

Abstract
Background
Chronic inflammation is an important mechanism for the development and progression of prostate cancer. To better understand the potential relationship between genes in the inflammation pathway and prostate cancer (PC) risk, we evaluated variants in 16 candidate genes.
Methods
A total of 143 tagging and amino acid altering single nucleotide polymorphisms (SNPs) were genotyped in Caucasian and African American men participating in one of two population-based, case-control studies (n = 1,458 cases and 1,351 controls). The relative risk of prostate cancer was estimated using logistic and polytomous regression models.
Results
Ten SNPs in seven genes (CXCL12, IL4, IL6, IL6ST, PTGS2, STAT3, and TNF) were nominally associated (p<0.05) with risk of PC in Caucasians. The most significant effect on risk was seen with rs11574783 in the IL6ST gene (odds ratio, OR=0.08, 95% CI 0.01–0.63). Cumulatively, four SNPs in genes IL4, IL6ST, PTGS2, and STAT3 conferred a three-fold elevation in PC risk among men carrying the maximum number of high-risk alleles (OR=2.97, 95% CI 1.41–6.25, ptrend = 0.0003). Risk estimates for seven SNPs varied significantly according to disease aggressiveness (phomogeneity<0.05), with SNPs in AKT1, PIK3R1 and STAT3 independently associated with more aggressive PC; OR=5.1 (95% CI 2.29–11.40, ptrend = 3.8×10−5) for carriers of all high-risk genotypes.
Conclusions
These results suggest that variants in genes within the inflammation pathway may play a role in the development of PC, however further studies are needed to replicate our findings.
Impact
These results underline the potential importance of the inflammation pathway in PC development and progression.
Keywords: prostate cancer, genetic association, inflammation, genetic variation
Introduction
Prostate cancer (PC) is the most frequently diagnosed cancer in men in the United States accounting for over 217,730 new cases and 32,050 deaths in 2010 (1). Twin studies, family-based linkage and segregation analyses all strongly suggest that there is a hereditary component for PC susceptibility (2). In addition, results from genome-wide association studies (GWAS) have suggested that multiple genes influence predisposition to PC and that pathway-based association studies are needed to fully understand the genetics of disease susceptibility (3).
In recent years, the role for inflammation in tumorigenesis has become more evident, although a direct causal relationship remains to be elucidated. Virchow first proposed a connection between inflammation and cancer (4) and since then a growing body of epidemiologic and molecular evidence has emerged to support the hypothesis that chronic inflammation promotes development and progression of ~20% of all human malignancies, including those of the colon, bladder, lung and prostate (5). The pro-tumorigenic effects of chronic inflammation include DNA damage, enhancement of cell proliferation, inhibition of apoptosis, and stimulation of angiogenesis. Foci of chronic inflammation are common in prostate tissue, with a prevalence of 79% reported among participants of one study (6). Epidemiological studies have shown an increase in risk of prostate cancer associated with a history of sexually transmitted infections (STIs) and prostatitis (7, 8), both of which may be associated with prostatic inflammation. Additional evidence implicating inflammation in the development of PC comes from studies of non-steroidal anti-inflammatory drugs (NSAIDs), which have been associated with a reduced risk of PC (9).
Genetic studies of hereditary and sporadic PC also suggest a possible role for inflammatory factors. Two putative PC susceptibility genes in the inflammatory pathway, ribonuclease L (RNASEL) and macrophage scavenger receptor I (MSR1), were initially identified from linkage studies (10). In addition, studies have identified associations with SNPs in genes involved in inflammation including the Toll-like receptor family of genes as well as the cyclooxygenase gene PTGS2 (11, 12). Finally, associations between PC risk and SNPs in several cytokines including IL1B, IL6, IL8, IL10, and TNF, each of which is involved in the initiation and maintenance of inflammation, have been reported (13, 14).
We have investigated the association between PC risk and genetic variants in selected inflammation pathway-related genes in a population-based, case-control study of Caucasians and African Americans. The genes considered have been implicated in underlying biology of both normal and neoplastic prostate tissues (15, 16) and we focused on selected genes in the pro-inflammatory cytokine IL6 signaling pathway (IL6, IL6R, IL6ST, STAT3, PI3K, and AKT) and downstream target genes such as NFκB (NFKB1), PTGS2, and VEGF (Figure 1). In addition to genes shown in Figure 1, we selected 2 genes (IL4, and PTGS1) based on previously published reports (17, 18). The association between SNPs in each of these genes and clinical characteristics of PC was also explored. Because the association of PC with SNPs in these genes may be modified by exposure to pro-inflammatory factors, we also considered whether associations varied by smoking history, body mass index (BMI), and history of STIs or prostatitis.
Figure 1. Chronic inflammation promotes the development of prostate cancer.
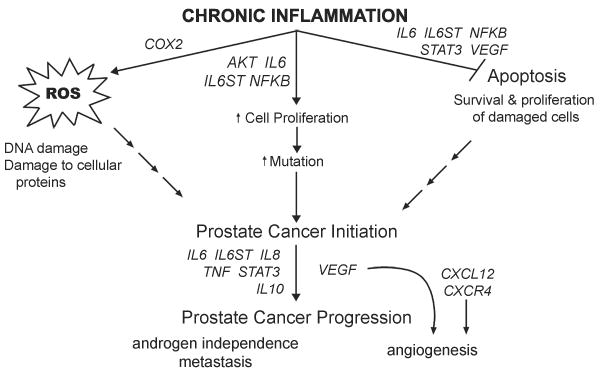
Selected genes involved in inflammation (1) increase formation of DNA and protein-damaging reactive oxygen species (ROS), and (2) inhibit apoptosis while (3) simultaneously promoting increased cellular proliferation in a reactive molecular environment. This increases survival and growth of damaged or mutation-bearing cells that may initiate carcinogenesis. Other gene products promote the nascent cancer by stimulating angiogenesis, progression to androgen independence and metastasis.
Methods and Materials
Study population
The study population consists of participants from one of two population-based, case-control studies conducted in King County, Washington (Study I and Study II), both of which included Caucasians and African Americans and have been described elsewhere (19, 20). Briefly, incident cases with histologically confirmed PC were ascertained from the Seattle-Puget Sound Surveillance, Epidemiology, and End Results cancer registry. Cases from Study I were aged 40 to 64 years at diagnosis, which occurred between January 1, 1993 and December 31, 1996. Cases from Study II were aged 35 to 74 years at diagnosis, which was between January 1, 2002 and December 31, 2005. Overall, 1,754 (78.2%) of 2,244 eligible PC patients were interviewed. Blood samples yielding sufficient DNA for genotyping were collected from 1,458 (83.1%) cases, each of whom completed the study interview.
A comparison group of Caucasian and African American controls residing in King County, Washington, without a self-reported physician’s diagnosis of PC was identified by random digit dialing. Controls were frequency matched to cases by five-year age groups and recruited evenly throughout each case ascertainment period. Of the 2,448 identified men who met the aforementioned eligibility criteria, 1,645 (67.2%) completed a study interview and DNA was prepared using standard protocols from blood samples drawn from 1,358 (82.6%) interviewed controls.
Data collection
Subjects in both studies completed in-person interviews conducted by trained male interviewers using standardized questionnaires that queried family structure, cancer history, medical history, social and demographic factors and environmental exposures. Exposure data were collected up to reference date (i.e., date of diagnosis for cases and a randomly assigned date for controls that approximated the distribution of cases’ diagnosis dates). Clinical information on cases (Gleason score, tumor stage, and serum prostate-specific antigen [PSA] level at diagnosis) was obtained from the cancer registry. Plasma PSA level in controls was measured using blood collected at interview. Studies I and II were approved by the Institutional Review Board (IRB) of the Fred Hutchinson Cancer Research Center and genotyping protocols were approved by the IRB of the National Human Genome Research Institute (NHGRI). Written, informed consent was obtained from all study participants.
SNP selection and genotyping
The majority of genotyped SNPs were selected using The Genome Variation Server (http://gvs-p.gs.washington.edu/GVS). Utilizing the HapMap CEU population (version 22; http://www.hapmap.org), tagSNP selection parameters were set to capture SNPs with a minor allele frequency ≥ 5% and an r2 of ≥ 0.8 in a region encompassing the gene of interest (5000 bp upstream and downstream). One hundred forty-six tagSNPs and six candidate SNPs were selected. Three PTGS1 non-synonymous SNPs (R8W, P17L and L237M) were added because they represented potentially functional amino acid substitutions (18). The Applied Biosystems (ABI) SNPlex Genotyping System was used to genotype SNPs and proprietary GeneMapper software was used for allele assignment (21). Discrimination of each specific SNP allele was carried out with the ABI 3730xl DNA Analyzer and was based on the presence of a unique sequence tag assigned to the allele-specific oligonucleotide. Quality control included genotyping of 140 blind duplicate samples distributed across all genotyping batches. Of the 152 SNPs attempted, three were monomorphic (rs5272, rs324509, and rs12079514), and six failed due to a low call rate (< 90%). The remaining 143 SNPs demonstrated >95% agreement between blind duplicates, and completeness of genotype calls ranged from 95.6% to 99.9%, with an average completeness of 99.2%. Nine SNPs were genotyped in samples from only one of the two studies: rs2206593, rs173702, and rs2431166 in Study I; rs2745557, rs20417, rs1119231, rs1029153, rs706716, and rs1800750 in Study II. Each batch of DNA aliquots genotyped incorporated similar numbers of case and control samples. Laboratory personnel were blinded to the case-control status of samples.
Statistical analysis
Departure from Hardy-Weinberg equilibrium (HWE) was assessed in controls for each SNP separately by race using a χ2 test, with raw genotyping data and allele separation manually reviewed for any SNP showing a significant departure from HWE. Pairwise linkage disequilibrium (LD) between SNPs was estimated in controls separately by race using the r2 statistic in the Haploview software v4.1. Unconditional logistic regression models were used to generate odds ratios (ORs), as an estimate of PC risk associated with SNP genotypes, and 95% confidence intervals (CI). Polytomous regression models were used to generate ORs and 95% CIs for the association between SNP genotypes and cases stratified by disease aggressiveness (less vs. more) compared to controls. Disease aggressiveness was defined as follows: less aggressive cases were those with a Gleason score ≤ 7 (3+4), local stage, and PSA level ≤20 ng/mL at diagnosis, and more aggressive disease was defined as Gleason score ≥ 7(4+3), or regional or distant stage, or a diagnostic PSA level >20 ng/mL. All models were adjusted for age at reference date. Potential confounding factors, including first-degree family history of PC and PC screening history (digital rectal examination and/or PSA testing within the five years prior to reference date), were examined to see if they altered risk estimates by >10%. Log-additive (trend), recessive and dominant genetic models were considered separately for each SNP genotype and the best-fitting model was selected for further analysis. Model fit was evaluated by comparing likelihood-ratio based statistics. The effect of multiple comparisons was accounted for using a permutation analysis, with pairs of case-control labels and ages permuted (1000 times) jointly to approximate the distribution of the age-adjusted p-values under the null hypothesis (22).
For each permutation, log-additive, recessive and dominant models were fit for all SNPs and the minimum of the p-values were kept for each SNP. The p-values were ordered to approximate the null distribution of the order statistics for the p-values. The original p-values were also ordered and permuted p-values were obtained by comparing ordered p-values to the null distribution for an appropriate order statistic. For SNPs that were found to be nominally significant, potential gene-environment and gene-gene interactions were evaluated by comparing the reduced model (main effects only) to the full model that also included an interaction term (gene by environment or gene by gene, assuming a dominant genetic model). All analyses were performed using SAS v9.1.3, or STATA v10.1.
The association between a SNP and PC risk was considered to be significant if the nominal p-value and the permuted p-value were both <0.05. In the results, we report nominal p-values. Nominally significant SNPs (p <0.05) were included together in a stepwise selection model using Akaike’s Information Criterion to identify the most parsimonious model (23). SNPs that were independently associated with PC were included in a final model where high-risk genotypes were counted according to the best-fitting genetic model.
Results
A total of 1,458 PC cases and 1,351 age-matched controls were included in the analyses and selected characteristics of these men are presented in Table 1. Cases and controls were predominantly Caucasian (89.8%) and approximately 77% of study participants were under the age of 65 years, which reflects the oversampling of younger cases in Studies I and II. Compared to controls, a higher proportion of cases reported first-degree relatives with PC, more frequent PSA screening within the 5-year period before reference date, and a history of prostatitis. There were no differences between cases and controls with respect to education, BMI, smoking status, or a history of STIs. The majority of genotyped cases had PSA values of 4.0 to 9.9 ng/mL, localized stage disease, and Gleason scores of 5 or 6 at diagnosis.
Table 1.
Selected characteristics of population-based prostate cancer (PC) cases and controls in King County, WA
| Characteristic | Cases | Controls | OR1 | 95% CI | |||
|---|---|---|---|---|---|---|---|
| n=1,458 | % | n=1,351 | % | ||||
| Age at reference date | |||||||
| 35–49 | 118 | 8.1 | 126 | 9.3 | |||
| 50–54 | 215 | 14.7 | 209 | 15.5 | |||
| 55–59 | 357 | 24.5 | 358 | 26.5 | |||
| 60–64 | 433 | 29.7 | 348 | 25.8 | |||
| 65–69 | 177 | 12.1 | 164 | 12.1 | |||
| 70–74 | 158 | 10.8 | 146 | 10.8 | |||
| Race | |||||||
| Caucasian | 1,309 | 89.8 | 1,266 | 93.7 | 1.00 | Reference | |
| African American | 149 | 10.2 | 85 | 6.3 | 1.74 | 1.32 | 2.30 |
| Family history of PC2 | |||||||
| No | 1,145 | 78.5 | 1,199 | 88.8 | 1.00 | Reference | |
| Yes | 313 | 21.5 | 152 | 11.3 | 2.19 | 1.77 | 2.71 |
| Body mass index (kg/m2) | |||||||
| Normal (<25) | 467 | 32.0 | 411 | 30.4 | 1.00 | Reference | |
| Overweight (25–29.9) | 707 | 48.5 | 650 | 48.1 | 0.95 | 0.80 | 1.13 |
| Obese (≥30) | 284 | 19.5 | 290 | 21.5 | 0.83 | 0.67 | 1.02 |
| Smoking status | |||||||
| Never | 586 | 40.2 | 572 | 42.3 | 1.00 | Reference | |
| Former | 676 | 46.4 | 591 | 43.8 | 1.11 | 0.95 | 1.30 |
| Current | 196 | 13.4 | 188 | 13.9 | 0.97 | 0.77 | 1.23 |
| Sexually transmissible infection3 | |||||||
| Never | 1,236 | 84.8 | 1,149 | 85 | 1.00 | Reference | |
| Ever | 221 | 15.2 | 202 | 15.0 | 1.00 | 0.81 | 1.24 |
| Missing | 1 | ||||||
| Prostatitis | |||||||
| Never | 1,268 | 87.0 | 1,241 | 91.9 | 1.00 | Reference | |
| Ever | 181 | 12.4 | 107 | 7.9 | 1.43 | 1.11 | 1.86 |
| Missing | 9 | 0.6 | 3 | 0.2 | |||
| Prostate cancer screening history4 | |||||||
| None | 157 | 10.8 | 182 | 13.5 | 1.00 | Reference | |
| Digital rectal examination only | 258 | 17.7 | 519 | 38.4 | 0.57 | 0.44 | 0.74 |
| PSA test | 1,043 | 71.5 | 650 | 48.1 | 1.94 | 1.52 | 2.47 |
| PSA value (ng/mL)5 | |||||||
| 0–3.9 | 189 | 12.9 | 1,253 | 92.8 | |||
| 4.0–9.9 | 814 | 55.8 | 80 | 5.9 | |||
| 10.0–19.9 | 208 | 14.2 | 16 | 1.2 | |||
| ≥ 20.0 | 138 | 9.47 | 2 | 0.1 | |||
| Missing | 109 | 7.47 | 0 | ||||
| Gleason score | |||||||
| 2–4 | 72 | 5.0 | |||||
| 5–6 | 741 | 51.0 | |||||
| 7 (3+4) | 408 | 28.1 | |||||
| 7 (4+3) | 91 | 6.3 | |||||
| 8–10 | 140 | 9.6 | |||||
| Missing | 6 | ||||||
| Stage of disease | |||||||
| Local | 1,141 | 78.3 | |||||
| Regional | 280 | 19.2 | |||||
| Distant | 37 | 2.5 | |||||
OR=odds ratio, adjusted for age and race, except for ‘race’, which was adjusted for age only.
First-degree family history of prostate cancer (PC).
STIs (gonorrhea, syphilis, infection or inflammation of urethra, genital herpes, genital warts, or Chlamydia)
Prostate cancer screening history in the five years prior to reference date.
Prostate-specific antigen (PSA) level at diagnosis (cases) or interview (controls).
All of the SNPs were in HWE in Caucasian controls (p > 0.05) with the exception of rs2839689 in CXCL12 and rs10940495 in IL6ST (P < 0.01), which were removed from subsequent analyses. Three SNPs (rs3918304, rs3093672, and rs5270) had low minor allele frequencies (MAF < 0.01) and were removed from further analysis in Caucasians. Two SNPs, rs18001157 (p = 6.4×10−28) and rs2839689 (p = 1.3×10−5) in CXCL12, did not fit HWE in African American controls. Moreover, six SNPs (rs5789, rs2206593, rs2069860, rs5272, rs13447446, and rs12079514) had low MAF (< 0.01) and were thus excluded from further analysis in African Americans. After considering the above, coverage of the underlying genetic variation in ten genes in Caucasians (AKT1, CXCL12, CXCR4, IL4, IL10, IL6ST, PTGS2, STAT3, TNF, and VEGF) remained greater than 85%. Coverage of PIK3R1 was 77% and only selected candidate SNPs based on previously published literature were genotyped in PTGS1, IL8, and NFκB. Because of differences in PC incidence and SNP allele frequencies between Caucasians and African Americans, which constitute a small group in our study, subsequent analyses focused on Caucasian men. However, results for African Americans are presented separately in Supplementary Table 1.
The associations between nominally significant SNPs in genes involved in the inflammation pathway and PC risk in Caucasians are presented in Table 2. Of the 143 SNPs, ten SNPs from seven genes showed suggestive associations with overall risk of disease. Homozygous carriers of the A allele of CXCL12 SNP rs2297630 had a 29% (95% CI 0.53–0.97) reduction in risk of PC relative to the other genotypes combined. The strongest association was observed for SNP rs11574783 in IL6ST; compared to men with the GA or AA genotypes, those with the GG genotype had a significant reduction in risk (OR=0.08; 95% CI 0.01–0.63). Three SNPs in PTGS2 were associated with PC (rs2206593: OR=1.69; 95% CI 1.14–2.50, rs2745557: OR=0.78; 95% CI 0.62–0.97, rs6685280: OR=1.16; 95% CI 1.01 – 1.33). Two SNPs in the STAT3 gene, rs744166 and rs12949918, were also associated with ~20% reductions in the relative risk of PC. However these two SNPs are in nearly perfect LD (r2=0.98) and thus, only one unique association was identified. The three remaining significant SNPs were in the IL4, IL6, and TNF genes. After adjusting for multiple comparisons, however, none of these associations remained significant in the Caucasian population. In addition, none of the genetic variants examined in AKT1, CXCR4, IL6R, IL8, IL10, NFκB, PIK3R1, PTGS1, or VEGF were associated with overall risk of PC.
Table 2.
Prostate cancer risk associated with selected SNPs in inflammation pathway genes among Caucasian men
| Gene | SNP1 | Chr. | Location2 | Allele3 | Genetic Model4 | Cases MAF55 | Controls MAF5 | OR6 | 95% CI | p-value7 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CXCL12 | rs2297630 | 10 | 44191554 | G/A | rec | 0.263 | 0.275 | 0.71 | 0.53 | 0.97 | 0.03 |
| IL4 | rs2243228 | 5 | 132032262 | A/C | rec | 0.079 | 0.092 | 0.27 | 0.09 | 0.84 | 0.03 |
| IL6 | rs1800797 | 7 | 22732746 | G/A | rec | 0.401 | 0.419 | 0.79 | 0.64 | 0.97 | 0.03 |
| IL6ST | rs11574783 | 5 | 55271763 | A/G | rec | 0.07 | 0.075 | 0.08 | 0.01 | 0.63 | 0.02 |
| PTGS2 | rs2206593 A | 1 | 184909052 | G/A | trend | 0.07 | 0.045 | 1.69 | 1.14 | 2.5 | 0.01 |
| rs2745557 B | 1 | 184915844 | G/A | dom | 0.181 | 0.207 | 0.78 | 0.62 | 0.97 | 0.03 | |
| rs6685280 | 1 | 184952441 | A/C | trend | 0.227 | 0.203 | 1.16 | 1.01 | 1.33 | 0.04 | |
| STAT3 | rs129499188 | 17 | 37779799 | T/C | dom | 0.402 | 0.423 | 0.81 | 0.68 | 0.96 | 0.01 |
| TNF | rs3093559 | 6 | 31655771 | G/A | dom | 0.027 | 0.019 | 1.22 | 1.01 | 1.48 | 0.04 |
Some SNPs were genotyped in only one study: ‘A’ for those genotyped in Study I only or ‘B’ in Study II only.
Chromosome coordinates based on Genome build 36.3.
Major/minor alleles based on frequencies in controls.
Genetic model selected based on the best fit in the regression model (dominant, recessive, or log-additive).
MAF=Minor allele frequency; Cases n=1,309 Controls n=1,265.
OR=odds ratio, adjusted for age.
Nominal p-values based on likelihood ratio-based test.
rs744166, in LD with rs12949918, (r2=0.98) was also genotyped and showed a similar association (OR=0.82, 95% CI 0.7–0.98; p=0.03)
We next evaluated the association of inflammation-related SNPs using a composite variable to compare disease aggressiveness (Table 3). Among cases, 34% were classified as having comparatively more aggressive PC. Eight SNPs were associated with risk of more aggressive disease and eight SNPs showed associations with less aggressive disease, with six SNPs reflecting associations previously observed in the overall PC risk analyses. Seven SNPs in five genes (CXCL12, IL6, PIK3R1, STAT3 and VEGF) demonstrated different relative risk estimates by disease aggressiveness (phomogeneity<0.05). Interestingly, variants in three genes (AKT1, PIK3R1, and VEGF) were associated only with more aggressive disease, with the strongest association observed for PIK3R1, with a risk estimate of 0.59 (95% CI 0.43–0.81) when the recessive genetic model was assumed.
Table 3.
Prostate cancer risk associated with selected SNPs in inflammation pathway genes among Caucasian men, by disease aggressiveness
| Gene | SNP | Alleles*** | Genetic Model††† | Controls MAF****†††† | Less Aggressive Cases‡‡‡ | More Aggressive Cases | phomogeneity§§§ | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MAF | OR‡‡‡‡ | 95% CI | MAF | OR | 95% CI | ||||||||
| AKT1 | rs2494738 | G/A | rec | 0.081 | 0.083 | 1.45 | 0.67 | 3.14 | 0.086 | 2.28 | 1.01 | 5.15 | 0.29 |
| rs1130214 | G/T | trend | 0.312 | 0.302 | 0.94 | 0.81 | 1.08 | 0.269 | 0.79 | 0.66 | 0.94 | 0.07 | |
| CXCL12 | rs2297630 | G/A | rec | 0.275 | 0.254 | 0.59 | 0.41 | 0.87 | 0.282 | 0.95 | 0.63 | 1.44 | 0.047 |
| IL4 | rs2243228 | A/C | rec | 0.092 | 0.074 | 0.26 | 0.07 | 0.94 | 0.089 | 0.20 | 0.03 | 1.50 | 0.8 |
| IL6 | rs2069840 | C/G | rec | 0.35 | 0.339 | 0.90 | 0.67 | 1.20 | 0.373 | 1.33 | 0.96 | 1.84 | 0.03 |
| IL6ST | rs11574783 | A/G | rec | 0.075 | 0.069 | 0.10 | 0.01 | 0.82 | 0.072 | - | - | ||
| PIK3R1 | rs706716 | C/T | dom | 0.265 | 0.264 | 0.98 | 0.78 | 1.25 | 0.204 | 0.68 | 0.50 | 0.94 | 0.03 |
| rs34309 | G/A | rec | 0.4 | 0.39 | 0.95 | 0.73 | 1.22 | 0.364 | 0.64 | 0.45 | 0.91 | 0.04 | |
| rs251408 | A/G | rec | 0.457 | 0.452 | 1.01 | 0.81 | 1.26 | 0.405 | 0.59 | 0.43 | 0.81 | 0.002 | |
| rs34306 | G/A | rec | 0.166 | 0.154 | 0.54 | 0.3 | 0.98 | 0.176 | 0.81 | 0.42 | 1.57 | 0.29 | |
| PTGS2 | rs2206593 | G/A | trend | 0.045 | 0.073 | 1.68 | 1.06 | 2.69 | 0.066 | 1.51 | 0.88 | 2.59 | 0.68 |
| rs6685280 | A/C | trend | 0.203 | 0.229 | 1.20 | 1.02 | 1.41 | 0.222 | 1.14 | 0.94 | 1.39 | 0.64 | |
| STAT3 | rs3809758§§§§ | G/A | rec | 0.201 | 0.184 | 1.11 | 0.68 | 1.81 | 0.233 | 1.96 | 1.19 | 3.23 | 0.04 |
| TNF | rs3093662 | A/G | trend | 0.06 | 0.072 | 1.34 | 1.02 | 1.74 | 0.067 | 1.21 | 0.87 | 1.68 | 0.56 |
| rs3093559 | G/A | dom | 0.019 | 0.027 | 1.29 | 1.04 | 1.61 | 0.026 | 1.24 | 0.95 | 1.62 | 0.77 | |
| VEGF | rs3025035 | C/T | trend | 0.065 | 0.07 | 1.08 | 0.84 | 1.39 | 0.091 | 1.48 | 1.11 | 1.98 | 0.04 |
Major/minor alleles based on frequencies in controls.
Genetic model selected based on the best fit in the regression model (dominant, recessive, log-additive).
Less aggressive prostate cancer (PC) defined as Gleason score 2–6 or 7(3+4) and local stage and PSA < 20 ng/mL at diagnosis; more aggressive disease as Gleason score 7(4+3) or 8–10 or regional/distant stage or PSA ≥ 20 ng/mL at diagnosis.
P-value testing Ho: no difference between risk estimates for less versus more aggressive PC.
MAF=Minor allele frequency.
Controls n=1,265, Cases: less aggressive n=874, more aggressive n=435.
OR=odds ratio, adjusted for age and prostate cancer screening in the 5 years prior to reference date
rs1053005, in LD with rs3809758 ( r2=0.93), was also genotyped and showed a similar association (OR=1.78, 95% CI 1.07–2.96; p=0.12)
In addition to single SNP analyses, we considered whether individual SNPs demonstrating an association with PC risk remained significant after adjustment for other SNPs. Four SNPs in each of IL4 (rs2243228), IL6ST (rs11574783), PTGS2 (rs6685280), and STAT3 (rs12949918) were independently associated with overall risk of PC, and three other SNPs were associated with more aggressive disease. These SNPs were then modeled together to evaluate a potential dose effect of increasing number of at-risk alleles (Table 4). There was a significant linear increase in risk of overall PC according to the number of at-risk alleles carried (ptrend = 0.0003, ppermuted=0.03). Men carrying the rs6685280 CC genotype (homozygote for risk allele) and the other at-risk alleles in each of rs2243228, rs11574783, and rs12949918, had an almost three-fold (95% CI 1.41–6.25) greater risk of PC than men with 0 to 2 at-risk alleles for these SNPs.
Table 4.
Prostate cancer risk associated with the number of at-risk SNP alleles carried among Caucasian men
| Cases***** | Controls | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Overall PC risk | n=1,130 | % | n=1,144 | % | OR††††† | 95% CI | OR‡‡‡‡‡ | 95% CI | ||
| No. at-risk alleles§§§§§ | ||||||||||
| 0–2 | 432 | 38.2 | 499 | 43.6 | 1.00 | Reference | 1.00 | Reference | ||
| 3 | 497 | 44.0 | 491 | 42.9 | 1.17 | 0.98 | 1.40 | 1.17 | 0.97 | 1.40 |
| 4 | 174 | 15.4 | 144 | 12.6 | 1.40 | 1.08 | 1.80 | 1.43 | 1.10 | 1.85 |
| 5 | 27 | 2.4 | 10 | 0.9 | 3.13 | 1.50 | 6.54 | 2.97 | 1.41 | 6.25 |
| Ppermuted ****** = 0.034 | ||||||||||
| Aggressive PC risk†††††† | n=390 | % | n=1,264 | % | OR‡‡‡‡‡‡ | 95% CI | OR§§§§§§ | 95% CI | ||
| No. at-risk | ||||||||||
| 0–1 | 44 | 11.3 | 217 | 18.2 | 1.00 | Reference | 1.00 | Reference | ||
| 2 | 159 | 40.8 | 498 | 41.8 | 1.69 | 1.16 | 2.46 | 1.76 | 1.20 | 2.57 |
| 3 | 172 | 44.1 | 460 | 38.6 | 1.94 | 1.94 | 2.82 | 1.98 | 1.36 | 2.89 |
| 4 | 15 | 3.8 | 16 | 1.3 | 4.75 | 2.15 | 10.5 | 5.11 | 2.29 | 11.4 |
| Ppermuted ******* = 2.5×10−3 | ||||||||||
Only cases and controls with complete genotype data for all SNPs are included.
OR=odds ratio, adjusted for age.
OR=odds ratio, adjusted for age and first-degree family history of PC.
Count of at-risk alleles in SNPs significantly associated with overall PC (rs6685280, rs2243228, rs11574783, rs12949918) or more aggressive PC (rs1130214, rs251408, rs3809758).
P-value for the number of associated alleles is based on the Cochran-Armitage test permutated 1000 times (ptrend = 0.0003).
Aggressive prostate cancer is defined as Gleason score ≥7(4+3) or regional/distant stage or PSA >20ng/mL at diagnosis.
OR adjusted for age and prostate cancer screening within the 5 years prior to reference date.
OR adjusted for age, prostate cancer screening history within the 5 years prior to reference date and first-degree family history of PC.
Similarly, we identified three SNPs that were independently associated with risk of aggressive PC in each of AKT1, PIK3R1, and STAT3. None of the SNPs associated with overall PC risk demonstrated a significant association with risk of more aggressive disease. The SNP identified in STAT3 in the aggressive disease analysis was in weak LD with the SNP that was associated with overall PC risk (r2 = 0.21). Three SNP genotypes, rs1130214 (AKT1), rs251408 (PIK3R1), and rs3809758 (STAT3), when considered together showed that risk of aggressive PC increased directly with the cumulative number of at-risk alleles carried (ptrend = 3.8×10−5, ppermuted=2.5×10−3) (Table 4). Men who carried four of the at-risk alleles had about a five-fold (95% CI 2.29–11.40) increased risk of aggressive disease compared to men with 0 or 1 at-risk alleles.
We also evaluated gene-gene interaction. Interestingly, the at-risk allele for SNP (rs6427627) in the promoter region of IL6R had a significant interaction with at-risk alleles of either of two SNPs that are in strong LD (r2=0.99), located in the promoter region of IL10 (pinteraction = 4.7 × 10−5 for rs1800871 and pinteraction = 5.3 × 10−5 for rs1800872), where both showed reduced risk estimates of PC (ORs = 0.67). In a single SNP analysis, these SNPs were not found to be associated with PC risk (rs6427627; OR=0.92, 95% CI 0.78–1.08, rs1800871; OR=1.05, 95% CI 0.9–1.24, and rs1800872; OR=1.07, 95% 0.9–1.24). These results should be interpreted with caution and future studies are needed to evaluate the observed gene-gene interaction in larger datasets.
Next, we considered potential gene-environment interactions. We evaluated associations between SNPs that were found to be nominally significant and PC risk after stratifying by several factors that may be associated with inflammation: BMI (<25.0, 25.0–29.9, and ≥30.0 kg/m2), smoking (non-smoker, former smoker, and current smoker), history of prostatitis, and history of STIs. Of the ten SNPs associated with overall risk of PC, two in PTGS2, rs2745557 (p = 0.01) and rs6685280 (p = 0.01) and two in STAT3, rs12949918 (p = 0.046) and rs744166 (p = 0.02) showed an interaction with smoking status. The at-risk allele for one SNP in PTGS2, rs2745557, showed different effects by smoking status when comparing the at-risk allele carriers relative to non-carriers in each strata of smoking (ORnon-smokers=0.65, 95% CI 0.46–0.91; ORformer-smokers=0.76, 95% CI 0.55–1.06; ORcurrent-smokers=2.43, 95% CI 1.13–5.25). Interestingly, rs6685280 in PTGS2 also showed variation by smoking status when comparing the at-risk allele for carriers to non carriers for each strata (ORnon-smokers=1.37, 95% CI 1.10–1.70; ORformer-smokers=1.15, 95% CI 0.94–1.41; ORcurrent-smokers=0.67, 95% CI 0.45–1.01).
In African Americans, modest associations with overall risk of PC were observed for five SNPs (rs2745557, rs4648276, rs2781551, rs4845623, and rs2228043) in four genes (PTGS2, IL6R, IL6ST, and CXCL12) (Supplementary Table 1). In PTGS2, carriers of the A allele at rs2745557 had a 2.2-fold greater risk (95% CI 1.03–4.86) in contrast to the significantly reduced relative risk observed for carriers of this same allele in Caucasians. A novel association was seen for rs4648276, with a risk estimate of 2.38 (95% CI 1.10–5.13) conferred by each additional C allele carried compared to those with the TT genotype. As the number of African Americans was limited (cases n=149, controls n=85), results should be interpreted with caution.
Discussion
Recent findings on pro-tumorigenic effects of inflammation suggest that chronic inflammation can affect tumor development and progression (24). Variants in genes known to be involved in inflammation therefore may be associated with risk of PC. In this study, we investigated 143 SNPs in 16 genes in the inflammation pathway for their association with PC. Although none of the single SNP-PC associations retrained statistical significance after adjustment for multiple comparisons, we observed suggestive evidence of associations for ten SNPs in seven genes in Caucasians: CXCL12, IL4, IL6, IL6ST, PTGS2, STAT3 and TNF. Moreover, evaluation of SNPs by disease aggressiveness showed that eight SNPs were associated with risk of comparatively more aggressive disease and eight SNPs were associated with less aggressive disease. Of the 22 SNPs that were nominally associated with PC risk, genotype data were available for 15 (11 SNPs were directly genotyped and 4 SNPs were in LD (r2 > 0.8) with genotyped SNPs)) in the Cancer Genetic Markers of Susceptibility (CGEMS) dataset. The CGEMS dataset confirmed our observed associations for SNPs in IL4, PIK3R1 and STAT3 (25, 26).
The strongest effect on overall PC risk was observed for a SNP, rs11574783, in IL6 signal transducer (IL6ST), where homozygous carriers of the minor G allele had a significant reduction in risk (OR=0.08, 95% CI 0.01–0.63; p = 0.016) compared to the GA and AA genotypes combined. IL6ST, also known as gp130, is the cell-surface signaling receptor subunit through which IL6 and other members of the IL6 family of cytokines exert their effects. A recent GWAS of genetic variants influencing protein expression found a nominally significant relation between the genotype at rs11574783 and IL6ST levels (27). To our knowledge, this is the first study to investigate the association between variants in this gene and PC risk.
A single locus in STAT3, defined by the SNPs rs744166 and rs12949918 (r2=0.98), was independently associated with overall PC risk (OR=0.83, 95% CI 0.70–0.98) in Caucasians, after adjusting for the effects of other risk-associated SNPs. Our result, if confirmed, may reflect a true association between rs744166, which is located in the first intron of STAT3, and PC as regulatory regions are commonly located in the promoter region and the first intron. The LD block that contains rs744166 extends for 54kb and contains the promoter region of STAT3. Examination of HapMap data reveals the presence of seven other SNPs that are modestly correlated with rs744166 and rs12949918 (0.65 ≤ r2 ≤ 0.72). Our evaluation of STAT3 SNPs with clinical characteristics revealed a second locus in STAT3, defined by rs1053005 and rs3809758 (r2=0.93), which was independently associated with more aggressive PC. In the CGEMS dataset SNP rs8074524, in perfect LD with rs3809758 (r2=1.00 in the Caucasian HapMap population), was associated with risk of more aggressive prostate cancer (OR=1.38, 95% CI 1.01–1.88, p=.04).
Although we are not aware of any prior studies that have investigated the association between genetic polymorphisms in STAT3 and PC, there is strong support for the involvement of this gene in the biology of both inflammation and PC (28). STAT3 protein is a key target of IL6 signaling and constitutively activated STAT3 has been implicated in PC cell proliferation and inhibition of apoptosis (29). Indeed, constitutively activated STAT3 is present in neoplastic prostate epithelia and tumors of higher grade and advanced stage have higher levels of activated protein (30). STAT3 protein has also been implicated in promoting migration of PC cells as well as in association with metastases (31).
In the genomic region of PTGS2, three SNPs were nominally associated with overall risk of PC, with rs6685280 remaining independently associated (p=0.03) after taking into account the other SNP genotypes. The cyclooxygenase genes are of interest, particularly PTGS2, also known as COX-2, and nine previous studies have investigated genetic variants in PTGS2 in relation to PC risk with inconsistent results (14, 32–34). Significant associations have been reported for SNPs in diverse regions of the gene, including promoter (34), intronic (12, 32, 35) and 3′ UTR regions (33). Few studies have evaluated the entire PTGS2 gene and many have analyzed non-tagging SNPs, which makes it difficult to compare results across studies. One of the largest studies combined men from the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial (PLCO) and the Cancer Prevention II (CPSII) Nutrition Cohort in a nested case-control study (12). In that study, rs5275 genotype was significantly associated with risk among PLCO participants (ptrend=0.02), but became non-significant when men from the CPSII were added to the analysis. Our results for rs2745557 replicate the finding by Cheng et al. with risk estimates of similar magnitude (32).
A recent study by the Breast and Prostate Cancer Cohort Consortium (BPC3), found no evidence that genetic variation in PTGS2 was associated with PC risk (14). Seven SNPs overlapped between the BPC3 study and ours, two of which, (rs2206593 and rs2745557), showed an association with PC risk in our study. Inconsistent results from BPC3 and our study could be due to differences in study populations or study design. Furthermore, conflicting results across studies may also be due to unmeasured effects of gene-environment or gene-gene interactions. There are several lines of evidence that suggest involvement of the PTGS2 protein in PC initiation. For instance, PTGS2 protein levels are increased in PC cell lines and tissues when compared to benign prostate cell lines or tissues (36, 37), and are positively correlated with Gleason grade (38). Also, an immediate downstream product of PTGS2 activity, prostaglandin E2 (PGE2), may have direct biological effects that promote tumor growth and progression, including increasing cellular proliferation, angiogenesis and decreasing immune surveillance (39).
Despite the functional data, none of the six known missense substitutions in PTGS2 has been associated with PC risk, which may suggest that variants associated with risk act by influencing some aspect of transcriptional or translational regulation. Interestingly, two SNPs, rs964570 and rs6685280, are located >30kb from the 5′ end of the PTGS2 transcript, and are highly correlated (r2>0.9) with variants located much farther (>20kb) upstream. Recently, an evolutionarily conserved, large intervening non-coding RNA (lincRNA) has been discovered 51 kb upstream from the PTGS2 transcription start site (40). The function of this RNA is unknown, but it is induced over 1000-fold in response to signaling through the master inflammatory regulator NF-κB. SNP rs6685280, which is correlated with other SNPs in the region of this lincRNA, was nominally significant (p=0.04), but only weakly associated with PC risk in our study (OR=1.16, 95% CI 1.01 – 1.33). Further analyses of genetic variation in PTGS2 should expand coverage to include variants in the far upstream region to explore the possibility that SNPs in the lincRNA play a role in PC susceptibility.
In interleukin 4 (IL4), a SNP located five kb upstream of the transcription start site, rs2243228, had a borderline association with PC in Caucasians after adjusting for other risk-associated SNPs (OR=0.3, 95% CI 0.1–1.0). Our SNP is in perfect LD with CGEMS SNP rs2243300 (r2=1) and CGEMS data confirm our finding (OR=0.76, 95% CI 0.60–0.96, p=0.02). IL4 has been shown to stimulate androgen-independent growth in LNCaP cells (17) and rs2243228 lies within 15 bases of an AP-1 transcription factor binding site (41). This is important as levels of IL4 have been shown to be significantly elevated in patients with hormone refractory PC (42), and receptors for this cytokine are found on prostate tumor cells. These observations suggest a potential role for IL4 in the progression of androgen-independent PC. To our knowledge, this is the first study to report on the association of SNPs in IL4 with PC risk.
Among Caucasians, three SNPs in AKT1, PIK3R1, and STAT3, were independently associated with aggressive PC in a multivariate model that included all three SNPs. STAT3 was described above, but two SNPs in AKT1 and three in PIK3R1 had no association with overall PC risk. For each SNP the strength of the association with PC risk increased and became nominally significant when more aggressive disease, i.e., a more extreme phenotype, was considered. One SNP, rs706716 in PIK3R1, was directly genotyped as part of the CGEMS GWAS and was found to be associated with both overall PC risk (OR=0.82, 95% CI 0.70–0.97, p=.02) and more aggressive disease (OR=0.82, 95% CI 0.67–0.99, p=0.04).
Biologically, both AKT1 and PIK3R1 are part of a major survival pathway central to the development and progression of cancer cells, including PC (43). Phosphatidylinositol 3-kinase (PI3K) lies upstream of AKT1 in the pathway and is activated as a consequence of IL6 binding at its receptor. Activated PI3K then promotes subsequent activation of AKT1 (44). Increased expression and protein activity of the AKT1 oncogene are correlated with more advanced PC and poor prognosis (45). Recently, PI3K pathway variants and PC risk were evaluated in 8,309 cases and 9,286 controls from the National Cancer Institute’s Breast and Prostate Cancer Consortium (46). Although variants in PIK3R1 were not included in that study, SNP rs7556371 in PIK3C2B showed a significant association with PC risk, especially for men who were diagnosed before 65 years. Our study is the first report on the risk of aggressive PC associated with polymorphisms in AKT1 or PIK3R1.
Finally, nominally significant associations among Caucasians in CXCL12 for rs2297630, IL6 for rs1800797 and TNF for rs3093559 did not remain significant in a stepwise model that considered all 10 SNPs initially associated with risk (Table 2). Other studies have also investigated the existence of associations with SNPs in CXCL12, IL6, IL8, IL10, TNF, and VEGF with mixed results (13, 47).
There are several strengths and limitations to our study that should be considered. Strengths include the population-based study design, the sample size for analysis of overall risk of PC, the availability of information on potential confounders and inflammation-related exposures, and clinical information on PC cases. However, the sample size yielded limited power for evaluating interactions. Much larger studies will be needed to examine gene-gene and gene-environment interactions. Finally, the small number of African Americans in our study limited our ability to interpret associations in this subset of men.
In summary, results reported here suggest that SNPs in four inflammation pathway-related genes (IL4, IL6ST, PTGS2, and STAT3) are significantly and independently associated with PC susceptibility. Three additional SNPs (in AKT1, PIK3R1, and STAT3) were associated with aggressive PC. Furthermore, we observed almost a three-fold increase in the relative risk of PC for men carrying the maximum number of five at-risk alleles, based on significantly associated SNPs being analyzed in a dose-response model. All associated SNPs identified in this study lie within genes that form part of the IL6 signaling pathway, with the exception of IL4 and PTGS2. Our findings are consistent with the biological evidence previously identified for the involvement of PTGS2 and the IL6 cytokine pathway in PC (15, 28). Novel PC associations with genetic variants in IL6ST, STAT4, AKT1, and PIK3R1 were observed, and each of these genes has a key role in the inflammatory pathway. Thus, while our results await confirmation from other studies, these data provide support for the hypothesis that genetic variation in inflammation pathway genes plays a role in the development and progression of PC.
Supplementary Material
Acknowledgments
We thank all the men who participated in the study for their time and effort and the study managers and interviewers for their help with data collection. We also thank Dr. Robert Vessella, Urology Department, University of Washington, whose laboratory completed the PSA assays for control subjects.
Support: This work was supported by grants RO1-CA 056678, RO1-CA092579, and R03-CA121871 (to J.L.S.) and Contract N01-CN-05230 from the National Cancer Institute, with additional support provided by the Fred Hutchinson Cancer Research Center and the Intramural Program of the National Human Genome Research Institute (E.A.O and E.M.K.). C.A.S. was supported by training grant PC06445 from the U.S. Department of Defense.
Footnotes
Disclosure Statement: None
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Johns LE, Houlston RS. A systematic review and meta-analysis of familial prostate cancer risk. BJU Int. 2003;91:789–94. doi: 10.1046/j.1464-410x.2003.04232.x. [DOI] [PubMed] [Google Scholar]
- 3.Cantor RM, Lange K, Sinsheimer JS. Prioritizing GWAS results: A review of statistical methods and recommendations for their application. Am J Hum Genet. 2010;86:6–22. doi: 10.1016/j.ajhg.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 5.Schottenfeld D, Beebe-Dimmer J. Chronic inflammation: a common and important factor in the pathogenesis of neoplasia. CA Cancer J Clin. 2006;56:69–83. doi: 10.3322/canjclin.56.2.69. [DOI] [PubMed] [Google Scholar]
- 6.Nickel JC, Roehrborn CG, O’Leary MP, Bostwick DG, Somerville MC, Rittmaster RS. Examination of the relationship between symptoms of prostatitis and histological inflammation: baseline data from the REDUCE chemoprevention trial. J Urol. 2007;178:896–900. doi: 10.1016/j.juro.2007.05.041. discussion 900–1. [DOI] [PubMed] [Google Scholar]
- 7.Huang WY, Hayes R, Pfeiffer R, Viscidi RP, Lee FK, Wang YF, Reding D, Whitby D, Papp JR, Rabkin CS. Sexually transmissible infections and prostate cancer risk. Cancer Epidemiol Biomarkers Prev. 2008;17:2374–81. doi: 10.1158/1055-9965.EPI-08-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dennis LK, Lynch CF, Torner JC. Epidemiologic association between prostatitis and prostate cancer. Urology. 2002;60:78–83. doi: 10.1016/s0090-4295(02)01637-0. [DOI] [PubMed] [Google Scholar]
- 9.Salinas CA, Kwon EM, Feng Z, Nelson PS, Ostrander EA, Peters U, Stanford JL. Use of Aspirin and Other Non-steroidal Anti-inflammatory Medications in Relation to Prostate Cancer Risk. American Journal of Epidemiology. 2010 doi: 10.1093/aje/kwq175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langeberg WJ, Isaacs WB, Stanford JL. Genetic etiology of hereditary prostate cancer. Front Biosci. 2007;12:4101–10. doi: 10.2741/2374. [DOI] [PubMed] [Google Scholar]
- 11.Sun J, Wiklund F, Zheng SL, Chang B, Balter K, Li L, Johansson JE, Li G, Adami HO, Liu W, Tolin A, Turner AR, Meyers DA, Isaacs WB, Xu J, Gronberg H. Sequence variants in Toll-like receptor gene cluster (TLR6-TLR1-TLR10) and prostate cancer risk. J Natl Cancer Inst. 2005;97:525–32. doi: 10.1093/jnci/dji070. [DOI] [PubMed] [Google Scholar]
- 12.Danforth KN, Hayes RB, Rodriguez C, Yu K, Sakoda LC, Huang WY, Chen BE, Chen J, Andriole GL, Calle EE, Jacobs EJ, Chu LW, Figueroa JD, Yeager M, Platz EA, Michaud DS, Chanock SJ, Thun MJ, Hsing AW. Polymorphic variants in PTGS2 and prostate cancer risk: results from two large nested case-control studies. Carcinogenesis. 2008;29:568–72. doi: 10.1093/carcin/bgm253. [DOI] [PubMed] [Google Scholar]
- 13.McCarron SL, Edwards S, Evans PR, Gibbs R, Dearnaley DP, Dowe A, Southgate C, Easton DF, Eeles RA, Howell WM. Influence of cytokine gene polymorphisms on the development of prostate cancer. Cancer Res. 2002;62:3369–72. [PubMed] [Google Scholar]
- 14.Dossus L, Kaaks R, Canzian F, Albanes D, Berndt SI, Boeing H, Buring J, Chanock SJ, Clavel-Chapelon F, Feigelson HS, Gaziano JM, Giovannucci E, Gonzalez C, Haiman CA, Hallmans G, Hankinson SE, Hayes RB, Henderson BE, Hoover RN, Hunter DJ, Khaw KT, Kolonel LN, Kraft P, Ma J, Le Marchand L, Lund E, Peeters PH, Stampfer M, Stram DO, Thomas G, Thun MJ, Tjonneland A, Trichopoulos D, Tumino R, Riboli E, Virtamo J, Weinstein SJ, Yeager M, Ziegler RG, Cox DG. PTGS2 and IL6 genetic variation and risk of breast and prostate cancer: results from the Breast and Prostate Cancer Cohort Consortium (BPC3) Carcinogenesis. 2010;31:455–61. doi: 10.1093/carcin/bgp307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hussain T, Gupta S, Mukhtar H. Cyclooxygenase-2 and prostate carcinogenesis. Cancer Lett. 2003;191:125–35. doi: 10.1016/s0304-3835(02)00524-4. [DOI] [PubMed] [Google Scholar]
- 16.Sun J, Turner A, Xu J, Gronberg H, Isaacs W. Genetic variability in inflammation pathways and prostate cancer risk. Urol Oncol. 2007;25:250–9. doi: 10.1016/j.urolonc.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Lee SO, Pinder E, Chun JY, Lou W, Sun M, Gao AC. Interleukin-4 stimulates androgen-independent growth in LNCaP human prostate cancer cells. Prostate. 2008;68:85–91. doi: 10.1002/pros.20691. [DOI] [PubMed] [Google Scholar]
- 18.Lee CR, Bottone FG, Jr, Krahn JM, Li L, Mohrenweiser HW, Cook ME, Petrovich RM, Bell DA, Eling TE, Zeldin DC. Identification and functional characterization of polymorphisms in human cyclooxygenase-1 (PTGS1) Pharmacogenet Genomics. 2007;17:145–60. doi: 10.1097/01.fpc.0000236340.87540.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stanford JL, Wicklund KG, McKnight B, Daling JR, Brawer MK. Vasectomy and risk of prostate cancer. Cancer Epidemiol Biomarkers Prev. 1999;8:881–6. [PubMed] [Google Scholar]
- 20.Agalliu I, Salinas CA, Hansten PD, Ostrander EA, Stanford JL. Statin use and risk of prostate cancer: results from a population-based epidemiologic study. Am J Epidemiol. 2008;168:250–60. doi: 10.1093/aje/kwn141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.http://www.appliedbiosystems.com
- 22.Edginton ES, Onghena P. Randomization Tests. New York: Marcel Dekker, Inc; 2007. [Google Scholar]
- 23.Li W, Nyholt DR. Marker selection by Akaike information criterion and Bayesian information criterion. Genet Epidemiol. 2001;21(Suppl 1):S272–7. doi: 10.1002/gepi.2001.21.s1.s272. [DOI] [PubMed] [Google Scholar]
- 24.De Marzo AM, Platz EA, Sutcliffe S, Xu J, Gronberg H, Drake CG, Nakai Y, Isaacs WB, Nelson WG. Inflammation in prostate carcinogenesis. Nat Rev Cancer. 2007;7:256–69. doi: 10.1038/nrc2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF, Hoover R, Hunter DJ, Chanock SJ, Thomas G. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 26.(http://cgems.cancer.gov/data/).
- 27.Melzer D, Perry JR, Hernandez D, Corsi AM, Stevens K, Rafferty I, Lauretani F, Murray A, Gibbs JR, Paolisso G, Rafiq S, Simon-Sanchez J, Lango H, Scholz S, Weedon MN, Arepalli S, Rice N, Washecka N, Hurst A, Britton A, Henley W, van de Leemput J, Li R, Newman AB, Tranah G, Harris T, Panicker V, Dayan C, Bennett A, McCarthy MI, Ruokonen A, Jarvelin MR, Guralnik J, Bandinelli S, Frayling TM, Singleton A, Ferrucci L. A genome-wide association study identifies protein quantitative trait loci (pQTLs) PLoS Genet. 2008;4:e1000072. doi: 10.1371/journal.pgen.1000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–12. doi: 10.1016/j.ejca.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 29.Barton BE, Karras JG, Murphy TF, Barton A, Huang HF. Signal transducer and activator of transcription 3 (STAT3) activation in prostate cancer: Direct STAT3 inhibition induces apoptosis in prostate cancer lines. Mol Cancer Ther. 2004;3:11–20. [PubMed] [Google Scholar]
- 30.Horinaga M, Okita H, Nakashima J, Kanao K, Sakamoto M, Murai M. Clinical and pathologic significance of activation of signal transducer and activator of transcription 3 in prostate cancer. Urology. 2005;66:671–5. doi: 10.1016/j.urology.2005.03.066. [DOI] [PubMed] [Google Scholar]
- 31.Abdulghani J, Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, Lisanti MP, Zellweger T, Alanen K, Mirtti T, Visakorpi T, Bubendorf L, Nevalainen MT. Stat3 promotes metastatic progression of prostate cancer. Am J Pathol. 2008;172:1717–28. doi: 10.2353/ajpath.2008.071054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng I, Liu X, Plummer SJ, Krumroy LM, Casey G, Witte JS. COX2 genetic variation, NSAIDs, and advanced prostate cancer risk. Br J Cancer. 2007;97:557–61. doi: 10.1038/sj.bjc.6603874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shahedi K, Lindstrom S, Zheng SL, Wiklund F, Adolfsson J, Sun J, Augustsson-Balter K, Chang BL, Adami HO, Liu W, Gronberg H, Xu J. Genetic variation in the COX-2 gene and the association with prostate cancer risk. Int J Cancer. 2006;119:668–72. doi: 10.1002/ijc.21864. [DOI] [PubMed] [Google Scholar]
- 34.Panguluri RC, Long LO, Chen W, Wang S, Coulibaly A, Ukoli F, Jackson A, Weinrich S, Ahaghotu C, Isaacs W, Kittles RA. COX-2 gene promoter haplotypes and prostate cancer risk. Carcinogenesis. 2004;25:961–6. doi: 10.1093/carcin/bgh100. [DOI] [PubMed] [Google Scholar]
- 35.Hedelin M, Chang ET, Wiklund F, Bellocco R, Klint A, Adolfsson J, Shahedi K, Xu J, Adami HO, Gronberg H, Balter KA. Association of frequent consumption of fatty fish with prostate cancer risk is modified by COX-2 polymorphism. Int J Cancer. 2007;120:398–405. doi: 10.1002/ijc.22319. [DOI] [PubMed] [Google Scholar]
- 36.Subbarayan V, Sabichi AL, Llansa N, Lippman SM, Menter DG. Differential expression of cyclooxygenase-2 and its regulation by tumor necrosis factor-alpha in normal and malignant prostate cells. Cancer Res. 2001;61:2720–6. [PubMed] [Google Scholar]
- 37.Lee LM, Pan CC, Cheng CJ, Chi CW, Liu TY. Expression of cyclooxygenase-2 in prostate adenocarcinoma and benign prostatic hyperplasia. Anticancer Res. 2001;21:1291–4. [PubMed] [Google Scholar]
- 38.Madaan S, Abel PD, Chaudhary KS, Hewitt R, Stott MA, Stamp GW, Lalani EN. Cytoplasmic induction and over-expression of cyclooxygenase-2 in human prostate cancer: implications for prevention and treatment. BJU Int. 2000;86:736–41. doi: 10.1046/j.1464-410x.2000.00867.x. [DOI] [PubMed] [Google Scholar]
- 39.Badawi AF. The role of prostaglandin synthesis in prostate cancer. BJU Int. 2000;85:451–62. doi: 10.1046/j.1464-410x.2000.00507.x. [DOI] [PubMed] [Google Scholar]
- 40.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL, Kolchanov NA. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26:362–7. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wise GJ, Marella VK, Talluri G, Shirazian D. Cytokine variations in patients with hormone treated prostate cancer. J Urol. 2000;164:722–5. doi: 10.1097/00005392-200009010-00024. [DOI] [PubMed] [Google Scholar]
- 43.Li L, Ittmann MM, Ayala G, Tsai MJ, Amato RJ, Wheeler TM, Miles BJ, Kadmon D, Thompson TC. The emerging role of the PI3-K-Akt pathway in prostate cancer progression. Prostate Cancer Prostatic Dis. 2005;8:108–18. doi: 10.1038/sj.pcan.4500776. [DOI] [PubMed] [Google Scholar]
- 44.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 45.Liao Y, Grobholz R, Abel U, Trojan L, Michel MS, Angel P, Mayer D. Increase of AKT/PKB expression correlates with gleason pattern in human prostate cancer. Int J Cancer. 2003;107:676–80. doi: 10.1002/ijc.11471. [DOI] [PubMed] [Google Scholar]
- 46.Koutros S, Schumacher FR, Hayes RB, Ma J, Huang WY, Albanes D, Canzian F, Chanock SJ, Crawford ED, Diver WR, Feigelson HS, Giovanucci E, Haiman CA, Henderson BE, Hunter DJ, Kaaks R, Kolonel LN, Kraft P, Le Marchand L, Riboli E, Siddiq A, Stampfer MJ, Stram DO, Thomas G, Travis RC, Thun MJ, Yeager M, Berndt SI. Pooled analysis of phosphatidylinositol 3-kinase pathway variants and risk of prostate cancer. Cancer Res. 2010;70:2389–96. doi: 10.1158/0008-5472.CAN-09-3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang MH, Helzlsouer KJ, Smith MW, Hoffman-Bolton JA, Clipp SL, Grinberg V, De Marzo AM, Isaacs WB, Drake CG, Shugart YY, Platz EA. Association of IL10 and other immune response- and obesity-related genes with prostate cancer in CLUE II. Prostate. 2009;69:874–85. doi: 10.1002/pros.20933. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.