

Abstract
Infection of cell cultures with Neisseria gonorrhoeae results in apoptosis that is mediated by the PorB porin. During the infection process porin translocates from the outer bacterial membrane into host cell membranes where its channel activity is regulated by nucleotide binding and voltage-dependent gating, features that are shared by the mitochondrial voltage-dependent anion channel (VDAC). Here we show that porin is selectively and efficiently transported to mitochondria of infected cells. Prevention of porin translocation also blocked the induction of apoptosis. Mitochondria of cells treated with porin both in vitro and in vivo were depleted of cytochrome c and underwent permeability transition. Overexpression of Bcl-2 blocked porin-induced apoptosis. The release of cytochrome c occurred independently of active caspases but was completely prevented by Bcl-2. Our data suggest that the Neisseria porin can, like its eukaryotic homologue, function at the mitochondrial checkpoint to mediate apoptosis.
Keywords: apoptosis/mitochondria/Neisseria/permeability transition/voltage-dependent anion channel (VDAC)-like porin
Introduction
Apoptosis is a special form of programmed cell death that plays a pivotal role during developmental morphogenesis and cell homeostasis. The basic mechanism of apoptosis regulation is evolutionarily conserved from nematodes to man; in fact, some major lessons were learned by studies of apoptosis in the nematode Caenorhabditis elegans (Metzstein et al., 1998). The major executioners of apoptosis are the caspases (cysteinyl aspartate-directed proteases), which comprise a family of 15 members exerting different functions in inflammation and apoptosis (Nicholson and Thornberry, 1997; Salvesen and Dixit, 1997).
The pathways by which caspases are activated have been pinned down to two major branches: one is initiated by cell surface receptors, the other by mitochondria. Ligation of receptors of the tumour necrosis factor (TNF) receptor family (Wallach et al., 1997) results in the recruitment and activation of initiator caspases, e.g. caspase-8 (Boldin et al., 1996; Muzio et al., 1996). Active caspase-8 is able to process and activate other caspases, which initiate apoptosis by cleavage of cellular substrates (Salvesen and Dixit, 1997). Substrates include the DNA-repair enzyme poly(ADP-ribose) polymerase (PARP) (Tewari et al., 1995) and the cytoskeleton-associated protein fodrin (Cryns et al., 1996; Janicke et al., 1998), both of which are cleaved by caspase-3.
For many of the pathways induced by diverse stimuli such as cytotoxic or environmental stress or toxic cell metabolites, the mitochondria appear to be the main integrators of apoptotic signalling. They respond to these stimuli with the release of caspases-2 and -9 (Susin et al., 1999a) or caspase-activating proteins like cytochrome c (Liu et al., 1996) and apoptosis initiator factor (AIF) (Susin et al., 1999b). Cytochrome c is required as cofactor in a complex with the adapter molecule Apaf-1 and dATP for caspase-9 activation (Zou et al., 1997). Like caspase-8, active caspase-9 cleaves and activates effector caspases that coordinate the execution phase of the death programme (Slee et al., 1999). Thus, although the initial phase of receptor-mediated and mitochondria-mediated apoptotic pathways appears to be rather different, a similar set of caspases is eventually activated and executes the death programme.
Although the major molecular effects of apoptotic activation of mitochondria have been elucidated, the trigger initiating the release of cytochrome c and other factors is still not well understood. Many investigators find a collapse of the mitochondrial inner membrane potential (Δψm) during apoptosis, indicating the opening of a large conductance channel generally known as the permeability transition (PT) pore complex (Zoratti and Szabo, 1995; Green and Reed, 1998). The complex involved in PT regulation consists of several factors including the mitochondrial porin, also known as voltage-dependent anion channel (VDAC) (Zoratti and Szabo, 1995; Beutner et al., 1998), the adenine nucleotide translocator (ANT) (Brustovetsky and Klingenberg, 1996), the peripheral benzodiazepine receptor (Pastorino et al., 1994) and probably kinases like hexokinase and/or creatine kinase (Beutner et al., 1998). Interestingly, PT pore opening is induced by several pro-apoptotic second messengers like Ca2+, pro-oxidants, nitric oxide, ceramide and caspases (Zamzami et al., 1995; Bernardi and Petronilli, 1996; Marzo et al., 1998a), suggesting a direct involvement of PT pore opening in apoptosis induction. Moreover, it is regulated by the anti-apoptotic members of the Bcl-2 family, Bcl-2 and Bcl-XL, which stabilize mitochondrial membranes (Decaudin et al., 1997; Kroemer, 1997a) and by the pro-apoptotic member, Bax, which induces PT (Xiang et al., 1996).
Recently, we demonstrated the induction of apoptosis during the infection of epithelial cells and phagocytes by Neisseria gonorrhoeae (Ngo), a human-specific bacterial pathogen (Müller et al., 1999). Gonococci attach to their target cells via pili, hair-like protein appendages (Swanson et al., 1987) or by Opa proteins, which in addition to adhesion induce the receptor-mediated uptake by the host cell (Makino et al., 1991; Gray-Owen et al., 1997; Hauck et al., 1998). Although adhesion or invasion is a prerequisite, neither pili nor Opa proteins, but the PorB porin, induces apoptosis of target cells, when added in its purified form (Müller et al., 1999). Porins form integral diffusion channels in the outer membrane of Gram-negative bacteria. Some porins contain binding sites for certain substrates found in the environment of these bacteria (Benz, 1995). The binding of nucleotides by porin is unusual in this respect (Rudel et al., 1996) since nucleotides are not a natural substrate for Neisseria. However, during an infection porin translocates into the membrane of target cells (Weel and van Putten, 1991), where its channel activity is tightly regulated by cytosolic nucleotides that increase the voltage-dependent gating of the porin channel (Rudel et al., 1996). The properties of PorB such as voltage dependence and nucleotide binding closely resemble mitochondrial VDAC functions (Benz, 1994) and are not found in other porins of Gram-negative bacteria.
Since PorB exhibits similar properties to mitochondrial VDACs and since both are involved in apoptosis induction, we investigated the influence of PorB on mitochondrial function during apoptosis induction. Here we show that infection or PorB treatment causes loss of membrane potential and the release of cytochrome c from mitochondria of intact cells. Surprisingly, PorB also induces the release of cytochrome c from purified mitochondria. The release of cytochrome c as well as PT and the induction of apoptosis is blocked by Bcl-2 in intact cells. Furthermore, in porin-treated cells as well as in infected cells PorB is specifically and efficiently targeted to the mitochondria.
Results
Porin induces structural and biochemical changes of mitochondria that are typical of apoptotic cells
When epithelial or immune cells are treated with porin purified from N.gonorrhoeae they undergo apoptosis (Müller et al., 1999). The underlying mechanism involves release of Ca2+ into the cytosol and the activation of proteases of the caspase and calpain families. Since mitochondria play a central integrative role in the regulation of apoptosis (reviewed by Kroemer et al., 1998) we investigated the effects that porin treatment has on the structural and biochemical integrity of mitochondria (Figure 1). Jurkat cells were treated with 7 µg/ml porin for 15 h, the mitochondria were isolated and the cytochrome c content was determined by western blot analysis. Mitochondria of untreated and buffer-treated cells (Figure 1A) contained the same amount of cytochrome c while mitochondria of porin-treated cells had lost all their cytochrome c (Figure 1A). To ensure equal loading of proteins the western blots were also developed with an antibody directed against cytochrome c oxidase. The same result was obtained with mitochondria isolated from porin-treated monocytic and epithelial cell lines (not shown). Redistribution of cytochrome c was also analysed microscopically by double staining Jurkat cells with an antibody directed against native cytochrome c and MitoTracker, a potential-sensitive dye specific for mitochondria (Figure 2). In control or buffer-treated cells both dyes colocalized completely, indicating an intact mitochondrial membrane potential and a cytochrome c distribution typical of healthy cells (Figure 2, vector control and vector + buffer). In contrast, upon porin treatment a large population of cells (80–90%) no longer stained with MitoTracker and in addition had lost the granular staining of cytochrome c (Figure 2, vector + porin). These cells also showed clear signs of apoptosis, i.e. apoptotic body formation, condensation of the cytoplasm and cell shrinkage. Similarly, infection of HeLa cells with N.gonorrhoeae strain N242 also resulted in PT, the release of cytochrome c from mitochondria and the typical morphological alterations of apoptotic cells (Figure 3). This occurred in ∼40–50% of the population.
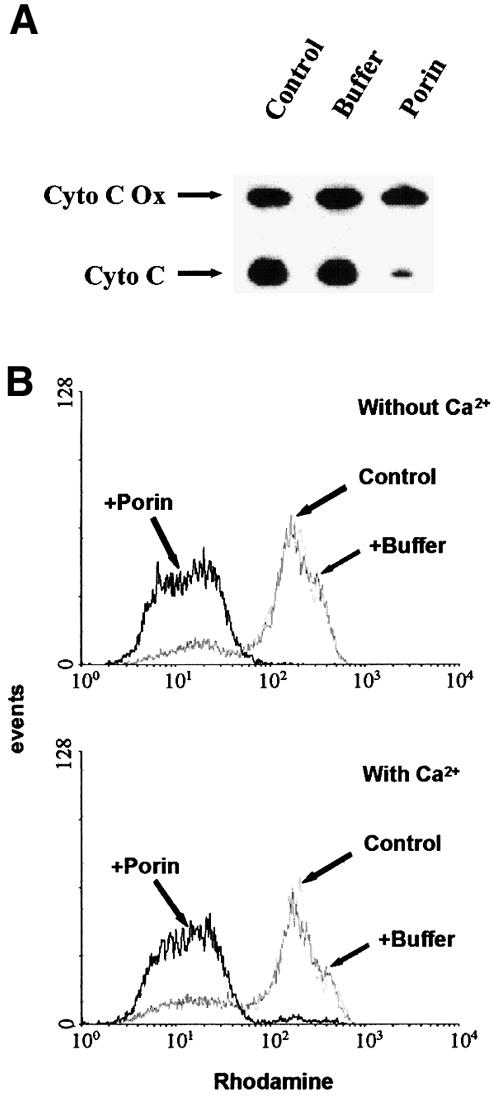
Fig. 1. Effects of porin on the release of cytochrome c and PT of mitochondria in vivo. (A) Mitochondria were isolated from Jurkat T cells treated with either 7 µg/ml purified porin or an equal volume of porin purification buffer for 15 h. Mitochondrial lysates were subjected to SDS–PAGE followed by western blotting using monoclonal antibodies against human cytochrome c oxidase subunit II (Cyto C Ox) and denatured human cytochrome c (Cyto C), respectively. (B) Jurkat T cells were treated with porin or an equal volume of porin purification buffer for 15 h in the presence and absence of 1 mM Ca2+ as indicated, stained with rhodamine 123 for 30 min at 37°C and analysed by flow cytometry. The histogram shows the analysis of 10 000 cells per sample.

Fig. 2. Bcl-2 overexpression and caspase inhibition block porin-induced apoptosis by different mechanisms. Jurkat T cells either expressing Bcl-2 or carrying an empty vector were treated with 7 µg/ml porin or purification buffer for 15 h or left untreated. They were then subjected to immunocytochemistry using an anti-cytochrome c-specific antibody and an Alexa-488-coupled secondary antibody. Cells were stained with MitoTracker before fixation. A phase contrast image, single colours and overlays are shown for every section. Cells in the lowest panel were treated with 50 µM zVAD-fmk (Bachem) for 1 h before addition of porin.

Fig. 3. Cytochrome c release and PT also occurs in HeLa cells infected with N.gonorrhoeae. HeLa cells were infected with gonococcal strain N242 for 15 h at an m.o.i. of 1. Cells were then subjected to immunocytochemistry using an anti-cytochrome c-specific antibody and an Alexa-488-coupled secondary antibody. MitoTracker staining was performed before fixation. A phase contrast image, single colours and overlays are shown for infected cells and uninfected control cells. Note the swollen mitochondria in infected, non-apoptotic cells. Loss of or diffuse cytochrome c staining in apoptotic cells is indicative of cytochrome c release.
We confirmed the loss of the mitochondrial membrane potential after porin treatment by staining live cells with the potential-sensitive dye rhodamine 123 for flow-cytometric analysis (Figure 1B). This dye is reportedly only incorporated into mitochondria with intact membrane potential (Van der Heiden et al., 1997). Cells that were treated with porin for 15 h showed a shift towards lower intensity (Figure 1B), the cells have therefore undergone PT. Since porin provokes a rapid influx of extracellular Ca2+ in treated cells (Müller et al., 1999) and Ca2+ alone is sufficient to induce PT in other systems in vitro (Marzo et al., 1998a), we tested whether Ca2+ is required for PT induced by porin. PT occurred with the same kinetics in cells treated with porin in Ca2+-free medium compared with the control with Ca2+ (Figure 1B), thus excluding a direct effect of Ca2+ on porin-induced PT.
Porin induces cytochrome c release and PT in purified mitochondria
Isolated mitochondria have been used previously to demonstrate apoptogenic effects of purified proteins such as Bax on cytochrome c content and membrane potential (Jürgensmeier et al., 1998). In the case of porin-induced apoptosis it is especially relevant to investigate the effects of the porin on isolated mitochondria because of its similarities to mitochondrial VDAC, which is known to participate in PT and cytochrome c release (Beutner et al., 1998; Shimizu et al., 1999). Purified mitochondria from 1 × 107 Jurkat T cells were treated with 2 µg of porin and cytochrome c release was monitored by western blotting (Figure 4A). Mitochondria treated with porin were completely depleted of cytochrome c, whereas addition of porin purification buffer alone did not trigger this release. Interestingly, amounts of porin (0.5 µg) that were insufficient to induce a cytochrome c release reproducibly had an opposite effect: the mitochondria were protected from the spontaneous loss of cytochrome c that is otherwise observed after extended incubation at 37°C (not shown). We also looked for PT as a putative response of isolated mitochondria to porin treatment. For this purpose, the mitochondria were stained with rhodamine 123 and fluorescence intensity was monitored by flow cytometry (Figure 4B). Indeed, a clear shift to lower intensity is observed in mitochondria treated with porin as compared with the control, which is indicative of loss of membrane potential. Also, purified mitochondria treated with porin increased in volume compared with an untreated control, which was monitored in a swelling assay over time (Figure 4C). These data therefore provide clear evidence that neisserial porin alone is sufficient for eliciting an in vitro mitochondrial response similar to that typically observed in the course of apoptosis in vivo.

Fig. 4. Effects of porin on the release of cytochrome c, PT and swelling of mitochondria in vitro. (A) Mitochondria were isolated from 5 × 107 Jurkat cells. Porin (2 µg) or an equal volume of porin purification buffer was applied for 30 min at 37°C in vitro. The pellet (P) and the supernatant (SN) of every sample were then analysed by western blotting using monoclonal antibodies against human cytochrome c oxidase subunit II (Cyto C Ox) and denatured human cytochrome c (Cyto C), respectively. (B) Mitochondria obtained as described in (A) were stained with rhodamine 123 for 30 min at 37°C and analysed by flow cytometry. (C) Mitochondria were purified from mouse liver and subjected to a swelling assay as described in Materials and methods. The increase in mitochondrial volume was monitored by determining the optical density at 600 nm. The increase in volume after addition of the stimulus is visible as a decline in the OD600 over 10 min. Note that the untreated mitochondria swell spontaneously, a process that is not increased by low amounts of porin. However, higher concentrations of porin lead to an increase in the volume of mitochondria similar to treatment of the mitochondria with Ca2+, a well known inducer of mitochondrial swelling.
Overexpression of Bcl-2 or Bcl-XL blocks porin-induced apoptosis
Bcl-2 and Bcl-XL are anti-apoptotic members of the Bcl family. They are localized to organelle membranes as a result of their C-terminal membrane anchor (Yang and Korsmeyer, 1996) and Bcl-XL in addition is able to form ion channels in synthetic lipid membranes (Antonsson et al., 1997; Minn et al., 1997). Overexpression of these proteins has repeatedly been shown to affect all the apoptotic phenotypes seen at the mitochondrial level, yet it remains unclear if this is due to a direct function or if it is a consequence of inhibition of an earlier step in the death pathway (Zamzami et al., 1996; Kluck et al., 1997). Jurkat T cells stably expressing Bcl-2 and the control cell line were treated with porin for 15 h. Control transfected Jurkat cells showed clear morphological signs of apoptosis including cell shrinkage and extensive apoptotic body formation (Figure 2, phase contrast). These features were completely abolished by overexpression of Bcl-2. In order to investigate whether the observed protection from apoptosis induction was due to an inhibition of cytochrome c release by Bcl-2, the cells were subjected to immunocytochemistry using an anti-cytochrome c antibody and additional staining of the mitochondria by MitoTracker (Figure 2). While the control cell line completely released cytochrome c upon porin treatment and had undergone PT, Jurkat–Bcl cells retained cytochrome c and membrane potential just like the untreated control (Figure 2). These results were confirmed with two additional cell lines, CEM-Bcl-XL and SKW6-Bcl-2, which both stably expressed anti-apoptotic Bcl proteins (data not shown).
Caspases are known inducers of cytochrome c release and PT (Marzo et al., 1998b) and are known to be activated during porin-induced apoptosis (Müller et al., 1999). We therefore assessed their role in porin-mediated mitochondrial damage. Interestingly, the inhibition of caspases by the broad range caspase inhibitor zVAD-fmk blocked all described morphological alterations but could not prevent the release of cytochrome c and PT in treated cells (Figure 2, zVAD + porin).
In order to quantify the anti-apoptotic effect of the Bcl proteins (Figure 5A, left panels) we measured phosphatidylserine (PS) exposure. The majority (>90%) of vector-transfected Jurkat cells treated with porin for 15 h bound annexin V, which is indicative of PS exposure on their surface (upper left panel), whereas no increase in the annexin V-positive population was observed with Jurkat–Bcl (lower left panel). These results were confirmed by quantifying DNA degradation via propidium iodide incorporation (not shown).

Fig. 5. Bcl-2 overexpression inhibits porin-induced PT, caspase activation and apoptosis. (A) Jurkat cells either expressing Bcl-2 or carrying an empty vector were treated with 7 µg/ml porin (as indicated in the histograms) for 15 h and split: one half was subjected to an annexin V binding protocol in order to determine PS exposure, the other was stained with rhodamine 123 to assess mitochondrial membrane potential. Both samples were analysed by flow cytometry. (B) Aliquots of the samples in (A) were lysed and subjected to western blotting for assessment of caspase activation. Cleavage of full-length fodrin (240 kDa) into signature fragments of 120 and 150 kDa was monitored by using a monoclonal antibody against fodrin that recognizes all three forms.
When the same cells were monitored for mitochondrial membrane potential by staining with rhodamine 123, no porin-induced shift towards lower fluorescence intensity was observed in cells expressing Bcl-2, indicating that PT did not occur in these cells (Figure 5A, right panels) and thereby confirming the conclusions drawn from microscopic analysis. Downstream caspases were not active in Jurkat–Bcl-2, as judged by the absence of substrate cleavage of PARP (data not shown) and fodrin (Figure 5B). Interestingly, whereas the generation of the 120 kDa caspase signature fragment of fodrin (Nath et al., 1996) was completely blocked, the 150 kDa calpain fragment did appear, indicating that calpains are active in the presence of overexpressed Bcl-2, but their activity is not sufficient to drive the cells into apoptosis.
Porin is targeted to the mitochondria
Due to the similarities between eukaryotic VDAC and neisserial porin it seemed a promising hypothesis that porin is targeted to the mitochondria where it then exerts its cytotoxic effects directly by inducing PT and cytochrome c release. When isolated mitochondria were treated with porin in vitro, the vast majority of the porin amount applied was detected in the mitochondrial pellet, whereas hardly any porin was found in the supernatant (not shown). Thus, porin associated with mitochondria in vitro. We then addressed the question of whether porin is localized to mitochondria of porin-treated cells. Cells were either treated with porin purification buffer or with porin for 15 h and washed extensively before preparation of whole cell lysates and mitochondria. We also treated cells for the same period of time with purified Staphylococcus aureus α-toxin which, like porin, consists of amphipathic β-barrels and induces apoptosis by forming pores in the cytoplasmic membrane of the cells (Jonas et al., 1994; Song et al., 1996). Purified gonococcal PilC adhesin, which binds to several human cell lines, served as additional control. Porin, as well as PilC and α-toxin, were present in whole cell lysates, indicating their insertion into or tight attachment to membranes (Figure 6A and B). However, large amounts of porin purified with the mitochondrial fraction whereas PilC and α-toxin were always absent from this compartment. Pro-cathepsin D (Figure 6A) and transferrin receptor (not shown) did not copurify with the mitochondrial compartment, suggesting the absence of endosomal and cytoplasmic membranes. To test whether porin is also transported to mitochondria during infection with N.gonorrhoeae, HeLa cells were infected with either the adherent and invasive Opa-expressing strain N242 (Figure 6C) or the adherent, piliated strain N138 (Figure 6D), both of which induce apoptosis in HeLa cells (Müller et al., 1999). Highly purified mitochondria from these cells contain large amounts of porin protein. Indeed, porin was the only neisserial antigen detectable in mitochondria from infected cells. Neither Opa outer membrane proteins (Figure 6C) nor PilC (Figure 6D) were found in the same mitochondrial preparations, suggesting a selective transport of porin to the mitochondria of infected cells. Porin was not present in mitochondria of HeLa cells infected with non-adherent gonococcal strains, e.g. N898 (Figure 6D) and commensal Neisseria spp. (not shown), which are unable to induce apoptosis. Confocal immunomicroscopy of porin-treated or infected cells using purified antiserum against porin revealed the presence of porin in mitochondria that appeared extremely enlarged (Figure 7). In summary, porin is efficiently and selectively transported to the mitochondria of porin-treated or infected cells undergoing apoptosis.

Fig. 6. Porin is targeted to the mitochondria of porin-treated and infected cells. (A) HeLa cells were treated with 7 µg/ml porin or respective amounts of staphylococcal α-toxin for 15 h. Lysates were prepared from an aliquot of every sample. In parallel, mitochondria were prepared and both cell lysates and mitochondrial preparations were subjected to western blotting using specific antibodies against porin, α-toxin, cathepsin D and cytochrome c oxidase. Detection was performed using the ECL system (Amersham) according to the manufacturer’s instructions. (B) HeLa cells were treated with 5 µg/ml purified PilC for 15 h. Cell lysates and mitochondria were prepared from treated and control cells and were subjected to western blotting using specific antibodies against PilC and cytochrome c oxidase. Detection was performed as described above. (C) HeLa cells were infected with gonococcal strain N242 for 15 h at an m.o.i. of 1. Lysates and mitochondria were prepared from infected and non-infected control cells and subjected to western blotting using specific antibodies against Opa proteins, porin, cathepsin D and cytochrome c oxidase. Detection was performed as described above. (D) HeLa cells were infected with piliated (N138) and non-piliated (N898), isogenic derivatives of gonococcal strain MS11 for 15 h at an m.o.i. of 1. Lysates and mitochondria were prepared from infected and non-infected control cells and subjected to western blotting using specific antibodies against PilC, porin and cathepsin D. Detection was performed as described above. L, lysate; M, mitochondria.

Fig. 7. Porin is targeted to the mitochondria of porin-treated and infected cells. HeLa cells were infected with the non-piliated, Opa-positive gonococcal strain VP1 (N242) for 3 h at an m.o.i. of 1 or treated with 7 µg/ml porin for the same time. Infected, treated and control cells were double stained with MitoTracker and a polyclonal, porin-specific antiserum followed by an Alexa-488-labelled secondary antibody. Single fluorescence pictures and overlays are shown. White arrows point to bacteria, blue arrows to mitochondria.
Interference with porin translocation prevents apoptosis
We noticed that induction of apoptosis by porin is highly sensitive to serum. In the presence of 10% heat-inactivated fetal calf serum (FCS) the strong pro-apoptotic effect of porin was completely blocked (Figure 8A). The inhibitory effect of serum was not due to degradation since the overall amount of porin was similar during incubation in the absence or presence of serum (data not shown). However, in the presence of serum, porin was no longer present in the cell lysate and therefore was also absent from mitochondria prepared from treated cells (Figure 8B). Thus, translocation of porin is an absolute prerequisite for its pro-apoptotic effect.

Fig. 8. FCS blocks apoptosis and porin insertion. (A) HeLa cells were treated with 7 µg/ml porin for 15 h in the presence or absence of 10% FCS. Cells were harvested and stained with annexin V. (B) HeLa cells were treated with porin for 15 h in the presence or absence of 10% FCS. Lysates and mitochondria were prepared and subjected to western blotting using a specific antibody against gonococcal porin variant P.IA.
Discussion
It has become increasingly apparent that mitochondria participate in the regulation of those forms of apoptotic cell death that do not involve activation of upstream caspases via signalling cascades triggered by death receptor ligation (Scaffidi et al., 1998; Yoshida et al., 1998). However, the significance of single events observed at the mitochondrial level for the overall regulatory process and especially the exact sequence of events is still controversially debated (for a review see Green and Reed, 1998). Here we describe a new mechanism of mitochondria-dependent apoptosis induction occurring naturally during infection of human cells by N.gonorrhoeae.
As in most other complex apoptosis scenarios, cells either infected with N.gonorrhoeae or treated with neisserial porin display a release of cytochrome c into the cytosol and a complete loss of the mitochondrial membrane potential (PT). We did not investigate the sequence of these events in detail and thus do not know whether PT and cytochrome c release are functionally connected. In such diverse systems as CD95(Fas/Apo-1)-induced apoptosis in Jurkat T cells (Van der Heiden et al., 1997) and UVB irradiation or staurosporine treatment of HeLa cells (Bossy-Wetzel et al., 1998), cytochrome c release was observed before membrane depolarization. Under the experimental conditions cited, PT does not take place until several hours after cytochrome c release, thereby ruling out the possibility that loss of cytochrome c results immediately in disconnection of the respiratory chain and membrane depolarization. PT here occurs rather as a result of caspase activation by the ‘apoptosome’, a cytosolic complex consisting of dATP, cytochrome c, Apaf-1 and caspase-9. This is in contrast to our observation of PT in porin-treated cells in the absence of caspase activity (Figure 2). It is currently not known whether porin-induced PT is a prerequisite for cytochrome c release. A remarkable example of PT playing a role in downstream caspase activation is given by the HIV-1 viral protein R (Vpr), which targets mitochondria if added as purified protein or after overexpression in transfected cells (Jacotot et al., 2000). Inhibition of Vpr-induced PT, which is accomplished via interaction with the ANT, also prevented caspase activation and apoptosis.
Overexpression of anti-apoptotic Bcl family members such as Bcl-2 or Bcl-XL completely inhibited porin-induced apoptosis as determined by morphological criteria and PS exposure. Active caspases were not detected and PT as well as the release of cytochrome c was completely blocked in cell lines stably expressing Bcl-2 or Bcl-XL upon porin treatment. These observations are in accordance with the results obtained in other systems, which suggest that anti-apoptotic Bcl family members act in mitochondria at the level of cytochrome c release (Kluck et al., 1997; Van der Heiden et al., 1997; Yang et al., 1997; Shimizu et al., 1999).
Several models attempting to explain the mechanism of the release of cytochrome c and other caspase-activating proteins are currently discussed. One hypothesis suggests that hyperpolarization-induced mitochondrial swelling results in outer membrane rupture followed by release of mitochondrial components from the intermembrane space into the cytosol (Van der Heiden et al., 1997). Another possibility is the formation of large pores by Bcl family proteins such as Bax (Antonsson et al., 2000) and still another model favours rapid loss of inner membrane potential followed by PT (Kroemer et al., 1997). All models are capable of explaining the massive leakage of not only cytochrome c and AIF, but also an increasing number of other constituents of the mitochondrial intermembrane space into the cytosol. Among these are active caspases-2 and -9 (Susin et al., 1999a), caspase-3 (Mancini et al., 1998; Samali et al., 1998), AIF (Susin et al., 1999b) and the mitochondrial chaperones HSP60 and HSP10 (Samali et al., 1999; Xanthoudakis et al., 1999). However, none of the models fully explains the phenotype we obtained. The model proposing mitochondrial swelling and outer membrane rupture is in agreement with the swelling we observed when isolated mitochondria were treated with porin in vitro or when cells were infected with N.gonorrhoeae. The effects of porin on isolated mitochondria argue that cytochrome c release occurs as a result of channel formation. The release of cytochrome c might involve the opening of a pre-existing channel, e.g. by direct binding to one of its components, as was recently described for an interaction of Bax and ANT (Marzo et al., 1998c) or Bax and VDAC (Shimizu et al., 1999).
Several caspases have been shown to induce the release of cytochrome c (Marzo et al., 1998b). However, since mitochondria of porin-treated cells completely lost their cytochrome c in the presence of the caspase inhibitor zVAD we can exclude this possibility.
Another completely different explanation for the observed mitochondrial changes involves elevations in cytosolic Ca2+. Porin induces a very rapid transient increase in intracellular Ca2+, thereby activating the Ca2+-dependent protease calpain (Müller et al., 1999). One can speculate that increased Ca2+ levels may also be sufficient to induce PT, as was shown previously in vitro (Marzo et al., 1998a) and in vivo (Bernardi and Petronilli, 1996). However, porin induced PT in the absence of extracellular Ca2+, indicating that the influx of Ca2+ is not the primary trigger for PT.
It is an intriguing feature of the neisserial porin that it translocates from the outer bacterial into artificial membranes and host cell membranes (Lynch et al., 1984; Weel and van Putten, 1991; Rudel et al., 1996). Biochemical analysis of purified mitochondria from porin-treated as well as infected cells clearly reveals that ∼50% of all porin found in the cell is localized to these organelles, whereas other neisserial antigens tested were absent. Among these were Opa proteins, very abundant integral proteins in the outer membrane of the gonococcal strains used in this study, which also form several amphipathic β-strands. PilC is found in pili (Rudel et al., 1995a) as well as in the outer membrane (Rudel et al., 1995b; Rahman et al., 1997). Both proteins can thus be considered adequate controls for elucidating artificial fusion of outer membrane vesicles with mitochondria. However, neither Opa nor PilC was found in the mitochondrial preparations of infected cells. To our knowledge, this is the first example ever of a bacterial antigen being transported to host cell mitochondria. Interestingly, the capability of a strain to translocate its porin into host cell membranes is dependent on adherence and correlates with its ability to induce apoptosis. This and the fact that FCS blocks both porin insertion and apoptosis induction argues for a functional connection between these processes.
While the pro-apoptotic effects of porin targeting on the organelles could be demonstrated, the mechanism of the targeting process is completely unknown. One might hypothesize that porin residing in the plasma membrane is somehow internalized by a process similar to recycling of surface receptors and, once inside, couples to the regular intracellular vesicle transport. This would require active participation of the cytoskeleton. However, inhibitors of actin reorganization (cytochalasin D) or microtubules (colchicin) were unable to block targeting of porin to mitochondria (not shown). Furthermore, cross-linking experiments revealed that porin located in mitochondria forms multimers consisting of two and three monomers, i.e. the same complexes that can also be observed in the outer membrane of gonococci. In addition, the process of intracellular trafficking seems to be very fast, since traces of porin were already present in purified mitochondria as early as 20 min post-infection. We therefore assume that the porin moves intracellularly either by means of a soluble intermediate or that it actually has the ability to ‘jump’ from one membrane to another by an as yet undefined mechanism. Recently, the M11L protein of myxoma virus was shown to target mitochondria via a C-terminal targeting signal sequence that conforms to a newly described consensus sequence present in Bcl family members (Everett et al., 2000). Experiments are currently under way to identify the essential epitopes for mitochondrial targeting of porin. The molecular basis of the insertion of mitochondrial porin into the outer mitochondrial membrane has only recently been elucidated (Schleiff et al., 1999). Integration into the lipid bilayer is accordingly mediated by the mitochondrial receptor Tom20 but bypasses the requirement for the channel-forming complex Tom40. It will be interesting to see if gene deletions in essential components of the mitochondrial import machinery have an effect on neisserial porin insertion similar to that on mitochondrial porin insertion (Sollner et al., 1989).
Although the precise composition of the PT complex is not yet defined it is remarkable that nearly all components identified so far have functional and structural homologues in the bacterial kingdom. The adenine-nucleotide translocator and the peripheral benzodiazepine receptor homologue are found in Rhodobacter capsulatus (Armstrong et al., 1989; Carmeli and Lifshitz, 1989). Interestingly, cytochrome c, one of the major effectors of mitochondria-mediated caspase activation, is generally found localized to the outer side of the inner membrane in aerobic bacteria. Furthermore, members of the Bcl-2 family exhibit structural homology to colicins, pore-forming toxins produced by Escherichia coli (Muchmore et al., 1996). Porins of Gram-negative bacteria (Mannella, 1998) exhibit structural and, in the case of the Neisseria porin (Rudel et al., 1996), also functional homologies to eukaryotic porins. Recently, the apoptosis-inducing factor was identified and shown to be homologous to bacterial oxidoreductases (Susin et al., 1999b). Thus, on the basis of these findings there is overwhelming support for the recently raised hypothesis that endosymbiosis of bacteria brought not only aerobic life to the early eukaryotes but also the basic machinery for programmed cell death, a critical step from unicellular to multicellular life (Frade and Michaelidis, 1997; Kroemer, 1997b).
The system of apoptosis induction described in this and our previous study (Müller et al., 1999) employs an infection model of human cells cocultured with the bacterial pathogen N.gonorrhoeae. The main advantage of this system for investigating basic questions related to mitochondrial function is that all of the observed morphological and biochemical effects linked to apoptosis induced by the pathogen can be achieved by a single bacterial factor, the neisserial porin. This factor is unique in that it acts directly on purified mitochondria, exerting the same effects (i.e. cytochrome c release and loss of membrane potential) as in vivo. Since the porin can be detected in the mitochondria of infected cells, this in vitro system simplifies the situation without rendering it completely artificial. In addition, the similarities between the neisserial porin and the mitochondrial porin, which is reportedly involved in mitochondrial regulation of apoptosis, invite speculations on aspects of the evolution of the apoptotic machinery.
Materials and methods
Cell cultures and bacterial strains
All cell lines were cultured in RPMI supplemented with 10% FCS. Bcl-2- and neomycin control vector (Neo)-transfected Jurkat cells additionally received 200 µg/ml G418. Strains N242 (P–, O+), N138 (P+, O–) and its non-piliated derivative N898 (P–, O–) have been described previously (Müller et al., 1999).
Purification of mitochondria
The procedure was modified from Scaffidi et al. (1998). In short, cells were harvested by centrifugation at 400 g and washed once with ice-cold phosphate-buffered saline (PBS). All subsequent centrifugation steps were performed at 4°C. After another wash with MB buffer [400 mM sucrose, 50 mM Tris, 1 mM EGTA, 5 mM β-mercaptoethanol, 0.2% bovine serum albumin (BSA), 10 mM KH2PO4 pH 7.6], the pellet was resuspended in 2 ml of the same buffer and incubated for 20 min on ice. The cells were then homogenized with 35 strokes of a Kontes douncer. Cell debris was removed by centrifugation at 4000 g for 1 min, then the supernatant was centrifuged for 10 min at 15 000 g to precipitate the mitochondria. The pellet was then resuspended in MSM buffer (10 mM KH2PO4, 0.3 mM mannitol, 0.1% BSA pH 7.2) and loaded on to a sucrose gradient consisting of 3 ml of SA (lower) buffer (1.6 M sucrose, 10 mM KH2PO4, 0.1% BSA pH 7.5) and 3 ml of SB (upper) buffer (1.2 M sucrose, 10 mM KH2PO4, 0.1% BSA pH 7.5). The gradient was centrifuged at 72 000 g in a swinging bucket rotor for 1 h. The mitochondria that had accumulated in the interphase were harvested, supplemented with 4 vols of MSM buffer to dilute the sucrose and pelleted by centrifugation at 45 000 g for 10 min. The resulting pellet was resuspended either in sample solution (for electrophoresis and immunoblotting) or in MSM buffer without BSA (for in vitro experiments).
Electrophoresis and immunoblotting
Proteins were separated under reducing conditions for 1 h at 160 V in 7, 10 or 12% SDS–polyacrylamide gels and then blotted overnight at 100 mA on to a PVDF membrane (Millipore). The membrane was blocked for 1 h in 3% BSA in Tris-buffered saline (TBS)–0.1% Tween, and then incubated with monoclonal antibodies against cytochrome c (clone 7H8.2C12; Pharmingen, San Diego, CA), cytochrome c oxidase subunit II (Molecular Probes, Eugene, OR), fodrin (Chemicon, Temecula, CA), α-toxin (a generous gift from S.Bhakdi, Mainz, Germany), Opa (a generous gift from M.Achtman, Berlin, Germany), gonococcal porin P.IA and polyclonal antisera against cathepsin D (DAKO, Copenhagen, Denmark) and gonococcal PilC. After three washes in TBS–0.1% Tween, the filter was incubated for 1 h with peroxidase-coupled secondary antibody and bound antibody was detected by enhanced chemiluminescence (Amersham).
Assessment of mitochondrial membrane potential
Cells were incubated with 7 µg/ml porin for 15 h and stained with 5 µg/ml rhodamine 123 (Molecular Probes, Eugene, OR), which was added to the culture medium for 30 min at 37°C. The stained cells were harvested, washed once with PBS and analysed on a FACS-Calibur flow cytometer (Becton Dickinson). Rhodamine fluorescence was monitored in channel 2.
Mitochondrial swelling assay
Mitochondrial swelling assays were performed as described (Uyemura et al., 1997). Briefly, mouse liver mitochondria were purified and resuspended in 125 mM sucrose, 65 mM KCl, 5 mM succinate, 5 µM rotenone, 20 mM CaCl2, 10 mM HEPES pH 7.5, at 0.3 mg/ml. Porin or Ca2+ was added to the mitochondria suspension and the OD600 of the suspension was measured every 30 s for 10 min. The decrease in the optical density correlates with the increase in mitochondrial volume. Since the untreated mitochondria swell spontaneously the same volume of low concentrated porin, which had no effect on swelling, was used as a buffer control. The comparison of the untreated or buffer-treated mitochondria and the porin or Ca2+-treated mitochondria indicates the relative potential of these substances to induce swelling.
In vitro assay for mitochondrial cytochrome c release and PT
Mitochondria were isolated from 5 × 107 cells as described and resuspended in MSM buffer without BSA. A stimulus (e.g. porin or buffer) was applied for 30 min at 37°C to an aliquot of the preparation. The mitochondria were then pelleted at 18 000 g for 5 min and resuspended in sample buffer, whereas the proteins contained in the supernatant were precipitated with trichloracetic acid before resuspension in sample buffer. SDS–PAGE was performed as described above. Alternatively, mitochondria were stained with 5 µg/ml rhodamine 123 and analysed by flow cytometry.
Quantification of PS exposure
The flipping of PS from the inner to the outer leaflet of the plasma membrane is assessed by in vitro binding of the serum factor annexin V, which is coupled to FITC for detection by flow cytometry. Cells were treated with porin for 15 h, harvested, washed twice in PBS and suspended in 100 µl of binding buffer (10 mM HEPES–NaOH pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). Annexin V–FITC was added and cells were incubated for 15 min in the dark. After one wash, cells were resuspended in 200 µl of binding buffer and counterstained with 1 µg/ml propidium iodide for determination of permeable (necrotic) cells. Ten thousand cells per sample were analysed with a Becton Dickinson FACS-Calibur equipped with a 15 mW, 488 nm air-cooled argon laser using Cell Quest software. Histograms showing the FITC fluorescence intensity in 1024 channels revealed the amount of annexin V–FITC-positive cells that were determined as apoptotic.
Immunofluorescence microscopy
Cells were seeded on coverslips, treated with porin for 15 h or infected with N.gonorrhoeae, and fixed in 3% paraformaldehyde. Fixed cells were permeabilized using 0.2% Triton X-100; non-specific binding was blocked by 30 min of incubation in goat serum, and staining was performed using a monoclonal antibody against native cytochrome c (Pharmingen, San Diego, CA) or a purified polyclonal anti-P.IA serum followed by an Alexa-488-coupled secondary antibody. Colocalization experiments required staining of live mitochondria with 150 nM MitoTracker (Molecular Probes, Eugene, OR) for 30 min at 37°C before fixation. In the case of suspension cells, the staining procedure described above was performed before cytospinning of the cells on to coverslips. Samples were analysed on a Leica confocal microscope using TCS software.
Acknowledgments
Acknowledgements
We thank Dr Marc Achtman for providing a monoclonal antibody against Opa and Dr Sucharit Bhakdi for donating purified staphylococcal α-toxin as well as the specific antibody. Purified PilC was kindly provided by Marieluise Kirchner. We are grateful to Drs A.Strasser and M.Peter for sending stable cell lines, to Dr M.Peter for helpful instructions on mitochondria purification protocols and to Dr Oliver Billker for critically reading the manuscript.
References
- Antonsson B. et al. (1997) Inhibition of Bax channel-forming activity by Bcl-2. Science, 277, 370–372. [DOI] [PubMed] [Google Scholar]
- Antonsson B., Montessuit,S., Lauper,S., Eskes,R. and Martinou,J.C. (2000) Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome C release from mitochondria. Biochem. J., 345, 271–278. [PMC free article] [PubMed] [Google Scholar]
- Armstrong G.A., Alberti,M., Leach,F. and Hearst,J.E. (1989) Nucleotide sequence, organization and nature of the protein products of the carotenoid biosynthesis gene cluster of Rhodobacter capsulatus. Mol. Gen. Genet., 216, 254–268. [DOI] [PubMed] [Google Scholar]
- Benz R. (1994) Permeation of hydrophilic solutes through mitochondrial outer membranes: review on mitochondrial porins. Biochim. Biophys. Acta, 1197, 167–196. [DOI] [PubMed] [Google Scholar]
- Benz R. (1995) Uptake of solutes through bacterial outer membranes. In Ghuysen,J.M. and Hakenbeck,R. (eds), Bacterial Cell Wall. Elsevier Science Publishers B.V., Amsterdam, The Netherlands, pp. 397–423. [Google Scholar]
- Bernardi P. and Petronilli,V. (1996) The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J. Bioenerget. Biomembr., 28, 131–138. [DOI] [PubMed] [Google Scholar]
- Beutner G., Ruck,A., Riede,B. and Brdiczka,D. (1998) Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim. Biophys. Acta, 1368, 7–18. [DOI] [PubMed] [Google Scholar]
- Boldin M.P., Goncharov,T.M., Goltsev,Y.V. and Wallach,D. (1996) Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/Apo-1 and TNF receptor-induced cell death. Cell, 85, 803–815. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E., Newmeyer,D.D. and Green,D.R. (1998) Mitochondrial cytochrome-c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J., 17, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N. and Klingenberg,M. (1996) Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry, 35, 8483–8488. [DOI] [PubMed] [Google Scholar]
- Carmeli C. and Lifshitz,Y. (1989) Nucleotide transport in Rhodobacter capsulatus. J. Bacteriol., 171, 6521–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns V.L., Bergeron,L., Zhu,H., Li,H. and Yuan,J. (1996) Specific cleavage of α-fodrin during Fas- and tumor necrosis factor-induced apoptosis is mediated by an interleukin-1β-converting enzyme/Ced-3 protease distinct from the poly(ADP-ribose) polymerase protease. J. Biol. Chem., 271, 31277–31282. [DOI] [PubMed] [Google Scholar]
- Decaudin D., Geley,S., Hirsch,T., Castedo,M., Marchetti,P., Macho,A., Kofler,R. and Kroemer,G. (1997) Bcl-2 and Bcl-XL antagonize the mitochondrial dysfunction preceding nuclear apoptosis induced by chemotherapeutic agents. Cancer Res., 57, 62–67. [PubMed] [Google Scholar]
- Everett H., Barry,M., Lee,S.F., Sun,X.J., Graham,K., Stone,J., Bleackley,R.C. and Mcfadden,G. (2000) M11L: a novel mitochondria-localized protein of myxoma virus that blocks apoptosis of infected leukocytes. J. Exp. Med., 191, 1487–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade J.M. and Michaelidis,T.M. (1997) Origin of eukaryotic programmed cell death—a consequence of aerobic metabolism. BioEssays, 19, 827–832. [DOI] [PubMed] [Google Scholar]
- Gray-Owen S.D., Dehio,C., Haude,A., Grunert,F. and Meyer,T.F. (1997) CD66 carcinoembryonic antigens mediate interactions between Opa-expressing Neisseria gonorrhoeae and human polymorphonuclear phagocytes. EMBO J., 16, 3435–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green D.R. and Reed,J.C. (1998) Mitochondria and apoptosis. Science, 281, 1309–1312. [DOI] [PubMed] [Google Scholar]
- Hauck C.R., Meyer,T.F., Lang,F. and Gulbins,E. (1998) Cd66-mediated phagocytosis of Opa(52) Neisseria gonorrhoeae requires a src-like tyrosine kinase-dependent and rac1-dependent signaling pathway. EMBO J., 17, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicke R.U., Ng,P., Sprengart,M.L. and Porter,A.G. (1998) Caspase-3 is required for α-fodrin cleavage but dispensable for cleavage of other death substrates in apoptosis. J. Biol. Chem., 273, 15540–15545. [DOI] [PubMed] [Google Scholar]
- Jacotot E. et al. (2000) The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med., 191, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas D., Walev,I., Berger,T., Liebetrau,M., Palmer,M. and Bhakdi,S. (1994) Novel path to apoptosis: small transmembrane pores created by staphylococcal α-toxin in T lymphocytes evoke internucleosomal DNA degradation. Infect. Immun., 62, 1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jürgensmeier J.M., Xie,Z.H., Deveraux,Q., Ellerby,L., Bredesen,D. and Reed,J.C. (1998) Bax directly induces release of cytochrome-c from isolated mitochondria. Proc. Natl Acad. Sci. USA, 95, 4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel,E., Green,D.R. and Newmeyer,D.D. (1997) The release of cytochrome C from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science, 275, 1132–1136. [DOI] [PubMed] [Google Scholar]
- Kroemer G. (1997a) The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nature Med., 3, 614–620. [DOI] [PubMed] [Google Scholar]
- Kroemer G. (1997b) Mitochondrial implication in apoptosis—towards an endosymbiont hypothesis of apoptosis evolution. Cell Death Differ., 4, 443–456. [DOI] [PubMed] [Google Scholar]
- Kroemer G., Zamzami,N. and Susin,S.A. (1997) Mitochondrial control of apoptosis. Immunol. Today, 18, 44–51. [DOI] [PubMed] [Google Scholar]
- Kroemer G., Dallaporta,B. and Rescherigon,M. (1998) The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol., 60, 619–642. [DOI] [PubMed] [Google Scholar]
- Liu X., Kim,C.N., Yang,J., Jemmerson,R. and Wang,X. (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell, 86, 147–157. [DOI] [PubMed] [Google Scholar]
- Lynch E.C., Blake,M.S., Gotschlich,E.C. and Mauro,A. (1984) Studies on porins spontaneously transferred from whole cells and from proteins of Neisseria gonorrhoeae and Neisseria meningitidis. Biophys. J., 45, 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S., van Putten,J.P. and Meyer,T.F. (1991) Phase variation of the opacity outer membrane protein controls invasion by Neisseria gonorrhoeae into human epithelial cells. EMBO J., 10, 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini M., Nicholson,D.W., Roy,S., Thornberry,N.A., Peterson,E.P., Casciolarosen,L.A. and Rosen,A. (1998) The caspase-3 precursor has a cytosolic and mitochondrial distribution—implications for apoptotic signaling. J. Cell Biol., 140, 1485–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannella C.A. (1998) Conformational changes in the mitochondrial channel protein, VDAC and their functional implications. J. Struct. Biol., 121, 207–218. [DOI] [PubMed] [Google Scholar]
- Marzo I. et al. (1998a) The permeability transition pore complex—a target for apoptosis regulation by caspases and bcl-2-related proteins. J. Exp. Med., 187, 1261–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I., Susin,S.A., Petit,P.X., Ravagnan,L., Brenner,C., Larochette,N., Zamzami,N. and Kroemer,G. (1998b) Caspases disrupt mitochondrial-membrane barrier function. FEBS Lett., 427, 198–202. [DOI] [PubMed] [Google Scholar]
- Marzo I. et al. (1998c) Bax and adenine-nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science, 281, 2027–2031. [DOI] [PubMed] [Google Scholar]
- Metzstein M.M., Stanfield,G.M. and Horvitz,H.R. (1998) Genetics of programmed cell-death in C.elegans—past, present and future. Trends Genet., 14, 410–416. [DOI] [PubMed] [Google Scholar]
- Minn A.J., Velez,P., Schendel,S.L., Liang,H., Muchmore,S.W., Fesik,S.W., Fill,M. and Thompson,C.B. (1997) Bcl-XL forms an ion channel in synthetic lipid membranes. Nature, 385, 353–357. [DOI] [PubMed] [Google Scholar]
- Muchmore S.W. et al. (1996) X-ray and NMR structure of human Bcl-XL, an inhibitor of programmed cell death. Nature, 381, 335–341. [DOI] [PubMed] [Google Scholar]
- Müller A., Günther,D., Düx,F., Naumann,M., Meyer,T.F. and Rudel,T. (1999) Neisserial porin (PorB) causes rapid calcium influx in target cells and induces apoptosis by the activation of cysteine proteases. EMBO J., 18, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzio M. et al. (1996) FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/Apo-1) death-inducing signaling complex. Cell, 85, 817–827. [DOI] [PubMed] [Google Scholar]
- Nath R. et al. (1996) Non-erythroid α-spectrin breakdown by calpain and interleukin 1-β-converting-enzyme-like protease(s) in apoptotic cells—contributory roles of both protease families in neuronal apoptosis. Biochem. J., 319, 683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson D.W. and Thornberry,N.A. (1997) Caspases—killer proteases. Trends Biochem. Sci., 22, 299–306. [DOI] [PubMed] [Google Scholar]
- Pastorino J.G., Simbula,G., Gilfor,E., Hoek,J.B. and Farber,J.L. (1994) Protoporphyrin IX, an endogenous ligand of the peripheral benzodiazepine receptor, potentiates induction of the mitochondrial permeability transition and the killing of cultured hepatocytes by rotenone. J. Biol. Chem., 269, 31041–31046. [PubMed] [Google Scholar]
- Rahman M., Kallstrom,H., Normark,S. and Jonsson,A.B. (1997) PilC of pathogenic Neisseria is associated with the bacterial cell surface. Mol. Microbiol., 25, 11–25. [DOI] [PubMed] [Google Scholar]
- Rudel T., Scheuerpflug,I. and Meyer,T.F. (1995a) Neisseria PilC protein identified as type-4 pilus tip-located adhesin. Nature, 373, 357–359. [DOI] [PubMed] [Google Scholar]
- Rudel T., Facius,D., Barten,R., Scheuerpflug,I., Nonnenmacher,E. and Meyer,T.F. (1995b) Role of pili and the phase-variable PilC protein in natural competence for transformation of Neisseria gonorrhoeae. Proc. Natl Acad. Sci. USA, 92, 7986–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudel T., Schmid,A., Benz,R., Kolb,H.A., Lang,F. and Meyer,T.F. (1996) Modulation of Neisseria porin (PorB) by cytosolic ATP/GTP of target cells: parallels between pathogen accommodation and mitochondrial endosymbiosis. Cell, 85, 391–402. [DOI] [PubMed] [Google Scholar]
- Salvesen G.S. and Dixit,V.M. (1997) Caspases—intracellular signaling by proteolysis. Cell, 91, 443–446. [DOI] [PubMed] [Google Scholar]
- Samali A., Zhivotovsky,B., Jones,D.P. and Orrenius,S. (1998) Detection of pro-caspase-3 in cytosol and mitochondria of various tissues. FEBS Lett., 431, 167–169. [DOI] [PubMed] [Google Scholar]
- Samali A., Cai,J.Y., Zhivotovsky,B., Jones,D.P. and Orrenius,S. (1999) Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of Jurkat cells. EMBO J., 18, 2040–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C., Fulda,S., Srinivasan,A., Friesen,C., Li,F., Tomaselli,K.J., Debatin,K.M., Krammer,P.H. and Peter,M.E. (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J., 17, 1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiff E., Silvius,J.R. and Shore,G.C. (1999) Direct membrane insertion of voltage-dependent anion-selective channel protein catalyzed by mitochondrial Tom20. J. Cell Biol., 145, 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S., Narita,M. and Tsujimoto,Y. (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome C by the mitochondrial channel VDAC. Nature, 399, 483–487. [DOI] [PubMed] [Google Scholar]
- Slee E.A. et al. (1999) Ordering the cytochrome c-initiated caspase cascade: Hierarchical activation of caspases-2, -3, -6, -7, -8 and -10 in a caspase-9-dependent manner. J. Cell Biol., 144, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollner T., Griffiths,G., Pfaller,R., Pfanner,N. and Neupert,W. (1989) MOM19, an import receptor for mitochondrial precursor proteins. Cell, 59, 1061–1070. [DOI] [PubMed] [Google Scholar]
- Song L., Hobaugh,M.R., Shustak,C., Cheley,S., Bayley,H. and Gouaux,J.E. (1996) Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science, 274, 1859–1866. [DOI] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo,H.K., Zamzami,N., Marzo,I., Brenner,C., Larochette,N., Prevost,M.C., Alzari,P.M. and Kroemer,G. (1999a) Mitochondrial release of caspase-2 and -9 during the apoptotic process. J. Exp. Med., 189, 381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A. et al. (1999b) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature, 397, 441–446. [DOI] [PubMed] [Google Scholar]
- Swanson J., Robbins,K., Barrera,O., Corwin,D., Boslego,J., Ciak,J., Blake,M.S. and Koomey,J.M. (1987) Gonococcal pilin variants in experimental gonorrhoea. J. Exp. Med., 165, 1344–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari M., Quan,L.T., O’Rourke,K., Desnoyers,S., Zeng,Z., Beidler,D.R., Poirier,G.G., Salvesen,G.S. and Dixit,V.M. (1995) Yama/CPP32 β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell, 81, 801–809. [DOI] [PubMed] [Google Scholar]
- Uyemura S.A., Santos,A.C., Mingatto,F.E., Jordani,M.C. and Curti,C. (1997) Diclofenac sodium and mefenamic acid: potent inducers of the membrane permeability transition in renal cortex mitochondria. Arch. Biochem. Biophys., 342, 231–235. [DOI] [PubMed] [Google Scholar]
- Van der Heiden M.G., Chandel,N.S., Williamson,E.K., Schumacker,P.T. and Thompson,C.B. (1997) Bcl-x(l) regulates the membrane-potential and volume homeostasis of mitochondria. Cell, 91, 627–637. [DOI] [PubMed] [Google Scholar]
- Wallach D., Boldin,M., Varfolomeev,E., Beyaert,R., Vandenabeele,P. and Fiers,W. (1997) Cell death induction by receptors of the TNF family—towards a molecular understanding. FEBS Lett., 410, 96–106. [DOI] [PubMed] [Google Scholar]
- Weel J.F. and van Putten,J.P. (1991) Fate of the major outer membrane protein P.IA in early and late events of gonococcal infection of epithelial cells. Res. Microbiol., 142, 985–993. [DOI] [PubMed] [Google Scholar]
- Xanthoudakis S. et al. (1999) Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J., 18, 2049–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J., Chao,D.T. and Korsmeyer,S.J. (1996) BAX-induced cell death may not require interleukin 1β-converting enzyme-like proteases. Proc. Natl Acad. Sci. USA, 93, 14559–14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E. and Korsmeyer,S.J. (1996) Molecular thanatopsis: a discourse on the BCL2 family and cell death. Blood, 88, 386–401. [PubMed] [Google Scholar]
- Yang J., Liu,X., Bhalla,K., Kim,C.N., Ibrado,A.M., Cai,J., Peng,T.I., Jones,D.P. and Wang,X. (1997) Prevention of apoptosis by Bcl-2: release of cytochrome C from mitochondria blocked. Science, 275, 1129–1132. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Kong,Y.Y., Yoshida,R., Elia,A.J., Hakem,A., Hakem,R., Penninger,J.M. and Mak,T.W. (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain-development. Cell, 94, 739–750. [DOI] [PubMed] [Google Scholar]
- Zamzami N., Marchetti,P., Castedo,M., Zanin,C., Vayssiere,J.L., Petit,P.X. and Kroemer,G. (1995) Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J. Exp. Med., 181, 1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N., Susin,S.A., Marchetti,P., Hirsch,T., Gomez-Monterrey,I., Castedo,M. and Kroemer,G. (1996) Mitochondrial control of nuclear apoptosis. J. Exp. Med., 183, 1533–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M. and Szabo,I. (1995) The mitochondrial permeability transition. Biochim. Biophys. Acta, 1241, 139–176. [DOI] [PubMed] [Google Scholar]
- Zou H., Henzel,W.J., Liu,X., Lutschg,A. and Wang,X. (1997) Apaf-1, a human protein homologous to C.elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell, 90, 405–413. [DOI] [PubMed] [Google Scholar]