

Abstract
The Raf kinase family serves as a central intermediate to relay signals from Ras to ERK. The precise molecular mechanism for Raf activation is still not fully understood. Here we report that phosphorylation of Thr598 and Ser601, which lie between kinase subdomains VII and VIII, is essential for B-Raf activation by Ras. Substitution of these residues by alanine (B-RafAA) abolished Ras-induced B-Raf activation without altering the association of B-Raf with other signaling proteins. Phosphopeptide mapping and immunoblotting with phospho-specific antibodies confirmed that Thr598 and Ser601 are in vivo phosphorylation sites induced by Ras. Furthermore, replacement of these two sites by acidic residues (B-RafED) renders B-Raf constitutively active. Con sistent with these data, B-RafAA and B-RafED exhibited diminished and enhanced ability, respectively, to stimulate ERK activation and Elk-dependent transcription. Moreover, functional studies revealed that B-RafED was able to promote NIH 3T3 cell transformation and PC12 cell differentiation. Since Thr598 and Ser601 are conserved in all Raf family members from Caenorhabditis elegans to mammals, we propose that phosphorylation of these two residues may be a general mechanism for Raf activation.
Keywords: B-Raf/kinase activity/phosphorylation/Ras
Introduction
The Raf proteins are a family of serine/threonine-specific kinases that serve as a central intermediate in transmitting extracellular signals to the mitogen-activated protein kinase (MAPK), also known as the extracellular signal regulated kinase (ERK) cascade, which controls cell growth, differentiation and survival (Marshall, 1995; Morrison and Cutler, 1997; English et al., 1999; Hagemann and Rapp, 1999). Activation of the small GTPase protein Ras is an initial step in the activation of Raf. Activated Raf proteins directly phosphorylate and activate the downstream dual specificity kinase MEK, which in turn phosphorylates ERK on threonine and tyrosine residues and results in a dramatic activation of ERK. Activated ERK is critical for numerous Ras-induced cellular responses, including Elk-dependent transcriptional activation of a number of genes (Hill and Treisman, 1995; Lewis et al., 1998).
Three isoforms of Raf proteins have been found in mammalian cells: Raf-1 (or C-Raf), A-Raf and B-Raf (Magnuson et al., 1994). Although the three Raf proteins share high homology in amino acid sequence, it has recently been found that they are differentially regulated and exert different functions (Hagemann and Rapp, 1999). For example, B-Raf has higher affinity and stronger stimulation towards MEK than C-Raf and A-Raf, while A-Raf has the lowest activity due to the weakest binding affinity with Ras (Catling et al., 1994; Reuter et al., 1995; Weber et al., 2000). Mice lacking B-Raf, rather than C-Raf or A-Raf, show disturbances in cell survival, indicating that B-Raf may possess specific functions in cell death regulation (Pritchard et al., 1996; Wojnowski et al., 1997). Indeed, B-Raf specifically promotes cell survival by activating the MAPK pathway (Erhardt et al., 1999). B-Raf is also differentially regulated by cAMP-dependent protein kinase A (PKA) activation through Rap1 GTPase, which specifically activates B-Raf but not C-Raf (Ohtsuka et al., 1996; Vossler et al., 1997; MacNicol and MacNicol, 1999).
Structurally, the Raf proteins can be divided into two functional regions: the N-terminal regulatory domains (CR1 and CR2) and the C-terminal kinase domain (CR3) (Daum et al., 1994). The CR1 region contains Ras binding domains, which are essential for Raf activation by Ras (Vojtek et al., 1993; Nassar et al., 1995; Gorman et al., 1996; Mott et al., 1996). When Ras is activated, Raf translocates from the cytosol to the plasma membrane where it binds to the active GTP-bound Ras and becomes activated. Although multiple steps have been involved in Raf activation, the molecular mechanism of Raf activation is not fully understood. Many of the studies were performed on C-Raf. It is postulated that phosphorylation of Raf is one of the critical steps for Raf activation. However, the effect of phosphorylation on Raf activity is complex because both inhibitory and stimulatory phosphorylation sites have been characterized (Morrison and Cutler, 1997). For example, phosphorylation of Ser43 (possibly by PKA) (Cook and McCormick, 1993; Morrison et al., 1993; Wu et al., 1993; Schramm et al., 1994) or Ser259 (possibly by PKB/AKT) (Michaud et al., 1995; Muslin et al., 1996; Rommel et al., 1999; Zimmermann and Moelling, 1999) exerts an inhibitory effect on C-Raf activity. In contrast, phosphorylation of Thr269 (possibly by CAP/KSR) (Yao et al., 1995; Zhang et al., 1997), Ser338 (possibly by Pak3) (King et al., 1998; Mason et al., 1999), Ser497/Ser499 (possibly by PKC) (Kolch et al., 1993; Carroll and May, 1994; Cai et al., 1997) or Ser621 (Michaud et al., 1995) has a positive effect on C-Raf activity. The effect of Ser621 phosphorylation is more complex in that it may also be involved in negative regulation of B-Raf by PKA (Mischak et al., 1996). Furthermore, tyrosine phosphorylation of Tyr341 activates C-Raf (Fabian et al., 1993; Jelinek et al., 1996; Mason et al., 1999). Using phospho-specific antisera, Mason et al. (1999) have found that active oncogenic Ras induces predominantly Ser338 phosphorylation, whereas activated Src gives predominantly Tyr341 phosphorylation. Moreover, a synergistic activation was observed when both sites are phosphorylated (Mason et al., 1999). However, the current model for C-Raf activation cannot explain the activation of other Raf family members due to the fact that Ser338 and Tyr341 of C-Raf are not completely conserved in other Raf proteins. For example, Caenorhabditis elegans lin-45 Raf contains an aspartate at the position corresponding to Ser338 of C-Raf, whereas Drosophila Raf contains an asparagine residue at the corresponding site of Tyr341. Thus, receptor tyrosine kinases (RTKs) and Ras may induce phosphorylation of other residues to activate C.elegans lin-45 and Drosophila Raf proteins.
Similarly, B-Raf is strongly activated by oncogenic Ras but not by Src, probably due to the replacement of the tyrosine residue by an aspartate at the site equivalent to Tyr341 of C-Raf. Moreover, Ser445 of B-Raf, corresponding to Ser338 of C-Raf, is not involved in Ras-induced B-Raf activation in that it is constitutively phosphorylated and not stimulated by Ras (Mason et al., 1999). Thus, mutation of B-Raf Ser445 to alanine has little effect on Ras-induced B-Raf activation, whereas the corresponding mutation in C-Raf abolishes Ras-dependent activation (Mason et al., 1999). These results demonstrate that the mechanism for regulation of B-Raf activation is different from that for C-Raf. However, a common unidentified biochemical activation event may be shared by all Raf family kinases because all Raf proteins can be activated by Ras and RTKs. Thus, genetic data have unequivocally demonstrated that the C.elegans and Drosophila Raf are activated by RTKs and Ras pathways (Sternberg and Han, 1998). Similarly, biochemical studies in mammalian cells have shown that oncogenic mutant Ras activates A-Raf, B-Raf and C-Raf.
We investigate Raf activation using B-Raf as a model because the C.elegans and Drosophila Raf are closer to B-Raf than C-Raf. Furthermore, B-Raf activation has been studied much less. In this report, we examined the potential phosphorylation sites in B-Raf responsible for Ras-induced activation. Using mutational analysis and phosphopeptide mapping, we demonstrate that Thr598 and Ser601 are the major phosphorylation sites in response to oncogenic Ras, and phosphorylation of these two residues is required for full activation of B-Raf. These two residues, which are located within the kinase activation loop between kinase subdomains VII and VIII of B-Raf, are conserved in C-Raf, but no data have been reported about the importance of these residues in Raf activation. Furthermore, functional studies reveal that phosphorylation of Thr598 and Ser601 is important for B-Raf to induce ERK activation, Elk-1-dependent transcription, NIH 3T3 transformation and PC12 differentiation. These results provide the first evidence that phosphorylation of Thr598 and Ser601 of B-Raf is responsible, at least in part, for oncogenic Ras-induced B-Raf activation. Since Thr598 and Ser601 are completely conserved in all Raf members including C.elegans lin-45, Drosophila Raf and mammalian C-Raf and A-Raf, our data suggest that phosphorylation of these two sites may be a general mechanism for activation of the Raf family kinases.
Results
Phosphorylation of both Thr598 and Ser601 is required for full activation of B-Raf induced by oncogenic Ras
Phosphorylation of serine and/or threonine residue(s) within kinase subdomains VII and VIII has been found to be responsible for the activation of a number of kinases including ERK (Payne et al., 1991), MEK (Alessi et al., 1994; Zheng and Guan, 1994; Resing et al., 1995), PKA (Shoji et al., 1983), PDK1 (Casamayor et al., 1999), glycogen synthase kinase (Hughes et al., 1993) and PKB/Akt (Alessi and Cohen, 1998). We wanted to identify the common activating phosphorylation sites in Raf in response to Ras. Amino acid sequences between kinase subdomains VII and VIII of Raf family members were aligned (Figure 1A). In B-Raf, there are five putative phosphorylation sites (Thr598, Ser601, Ser604, Ser606 and Ser613) within this region, while Ser615 is highly conserved in almost all of the kinases and is unlikely to be a target for regulation. It has been reported that Ser497 and Ser499 in C-Raf, which correspond to Ser604 and Ser606 in B-Raf, are the PKC phosphorylation sites. Therefore, we examined, using mutational analysis, whether other serine and threonine residues within this region in B-Raf are potential phosphorylation sites in response to Ras, and whether the phosphorylation contributes to B-Raf activation. The following N-terminal hemagglutinin (HA)-tagged B-Raf mutations were created: B-RafT598A, B-RafS601A and B-RafS613A (Figure 1A). COS1 cells were transfected with HA-tagged wild-type B-Raf (B-Raf) or the above individual mutants in the presence or absence of an active version of Ras, HRasV12. HA-B-Raf was immunoprecipitated and its activity was assayed using the coupled kinase assay method. As expected, HRasV12 greatly increased the B-Raf activity reflected by Elk phosphorylation by ∼6-fold compared with non-stimulated cells expressing B-Raf only (Figure 1B, compare lanes 2 and 3). This increase in Elk1 phosphorylation by oncogenic Ras is substrate MEK dependent, indicating that our kinase assay is B-Raf specific (Figure 1B, lane 4). When the mutants were tested, we observed that B-RafT598A and S601A decreased the oncogenic Ras-induced B-Raf activity to ∼10 and 60% of the wild type, respectively (Figure 1B, compare lanes 6, 8 and 3), whereas S613A did not alter the kinase activity (Figure 1B, lane 10). These results suggest that both T598 and S601 are essential for full activation of B-Raf. To examine this, we substituted both T598 and S601 by alanine (B-RafAA). Interestingly, compared with the wild type, basal B-Raf activity of this double mutant was reduced by ∼40% and the oncogenic Ras-induced kinase activation was abolished (Figure 1B, compare lanes 11, 12 and 2, 3).


Fig. 1. Activation of B-Raf by oncogenic Ras requires both Thr598 and Ser601 phosphorylation. (A) Sequence alignment of Raf kinase activation loop (residues 593–622 for human B-Raf). Residues subjected to site-directed mutagenesis are indicated by arrows. An asterisk denotes the protein kinase C phosphorylation sites. (B) Oncogenic Ras-induced kinase activation of B-Raf and its mutants. Fifty nanograms of HA-tagged pcDNA3 (vector control, lane 1), HA-tagged B-Raf (lanes 2--4), B-RafT598A (lane 5 and 6), B-RafS601A (lanes 7 and 8), B-RafS613A (lanes 9 and 10), B-RafAA (lanes 11 and 12) or B-RafED (lanes 13 and 14) were transiently transfected in COS cells alone (open bars) or with 100 ng of oncogenic Ras (HRasV12, solid bars), as indicated at the top of the panel. The kinase was immunoprecipitated and its activity, reflected by phosphorylation of GST–Elk1 (pElk1), was measured using the coupled assay (see Materials and methods). The upper panel shows autoradiographs of pElk1 that are representative of five independent experiments. The intensity of pElk1 bands was quantified by PhosphorImager (lower panel); results were subtracted from the reading of lane 1 (background) and expressed as fold increase with respect to the cells transfected with wild-type B-Raf without stimulation (lane 2). Lane 4* denotes that GST–MEK was omitted in the assay as a control. Results are mean ± SD from three independent experiments. Western blot of the immunoprecipitated kinase is shown in the middle panel, indicating equivalent protein loadings. (C) Activation of B-RafED by oncogenic Ras. COS cells were transfected with vector (lane 1), 50 ng of wild-type B-Raf (lanes 2 and 3), 5 ng of B-RafED (lanes 4 and 5), 25 ng of B-RafED (lanes 6 and 7) or 50 ng of B-RafED (lanes 8 and 9) in the presence or absence of HRasV12, as indicated at the top of the panel. Results of kinase assay and western blotting of B-Raf using α-HA were representative of two separate experiments. (D) Carbachol-induced kinase activation of B-Raf and its mutants. Cells were co-transfected with the above DNA constructs and hM3. After 24 h, cells were starved in FBS-free medium for 5 h followed by stimulation with carbachol for 5 min. Kinase immunoprecipitation, kinase assay and quantitation of results are as described in (B).
To test further that T598 and S601 are the potential Ras phosphorylation sites that contribute to B-Raf activity, we made substitutions of T598 and S601 with acidic residues (B-RafED, T598 to glutamic acid and S601 to aspartic acid) and found that B-RafED became constitutively active with a kinase activity comparable to that of the wild-type B-Raf induced by oncogenic Ras (Figure 1B, compare lanes 13 and 3). The kinase activity of B-RafED was further slightly increased by oncogenic Ras. To test whether an additional phosphorylation event is involved in B-Raf activaiton, we reduced the amount of B-RafED in the transfection to avoid potential saturation of the substrates for the in vitro coupled kinase assay. Cells were transfected with 5 ng (Figure 1C, lanes 4 and 5), 25 ng (Figure 1C, lanes 6 and 7) or 50 ng (Figure 1C, lanes 8 and 9) of B-RafED in the presence or absence of HRasV12. Indeed, results in Figure 1C clearly demonstrated that B-RafED could be further stimulated by oncogenic Ras, suggesting that there may be an additional phosphorylation mechanism for Ras-induced B-Raf activation. However, the magnitute of Ras stimulation on B-RafED was significantly reduced. Western blotting results demonstrated that equal amounts of protein were used for the kinase assays (Figure 1B and C, middle panel). Phosphorylation of Elk1 was quantified and is presented in the bottom panel of Figure 1B and C; all results were substracted from the background (lane 1) and are expressed as the fold increase compared with cells transfected with wild-type B-Raf only (lane 2). The above results demonstrate that Thr598 and Ser601 are putative phosphorylation sites important for B-Raf activation in response to active Ras.
Next we asked whether the phosphorylation of T598 and S601 is also responsible for B-Raf activation induced by G-protein-coupled receptors. COS1 cells were co-transfected with B-Raf or B-Raf mutants and human muscarinic receptor, hM3, followed by stimulation with 500 µM carbachol for 5 min. Similar to the results induced by oncogenic Ras, phosphorylation of T598 and S601 is also essential for G-protein-coupled receptor-induced B-Raf activation (Figure 1D).
Phosphopeptide mapping of B-Raf
To provide further evidence that T598 and S601 of B-Raf are the Ras-dependent phosphorylation sites, in vivo metabolic labeling with [32P]orthophosphate and two-dimensional phosphopeptide mapping were performed. Figure 2A shows the signals of 32P-labeled B-Raf immunoprecipitates resolved by SDS–PAGE. The phosphorylation of B-Raf was increased after stimulation with oncogenic Ras, as compared with the basal phosphorylation without Ras (Figure 2A, lanes 1 and 2). The hyperphosphorylation induced by Ras resulted in a mobility shift of B-Raf. However, this increase in kinase phosphorylation was reduced in B-RafAA, and a lesser degree of the mobility shift was seen under the same condition of Ras stimulation (Figure 2A, lane 3). These results suggest that phosphorylation of T598 and S601 represents a significant portion of the total B-Raf phosphorylation induced by oncogenic Ras. The changes in B-Raf phosphorylation were not due to the protein loading, since western blotting indicated equal loading of each sample (Figure 2A, bottom).

Fig. 2. Tryptic phosphopeptide mapping of B-Raf. (A) Metabolic labeling and immunoprecipitation of B-Raf and B-RafAA. COS1 cells grown on a 60 mm plate were transfected with wild-type B-Raf (lane 1), B-Raf + HRasV12 (lane 2) or B-RafAA + HRasV12 (lane 3), labeled with [32P]orthophosphate, and immunoprecipitated with anti-HA antibody. Immunocomplexes were separated by SDS–PAGE and visualized by antoradiography. Positions of size markers are indicated in kilodaltons (KD) on the right. An immunoblot of the above immunoprecipitation using anti-HA antibody is shown at the bottom. (B, C and D) Phosphopeptide mappings for wild-type B-Raf, B-Raf + HRasV12 and B-RafAA + HRasV12, respectively. The 32P-labeled bands were excised from the membrane, digested with trypsin and analyzed by two-dimensional thin-layer electrophoresis. The directions for electrophoresis (E, from cathode to anode) and TLC (T) are indicated in the lower left corner of each panel. Arrows point to positions of phosphopeptides for references between different panels. Dashed circles indicate phosphopeptides present in (C) that are missing in (D). Open arrowheads indicate the Ras-induced phosphopeptide not affected in B-RafAA. (E) Phosphorylation of Thr598 and Ser601 determined by specific anti-phospho Thr598 and Ser601 antibodies. COS cells were transfected with pcDNA (vector, lane 1), wild-type B-Raf (lane 2), co-transfected with wild-type B-Raf and HRasV12 (lane3), B-RafAA (lane 4) or co-transfected B-RafAA and HRasV12 (lane 5). Immunoprecipitates of B-Raf and B-RafAA were isolated and resolved by SDS–PAGE, transferred to membrane, and blotted with anti-phospho Thr598 (top panel) or anti-phospho Ser601 antibody (middle panel). Protein loading was examined by blotting using HA antibody (bottom panel).
The labeled bands were excised, digested with trypsin and analyzed by two-dimensional phosphopeptide mapping. Compared with the phosphopeptide pattern of unstimulated wild-type B-Raf (Figure 2B), wild-type B-Raf stimulated with oncogenic Ras displayed two additional phosphopeptides, as circled in Figure 2C. However, these two phosphopeptides were missing in the oncogenic Ras-stimulated B-RafAA sample (Figure 2D), suggesting that these two phosphopeptides are derived from phosphorylation of T598 and S601. In contrast, another Ras-induced phosphopeptide (indicated by open arrowheads) was not affected by the B-RafAA mutation (Figure 2C and D). Reference phosphopeptides, which were not altered by Ras stimulation, are indicated by arrows. These results confirmed that phosphorylation of Thr598 and Ser601 is stimulated by Ras.
To examine directly that Thr598 and Ser601 are phos phorylated by oncogenic Ras, we generated anti-phospho Thr598 (pThr598) and Ser601 (pSer601) antibodies, which specifically recognize phosphorylated Thr598 and Ser601 in B-Raf, respectively. Cells were transfected with vector (pcDNA3), wild-type B-Raf or B-RafAA in the presence or absence of HRasV12. Immunoprecipitates of B-Raf and B-RafAA were botted with pThr598 or pSer601 and the western blotting results are shown in Figure 2E. Only wild-type B-Raf but not B-RafAA was phosphorylated at both Thr598 and Ser601 after stimulation of HRasV12, as determined by pThr598 and pSer601 (compare lanes 3 and 5 in the top and middle panels in Figure 2E). These results support the view that the pThr598 and pSer601 antibodies selectively recognize the phosphorylated B-Raf. We have also performed experiments with B-RafT598A or B-RafS601A single mutants, and similar results were obtained. Thus, B-RafT598A eliminated the recognition by pThr598 but not by pSer601 antibody in response to HRasV12 stimulation (data not shown). Conversely, the B-RafS601A mutant eliminated the recognition by pSer601 but not by pThr598 antibody (data not shown). These data provided the direct evidence that B-Raf was indeed phosphorylated at Thr598 and Ser601 in response to oncogenic Ras stimulation. Furthermore, our results indicate that phosphorylation of Thr598 and Ser601 can occur independently.
B-RafT598A, B-RafS601A or B-RafAA does not alter the association of B-Raf with 14-3-3, HSP90 or MEK
Raf kinase activation is also regulated by interaction with other signaling components including 14-3-3 proteins and heat shock protein 90 (HSP90) (Morrison and Cutler, 1997; Roy et al., 1998; Tzivion et al., 1998; Grammatikakis et al., 1999). B-Raf also directly binds to and activates MEK. To exclude the possibility that the decreased kinase activity in the mutants is due to global conformational changes and disruption of protein–protein interactions, we conducted experiments examining the interactions of B-Raf, B-RafT598A, B-RafS601 or B-RafAA with endogenous 14-3-3 protein, HSP90 or MEK. Immunoprecipitates of overexpressed B-Raf and mutants were isolated and probed with antibodies against 14-3-3, HSP90 or MEK. The results in Figure 3 clearly showed that either wild-type B-Raf or mutants were able to bind to all three endogenous signaling proteins, suggesting that the decreased kinase activity is not a result of disrupted protein–protein interactions.
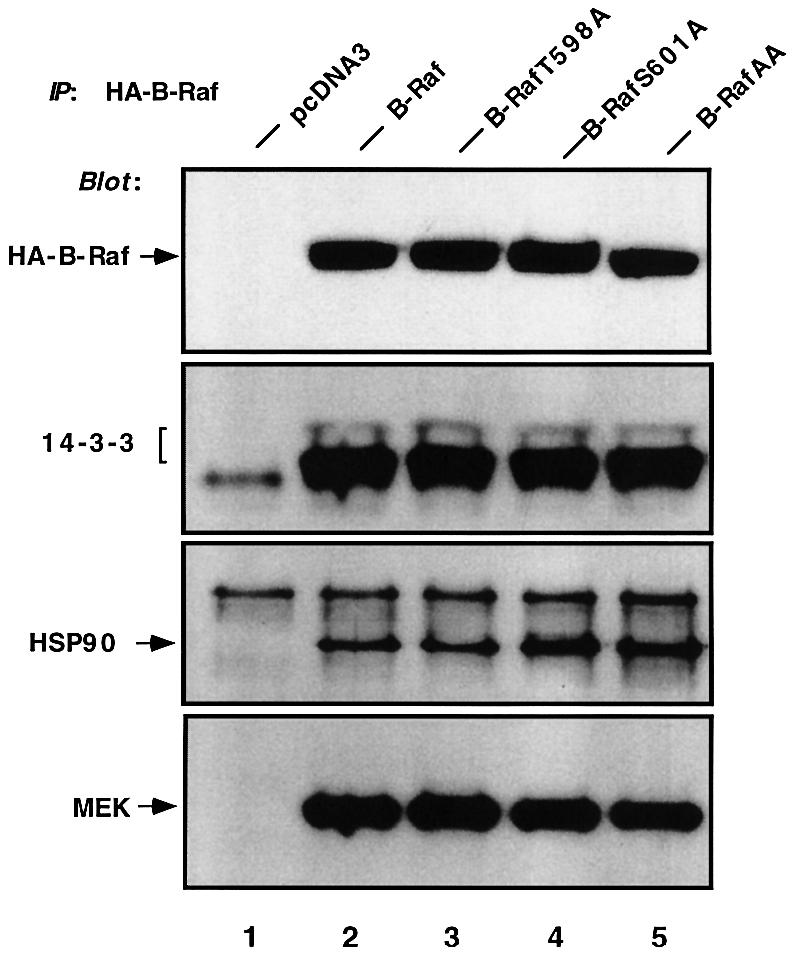
Fig. 3. B-RafT598A, B-RafS601A or B-RafAA does not alter the association with 14-3-3, HSP90 or MEK. COS cells were transiently transfected with pcDNA3 (vector, lane 1), HA-B-Raf (lane 2), HA-B-RafT598A (lane 3), HA-B-RafS601A (lane 4) or HA-B-RafAA (lane 5). Cells were lysed and the HA-tagged B-Raf was immuno precipitated (IP) with anti-HA and western blotted with anti-HA (B-Raf), anti-14-3-3, anti-HSP90 or anti-MEK antibodies.
Modulation of kinase activity by B-RafAA or B-RafED affects ERK activation and Elk-dependent transcription
Next, we wished to test the effects of B-RafAA and B-RafED on activation of ERK in vivo. Cells were co-transfected with HA-tagged B-Raf, B-RafAA or B-RafED and Myc-tagged ERK. Myc-ERK was immunoprecipitated by anti-Myc antibody and its kinase activity was determined. As expected and consistent with the in vitro B-Raf kinase assay results, mutant B-RafAA reduced the ability to activate ERK kinase by ∼40% compared with wild-type B-Raf (Figure 4A, compare lanes 4 and 3), whereas constitutively active B-RafED induced a large increase in ERK activity, at a level similar to oncogenic Ras-induced ERK activity (Figure 4A, compare lanes 5 and 2). Immunoblotting demonstrated equivalent levels of protein loading detected by anti-Myc or anti-HA (Figure 4A, middle or bottom panel). It is noteworthy that wild-type B-Raf exerts high basal activity even under the serum-starved condition, leading to a significant activation of ERK (Figure 4A, compare lanes 3 and 1).
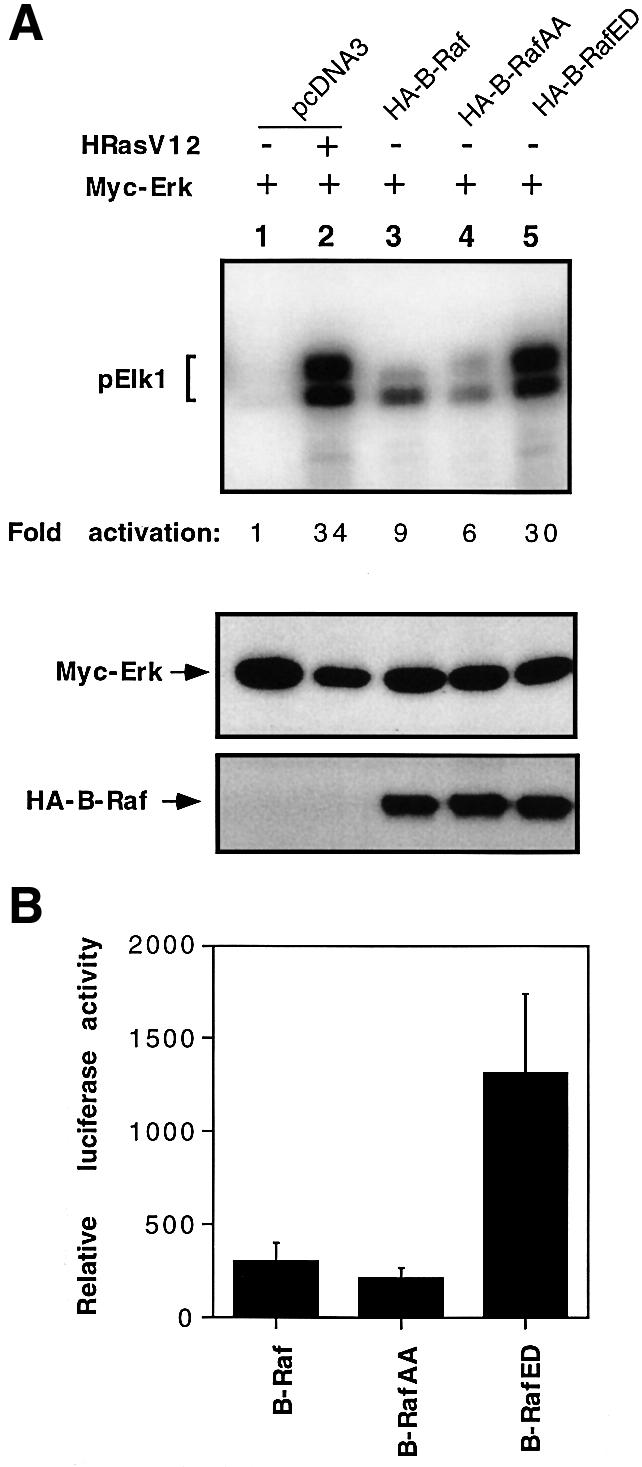
Fig. 4. Effects of B-Raf, B-RafAA or B-RafED on ERK activity and Elk-dependent transcription. (A) Activation of ERK by B-Raf, B-RafAA or B-RafED. Myc-ERK was co-transfected with pcDNA3, HA-tagged B-Raf, B-RafAA or B-RafED. Co-transfection of Myc-ERK and HRasV12 serves as a positive control (lane 2). Cells were cultured for 24 h followed by starvation for 15 h. Myc-ERK was immuno precipitated with anti-Myc monoclonal antibody 9E10 and assayed for kinase activity using GST–Elk1 as a substrate. ERK kinase activity is shown in the top panel and fold activation was determined by PhosphorImager analysis. Immunoblot detection of Myc-ERK or HA-B-Raf is shown in the middle or bottom panel. (B) Elk1-dependent transcription activation by B-Raf, B-RafAA or B-RafED. COS cells were transfected with Gal4-Elk1 and Gal4-LUC and B-Raf, B-RafAA or B-RafED. A pCMV-LacZ plasmid was co-transfected as an internal control for variations in transfection efficiency. Cells were cultured for 24 h followed by 15 h starvation. Luciferase activity was measured and normalized against the co-transfected β-galactosidase activity and results are mean ± SD from three independent experiments.
The concerted activation of the Ras/Raf/MEK/ERK pathway is known to be involved in many cell functions, including proliferation, differentiation and survival. Activated ERK is able to phosphorylate numerous transcription factors and thus regulate gene expression. One of the best characterized substrates for ERK is Elk1, a member of the ternary complex factor family. To examine whether the manipulation of B-Raf phosphorylation (mutants B-RafAA and B-RafED) affects Elk-dependent gene transcription, we measured the transcriptional activity of Elk using a luciferase reporter assay. The activity of a Gal4–Elk1 reporter (Sugimoto et al., 1998), which contains the C-terminal ERK-responsive domain of Elk1 fused to the DNA binding domain of Gal4, was tested. Consistent with the data on ERK activation shown in Figure 4A, Gal4–Elk1 activation in B-RafAA-expressing cells was reduced by 40%, whereas it was increased ∼4-fold in B-RafED-expressing cells as compared with the cells expressing wild-type B-Raf (Figure 4B).
B-RafED induces NIH 3T3 cell transformation and PC12 cell differentiation
Activation of Raf has been shown to be involved in diverse biological functions such as cell transformation and cell differentiation. To examine the effect of phosphorylation of Thr598 and Ser601 on cell transformation, NIH 3T3 cells were co-transfected with wild-type B-Raf or B-RafED, and pCMV-lacZ. Transfection efficiency was monitored by β-galactosidase activity. We observed that cells transfected with B-RafED reproducibly induced cell transformation, although the numbers of foci were low compared with those induced by oncogenic Ras. In contrast, at the same transfection efficiency, there was no focus formed in cells transfected with wild-type B-Raf. Three individual foci were cloned in medium containing G418 to establish stably transfected cells expressing B-RafED. The morphology of cells stably expressing wild-type HA-B-Raf or HA-B-RafED is shown in Figure 5A. The B-RafED cells showed morphological changes typical of transformed cells: cells were more refractile and less flattened. In addition, the B-RafED clones lost cell contact inhibition of growth. Western blotting using anti-HA antibody confirmed the expression of wild-type B-Raf and B-RafED in the above stably transfected cells.



Fig. 5. B-RafED induces NIH 3T3 cell transformation and PC12 cell differentiation. (A) Morphology of NIH 3T3 cells stably expressing wild-type B-Raf or B-RafED. NIH 3T3 cells were transfected with wild-type B-Raf or B-RafED. Stably transfected cells were selected as described in Materials and methods. Note that morphology for cells expressing B-RafED is from three individual cell colonies. The western blotting for B-Raf expression in stably transfected cells is shown in the lower panel. (B) PC12 cell differentiation induced by B-Raf3A, B-Raf-ED and B-Raf3AED. PC12 cells were co-transfected with pEGFP vector and pcDNA3 (vector control), wild-type B-Raf (WT-B-Raf), B-Raf3A, B-RafED or B-Raf3AED. Two days after transfection, cells were shifted to NGF-minus differentiation medium (DMEM supplemented with 2% horse serum and 1% FBS medium) for 3 days. Cells were examined under the fluorescence microscope for visualization of transfected cells (by GFP) and differentiation (by the presence of neurites). Shown are representative fluorescence images (left) and the corresponding Nomarski images (right) (×200). (C) Kinase activity of B-Raf3A, B-RafED and B-Raf3AED. COS cells were transfected with pcDNA3, WT-B-Raf, B-Raf3A, B-RafED or B-Raf3AED in the presence or absence of HRasV12. Kinase activity (upper panel) and western blotting for B-Raf expression (lower panel) were determined. (D) Quantitation of PC12 cell differentiation induced by B-Raf and its mutants. Results were expressed as a percentage of green cells exhibiting neurite extensions of >2 cell body lengths in total green cells. At least 200 transfected cells were counted.
B-Raf is a major neuronal Raf isoform and is highly expressed in the central nervous system and PC12 cells (Hagemann and Rapp, 1999). Activation of B-Raf induces cell differentiation in PC12 cells. Given the evidence that Thr 598 and S601 are the major sites for oncogenic Ras-induced phosphorylation and that phosphorylation of Thr598 and Ser601 is essential for B-Raf activation, we hypothesized that B-RafED should stimulate PC12 cell differentiation. To test this, we transfected PC12 cells with wild-type B-Raf or B-RafED, and cell differentiation was determined by neurite growth. Transfected cells were visualized by expression of a co-transfected pEGFP gene using fluorescence microscopy. In cells transfected with vector or wild-type B-Raf, no cells with neurite growth were observed (Figure 5B and D). However, B-RafED induced significant PC12 cell differentiation (17% of transfected cells were differentiated) (Figure 5B and D).
Previous studies have shown that B-Raf kinase activity is regulated by interaction of the N-terminal regulatory domain and the C-terminal kinase activation domain (Cutler et al., 1998). Thus, deletion of the N-terminal domain renders the C-terminal kinase domain constitutively active, although the mechanism underlying this phenomenon is not clear. Recently, we have found that phosphorylation of three residues (Ser364, Ser428 and Thr439) within the N-terminal domain exerts inhibitory effects on B-Raf kinase activity. Substitutions of these three residues by alanines (B-Raf3A) results in an increase in B-Raf activity (A.Vojtek and K.L.Guan, unpublished data; Figure 5C). We constructed a mutant B-Raf (B-Raf3AED) containing B-Raf3A and B-RafED, and found that the kinase activity of B-Raf3AED is further increased as compared with individual B-Raf3A or B-RafED (Figure 5C). Interestingly, B-Raf3AED induced 38% differentiation of tranfected cells (Figure 5D). However, although B-Raf3A has a high basal activity, it only induced 4% cell differentiation (Figure 5D). These results suggest that the biological effects induced by B-Raf are dependent on the phosphorylation of Thr598 and Ser601 in the kinase activation domain and phosphorylation of other sites in the kinase regulatory domain. Phosphorylation of the N-terminal regulatory domain cooperates with the kinase domain to induce Raf activation and biological functions such as cell differentiation.
Discussion
Activation of MEK and ERK by phosphorylation has been clearly elucidated while the biochemical mechanism of Raf activation is not fully understood. Activation of Raf involves multiple steps including membrane translocation, protein association and phosphorylation. Regulation of C-Raf by phosphorylation has been studied extensively. The current model, mainly based on data from C-Raf, indicates that phosphorylation of Ser338 is a major event in Raf activation in response to RTKs and Ras (Mason et al., 1999). In addition, phosphorylation of Tyr341 also plays an important role in C-Raf activation (Mason et al., 1999). However, the current model of C-Raf activation is not applicable to other Raf family members such as C.elegans Raf lin-45 and Drosophila Raf, which are also regulated by RTKs and Ras signaling pathways, because lin-45 Raf contains an aspartate at the position corresponding to Ser338 while the Drosophila Raf contains an asparagine residue at the corresponding site of C-Raf Tyr341.
Compared with C-Raf, little is known about how B-Raf is activated by phosphorylation, yet B-Raf is the major MEK activator in many cell types. The known phosphorylation mechanism for C-Raf regulation is not entirely applicable to B-Raf. For example, the site in B-Raf (Ser445) corresponding to the major phosphorylation site (Ser338) in C-Raf is not stimulated by Ras, but is constitutively phosphorylated. Mutation of Ser445 to alanine has a minor effect on activation of B-Raf by Ras, while corresponding mutation of Ser338 in C-Raf eliminates Ras-dependent activation. Furthermore, phosphorylation of Tyr341 in C-Raf is important for C-Raf activation while this residue is not conserved at the corresponding position of B-Raf. Therefore, activation of B-Raf by Ras is likely to be mediated by as yet unidentified phosphorylation sites.
In this report, we studied the mechanism of phosphorylation in B-Raf activation. Phosphorylation of residues in the kinase activation loop has been reported to be responsible for activation of many protein kinases including MEK (Alessi et al., 1994; Zheng and Guan, 1994; Resing et al., 1995) and ERK (Payne et al., 1991). The kinase activation loop lies between the kinase subdomains VII and VIII and is highly conserved in all Raf family kinases (Figure 1A). However, to date, there are no data showing that Raf activation requires phosphorylation of residues within the activation loop except that PKC may phosphorylate Ser497 and Ser499 in C-Raf. In the present study, we have demonstrated that phosphorylation of both Thr598 and Ser601 within the B-Raf kinase activation loop is essential for kinase activation. These two residues, corresponding to Thr491 and Ser494 in C-Raf, have not been previously reported as regulatory phosphorylation sites in C-Raf. We propose that phosphorylation of these residues within the kinase activation loop is a general mechanism for Raf phosphorylation and activation. Multiple lines of evidence support this hypothesis. First, both Thr598 and Ser601 are required for B-Raf activation in response to different stimuli, including oncogenic Ras and G-protein-coupled receptors (Figure 1B and D). Secondly, phosphorylation of both residues is essential for biological function. Thus, the B-RafED mutant can induce NIH 3T3 cell transformation and PC12 cell differentiation. Thirdly, these two residues are highly conserved in all members of the Raf family (Figure 1A). Indeed, mutations of the corresponding residues (Thr491 and Ser494) to alanine in C-Raf also decreased oncogenic Ras-induced C-Raf kinase activation (H.Chong and K.L.Guan, unpublished data). Furthermore, substitution of the corresponding residues by acidic residues in lin-45 results in multi-vulval induction in C.elegans (J.Lee and K.L.Guan, unpublished data). Therefore, phosphorylation of these conserved residues in the activation loop may be a common molecular mechanism for Raf family kinase activation.
Although Thr598 and Ser601 are the major phosphorylation sites in response to oncogenic Ras, phosphorylation of these two sites may not be the sole mechanism for Raf activation. Previous studies have shown that phosphorylation of the N-terminal regulatory domain inhibits C-Raf activity (Morrison et al., 1993; Schramm et al., 1994; Rommel et al., 1999; Zimmermann and Moelling, 1999). We have also observed that phosphorylation of Ser364, Ser428 and Thr439 exhibited an inhibitory effect on B-Raf activity (A.Vojtek and K.L.Guan, unpublished data). Furthermore, although phosphorylation of Thr598 and Ser601 represents a major event for B-Raf activation, B-RafED can be further activated by RasV12 (Figure 1C), suggesting that additional phosphorylation mechanisms may be involved in B-Raf activation. However, phosphorylation of other sites alone is not sufficient for activation of B-Raf because alanine replacement of Thr598 and Ser601 abolished the oncogenic Ras-induced B-Raf activation (Figure 1B). Importantly, phosphorylation of Thr598 and Ser601 is sufficient to activate B-Raf as the B-RafED displays high activity.
Activation by autophosphorylation has been found in many kinases such as AKT (Toker and Newton, 2000), RTKs (Schlessinger and Ullrich, 1992) and PDK1 (Casamayor et al., 1999). Interestingly, it has recently been reported that activation of C-Raf requires dimerization of Ras, suggesting that trans-phosphorylation may be involved in Raf activation (Inouye et al., 2000). We therefore examined whether phosphorylation of Thr598 and Ser601 in B-Raf is induced by autophosphorylation and/or trans-phosphorylation. However, results from phosphopeptide mapping and in vitro kinase assays do not support this hypothesis (data not shown). Therefore, identification of downstream kinases of Ras responsible for phosphorylation of T598 and S601 in B-Raf is required for further understanding of the regulation of B-Raf kinase activation.
Materials and methods
Plasmid construction
A cDNA construct containing the full-length open reading frame of wild-type human B-Raf (generously provided by Dr A.Vojtek, University of Michigan) was subcloned into the HA-tagged mammalian expression vector pcDNA3 (Sugimoto et al., 1998). Mutant constructs of HA-B-Raf were created by PCR mutagenesis and verified by DNA sequencing. Thr598, Ser601 and Ser613 were independently mutated to alanine (T598A, S601A and S613A, respectively). A double mutant (B-RafAA) including T598A and S601A, and a double mutant (B-RafED) in which T598 and S601 were substituted by the acidic residues glutamic acid (E) and aspartic acid (D), respectively, were also constructed. A constitutively active HA-tagged B-Raf mutant (B-Raf3A) in which three residues at positions Ser364, Ser428 and Thr439 are all mutated to alanine was kindly provided by Dr A.Vojtek (University of Michigan). Human subtype 3 muscarinic receptor (hM3) was generously provided by Dr C.Lodgston (University of Michigan). Other DNA constructs including HRasV12, Myc-ERK, Gal4–Elk, Gal4–LUC and pCMV-lacZ have been described elsewhere (Sugimoto et al., 1998).
Cell culture and transfection
COS1 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS). NIH 3T3 cells were grown in DMEM containing 10% calf serum and PC12 cells were maintained in DMEM plus 10% horse serum and 5% FBS. Cells were transfected using the LipofectAMINE (Life Technologies, Inc.) method, as recommended by the manufacturer.
In vitro kinase assays
For B-Raf kinase assay, COS1 cells grown in six-well plates were transfected with 50 ng of HA-tagged wild-type B-Raf, B-RafT598A, B-RafS601A, B-RafS613A, B-RafAA or B-RafED in the presence or absence of 100 ng of HRasV12. Twenty-four hours later, cells were washed twice in cold phosphate-buffered saline (PBS) and lysed in lysis buffer [25 mM HEPES pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5 mM EDTA, 0.025% mercaptoethanol, 1 mM NaF, 200 µM Na3VO4, 200 µM phenylmethylsulfonyl fluoride (PMSF), 10 µg/ml leupeptin, 10 µg/ml aprotinin], and lysates were clarified by centrifugation. HA epitope-tagged kinase was immunoprecipitated with anti-HA monoclonal antibody (Babco). Immune complexes were collected by binding to protein G–Sepharose, washed three times with lysis buffer, followed by once with PBS. B-Raf kinase activity was measured by coupled assay using glutathione S-transferase (GST)–MEK, GST–ERK and GST–Elk as sequential substrates. In brief, immunoprecipitated kinase was incubated for 30 min at 30°C with 4 µg/ml GST–MEK in kinase assay buffer (10 mM HEPES pH 8.0, 10 mM MgCl2, 1 mM dithiothreitol, 50 µM ATP) in a volume of 20 µl. Fifteen microliters of the supernatant were transferred to the fresh tube and incubated for 15 min at 30°C after addition of 5 µl of kinase buffer containing 20 µg/ml GST–ERK. GST–Elk (400 µg/ml) in 10 µl of kinase buffer containing 10 µCi of [γ-32PO4]ATP (ICN) was added to the mixture and incubated for a further 20 min at 30°C. Reactions were terminated by adding 10 µl ×4 SDS sample buffer, followed by boiling for 5 min. The reaction products were analyzed by SDS–PAGE and visualized by autoradiography. Fold activation was determined with a Storm 860 PhosphorImager in combination with ImageQuant version 1.1 software (Molecular Dynamics). In a separate set of experiments, cells were co-transfected with HA-tagged B-Raf or its mutants and hM3. Twenty-four hours later, cells were starved in FBS-free medium for 5 h, followed by stimulation with carbachol for 5 min. Cells were then lysed and the kinase assay was performed.
For the ERK kinase assay, cells were co-transfected with 100 ng of Myc-ERK and 50 ng of HA-tagged wild-type B-Raf, B-RafAA or B-RafED. Cells were maintained in 10% FBS medium for 24 h and starved in 0.1% FBS for 15 h. Cells were lysed in lysis buffer and Myc-ERK was immunoprecipitated with monoclonal antibody 9E10 (anti-Myc; Babco). Immune complexes were collected by incubation with protein G–Sepharose, washed and then assayed for 20 min at 30°C in kinase assay buffer containing 10 µCi of [γ-32PO4]ATP and 400 µg/ml GST–Elk. GST–MEK, GST–ERK and GST–Elk were expressed in Escherichia coli and purified as described previously (Sugimoto et al., 1998).
Immunoblot analysis
To check the equal loading of HA-B-Raf or Myc-ERK for kinase activity, cell lysates or immunoprecipitates were subjected to SDS–PAGE and transferred to a PVDF membrane (Millipore). Membranes were blotted and detected as published elsewhere (Sugimoto et al., 1998). For co-immunoprecipitation experiments, COS1 cells were grown on 10 cm plates and transfected with 1 µg of HA-tagged wild-type B-Raf, B-RafT598A, B-RafS601A or B-RafAA. Forty-eight hours after transfection, cells were washed twice with ice-cold PBS and lysed in hypotonic lysis buffer (10 mM HEPES pH 7.5, 50 mM NaCl, 1% Triton X-100, 2 mM EDTA, 0.1% mercaptoethanol, 50 mM NaF, 1 mM Na3VO4, 200 µM PMSF, 10 µg/ml leupeptin, 10 µg/ml aprotinin). HA-tagged B-Raf was immunoprecipitated, subjected to SDS–PAGE and transferred to a membrane. Membranes were blotted with the following primary antibodies: anti-MEK (Stewart et al., 1999), anti-14-3-3 (Santa Cruz Biotechnology) or anti-HSP90 (Transduction Laboratories). Antibodies specifically recognizing Thr598 or Ser601 phosphorylated B-Raf were prepared by immunizing rabbits with synthetic phosphopeptides (Cell Signaling Technologies, Boston, MA). For determining phosphorylation of T598 and S601 using anti-phospho-T598 and S601 antibodies, transfected cells were lysed in lysis buffer supplemented with 25 mM glycerophosphate and 10 mM sodium pyrophosphate. HA-B-Raf was immunoprecipitated and resolved by SDS–PAGE. Transferred membrane was blotted with anti-phospho-Thr598 or anti-phospho-Ser601 antibody, or HA antibody to check the protein loading.
Metabolic labeling and two-dimensional phosphopeptide mapping
COS1 cells were transfected with HA-tagged wild-type B-Raf, or co-transfected with wild-type B-Raf or B-RafAA with HRasV12. Forty-eight hours after transfection, COS cells were washed twice with phosphate-free medium and incubated with 0.5 mCi/ml 32P-labeled orthophosphate (ICN) at 37°C for 4 h. Cells were washed with PBS extensively and lysed in lysis buffer. The HA-tagged kinase was immunoprecipitated, resolved by SDS–PAGE and transferred to a membrane. Phosphorylated B-Raf was detected by autoradiography. Phosphopeptide mapping was performed as described previously (Zheng and Guan, 1994). In brief, phosphorylated B-Raf bands were excised, fixed in methanol and incubated in 0.5 ml of 0.5% polyvinylpyrrolidone-40 dissolved in 100 mM acetic acid for 30 min at 37°C. The samples were then digested with 20 µg of TPCK-treated trypsin (Sigma) at 37°C in 75 mM ammonium bicarbonate buffer pH 8.0 containing 5% acetonitrile. After digestion, samples were spin dried under vacuum and suspended in 6 µl of water. Samples were spotted onto a cellulose plate (20 × 20, Kodak) and first dimensional electrophoresis was performed using 1% ammonium bicarbonate pH 8.9. The plates were chromatographed in the second dimension in chromatography buffer (n-butanol/pyridine/acetic acid/water, 20:24:6:30). The plates were dried, and phosphopeptides were visualized by PhosphorImager.
Reporter analysis
Luciferase reporter analysis was performed as described previously (Sugimoto et al., 1998). COS cells were co-transfected with Gal4–Elk1, Gal4–luciferase (Gal4–LUC), pCMV-lacZ and HA-tagged wild-type B-Raf, B-RafAA or B-RafED. Twenty-four hours after transfection, cells were starved in 0.1% FBS for 24 h. Luciferase activity was determined and normalized against the co-transfected β-galactosidase activity, as described elsewhere (Sugimoto et al., 1998).
Transformation and differentiation assays
Cell transformation was assessed by focus formation assay. NIH 3T3 mouse fibroblasts were co-transfected with pCMV-lacZ gene and wild-type B-Raf, B-Raf mutants or oncogenic Ras (positive control) and grown in 10% calf serum medium. Twenty-four hours post-transfection, cells were trypsinized and 1/10 of the cells were taken for β-galactosidase activity assay to check the transfection efficiency among culture plates. The remaining cells were plated onto 10 cm dishes and maintained in 5% calf serum medium with medium change every 3 days. Fourteen days later, cells were stained with crystal violet and the number of transformed foci was counted. To select stably transfected cells expressing wild-type B-Raf, G418 (500 µg/ml) was added to the medium for 14 days. Individual colonies were cloned, expanded, and analyzed by western blot analysis. To select stably transfected cells expressing B-Raf ED, 14 days post-transfection, cell foci were isolated and cultured individually in 10% calf medium containing G418 (500 µg) for 10 days. For the cell differentiation assay, PC12 cells were co-transfected with green fluorescent protein (GFP) vector (pEGFP-C1) to visualize the transfected cells. After transfection, cells were cultured in low serum medium containing 2% horse serum and 1% FBS in order to maximize their differentiation responses (MacNicol et al., 2000). Green cells with one or more growth cone tipped neurites >2 cell bodies in length were counted under a fluorescence microscope from at least 10 different microscope fields. Cell differentiation was estimated by the percentage of differentiated cells in total green cells.
Acknowledgments
Acknowledgements
We thank Drs A.Vojtek and C.Lodgston for reagents, Dr G.Smith for assistance with the fluorescence microscope, members of the Guan laboratory, especially Dr T.Lanigan, H.Vikis and E.D.Tang, for helpful discussions and critical reading of the manuscript, T.Zhu for excellent technical assistance, and R.Fischer for constructing some of the Raf mutants. The authors gratefully acknowledge the assistance of Drs Yi Tan and Hong Ruan in raising anti-phospho-B-Raf antibodies. B.H.Z. is a recipient of a C.J.Martin Postdoctoral Fellowship from the Australian National Health and Medical Research Council (NHMRC). This work was supported by grants from NIH, American Cancer Society and Walther Cancer Institute, and the MacArthur Fellowship to K.L.G.
References
- Alessi D.R. and Cohen,P. (1998) Mechanism of activation and function of protein kinase B. Curr. Opin. Genet. Dev., 8, 55–62. [DOI] [PubMed] [Google Scholar]
- Alessi D.R., Saito,Y., Campbell,D.G., Cohen,P., Sithanandam,G., Rapp,U., Ashworth,A., Marshall,C.J. and Cowley,S. (1994) Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J., 13, 1610–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H., Smola,U., Wixler,V., Eisenmann-Tappe,I., Diaz-Meco,M.T., Moscat,J., Rapp,U. and Cooper,G.M. (1997) Role of diacylglycerol-regulated protein kinase C isotypes in growth factor activation of the Raf-1 protein kinase. Mol. Cell. Biol., 17, 732–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll M.P. and May,W.S. (1994) Protein kinase C-mediated serine phosphorylation directly activates Raf-1 in murine hematopoietic cells. J. Biol. Chem., 269, 1249–1256. [PubMed] [Google Scholar]
- Casamayor A., Morrice,N.A. and Alessi,D.R. (1999) Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo. Biochem. J., 342, 287–292. [PMC free article] [PubMed] [Google Scholar]
- Catling A.D., Reuter,C.W., Cox,M.E., Parsons,S.J. and Weber,M.J. (1994) Partial purification of a mitogen-activated protein kinase kinase activator from bovine brain. Identification as B-Raf or a B-Raf-associated activity. J. Biol. Chem., 269, 30014–30021. [PubMed] [Google Scholar]
- Cook S.J. and McCormick,F. (1993) Inhibition by cAMP of Ras-dependent activation of Raf [see comments]. Science, 262, 1069–1072. [DOI] [PubMed] [Google Scholar]
- Cutler R.E. Jr, Stephens,R.M., Saracino,M.R. and Morrison,D.K. (1998) Autoregulation of the Raf-1 serine/threonine kinase. Proc. Natl Acad. Sci. USA, 95, 9214–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G., Eisenmann-Tappe,I., Fries,H.W., Troppmair,J. and Rapp,U.R. (1994) The ins and outs of Raf kinases. Trends Biochem. Sci., 19, 474–480. [DOI] [PubMed] [Google Scholar]
- English J., Pearson,G., Wilsbacher,J., Swantek,J., Karandikar,M., Xu,S. and Cobb,M.H. (1999) New insights into the control of MAP kinase pathways. Exp. Cell Res., 253, 255–270. [DOI] [PubMed] [Google Scholar]
- Erhardt P., Schremser,E.J. and Cooper,G.M. (1999) B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol. Cell. Biol., 19, 5308–5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian J.R., Daar,I.O. and Morrison,D.K. (1993) Critical tyrosine residues regulate the enzymatic and biological activity of Raf-1 kinase. Mol. Cell. Biol., 13, 7170–7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman C., Skinner,R.H., Skelly,J.V., Neidle,S. and Lowe,P.N. (1996) Equilibrium and kinetic measurements reveal rapidly reversible binding of Ras to Raf. J. Biol. Chem., 271, 6713–6719. [DOI] [PubMed] [Google Scholar]
- Grammatikakis N., Lin,J.H., Grammatikakis,A., Tsichlis,P.N. and Cochran,B.H. (1999) p50(cdc37) acting in concert with Hsp90 is required for Raf-1 function. Mol. Cell. Biol., 19, 1661–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemann C. and Rapp,U.R. (1999) Isotype-specific functions of Raf kinases. Exp. Cell Res., 253, 34–46. [DOI] [PubMed] [Google Scholar]
- Hill C.S. and Treisman,R. (1995) Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell, 80, 199–211. [DOI] [PubMed] [Google Scholar]
- Hughes K., Nikolakaki,E., Plyte,S.E., Totty,N.F. and Woodgett,J.R. (1993) Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J., 12, 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouye K., Mizutani,S., Koide,H. and Kaziro,Y. (2000) Formation of the Ras dimer is essential for Raf-1 activation. J. Biol. Chem., 275, 3737–3740. [DOI] [PubMed] [Google Scholar]
- Jelinek T., Dent,P., Sturgill,T.W. and Weber,M.J. (1996) Ras-induced activation of Raf-1 is dependent on tyrosine phosphorylation [published erratum appears in Mol. Cell. Biol., 17, 2971, 1997]. Mol. Cell. Biol., 16, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A.J., Sun,H., Diaz,B., Barnard,D., Miao,W., Bagrodia,S. and Marshall,M.S. (1998) The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature, 396, 180–183. [DOI] [PubMed] [Google Scholar]
- Kolch W., Heidecker,G., Kochs,G., Hummel,R., Vahidi,H., Mischak,H., Finkenzeller,G., Marme,D. and Rapp,U.R. (1993) Protein kinase C α activates RAF-1 by direct phosphorylation. Nature, 364, 249–252. [DOI] [PubMed] [Google Scholar]
- Lewis T.S., Shapiro,P.S. and Ahn,N.G. (1998) Signal transduction through MAP kinase cascades. Adv. Cancer Res., 74, 49–139. [DOI] [PubMed] [Google Scholar]
- MacNicol M.C. and MacNicol,A.M. (1999) Nerve growth factor-stimulated B-Raf catalytic activity is refractory to inhibition by cAMP-dependent protein kinase. J. Biol. Chem., 274, 13193–13197. [DOI] [PubMed] [Google Scholar]
- MacNicol M.C., Muslin,A.J. and MacNicol,A.M. (2000) Disruption of the 14-3-3 binding site within the B-Raf kinase domain uncouples catalytic activity from PC12 cell differentiation. J. Biol. Chem., 275, 3803–3809. [DOI] [PubMed] [Google Scholar]
- Magnuson N.S., Beck,T., Vahidi,H., Hahn,H., Smola,U. and Rapp,U.R. (1994) The Raf-1 serine/threonine protein kinase. Semin. Cancer Biol., 5, 247–253. [PubMed] [Google Scholar]
- Marshall C.J. (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell, 80, 179–185. [DOI] [PubMed] [Google Scholar]
- Mason C.S., Springer,C.J., Cooper,R.G., Superti-Furga,G., Marshall,C.J. and Marais,R. (1999) Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J., 18, 2137–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud N.R., Fabian,J.R., Mathes,K.D. and Morrison,D.K. (1995) 14-3-3 is not essential for Raf-1 function: identification of Raf-1 proteins that are biologically activated in a 14-3-3- and Ras-independent manner. Mol. Cell. Biol., 15, 3390–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischak H., Seitz,T., Janosch,P., Eulitz,M., Steen,H., Schellerer,M., Philipp,A. and Kolch,W. (1996) Negative regulation of Raf-1 by phosphorylation of serine 621. Mol. Cell. Biol., 16, 5409–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison D.K. and Cutler,R.E. (1997) The complexity of Raf-1 regulation. Curr. Opin. Cell Biol., 9, 174–179. [DOI] [PubMed] [Google Scholar]
- Morrison D.K., Heidecker,G., Rapp,U.R. and Copeland,T.D. (1993) Identification of the major phosphorylation sites of the Raf-1 kinase. J. Biol. Chem., 268, 17309–17316. [PubMed] [Google Scholar]
- Mott H.R., Carpenter,J.W., Zhong,S., Ghosh,S., Bell,R.M. and Campbell,S.L. (1996) The solution structure of the Raf-1 cysteine-rich domain: a novel ras and phospholipid binding site. Proc. Natl Acad. Sci. USA, 93, 8312–8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslin A.J., Tanner,J.W., Allen,P.M. and Shaw,A.S. (1996) Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell, 84, 889–897. [DOI] [PubMed] [Google Scholar]
- Nassar N., Horn,G., Herrmann,C., Scherer,A., McCormick,F. and Wittinghofer,A. (1995) The 2.2 Å crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature, 375, 554–560. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T., Shimizu,K., Yamamori,B., Kuroda,S. and Takai,Y. (1996) Activation of brain B-Raf protein kinase by Rap1B small GTP-binding protein. J. Biol. Chem., 271, 1258–1261. [DOI] [PubMed] [Google Scholar]
- Payne D.M., Rossomando,A.J., Martino,P., Erickson,A.K., Her,J.H., Shabanowitz,J., Hunt,D.F., Weber,M.J. and Sturgill,T.W. (1991) Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J., 10, 885–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard C.A., Bolin,L., Slattery,R., Murray,R. and McMahon,M. (1996) Post-natal lethality and neurological and gastrointestinal defects in mice with targeted disruption of the A-Raf protein kinase gene. Curr. Biol., 6, 614–617. [DOI] [PubMed] [Google Scholar]
- Resing K.A., Mansour,S.J., Hermann,A.S., Johnson,R.S., Candia,J.M., Fukasawa,K., Vande Woude,G.F. and Ahn,N.G. (1995) Determination of v-Mos-catalyzed phosphorylation sites and autophosphorylation sites on MAP kinase kinase by ESI/MS. Biochemistry, 34, 2610–2620. [DOI] [PubMed] [Google Scholar]
- Reuter C.W., Catling,A.D., Jelinek,T. and Weber,M.J. (1995) Biochemical analysis of MEK activation in NIH 3T3 fibroblasts. Identification of B-Raf and other activators. J. Biol. Chem., 270, 7644–7655. [DOI] [PubMed] [Google Scholar]
- Rommel C., Clarke,B.A., Zimmermann,S., Nunez,L., Rossman,R., Reid,K., Moelling,K., Yancopoulos,G.D. and Glass,D.J. (1999) Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science, 286, 1738–1741. [DOI] [PubMed] [Google Scholar]
- Roy S., McPherson,R.A., Apolloni,A., Yan,J., Lane,A., Clyde-Smith,J. and Hancock,J.F. (1998) 14-3-3 facilitates Ras-dependent Raf-1 activation in vitro and in vivo. Mol. Cell. Biol., 18, 3947–3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger J. and Ullrich,A. (1992) Growth factor signaling by receptor tyrosine kinases. Neuron, 9, 383–391. [DOI] [PubMed] [Google Scholar]
- Schramm K., Niehof,M., Radziwill,G., Rommel,C. and Moelling,K. (1994) Phosphorylation of c-Raf-1 by protein kinase A interferes with activation. Biochem. Biophys. Res. Commun., 201, 740–747. [DOI] [PubMed] [Google Scholar]
- Shoji S., Ericsson,L.H., Walsh,K.A., Fischer,E.H. and Titani,K. (1983) Amino acid sequence of the catalytic subunit of bovine type II adenosine cyclic 3′,5′-phosphate dependent protein kinase. Biochemistry, 22, 3702–3709. [DOI] [PubMed] [Google Scholar]
- Sternberg P.W. and Han,M. (1998) Genetics of RAS signaling in C.elegans. Trends Genet., 14, 466–472. [DOI] [PubMed] [Google Scholar]
- Stewart S., Sundaram,M., Zhang,Y., Lee,J., Han,M. and Guan,K.L. (1999) Kinase suppressor of Ras forms a multiprotein signaling complex and modulates MEK localization. Mol. Cell. Biol., 19, 5523–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto T., Stewart,S., Han,M. and Guan,K.L. (1998) The kinase suppressor of Ras (KSR) modulates growth factor and Ras signaling by uncoupling Elk-1 phosphorylation from MAP kinase activation. EMBO J., 17, 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker A. and Newton,A.C. (2000) Akt/protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J. Biol. Chem., 275, 8271–8274. [DOI] [PubMed] [Google Scholar]
- Tzivion G., Luo,Z. and Avruch,J. (1998) A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature, 394, 88–92. [DOI] [PubMed] [Google Scholar]
- Vojtek A.B., Hollenberg,S.M. and Cooper,J.A. (1993) Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell, 74, 205–214. [DOI] [PubMed] [Google Scholar]
- Vossler M.R., Yao,H., York,R.D., Pan,M.G., Rim,C.S. and Stork,P.J. (1997) cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell, 89, 73–82. [DOI] [PubMed] [Google Scholar]
- Weber C.K., Slupsky,J.R., Herrmann,C., Schuler,M., Rapp,U.R. and Block,C. (2000) Mitogenic signaling of Ras is regulated by differential interaction with Raf isozymes. Oncogene, 19, 169–176. [DOI] [PubMed] [Google Scholar]
- Wojnowski L., Zimmer,A.M., Beck,T.W., Hahn,H., Bernal,R., Rapp,U.R. and Zimmer,A. (1997) Endothelial apoptosis in Braf-deficient mice [see comments]. Nature Genet., 16, 293–297. [DOI] [PubMed] [Google Scholar]
- Wu J., Dent,P., Jelinek,T., Wolfman,A., Weber,M.J. and Sturgill,T.W. (1993) Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3′,5′-monophosphate [see comments]. Science, 262, 1065–1069. [DOI] [PubMed] [Google Scholar]
- Yao B., Zhang,Y., Delikat,S., Mathias,S., Basu,S. and Kolesnick,R. (1995) Phosphorylation of Raf by ceramide-activated protein kinase. Nature, 378, 307–310. [DOI] [PubMed] [Google Scholar]
- Zhang Y. et al. (1997) Kinase suppressor of Ras is ceramide-activated protein kinase. Cell, 89, 63–72. [DOI] [PubMed] [Google Scholar]
- Zheng C.F. and Guan,K.L. (1994) Activation of MEK family kinases requires phosphorylation of two conserved Ser/Thr residues. EMBO J., 13, 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S. and Moelling,K. (1999) Phosphorylation and regulation of Raf by Akt (protein kinase B). Science, 286, 1741–1744. [DOI] [PubMed] [Google Scholar]