC11 and p1ΔC43. The latter was used taking into account that the N-terminal 42 amino acids of protein p1 are sufficient for the interference phenotype (see above). Various viral replication proteins were tested, but we only obtained positive results with primer TP (Figure 5). The truncated p1 protein preparations used in these experiments contained maltose-binding protein (MalE; 42.4 kDa) due to the purification procedure (see Materials and methods). A specific cross-linked product with the mobility expected for a complex of p1ΔC11 (8.7 kDa) and primer TP (31 kDa) was detected when both proteins were together but not when only one of them was present in the reaction (Figure 5A). A specific cross-linked product was also observed when p1ΔC43 (5.1 kDa) and primer TP were together. In both cases, the cross-linked product was recognized by anti-TP and anti-protein p1 polyclonal antibodies (Figure 5B). We conclude, therefore, that both p1ΔC11 and p1ΔC43 can interact in vitro with the primer TP.

Fig. 5. Cross-linking of C-terminal truncated p1 proteins and primer TP. Protein samples were treated with DTSSP and separated by SDS–Tricine–PAGE (Schagger and Jagow, 1987). The gel was either (A) stained with silver (Morrissey, 1981) or (B) processed for western blotting. The position of MalE (42.4 kDa), TP (31 kDa), p1ΔC11 (8.7 kDa) and p1ΔC43 (5.1 kDa) is indicated. The arrow shows the position of the p1ΔC11/TP and p1ΔC43/TP cross-linked products.
p1ΔC11 and p1ΔC43, when present in excess, interfere with the formation of the TP⋅dAMP complex in vitro
It has been demonstrated that a free molecule of TP (primer TP) becomes covalently bound to 5′-dAMP (TP⋅dAMP complex) in a reaction catalyzed by the φ29 DNA polymerase in the presence of 5′-dATP and φ29 TP–DNA as template (DNA-dependent initiation reaction) (Blanco and Salas, 1985). To understand p1-mediated in vivo interference, we studied the effect of both p1ΔC11 and p1ΔC43 on the in vitro formation of the TP⋅dAMP initiation complex (Figure 6A). We used increasing concentrations of the corresponding truncated p1 protein and low concentrations of primer TP and φ29 DNA polymerase (1.2 nM). The truncated p1 protein was added to the reaction before primer TP and φ29 DNA polymerase. As shown in Figure 6A, truncated p1 protein reduced the amount of TP⋅dAMP complexes in a dosage-dependent manner. A similar result was obtained when a 259 bp DNA fragment from the φ29 left end (oriL) lacking the parental TP was used as template (Figure 6B). These results indicate that both p1ΔC11 and p1ΔC43, when present in excess, interfere with formation of the TP⋅dAMP initiation complex in vitro. This interference is independent of the presence of the parental TP.
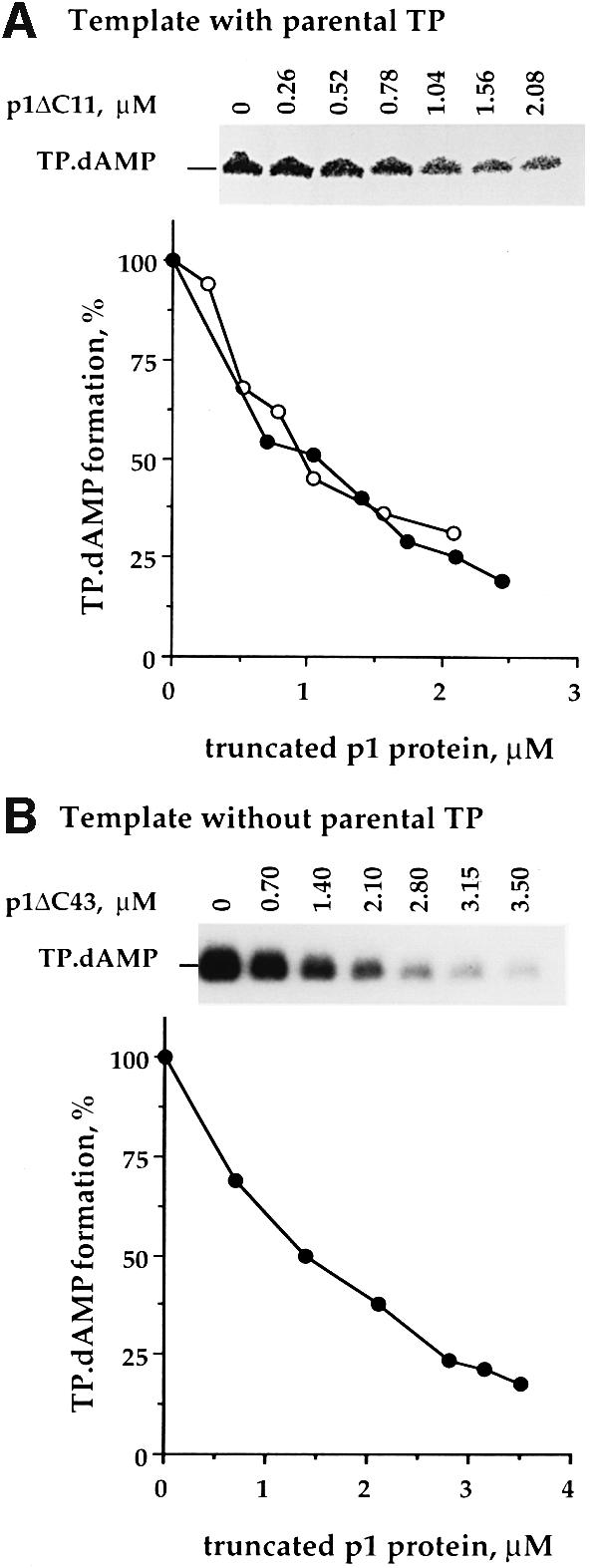
Fig. 6. Effect of C-terminal truncated p1 proteins on the in vitro formation of the TP⋅dAMP initiation complex. (A) The assay was carried out in the presence of 0.25 µM [α-32P]dATP (1 µCi), φ29 TP–DNA (0.08 nM), DNA polymerase (1.2 nM), primer TP (1.2 nM) and the indicated amounts of p1ΔC11 (open circles) or p1ΔC43 (filled circles) as described in Materials and methods. MgCl2 (10 mM) was used as a metal activator. (B) The assay was performed by incubation of 0.25 µM [α-32P]dATP (1 µCi), DNA polymerase (3 nM), primer TP (3 nM), a 259 bp DNA fragment from the φ29 left end (0.6 nM) and the indicated amounts of p1ΔC43. MnCl2 (1 mM) was used as a metal activator.
We next studied the formation of the TP⋅dAMP complex in the absence or presence of protein p1ΔC11 as a function of φ29 TP–DNA template concentration (Figure 7A). Again, this experiment was carried out using low concentrations of both primer TP and φ29 DNA polymerase (1.2 nM). In the absence of p1ΔC11, the initiation reaction increased linearly when the TP–DNA concentration was increased from 0.08 to 0.32 nM; in contrast, in the presence of an excess of p1ΔC11 (2 µM), the reaction did not increase significantly with the amount of TP–DNA. We conclude that p1ΔC11-mediated inhibition is not due to a reduction in the amount of functional template molecules (active origins).

Fig. 7. Inhibition of the in vitro initiation reaction by p1ΔC11. The TP⋅dAMP complex was formed in the absence of p1ΔC11 (open circles) and in its presence (filled circles) using (A) DNA polymerase (1.2 nM), primer TP (1.2 nM) and different amounts of φ29 TP–DNA; (B) DNA polymerase (1.2 nM), φ29 TP–DNA (0.08 nM) and different amounts of primer TP; (C) primer TP (1.2 nM), φ29 TP–DNA (0.08 nM) and different amounts of DNA polymerase.
p1ΔC11-mediated inhibition is suppressed by increasing the concentration of primer TP or φ29 DNA polymerase
Formation of the TP⋅dAMP initiation complex requires the efficient formation of a primer TP–φ29 DNA polymerase heterodimer (Blanco et al., 1987). Since protein p1ΔC11, like p1ΔC43, was able to bind primer TP in the in vitro cross-linking assay (Figure 5), it is possible that an excess of p1ΔC11 reduces the amount of primer TP molecules available to interact with φ29 DNA polymerase. This in turn would reduce the amount of primer TP–φ29 DNA polymerase heterodimers, resulting in a decrease in the amount of TP⋅dAMP complexes. If this were the case, it would be possible to suppress p1ΔC11-mediated inhibition simply by increasing the concentration of primer TP or φ29 DNA polymerase. To test this hypothesis, TP–DNA-dependent initiation reactions were performed in the presence of 0 or 1 µM p1ΔC11 as a function of primer TP concentration (Figure 7B). In both cases, the initiation reaction increased linearly when the primer TP concentration was increased from 0.5 to 6.25 nM. Interestingly, as the primer TP concentration increased, the ratio between the amount of TP⋅dAMP complexes formed in the absence of p1ΔC11 and the amount that formed in its presence (inhibitory effect) decreased (Table I). Moreover, the maximum reaction obtained in the presence of p1ΔC11 was only slightly lower (1.2-fold) than that obtained in its absence.
Table I. Effect of protein p1ΔC11 (1 µM) on the DNA-dependent initiation reaction.
| DNA polymerase (1.2 nM) |
TP (1.2 nM) |
||
|---|---|---|---|
| TP (nM) | Inhibitory effecta | DNA polymerase(nM) | Inhibitory effecta |
| 0.50 | 3.5 | 0.60 | 4.5 |
| 1.28 | 2.5 | 1.20 | 2.5 |
| 2.50 | 1.5 | 2.40 | 1.3 |
| 6.25 | 1.2 | 4.80 | 0.9 |
| 12.50 | 1.2 | 9.60 | 0.8 |
aThe inhibitory effect refers to the ratio between the amount of initiation complexes formed in the absence of p1ΔC11 and that formed in its presence. It was calculated from data presented in Figure 7.
We next carried out TP–DNA-dependent initiation reactions in the absence or presence of p1ΔC11 as a function of φ29 DNA polymerase concentration (Figure 7C). In this assay, in which the concentration of primer TP was 1.2 nM, the p1ΔC11-mediated inhibitory effect decreased as the DNA polymerase concentration increased (Table I). In the presence of p1ΔC11, a higher DNA polymerase concentration was required to reach the saturation of the initiation reaction, and the maximum reaction obtained was similar to that obtained in the absence of p1ΔC11.
We conclude that the inhibition of the initiation reaction mediated by C-terminal truncated p1 proteins can be explained by competition between these proteins and the φ29 DNA polymerase for binding to the primer TP. Thus, the initiation reaction in the presence of p1ΔC11 or p1ΔC43 can be schematized as follows:

where formation of the primer TP/φ29 DNA polymerase (pol) heterodimer (reaction a) is greatly favored under the experimental conditions used (Blanco et al., 1987); in the presence of 5′-dATP and φ29 TP–DNA, DNA polymerase catalyzes the linkage of dAMP to the primer TP (Blanco and Salas, 1985) (reaction b); and the C-terminal truncated p1 protein (Δp1) binds to the primer TP with very low affinity, leading to the formation of a non-functional complex (reaction c).
Discussion
Protein p1 is membrane associated in B.subtilis-infected cells, both during the synthesis of viral DNA and after blocking initiation of viral DNA replication (Bravo and Salas, 1997). Moreover, the C-terminal 52 amino acid residues of p1 are sufficient for assembly into protofilament sheets (Bravo and Salas, 1998). The results of this study, using chemical cross-linking techniques, show that truncated p1 proteins that retain the N-terminal 42 amino acids (p1ΔC11 and p1ΔC43) bind to primer TP in vitro. Although the formation of p1ΔC11–primer TP and p1ΔC43–primer TP complexes was not greatly favored under the experimental conditions used, the existence of this interaction is supported by in vivo and in vitro functional studies. First, an excess of protein p1 at the beginning of the infective process reduced the rate of viral DNA replication. A similar dose-dependent interference was observed in cells producing p1ΔC43, a truncated p1 protein that neither binds to membranes in vivo (Bravo and Salas, 1997) nor self-associates in vitro. Hence, the in vivo interference mediated by protein p1 is probably related to its ability to interact with primer TP. Secondly, both p1ΔC11 and p1ΔC43, when present in excess, interfered with the in vitro formation of the TP⋅dAMP initiation complex in a reaction that requires the efficient formation of a primer TP–φ29 DNA polymerase heterodimer. This in vitro interference was suppressed by increasing the concentration of primer TP or φ29 DNA polymerase. Thus, the inhibition of the initiation reaction imposed by a high concentration of the C-terminal truncated p1 protein can be explained by competition between such a protein and the φ29 DNA polymerase for binding to the primer TP.
Why does an excess of plasmid-encoded p1 inhibit φ29 DNA replication? Initiation of viral DNA replication, like any biological process, needs an ordered assembly of the different components involved in the process. This can be efficiently achieved if the intracellular concentration of each component is maintained precisely. In fact, the molar ratio of protein p1 to primer TP and to φ29 DNA polymerase remains constant throughout the course of infection (Bravo and Salas, 1997). Therefore, we explain the p1-mediated in vivo interference as a consequence of the increase in the molar ratio of protein p1 with respect to primer TP and φ29 DNA polymerase. Although we do not know in which order these three proteins interact to initiate phage DNA replication, the interference caused by p1 when it is accumulated before φ29 infection suggests that the first assembly step could be the formation of the primer TP–φ29 DNA polymerase complex. These heterodimers, which can be isolated from φ29-infected cells (Matsumoto et al., 1984), could further interact with protein p1. According to this view, it is conceivable that a high intracellular concentration of p1 (or p1ΔC43) at the beginning of the infective process sequesters the phage-encoded primer TP, leading to the formation of non-functional complexes. As a result, assembly of primer TP–φ29 DNA polymerase heterodimers would be impaired and the efficiency of the viral DNA replication process would decrease. This interpretation is supported by the in vitro results, which showed that a high concentration of protein p1ΔC43 reduces the amount of TP⋅dAMP initiation complexes, probably due to a reduction in the amount of primer TP–φ29 DNA polymerase heterodimers.
Some lines of evidence, both in prokaryotes and eukaryotes, suggest a model for chromosomal DNA replication in which DNA polymerases are immobilized by attachment to larger structures (stationary replisome model) (for review see Cook, 1999). This model necessarily implies direct interactions between the DNA replication machinery and the cellular substructure. The results presented here are of particular relevance because they support a comprehensive model for the initiation of in vivo φ29 DNA replication (Figure 8). The model postulates that the viral replisome, constituted at least by the primer TP–φ29 DNA polymerase heterodimer, assembles on a membrane-associated p1-based viral structure. Proper assembly of this multiprotein complex would demand a very precise balance in the intracellular concentration of each component. Attachment of the φ29 replisome to the membrane-associated p1 structure would be achieved by protein–protein interactions between primer TP and p1. In addition, direct contacts between primer TP and the bacterial membrane could take place because, in the absence of other viral components, primer TP has shown affinity for membranes (Bravo and Salas, 1997). Once the replisome is anchored in place, it would specifically interact with the replication origins, located at both ends of the φ29 genome. Then, initiation of viral DNA replication would occur by a protein-priming mechanism in which the viral DNA polymerase catalyzes the linkage of dAMP to the primer TP (for review see Salas et al., 1996). Since the primer TP molecule remains covalently bound to the newly synthesized DNA, the model accounts for the specific attachment of the parental φ29 DNA molecule to the membrane-associated p1 structure when it is replicated. The early viral protein, p16.7, an integral membrane protein that has non-specific DNA-binding affinity, could also contribute to the membrane localization of phage DNA replication (Meijer et al., 2000; W.J.J.Meijer, A.Serna-Rico and M.Salas, submitted). Our model also explains previous results showing that (i) parental φ29 DNA–membrane complexes are formed near the onset of viral DNA synthesis, and an apparent maximum is reached by the middle of the DNA synthesis period; (ii) formation of these complexes requires the synthesis of early viral-encoded proteins (Ivarie and Pène, 1973); (iii) membrane association of viral DNA encoding a thermosensitive DNA polymerase does not occur at the restrictive temperature unless cells are co-infected with the wild-type virus (Ivarie and Pène, 1973; Mellado et al., 1976); and (iv) large amounts of φ29 DNA polymerase are sequestered in membranes, in addition to protein p1 and primer TP, when initiation of in vivo φ29 DNA replication is blocked due to the lack of the viral protein p6 (Bravo and Salas, 1997).
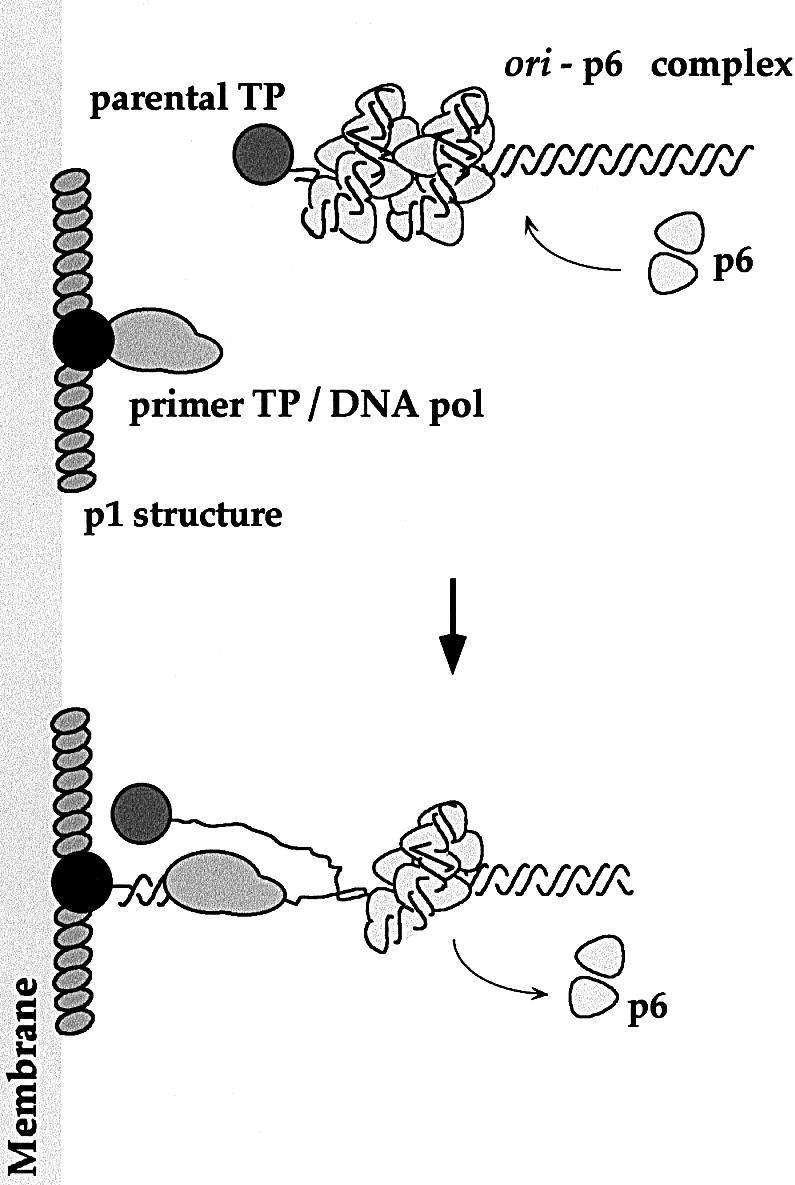
Fig. 8. Model for initiation of in vivo φ29 DNA replication. For simplicity, only one φ29 DNA end (replication origin) is represented. Viral DNA replication would start with the assembly of a protein p1-based structure on the bacterial membrane. This structure would provide an anchoring site for the viral replisome, constituted at least by the primer TP–φ29 DNA polymerase heterodimer. The membrane-associated replisome would further interact with the replication origins, located at both ends of the parental φ29 TP–DNA molecule. This stage is activated by the viral protein p6, which forms a multimeric nucleoprotein complex at the replication origins. This activation requires specific recognition of the protein p6 nucleoprotein complex by the primer TP–φ29 DNA polymerase heterodimer (Freire et al., 1996). Protein p6 is absolutely required for φ29 DNA replication in vivo (Carrascosa et al., 1976; Mellado et al., 1980). See Discussion for more details.
In conclusion, protein p1, in addition to its affinity for membranes in vivo and its ability to polymerize in vitro, has the capacity to interact with primer TP. This previously unknown function is located within the N-terminal 42 amino acid residues of the protein. These findings support a model for initiation of in vivo φ29 DNA replication in which the φ29 replisome is attached to a membrane-associated p1-based viral structure. Therefore, the viral replication components would be located at specific sites within the cell.
Materials and methods
Bacterial strains, bacteriophages, plasmids and growth conditions
Bacillus subtilis 110NA (Moreno et al., 1974) was used to grow the φ29 and Nf phages, and as a host for pUB110-based plasmids. Escherichia coli XL1-Blue (Stratagene) was used as a host for plasmids encoding MalE fusion proteins. Phage stocks were prepared essentially as reported previously (Talavera et al., 1971), except that the phage particles were concentrated as described (Bravo et al., 1990). To construct plasmids pw21 and pow3, the 114 bp SacI–SphI restriction fragment of the pUB110-based replicon, pPR53 (Bravo and Salas, 1997), was replaced by different φ29 DNA sequences: pw21 carries a HindIII–ScaI DNA fragment (coordinates 2899–706; Yoshikawa and Ito, 1982) and pow3 contains a DsaI–HindIII DNA fragment (coordinates 3773–2899), which contains most of gene 3 except for the last 15 nucleotides. Plasmid pA1IVΔ1 was constructed in the same way as pw21, but DNA sequencing revealed a spontaneous mutation in gene 1. This mutant gene (gene Δ1) has a deletion of one nucleotide that leads to a truncated p1 protein of 45 amino acids, conserving the 42 N-terminal residues (Bravo and Salas, 1997). A HpaII–ScaI DNA fragment (coordinates 1186–706), which contains gene Δ1 but not promoter PA1IV, was inserted into the expression vector pPR53 (Bravo and Salas, 1997) under control of the bacteriophage λ PR promoter (plasmid pPR53Δ1) and in the opposite orientation (plasmid pPR53oΔ1). To purify the C-terminally truncated p1 proteins p1ΔC11 and p1ΔC43, plasmids pMalE-p1ΔC11 and pMalE-p1ΔC43 were constructed. To construct pMalE-p1ΔC11, φ29 DNA sequences (coordinates 1130–873) were amplified by polymerase chain reaction (PCR) using the oligonucleotides SmaI (5′-GGAGATGTTTGTACCCGGGAAAATCTTCG-3′) and XbaIΔC11 (5′-GGAGCACCTCTAGATAACTACTTTATAACC-3′) as primers and plasmid pw21 as template. To construct plasmid pMalE-p1ΔC43, φ29 DNA sequences (coordinates 1130–809) were amplified by PCR using the oligonucleotides SmaI (see above) and XbaI (5′-CCGTCATCATCTAGAAGCAAC-3′) as primers and plasmid pPR53Δ1 as template. In both constructions, PCR fragments were digested with SmaI and XbaI, and ligated into the XmnI and XbaI sites of the E.coli plasmid pMAL-c2 (New England Biolabs). The nucleotide sequence of the inserts was confirmed by DNA sequencing using the dideoxynucleotide chain-termination method (Sanger et al., 1977). Bacteria were grown in LB broth (Sambrook et al., 1989) supplemented with 5 mM MgSO4 in phage infection experiments. Plasmid-containing B.subtilis cells were grown in LB medium supplemented with kanamycin (5 µg/ml) or phleomycin (0.8 µg/ml). Plasmid-containing E.coli cells were grown in LB medium supplemented with ampicillin (100 µg/ml).
Phage growth under one-step conditions
Bacillus subtilis 110NA cells carrying plasmid were exponentially grown at 30°C to ∼108 viable cells/ml, and then infected with the indicated phage at a multiplicity of infection of 5. After 10 min of incubation with gentle shaking, unadsorbed phages were eliminated by centrifugation of the infected culture. Cells were resuspended in the same volume of pre-warmed medium and incubated with vigorous shaking at the same temperature. These conditions ensure that the whole cell population is simultaneously infected and, consequently, that the infection process takes place synchronously and kinetic analyses can be carried out. At different times, aliquots were taken and the production of infective particles [measured as plaque-forming units (p.f.u.)/ml] was determined by plating on 110NA cells without plasmid. The time of phage addition was denoted time zero.
Synthesis of viral DNA under one-step conditions
The method has been described previously (Bravo et al., 1994). Basically, total intracellular DNA was isolated at different times after infection and analyzed by agarose gel electrophoresis. Gels were stained with ethidium bromide and photographed under UV light illumination. Bands corresponding to unit-length φ29 DNA were quantified by densitometry of the negatives in a Molecular Dynamics 300A densitometer. The relative amount of viral DNA refers to the intensity of the intracellular φ29 DNA band with respect to that of proteinase K-treated φ29 DNA used as internal marker.
Western blots
Plasmid-containing cells were exponentially grown at 30°C to ∼108 viable cells/ml. Before cell disruption by sonication, bacteria were concentrated 10- to 20-fold in loading buffer (60 mM Tris–HCl pH 6.8, 2% SDS, 5% β-mercaptoethanol, 30% glycerol). Aliquots of the cell extracts were loaded on to an SDS–polyacrylamide gradient gel (10–20%). After electrophoresis, proteins were transferred electrophoretically using a Trans Blot apparatus (Bio-Rad) at 100 mA and 4°C for 35 min as described (Bravo and Salas, 1997). Immobilon-P membranes (Millipore) were probed with the indicated serum. Antigen–antibody complexes were detected using anti-rabbit horseradish peroxidase-conjugated antibodies and ECL western blotting detection reagents (Amersham). In in vitro cross-linking experiments, proteins were transferred electrophoretically using a Mini Trans Blot (Bio-Rad) at 100 mA and 4°C for 80 min.
Protein purification
φ29 DNA polymerase was purified essentially as described by Lázaro et al. (1995). φ29 TP was purified as reported (Zaballos et al., 1989). To purify p1ΔC11 and p1ΔC43, we used a protein fusion system based on MalE (Kellerman and Ferenci, 1982). For this, plasmids pMalE-p1ΔC11 and pMalE-p1ΔC43 were constructed (see above). MalE fusion proteins were purified as described (Bravo and Salas, 1998) and concentrated to ≥0.3 mg/ml using a Microcon microconcentrator 10 (Amicon). To separate the MalE and p1 domains, 1 µg of protease factor Xa (New England Biolabs) was added to 25–100 µg of fusion protein. The reaction was incubated at 4°C overnight. After total digestion, the reaction mixture was loaded on to a 5 ml glycerol gradient (15–30% in dialysis buffer: 20 mM Tris–HCl pH 7.5, 200 mM NaCl, 10 mM β-mercaptoethanol) prepared in a Beckman polyallomer centrifuge tube (13 × 51 mm). Centrifugation was performed at 62 000 r.p.m. and 4°C in a Beckman SW65 rotor for 20 h. After centrifugation, 170 µl fractions were collected by puncturing the bottom of the tubes. Aliquots from each fraction were analyzed by SDS–Tricine–PAGE (Schagger and Jagow, 1987). Fractions containing the truncated p1 protein were pooled and dialyzed against dialysis buffer (see above). Protein preparations were concentrated using a Microcon microconcentrator.
In vitro formation of the TP⋅dAMP complex (initiation reaction)
The reaction mixtures (25 µl) contained 50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1 mM dithiothreitol, 20 mM ammonium sulfate, 4% glycerol, 0.1 mg/ml bovine serum albumin, 0.25 µM [α-32P]dATP (1 µCi), 25 ng (2 fmol) of φ29 TP–DNA as template and the indicated amounts of primer TP, viral DNA polymerase and truncated p1 protein. When indicated, 2.5 ng (15 fmol) of a 259 bp DNA fragment from the φ29 left end (oriL) were used as template. In this case, 1 mM MnCl2 was used as metal activator. After incubation for 3 min at 30°C, the reactions were stopped by adding EDTA to 10 mM and SDS to 0.1%. Samples were filtered through Sephadex G-50 spin columns in the presence of 0.1% SDS. The excluded volume was analyzed by SDS–PAGE. The relative amounts of incorporated [α-32P]dAMP were calculated by PhosphorImager densitometric scans of an exposed Fuji BAS-IIIs imaging plate.
Cross-linking of proteins
Protein preparations were dialyzed against 20 mM sodium phosphate buffer, 0.15 M NaCl, pH 7.5. The cross-linker DTSSP (Pierce) was dissolved in the same buffer just before use. The reaction mixture (25 µl) contained 5 mM DTSSP, 880 ng of φ29 primer TP and 640 ng of p1ΔC11 or p1ΔC43. After incubation at room temperature for 30 min, Tris–HCl pH 7.5 was added at a final concentration of 30 mM to quench the reaction. The reaction mixture was incubated for an additional 15 min. Samples were analyzed by SDS–Tricine–PAGE in the absence of β-mercaptoethanol.
Acknowledgments
Acknowledgements
We are grateful to J.M.Lázaro for purification of the φ29 DNA polymerase and to L.Villar for purification of the φ29 terminal protein. We thank A.Abril for helpful comments. This work was supported by the National Institutes of Health (grant 5R01 GM27242-20), by the Dirección General de Investigación Científica y Técnica (grant PB98-0645) and by the European Union (BIO4-CT98-0250 and ERBFMX-CT97-0125). The institutional help of Fundación Ramón Areces is acknowledged.
References
- Ahola T., Lampio,A., Auvinen,P. and Kääriäinen,L. (1999) Semliki Forest virus mRNA capping enzyme requires association with anionic membrane phospholipids for activity. EMBO J., 18, 3164–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthelemy I., Salas,M. and Mellado,R.P. (1986) In vivo transcription of bacteriophage φ29 DNA: transcription initiation sites. J. Virol., 60, 874–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco L. and Salas,M. (1985) Replication of phage φ29 DNA with purified terminal protein and DNA polymerase: synthesis of full-length φ29 DNA. Proc. Natl Acad. Sci. USA, 82, 6404–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco L., Prieto,I., Gutiérrez,J., Bernad,A., Lázaro,J.M., Hermoso,J.M. and Salas,M. (1987) Effect of NH4+ ions on φ29 DNA–protein p3 replication: formation of a complex between the terminal protein and the DNA polymerase. J. Virol., 61, 3983–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo A. and Salas,M. (1997) Initiation of bacteriophage φ29 DNA replication in vivo: assembly of a membrane-associated multiprotein complex. J. Mol. Biol., 269, 102–112. [DOI] [PubMed] [Google Scholar]
- Bravo A. and Salas,M. (1998) Polymerization of bacteriophage φ29 replication protein p1 into protofilament sheets. EMBO J., 17, 6096–6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo A., Alonso,J.C. and Trautner,T.A. (1990) Functional analysis of the Bacillus subtilis bacteriophage SPP1 pac site. Nucleic Acids Res., 18, 2881–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo A., Hermoso,J.M. and Salas,M. (1994) A genetic approach to the identification of functional amino acids in protein p6 of Bacillus subtilis phage φ29. Mol. Gen. Genet., 245, 529–536. [DOI] [PubMed] [Google Scholar]
- Brosius J., Dull,T.J., Sleeter,D.D. and Noller,H.F. (1981) Gene organization and primary structure of a ribosomal RNA operon from Escherichia coli. J. Mol. Biol., 148, 107–127. [DOI] [PubMed] [Google Scholar]
- Buck K.W. (1996) Comparison of the replication of positive-stranded RNA viruses of plants and animals. Adv. Virus Res., 47, 159–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrascosa J.L., Camacho,A., Moreno,F., Jiménez,F., Mellado,R.P., Viñuela,E. and Salas,M. (1976) Bacillus subtilis phage φ29: characterization of gene products and functions. Eur. J. Biochem., 66, 229–241. [DOI] [PubMed] [Google Scholar]
- Cook P.R. (1999) The organization of replication and transcription. Science, 284, 1790–1795. [DOI] [PubMed] [Google Scholar]
- de Jong R.N. and van der Vliet,P.C. (1999) Mechanism of DNA replication in eukaryotic cells: cellular host factors stimulating adenovirus DNA replication. Gene, 236, 1–12. [DOI] [PubMed] [Google Scholar]
- Freire R., Serrano,M., Salas,M. and Hermoso,J.M. (1996) Activation of replication origins in φ29-related phages requires the recognition of initiation proteins to specific nucleoprotein complexes. J. Biol. Chem., 271, 31000–31007. [DOI] [PubMed] [Google Scholar]
- Ivarie R.D. and Pène,J.J. (1973) DNA replication in bacteriophage φ29: the requirement of a viral-specific product for association of φ29 DNA with the cell membrane of Bacillus amyloliquefaciens. Virology, 52, 351–362. [DOI] [PubMed] [Google Scholar]
- Kellermann O.K. and Ferenci,T. (1982) Maltose-binding protein from Escherichia coli. Methods Enzymol., 90, 459–463. [DOI] [PubMed] [Google Scholar]
- Lázaro J.M., Blanco,L. and Salas,M. (1995) Purification of bacteriophage φ29 DNA polymerase. Methods Enzymol., 262, 42–49. [DOI] [PubMed] [Google Scholar]
- Lemon K.P. and Grossman,A.D. (1998) Localization of bacterial DNA polymerase: evidence for a factory model of replication. Science, 282, 1516–1519. [DOI] [PubMed] [Google Scholar]
- Lewis P.J., Thaker,S.D. and Errington,J. (2000) Compartmentalization of transcription and translation in Bacillus subtilis. EMBO J., 19, 710–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K., Saito,T., Kim,C.I., Ando,T. and Hirokawa,H. (1984) Bacteriophage φ29 DNA replication in vitro: participation of the terminal protein and the gene 2 product in elongation. Mol. Gen. Genet., 196, 381–386. [DOI] [PubMed] [Google Scholar]
- McKenzie T., Hoshino,T., Tanaka,T. and Sueoka,N. (1986) The nucleotide sequence of pUB110: some salient features in relation to replication and its regulation. Plasmid, 15, 93–103. [DOI] [PubMed] [Google Scholar]
- Meijer W.J.J., Lewis,P.J., Errington,J. and Salas,M. (2000) Dynamic relocalization of phage φ29 DNA during its replication cycle in Bacillus subtilis and the role of the viral protein p16.7. EMBO J., 19, 4182–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellado R.P., Moreno,F., Viñuela,E., Salas,M., Reilly,B.E. and Anderson,D.L. (1976) Genetic analysis of bacteriophage φ29 of Bacillus subtilis phage φ29: integration and mapping of reference mutants of two collections. J. Virol., 19, 495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellado R.P., Peñalva,M.A., Inciarte,M.R. and Salas,M. (1980) The protein covalently linked to the 5′ termini of the DNA of Bacillus subtilis phage φ29 is involved in the initiation of DNA replication. Virology, 104, 84–96. [DOI] [PubMed] [Google Scholar]
- Moreno F., Camacho,A., Viñuela,E. and Salas,M. (1974) Suppressor-sensitive mutants and genetic map of Bacillus subtilis bacteriophage φ29. Virology, 62, 1–16. [DOI] [PubMed] [Google Scholar]
- Morrissey J.H. (1981) Silver stain for proteins in polyacrylamide gels: a modified procedure with enhanced uniform sensitivity. Anal. Biochem., 117, 307–310. [DOI] [PubMed] [Google Scholar]
- Newport J. and Yan,H. (1996) Organization of DNA into foci during replication. Curr. Opin. Cell Biol., 8, 365–368. [DOI] [PubMed] [Google Scholar]
- Pombo A., Ferreira,J., Bridge,E. and Carmo-Fonseca,M. (1994) Adenovirus replication and transcription sites are spatially separated in the nucleus of infected cells. EMBO J., 13, 5075–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas M. (1991) Protein-priming of DNA replication. Annu. Rev. Biochem., 60, 39–71. [DOI] [PubMed] [Google Scholar]
- Salas M. and Rojo,F. (1993) Replication and transcription of bacteriophage φ29 DNA. In Sonenshein,A.L., Hoch,J.A. and Losick,R. (eds), Bacillus subtilis and other Gram-positive Bacteria: Biochemistry, Physiology, and Molecular Genetics., American Society for Microbiology, Washington, DC, pp. 843–857. [Google Scholar]
- Salas M., Miller,J.T., Leis,J. and DePamphilis,M.L. (1996) Mechanisms for priming DNA synthesis. In DePamphilis,M.L. (ed.), DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 131–176. [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Sanger F., Nicklen,S. and Coulson,A.R. (1977) DNA sequencing with chain-terminating inhibitors. Proc. Natl Acad. Sci. USA, 74, 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger H. and Jagow,G. (1987) Tricine–sodium dodecyl sulfate–polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem., 166, 368–379. [DOI] [PubMed] [Google Scholar]
- Soengas M.S., Esteban,J.A., Salas,M. and Gutiérrez,C. (1994) Complex formation between phage φ29 single-stranded DNA binding protein and DNA. J. Mol. Biol., 239, 213–226. [DOI] [PubMed] [Google Scholar]
- Sogo J.M., Lozano,M. and Salas,M. (1984) In vitro transcription of the Bacillus subtilis phage φ29 DNA by Bacillus subtilis and Escherichia coli RNA polymerases. Nucleic Acids Res., 12, 1943–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera A., Jiménez,F., Salas,M. and Viñuela,E. (1971) Temperature-sensitive mutants of bacteriophage φ29. Virology, 46, 586–595. [DOI] [PubMed] [Google Scholar]
- Yoshikawa H. and Ito,J. (1982) Nucleotide sequence of the major early region of bacteriophage φ29. Gene, 17, 323–335. [DOI] [PubMed] [Google Scholar]
- Zaballos A., Lázaro,J.M., Méndez,E., Mellado,R.P. and Salas,M. (1989) Effects of internal deletions on the priming activity of the phage φ29 terminal protein. Gene, 83, 187–195. [DOI] [PubMed] [Google Scholar]