

Abstract
Mitochondrial dysfunction and endoplasmic reticulum (ER) stress are closely associated with β-cell dysfunction and peripheral insulin resistance. Thus, each of these factors contributes to the development of type 2 diabetes mellitus (DM). The accumulated evidence reveals structural and functional communications between mitochondria and the ER. It is now well established that ER stress causes apoptotic cell death by disturbing mitochondrial Ca2+ homeostasis. In addition, recent studies have shown that mitochondrial dysfunction causes ER stress. In this paper, we summarize the roles that mitochondrial dysfunction and ER stress play in the pathogenesis of type 2 DM. Structural and functional communications between mitochondria and the ER are also discussed. Finally, we focus on recent findings supporting the hypothesis that mitochondrial dysfunction and the subsequent induction of ER stress play important roles in the pathogenesis of type 2 DM.
1. Introduction
Type 2 diabetes mellitus (DM) is characterized by impaired insulin secretion from pancreatic β-cells. In addition, insulin-responsive tissues, such as muscle, liver, and adipose tissue, exhibit insulin resistance. A number of findings suggest that both of these major features of type 2 DM are associated with mitochondrial dysfunction and/or endoplasmic reticulum (ER) stress [1–4]. Recently, it was shown that mitochondria and the ER interact both physically and functionally [5, 6]. In this paper, we will focus on the roles that mitochondrial dysfunction and ER stress play in the pathogenesis of type 2 DM. Particular emphasis will be placed on recent findings elucidating the interaction between mitochondria and the ER.
2. Role of Mitochondrial Dysfunction in Type 2 DM
2.1. Mitochondria
The mitochondrion is an intracellular double-membraned organelle found in most eukaryotic cells [7]. Mitochondria are well known to be power stations within cells, as one of their major functions is production of ATP [8]. In addition, mitochondria play essential roles in intracellular reactive oxygen species (ROS) production [9], regulation of apoptosis [10], and Ca2+ storage [11].
2.2. Mitochondrial and Pancreatic β-Cell Dysfunction
Insulin-resistant patients can develop overt type 2 DM when pancreatic β-cells are unable to produce enough insulin to maintain normoglycemia. Pancreatic β-cells from patients with type 2 DM cannot sense glucose properly, and this contributes to impairment of insulin secretion. Interestingly, glucose sensing by β-cells appears to be controlled by mitochondrial metabolism. Reduced forms of nicotinamide adenine dinucleotide (NADH) or flavin adenine dinucleotide (FADH2) are generated during glucose metabolism via both glycolysis and the tricarboxylic acid (TCA) cycle. Electron transfer to the mitochondrial electron-transport chain (ETC) by NADH and FADH2 leads to production of ATP via the process of oxidative phosphorylation (OXPHOS). Increases in the ATP/ADP ratio in β-cells inhibit ATP-sensitive potassium channels (KATP), in turn inducing depolarization of plasma membranes. The opening of voltage-sensitive Ca2+ channels allows Ca2+ uptake by β-cells, thereby contributing to secretion of insulin. Thus, mitochondrial dysfunction can impair glucose-stimulated insulin secretion by reducing the ATP/ADP ratio within β-cells (Figure 1) [12].
Figure 1.
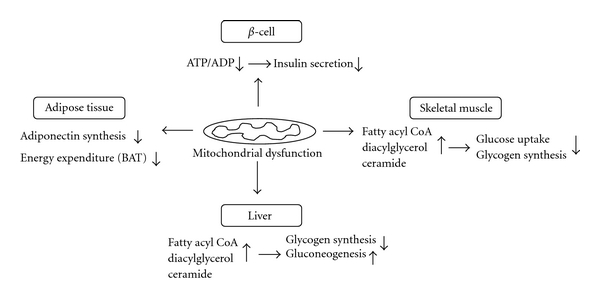
Roles of mitochondrial dysfunction in the pathogenesis of β-cell dysfunction and insulin resistance.
2.3. Mitochondrial Dysfunction and Skeletal Muscle Insulin Resistance
Defective mitochondrial fatty acid metabolism in skeletal muscle is thought to affect insulin signaling pathways, thereby leading to insulin resistance [13–15]. Impairment of mitochondrial fatty acid β-oxidation, either alone or in conjunction with increased delivery of free fatty acids (FFAs) from plasma, leads to elevated levels of intracellular fatty acid metabolites such as fatty acyl CoA, diacylglycerol, and ceramide [16–18]. Metabolites formed under such circumstances activate serine/threonine kinases including protein kinase C (PKC), leading to phosphorylation of serine sites on insulin receptor substrate-1 (IRS-1) [19, 20]. Increased serine phosphorylation of IRS-1 inhibits the tyrosine kinase activity of the insulin receptor on IRS-1 and the activity of insulin-stimulated phosphatidylinositol 3-kinase (PI 3-kinase), resulting in decreased activity of insulin-stimulated protein kinase B (PKB, also known as AKT). Reduced AKT activity leads to suppression of insulin-stimulated glucose transporter 4 (GLUT4) translocation and subsequent reduction of glycogen synthesis (Figure 1).
2.4. Mitochondrial Dysfunction and Hepatic Insulin Resistance
The liver plays a crucial role in the development of insulin resistance and type 2 DM [21]. Several lines of evidence indicate that defects in liver mitochondrial oxidative function can induce hepatic insulin resistance [14, 15, 22, 23]. For example, reduced levels of mitochondrial fatty acid β-oxidation in the liver, as in skeletal muscle, lead to accumulation of intracellular fatty acid metabolites [24, 25]. Note that similar results were observed either when de novo hepatic lipogenesis rose or when delivery of FFAs from the plasma increased. Under either circumstance, the metabolites adversely affected intracellular insulin signaling, leading to reduced insulin stimulation of glycogen synthesis and increased hepatic gluconeogenesis (Figure 1) [19].
2.5. Mitochondrial Dysfunction and Adipose Tissue
Adipose tissue has been described as an endocrine organ that plays a central role in fuel metabolism [26]. Adipocytokines such as leptin, adiponectin, resistin, and tumor necrosis factor-α (TNF-α) are released by adipose tissue, and these cytokines regulate fuel metabolism [27]. Adiponectin is known to have insulin-sensitizing effects. However, in contrast to other adipocytokines, the plasma levels of adiponectin are significantly decreased in obese subjects and in type 2 DM patients [28, 29]. Recently, we reported that the levels of adiponectin in plasma and adipose tissue were significantly lowered in obese mice; an associated reduction of mitochondrial content and function in adipose tissue was also documented [30]. Rosiglitazone, a peroxisome proliferator-activated receptor γ (PPARγ) agonist, reversed decreases in plasma adiponectin levels and adiponectin expression in obese mice, and elevated mitochondrial content and function in adipose tissue. These findings suggest that mitochondrial dysfunction in adipose tissue leads to decreased plasma adiponectin levels in obese subjects (Figure 1).
Many studies on rodents have shown that the capacity of mitochondria for oxidizing fatty acids in brown adipose tissue (BAT) plays a critical role in the regulation of adaptive thermogenesis, energy balance, and body weight [31, 32]. The presence of BAT was considered to be relevant only in human newborn and small mammals. However, recent studies using positron-emission tomography and computed tomography (PET-CT) demonstrated that adult humans possess active BAT [33, 34]. Thus, mitochondrial dysfunction in BAT appears to be linked to impaired thermogenesis and energy expenditure, contributing to the development of obesity and insulin resistance in adult humans (Figure 1) [35].
3. Role of ER Stress in Type 2 DM
3.1. ER
The ER is a complex organelle that is found in all eukaryotic cells. Structurally, the ER is formed by an interconnected network of cisternae and microtubules. From a functional viewpoint, the ER plays a central role in protein folding and in quality control of newly synthesized proteins [36]. The ER also serves as an essential site for synthesis of lipids [37] and for high-capacity buffering of intracellular Ca2+ [38].
3.2. ER Stress
If proteins are to be folded properly within the ER, a balance must be struck between the ER protein load and ER folding capacity. A number of conditions can disrupt ER homeostasis, leading to accumulation of misfolded proteins within the lumen of the ER [4, 39]. Such conditions include a large biosynthetic load, defects in folding machinery, and disturbances in the handling of Ca2+. Accumulation of misfolded proteins in the ER causes ER stress, and this activates an elaborative adaptive process termed the unfolded protein response (UPR) [40].
The UPR is triggered by three ER transmembrane proteins: protein kinase R-like ER kinase (PERK), inositol-requiring enzyme 1 (IRE1), and activating transcription factor 6 (ATF6). In unstressed conditions, ER luminal domain of these proteins are bound by the chaperone Bip, maintaining them in an inactive state until ER stress is present [41]. During ER stress, misfolded proteins sequester, Bip, leading to free PERK and IRE1 monomers to oligomerize and trans-autophosphorylate. Activated PERK mediates inhibition of protein translation via phosphorylation of eukaryotic translation initiation factor 2α (eIF2α), resulting in reduced global protein synthesis in an attempt to decrease the protein-folding load in the ER lumen [42]. PERK-mediated eIF2α phosphorylation also contributes to the activation of a subset of translational targets including activating transcription factor 4 (ATF4). ATF4 activates transcriptionally the proapoptotic transcription factor CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP) [43].
Activation of IRE1, which has endoribonuclease activity, leads to splicing of X-box binding protein-1 (XBP1) mRNA and translation of the active form (XBP1s) [44]. XBP1s translocates to the nucleus and regulates expression of ER chaperones and proteins involved in ER-associated degradation (ERAD) [45]. In addition, the cytosolic domain of IRE1 can associate with TNF receptor-associated factor 2 (TRAF2) to activate the apoptosis signal-regulating kinase 1 (ASK1) and c-Jun N-terminal kinase (JNK) pathway, independently with its endoribonuclease activity [46, 47].
In response to ER stress, ATF6, released from Bip, translocates to the Golgi where it is cleaved by proteases into an active amino-terminal form [48]. N-terminal ATF6 in turn moves to the nucleus to stimulate expression of ER chaperones and proteins involved in ERAD.
3.3. ER Stress and β-Cell Dysfunction
ER stress plays an important role in the pathogenesis of type 2 DM, as such stress contributes to pancreatic β-cell dysfunction and insulin resistance [4, 49]. When the demand for insulin overwhelms the folding capacity of the ER, the UPR becomes chronically activated. Several stimuli have been shown to cause sustained accumulation of misfolded proteins within the ER lumen of β-cells [4]. These include high levels of FFA (caused by either a high-fat diet or obesity) and glucose (chronic hyperglycemia), as well as aggregation of islet amyloid polypeptide. Accumulation of misfolded proteins triggers chronic activation of the UPR, inducing β-cell dysfunction and apoptosis [50, 51].
Several components of the UPR that contribute to β-cell apoptosis have been shown (Figure 2). ER stress can induce β-cell apoptosis through prolonged activation of IRE1-TRAF2-ASK1 cascade and JNK pathway [52]. CHOP also plays a crucial role in the induction of ER stress-mediated β-cell apoptosis [53].
Figure 2.
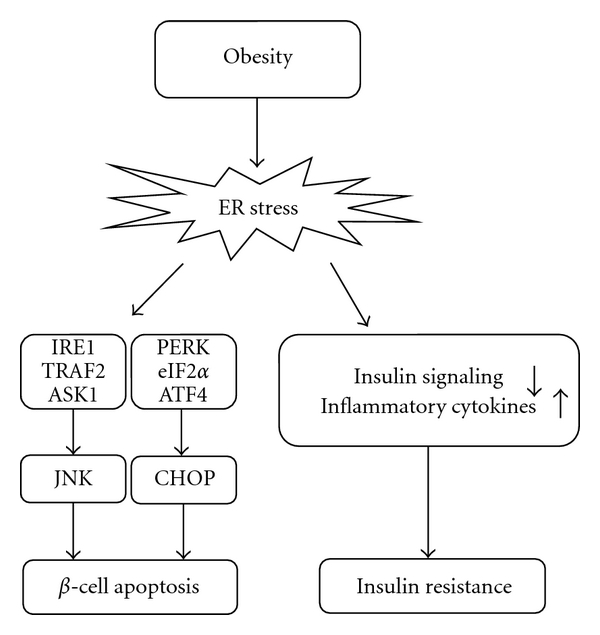
Roles of ER stress in the pathogenesis of β-cell apoptosis and insulin resistance.
3.4. ER Stress and Insulin Resistance
In addition to β-cell dysfunction, ER stress is involved in peripheral insulin resistance (Figure 2). Obesity results in chronic stimulation of ER stress, leading to continuous activation of the UPR. Recent studies have suggested that this may, in fact, be the main mechanism of peripheral insulin resistance and type 2 DM [3, 54]. In obese mice, the levels of ER stress markers are increased in the liver and adipose tissue [3]. Obesity-induced ER stress inhibits insulin signaling, and this leads to insulin resistance. ER stress can also activate nuclear factor-κB (NF-κB) signaling in the liver [55], thereby increasing production of proinflammatory cytokines and causing development of insulin resistance [56]. A recent study showed that treatment of obese diabetic mice with the chemical chaperones 4-phenyl butyric acid (PBA) and taurine-conjugated ursodeoxycholic acid (TUDCA) improved peripheral insulin sensitivity by alleviating ER stress [57]. TUDCA therapy also improved insulin sensitivity in the liver and muscle of obese subjects [58].
4. Structural Communication between Mitochondria and the ER
A number of studies have shown structural communication between the mitochondria and the ER. The evidence includes cosedimentation of ER particles with mitochondria, as well as electron microscopic observation of a close physical apposition between mitochondria and the ER [59, 60]. More recently, high-resolution three-dimensional images have been obtained showing an interaction between mitochondria and the ER; specific color labels were employed to this end [61]. A recent study using electron tomography also demonstrated that the outer mitochondrial membrane (OMM) and the ER are joined by tethers, enabling ER proteins to associate directly with proteins and lipids of the OMM [62].
The structural membrane hat bridges between mitochondria and the ER is known as the mitochondria-associated membrane (MAM) [63]. The MAM plays an essential role in several cellular functions, including lipid transport, Ca2+ signaling, and apoptosis [64]. A number of mitochondrial or ER-bound proteins are important for maintaining structural communication between the two organelles at the MAM [64, 65]. In particular, communication between the organelles is modulated by a family of chaperone proteins. The voltage-dependent anion channel (VDAC) is physically linked to the inositol 1,4,5-triphosphate receptor (IP3R) via the molecular chaperone grp75 [66]. Overexpression of the cytosolic form of grp75 selectively increases IP3-induced Ca2+ uptake into the mitochondrial matrix, whereas overexpression of the mitochondrial form of the protein does not have this effect. Another protein that modulates interaction between mitochondria and the ER is phosphofurin acidic cluster sorting protein 2 (PACS-2), which is known to integrate ER-mitochondrial communication and apoptosis signaling [67]. Accordingly, PACS-2 depletion induces mitochondrial fragmentation, dissociates the ER from mitochondria, and blocks apoptosis signaling. More recently, Sigma-1 receptors have been shown to be located at the MAM of the ER, where they form complexes with Bip [68]. Sigma-1 receptors dissociate from Bip and bind to type-3 IP3Rs under conditions of ER Ca2+ depletion. Thus, type-3 IP3Rs are not degraded by proteasomes. Ca2+ depletion appears to induce a prolonged Ca2+ signaling event from the ER to the mitochondria, via IP3Rs. Together, the data suggest that Sigma-1 receptors are involved in maintaining normal Ca2+ signaling from the ER to mitochondria.
Structural communication between mitochondria and the ER is also modulated by fission and fusion of mitochondria. Fission and fusion are regulated by a family of mitochondrion-shaping proteins including dynamin-related protein 1 (DRP1), mitofusin 1, and mitofusin 2 [69]. Mitofusin-2 is a mitochondrial transmembrane GTPase that regulates mitochondrial fusion [70], and this protein is enriched at MAMs [71]. Mitofusin-2 tethers the ER to mitochondria via formation of both homotypic and heterotypic complexes. For example, ER mitofusin-2 interacts with either mitofusin-2 or mitofusin-1 on mitochondria. The tethering effect of mitofusin-2 appears to play a role in the control of Ca2+ flow between mitochondria and the ER [71].
5. Functional Communication between Mitochondria and the ER
5.1. Role of ER Stress in Induction of Mitochondrial Dysfunction
Mitochondrial dysfunction and ER stress have each been recognized to play crucial roles in the pathogenesis of type 2 DM. However, the individual stressors appear to act sequentially in various tissues. For example, accumulating evidence has shown that ER stress induces mitochondrial dysfunction, thereby leading to disruption of various physiological responses within cells [5, 6].
Interactions between mitochondria and the ER facilitate control of Ca2+ signaling and Ca2+-dependent cellular processes such as apoptosis [72, 73]. Prolonged ER stress leads to release of Ca2+ from the ER lumen at the MAM. In contrast, such stress leads to increased Ca2+ uptake into the mitochondrial matrix. Elevated Ca2+ uptake induces an imbalance between mitochondrial Ca2+ load and the buffering capacity of the matrix, and such imbalance ultimately leads to a prolonged episode of massive mitochondrial Ca2+ accumulation. Sustained Ca2+ accumulation triggers opening of the mitochondrial permeability transition pore (mtPTP). Ultimately, this results in swelling of the organelle, rupture of the OMM, and release of proapoptotic proteins into the cytosol [74].
ROS are thought to act as local messengers between the ER and mitochondria [6]. Many ROS sources and targets are localized to the ER and mitochondria [75, 76]. Disulfide bond formation is a critical step in folding of newly synthesized proteins, and this is mediated by members of the ER oxidoreductin 1 (Ero1) family [77]. Importantly, ROS are concomitantly produced by Ero1. Previous studies have shown that Ero1 can be activated under conditions of ER stress [78, 79]. Thus, conditions that trigger such stress may lead to excessive production of ROS in the ER. Such elevated ROS levels inactivate the sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) and activate IP3R via oxidation [80, 81]. Modulation of Ca2+ channel activity by ROS increases the level of Ca2+ on the cytosolic face of the ER and also promotes Ca2+ uptake into the mitochondrial matrix. Therefore, ROS production mediated by Ero1 provides an additional mechanism by which ER stress can induce mitochondrial dysfunction.
5.2. Role of Mitochondrial Dysfunction in Induction of ER Stress
5.2.1. NO-Mediated Induction of the ER Stress Response via Inhibition of Mitochondrial Respiration
Protein folding processes and the handling of Ca2+ within the ER each require large amounts of ATP. Accordingly, ATP depletion is one of the best-known mechanisms by which ER stress may be induced [82]. Such observations have raised significant questions regarding the modes by which changes in mitochondrial function affect processes within the ER. It is widely accepted that ER stress induces mitochondrial dysfunction. However, it appears that this is not a one-way process; Xu et al. have shown that the ER stress response can be induced following disruption of the mitochondrial respiratory chain by nitric oxide [83].
NO can bind to cytochrome c oxidase and inhibit the enzyme, in competition with oxygen [84]. Thus, the respiratory chain is disrupted in NO-generating cells [83]. Because this process is accompanied by mitochondrial Ca2+ flux, disruption of electron transfer by cytochrome c oxidase may result in changes in the extent of Ca2+ flux between the mitochondria and the ER. NO-mediated changes in Ca2+ flux between these organelles increase expression of ER stress-responsive genes such as glucose-regulated protein 78 (Grp78), elevated levels of which provide significant cytoprotection against thapsigargin, a selective ER Ca2+ ATPase inhibitor. Interestingly, chemical disruption of mitochondrial Ca2+ flux has been shown to reverse NO-mediated cytoprotection. In addition, the NO-mediated ER stress response was diminished in rhoo cells devoid of mitochondrial DNA [83]. Together, these results suggest that NO signals the ER stress response via inhibition of mitochondrial respiration.
5.2.2. Mitochondrial Dysfunction Induces ER Stress and Decreases Adiponectin Synthesis
Recently, we showed that impairment of mitochondrial function increases the levels of ER stress markers [30]. Adenovirus-mediated overexpression of nuclear respiratory factor-1 (NRF-1), a transcription factor that regulates the expression of nuclear-encoded mitochondrial genes, reduced the upregulation of ER stress markers associated with mitochondrial dysfunction. Previous studies showed that JNK and activating transcription factor 3 (ATF3) were activated by ER stress [3, 85]. Further, impairment of mitochondrial function sequentially activated JNK and ATF3. However, inhibition of JNK and ATF3 reversed the reduction in adiponectin transcription that was induced by mitochondrial dysfunction [30]. Together, the data suggest that mitochondrial dysfunction induces ER stress. This, in turn, activates signaling cascades involving JNK and ATF3, thereby decreasing adiponectin synthesis in adipose tissue.
5.2.3. Induction of ER Stress by Mitochondrial Dysfunction and Hepatic Insulin Resistance
Mitochondrial dysfunction induces ER stress, and this, in turn, causes hepatic insulin resistance [86]. In human liver cell lines, inhibition of mitochondrial function by oligomycin disturbs insulin signaling. In contrast, hepatic gluconeogenesis is abnormally increased. The levels of ER stress markers were elevated in cells containing functionally inactivated mitochondria. However, this rise was reversed by decreasing the level of cytosolic-free Ca2+. Importantly, mitochondrial dysfunction elevated the level of cytosolic-free Ca2+, which in turn promoted an increase in the concentrations of the ER Ca2+ channels IP3Rs and the ryanodine receptor-2 (RyR-2). Elevated levels of these channels induced Ca2+ depletion within the lumen of the ER. Disturbances in Ca2+ homoeostasis in the ER are also known to trigger the ER stress response, leading to activation of p38 mitogen-activated protein kinase (MAPK), as well as increasing phosphoenolpyruvate carboxykinase (PEPCK) expression [87, 88]. Abnormal activation of JNK by mitochondrial dysfunction also increased PEPCK expression by affecting insulin signaling and forkhead box protein O1 (FOXO1) activity [86]. Together, the results suggest that mitochondrial dysfunction induces ER stress in a Ca2+-dependent manner, leading to disturbance of insulin signaling and an abnormal rise in gluconeogenesis within hepatocytes.
5.2.4. Induction of ER Stress by Mitochondrial Dysfunction and Local ATP Depletion
A number of events may contribute to the linking of mitochondrial dysfunction and ER stress. For example, local ATP pools in the mitochondria and the adjacent ER may be essential to supply the energy required by SERCA to import Ca2+ into the lumen of the ER. In agreement with this idea, inhibition of OXPHOS was shown to cause a prolonged delay in uptake of Ca2+ into the lumen of the ER; in addition, Ca2+ levels within the ER fell [89]. Inhibition of OXPHOS caused rapid local ATP depletion in mitochondria and the ER, although global cytosolic ATP levels decreased at a much later time. These results suggest that local ATP depletion in the region in which SERCA is active may reduce the uptake of Ca2+ into the lumen of the ER. This would cause Ca2+ depletion within the ER, which may trigger the ER stress response. Whether this mechanism is operative in pancreatic β-cells and/or insulin-responsive tissues remains to be determined.
6. Conclusions
We have provided a brief overview of the interaction between mitochondrial dysfunction and ER stress. In particular, we examined the role played by such interaction in the pathogenesis of type 2 DM. Mitochondrial dysfunction and ER stress are essential for β-cell dysfunction and peripheral insulin resistance. To date, substantial progress has been made in understanding structural and functional communications between mitochondria and the ER. We now know that ER stress can induce mitochondrial dysfunction. Thus, such stress plays a central role in apoptosis signaling via Ca2+- and/or ROS-dependent mechanisms. Together with recent findings linking mitochondrial dysfunction and ER stress, it appears that bidirectional communication exists between these two organelles (Figure 3). Characterization of interactions between mitochondria and the ER is a dynamic and growing area of interest; future research will carefully dissect such processes. Hopefully, the studies will help us to gain a better understanding of the pathogenesis underlying type 2 DM. Therapeutic approach aimed at restoring mitochondria function will prevent or treat insulin resistance and type 2 DM through suppression of ER stress.
Figure 3.
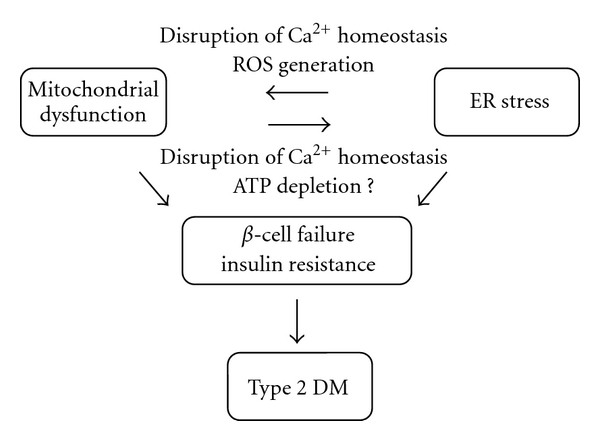
Bidirectional communication between dysfunctional mitochondria and the ER under stress contributes to the development of type 2 DM.
Acknowledgment
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grants funded by the Ministry of Science and Technology (2009-0091988), Seoul, Korea.
References
- 1.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 2.Patti ME, Corvera S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocrine Reviews. 2010;31(3):364–395. doi: 10.1210/er.2009-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Özcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 4.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocrine Reviews. 2008;29(1):42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 5.Giorgi C, De Stefani D, Bononi A, Rizzuto R, Pinton P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. International Journal of Biochemistry and Cell Biology. 2009;41(10):1817–1827. doi: 10.1016/j.biocel.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Csordás G, Hajnóczky G. SR/ER-mitochondrial local communication: calcium and ROS. Biochimica et Biophysica Acta. 2009;1787(11):1352–1362. doi: 10.1016/j.bbabio.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henze K, Martin W. Essence of mitochondria. Nature. 2003;426(6963):127–128. doi: 10.1038/426127a. [DOI] [PubMed] [Google Scholar]
- 8.Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283(5407):1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- 9.Stowe DF, Camara AKS. Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxidants and Redox Signaling. 2009;11(6):1373–1414. doi: 10.1089/ars.2008.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305(5684):626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 11.Murgia M, Giorgi C, Pinton P, Rizzuto R. Controlling metabolism and cell death: at the heart of mitochondrial calcium signalling. Journal of Molecular and Cellular Cardiology. 2009;46(6):781–788. doi: 10.1016/j.yjmcc.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic β-cells. Nature. 2001;414(6865):807–812. doi: 10.1038/414807a. [DOI] [PubMed] [Google Scholar]
- 13.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51(10):2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- 14.Petersen KF, Befroy D, Dufour S, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300(5622):1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. The New England Journal of Medicine. 2004;350(7):664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu C, Chen Y, Cline GW, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. Journal of Biological Chemistry. 2002;277(52):50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 17.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α . Diabetes. 2002;51(7):2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 18.Won JC, Park JY, Kim YM, et al. Peroxisome proliferator-activated receptor-γ coactivator 1-α overexpression prevents endothelial apoptosis by increasing ATP/ADP translocase activity. Arteriosclerosis, Thrombosis, and Vascular Biology. 2010;30(2):290–297. doi: 10.1161/ATVBAHA.109.198721. [DOI] [PubMed] [Google Scholar]
- 19.Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55(2):S9–S15. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koh EH, Lee WJ, Kim MS, Park JY, Lee IK, Lee KU. Intracellular fatty acid metabolism in skeletal muscle and insulin resistance. Current Diabetes Reviews. 2005;1(3):331–336. doi: 10.2174/157339905774574347. [DOI] [PubMed] [Google Scholar]
- 21.Kotronen A, Seppälä-Lindroos A, Bergholm R, Yki-Järvinen H. Tissue specificity of insulin resistance in humans: fat in the liver rather than muscle is associated with features of the metabolic syndrome. Diabetologia. 2008;51(1):130–138. doi: 10.1007/s00125-007-0867-x. [DOI] [PubMed] [Google Scholar]
- 22.Pérez-Carreras M, Del Hoyo P, Martín MA, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38(4):999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 23.Savage DB, Cheol SC, Samuel VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. Journal of Clinical Investigation. 2006;116(3):817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samuel VT, Liu ZX, Qu X, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. Journal of Biological Chemistry. 2004;279(31):32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 25.Zhang D, Liu ZX, Cheol SC, et al. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(43):17075–17080. doi: 10.1073/pnas.0707060104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scherer PE. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes. 2006;55(6):1537–1545. doi: 10.2337/db06-0263. [DOI] [PubMed] [Google Scholar]
- 27.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444(7121):847–853. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kern PA, Di Gregorio GB, Lu T, Rassouli N, Ranganathan G. Adiponectin expression from human adipose tissue: relation to obesity, insulin resistance, and tumor necrosis factor-α expression. Diabetes. 2003;52(7):1779–1785. doi: 10.2337/diabetes.52.7.1779. [DOI] [PubMed] [Google Scholar]
- 29.Hotta K, Funahashi T, Arita Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20(6):1595–1599. doi: 10.1161/01.atv.20.6.1595. [DOI] [PubMed] [Google Scholar]
- 30.Koh EH, Park JY, Park HS, et al. Essential role of mitochondrial function in adiponectin synthesis in adipocytes. Diabetes. 2007;56(12):2973–2981. doi: 10.2337/db07-0510. [DOI] [PubMed] [Google Scholar]
- 31.Guerra C, Koza RA, Walsh K, Kurtz DM, Wood PA, Kozak LP. Abnormal nonshivering thermogenesis in mice with inherited defects of fatty acid oxidation. Journal of Clinical Investigation. 1998;102(9):1724–1731. doi: 10.1172/JCI4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozak LP, Koza RA, Anunciado-Koza R. Brown fat thermogenesis and body weight regulation in mice: relevance to humans. International Journal of Obesity. 2010;34(1):S23–S27. doi: 10.1038/ijo.2010.179. [DOI] [PubMed] [Google Scholar]
- 33.Nedergaard J, Bengtsson T, Cannon B. Unexpected evidence for active brown adipose tissue in adult humans. American Journal of Physiology. 2007;293(2):E444–E452. doi: 10.1152/ajpendo.00691.2006. [DOI] [PubMed] [Google Scholar]
- 34.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, et al. Cold-activated brown adipose tissue in healthy men. The New England Journal of Medicine. 2009;360(15):1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 35.Enerbäck S. Brown adipose tissue in humans. International Journal of Obesity. 2010;34(1):S43–S46. doi: 10.1038/ijo.2010.183. [DOI] [PubMed] [Google Scholar]
- 36.Chevet E, Cameron PH, Pelletier MF, Thomas DY, Bergeron JJM. The endoplasmic reticulum: integration of protein folding, quality control, signaling and degradation. Current Opinion in Structural Biology. 2001;11(1):120–124. doi: 10.1016/s0959-440x(00)00168-8. [DOI] [PubMed] [Google Scholar]
- 37.Espenshade PJ, Hughes AL. Regulation of sterol synthesis in eukaryotes. Annual Review of Genetics. 2007;41:401–427. doi: 10.1146/annurev.genet.41.110306.130315. [DOI] [PubMed] [Google Scholar]
- 38.Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiological Reviews. 2005;85(1):201–279. doi: 10.1152/physrev.00004.2004. [DOI] [PubMed] [Google Scholar]
- 39.Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiological Reviews. 2006;86(4):1133–1149. doi: 10.1152/physrev.00015.2006. [DOI] [PubMed] [Google Scholar]
- 40.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology. 2007;8(7):519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 41.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nature Cell Biology. 2000;2(6):326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 42.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 43.Harding HP, Novoa I, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular Cell. 2000;6(5):1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 45.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Molecular and Cellular Biology. 2003;23(21):7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes and Development. 2002;16(11):1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 48.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Developmental Cell. 2002;3(1):99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 49.Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with β-cell failure and diabetes. Endocrine Reviews. 2008;29(3):317–333. doi: 10.1210/er.2007-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fonseca SG, Urano F, Burcin M, Gromada J. Stress hypERactivation in the β-cell. Islets. 2010;2(1):1–9. doi: 10.4161/isl.2.1.10456. [DOI] [PubMed] [Google Scholar]
- 51.Volchuk A, Ron D. The endoplasmic reticulum stress response in the pancreatic β-cell. Diabetes, Obesity and Metabolism. 2010;12(supplement 2):48–57. doi: 10.1111/j.1463-1326.2010.01271.x. [DOI] [PubMed] [Google Scholar]
- 52.Cunha DA, Hekerman P, Ladrière L, et al. Initiation and execution of lipotoxic ER stress in pancreatic β-cells. Journal of Cell Science. 2008;121(14):2308–2318. doi: 10.1242/jcs.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. Journal of Clinical Investigation. 2002;109(4):525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsiotra PC, Tsigos C. Stress, the endoplasmic reticulum, and insulin resistance. Annals of the New York Academy of Sciences. 2006;1083:63–76. doi: 10.1196/annals.1367.007. [DOI] [PubMed] [Google Scholar]
- 55.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. Journal of Clinical Investigation. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nature Medicine. 2005;11(2):183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Özcan U, Yilmaz E, Özcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313(5790):1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kars M, Yang L, Gregor MF, et al. Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 2010;59(8):1899–1905. doi: 10.2337/db10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shore GC, Tata JR. Two fractions of rough endoplasmic reticulum from rat liver. I. Recovery of rapidly sedimenting endoplasmic reticulum in association with mitochondria. Journal of Cell Biology. 1977;72(3):714–725. doi: 10.1083/jcb.72.3.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pickett CB, Montisano D, Eisner D, Cascarano J. The physical association between rat liver mitochondria and rough endoplasmic reticulum. I. Isolation, electron microscopic examination and sedimentation equilibrium centrifugation analyses of rough endoplasmic reticulum-mitochondrial complexes. Experimental Cell Research. 1980;128(2):343–352. doi: 10.1016/0014-4827(80)90070-1. [DOI] [PubMed] [Google Scholar]
- 61.Rizzuto R, Pinton P, Carrington W, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280(5370):1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 62.Csordás G, Renken C, Várnai P, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. Journal of Cell Biology. 2006;174(7):915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. Journal of Biological Chemistry. 1990;265(13):7248–7256. [PubMed] [Google Scholar]
- 64.Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends in Cell Biology. 2009;19(2):81–88. doi: 10.1016/j.tcb.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Simmen T, Lynes EM, Gesson K, Thomas G. Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM) Biochimica et Biophysica Acta. 2010;1798(8):1465–1473. doi: 10.1016/j.bbamem.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Szabadkai G, Bianchi K, Várnai P, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. Journal of Cell Biology. 2006;175(6):901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simmen T, Aslan JE, Blagoveshchenskaya AD, et al. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. The EMBO Journal. 2005;24(4):717–729. doi: 10.1038/sj.emboj.7600559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell. 2007;131(3):596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 69.Westermann B. Mitochondrial fusion and fission in cell life and death. Nature Reviews Molecular Cell Biology. 2010;11(12):872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- 70.Mozdy AD, Shaw JM. A fuzzy mitochondrial fusion apparatus comes into focus. Nature Reviews Molecular Cell Biology. 2003;4(6):468–478. doi: 10.1038/nrm1125. [DOI] [PubMed] [Google Scholar]
- 71.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456(7222):605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 72.Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology. 2008;23(2):84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- 73.Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27(50):6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Deniaud A, Sharaf El Dein O, Maillier E, et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene. 2008;27(3):285–299. doi: 10.1038/sj.onc.1210638. [DOI] [PubMed] [Google Scholar]
- 75.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxidants and Redox Signaling. 2007;9(12):2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 76.Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Frontiers in Bioscience. 2009;14:1197–1218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tavender TJ, Bulleid NJ. Molecular mechanisms regulating oxidative activity of the Ero1 family in the endoplasmic reticulum. Antioxidants and Redox Signaling. 2010;13(8):1177–1187. doi: 10.1089/ars.2010.3230. [DOI] [PubMed] [Google Scholar]
- 78.Harding HP, Zhang Y, Zeng H, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular Cell. 2003;11(3):619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 79.Marciniak SJ, Yun CY, Oyadomari S, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes and Development. 2004;18(24):3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Adachi T, Weisbrod RM, Pimentel DR, et al. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nature Medicine. 2004;10(11):1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 81.Li G, Mongillo M, Chin KT, et al. Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. Journal of Cell Biology. 2009;186(6):783–792. doi: 10.1083/jcb.200904060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. Journal of Clinical Investigation. 2002;110(10):1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xu W, Liu L, Charles IG, Moncada S. Nitric oxide induces coupling of mitochondrial signalling with the endoplasmic reticulum stress response. Nature Cell Biology. 2004;6(11):1129–1134. doi: 10.1038/ncb1188. [DOI] [PubMed] [Google Scholar]
- 84.Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochimica et Biophysica Acta. 2001;1504(1):46–57. doi: 10.1016/s0005-2728(00)00238-3. [DOI] [PubMed] [Google Scholar]
- 85.Jiang HY, Wek SA, McGrath BC, et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Molecular and Cellular Biology. 2004;24(3):1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lim JH, Lee HJ, Ho Jung M, Song J. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: a molecular mechanism leading to hepatic insulin resistance. Cellular Signalling. 2009;21(1):169–177. doi: 10.1016/j.cellsig.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 87.Cheong J, Coligan JE, Shuman JD. Activating transcription factor-2 regulates phosphoenolpyruvate carboxykinase transcription through a stress-inducible mitogen-activated protein kinase pathway. Journal of Biological Chemistry. 1998;273(35):22714–22718. doi: 10.1074/jbc.273.35.22714. [DOI] [PubMed] [Google Scholar]
- 88.Qiao L, MacDougald OA, Shao J. CCAAT/enhancer-binding protein α mediates induction of hepatic phosphoenolpyruvate carboxykinase by p38 mitogen-activated protein kinase. Journal of Biological Chemistry. 2006;281(34):24390–24397. doi: 10.1074/jbc.M603038200. [DOI] [PubMed] [Google Scholar]
- 89.Dumollard R, Marangos P, Fitzharris G, Swann K, Duchen M, Carroll J. Sperm-triggered [Ca2+] oscillations and Ca2+ homeostasis in the mouse egg have an absolute requirement for mitochondrial ATP production. Development. 2004;131(13):3057–3067. doi: 10.1242/dev.01181. [DOI] [PubMed] [Google Scholar]