

Abstract
RNA localization is a widespread mechanism to achieve localized protein synthesis. In budding yeast, localization of ASH1 mRNA controls daughter cell-specific accumulation of the transcriptional regulator Ash1p, which determines mating type switching. ASH1 mRNA localization depends on four independently acting sequences (‘zipcodes’) within the mRNA. In addition, the class V myosin Myo4p and a set of She proteins with as yet unknown function are essential for ASH1 localization. Here we show that She2p is a novel RNA-binding protein that binds specifically to ASH1 mRNA in vivo and to ASH1 RNA zip codes in vitro. She2p can interact with She3 protein via She3p’s C-terminus and becomes localized to the daughter cell tip upon ASH1 expression. The N-terminal coiled-coil domain of She3p is required to form an RNA-independent complex with the heavy chain of the myosin motor protein Myo4p. She2p and She3p are the first examples of adapters for tethering a localized mRNA to the motor protein and might serve as prototypes for RNA–motor protein adapters.
Keywords: cytoskeleton/myosin/RNA localization/Saccharomyces cerevisiae/yeast
Introduction
mRNA localization is a widespread mechanism by which cells generate asymmetry. It occurs in organisms as diverse as amoeba, yeast, plants, insects and vertebrates (for reviews see Bassell et al., 1999; Lasko, 1999; Mowry, 1999; Kiebler and DesGroseillers, 2000). Site-specific mRNA localization can be achieved by local protection, site-specific mRNA anchoring or mRNA transport (for an overview see Bashirullah et al., 1998).
RNA localization depends on signals within the mRNA as well as on proteins that recognize these signals. In the majority of cases, localization signals (also named zipcodes; Singer, 1993) reside in the 3′ untranslated region (3′UTR) of the mRNA. Only a few mRNAs have been reported with signals in the 5′UTR or in the coding region (Capri et al., 1997; Chartrand et al., 1999; Gonzalez et al., 1999; Thio et al., 2000). Proteins that recognize zipcodes have been isolated from various organisms (see Lasko, 1999; Mowry, 1999; Schnapp, 1999 for recent reviews). Most of them contain RNA-binding motifs like the double-stranded RNA-binding domain (Bycroft et al., 1995), the RNA recognition motif (RRM) domain (Query et al., 1989) or the hnRNP K homology (KH) domain (Adinolfi et al., 1999). In addition to such RNA-specific proteins, active transport of mRNAs requires a functional cytoskeleton and motor proteins that move along cytoskeletal filaments. Both actin-dependent myosin and microtubule-dependent kinesin motor proteins have been implicated in mRNA transport (Carson et al., 1997; Long et al., 1997; Takizawa et al., 1997). How the messenger ribonucleoprotein (mRNP) complex docks to the corresponding motor proteins is largely unknown but binding might require the function of adapter proteins that bridge between the mRNP proteins and the motors.
The only known actin-dependent motor protein with a specific role in mRNA localization is Myo4p, a class V myosin in Saccharomyces cerevisiae (Haarer et al., 1994; Long et al., 1997; Takizawa et al., 1997). Myo4p is essential for the localization of ASH1 mRNA to daughter cells in yeast. ASH1 encodes a transcriptional repressor that determines proper mating type switching by differentially regulating expression of the HO endonuclease (Bobola et al., 1996; Cosma et al., 1999). ASH1 mRNA is expressed at the end of anaphase and subsequently localized to the tip of the maturing daughter cell (Long et al., 1997; Takizawa et al., 1997). Localization depends on signals in the coding region and 3′UTR of ASH1 mRNA (Chartrand et al., 1999; Gonzalez et al., 1999). Five proteins (She1p–She5p) have been identified that are essential for ASH1 mRNA localization (Jansen et al., 1996; Long et al., 1997). One of them, She1p, is identical to Myo4p. Whereas two of the She proteins, She4p and She5p/Bni1p, are required for diverse cellular processes (Wendland et al., 1996; Evangelista et al., 1997), She1p/Myo4p, She2p and She3p appear to be specific for mRNA transport. However, the function of She2p and She3p during localization has not yet been understood. It has previously been shown that She3p and Myo4p colocalize with RNP particles that contain ASH1 RNA and that they can coprecipitate ASH1 mRNA from cell extracts (Bertrand et al., 1998; Münchow et al., 1999; Takizawa and Vale, 2000). Both proteins appear to associate with ASH1 RNP particles that have been observed to move from the mother to the daughter cell in live cells (Bertrand et al., 1998; Takizawa and Vale, 2000). However, it is not yet clear how the localization machinery recognizes the zipcode signals within ASH1 RNA and which proteins tether the RNA to the motor protein.
In order to understand the molecular details of this tethering we investigated the binding of She2p, She3p, Myo4p and ASH1 mRNA to each other in vivo and in vitro. Here we report that She2p binds ASH1 directly via its zipcode elements and that She3p serves as a linker to connect She2p to the motor Myo4p. She2p binding of ASH1 mRNA reinforces recruitment of She2p–ASH1 RNA to the She3p–Myo4p complex.
Results
She3p can bind to both Myo4p and She2p
Previous work has shown that the She3 protein has an essential role in ASH1 mRNA localization and colocalizes with both ASH1 mRNA and the motor protein Myo4p in particles (Bertrand et al., 1998; Takizawa and Vale, 2000). Colocalization with Myo4p is independent of ASH1 mRNA since it is also seen at stages where the RNA is not present (Jansen et al., 1996), suggesting a direct interaction of She3p with Myo4p. In order to test this and to identify the region of She3p that is responsible for its interaction with Myo4p, we followed two approaches. In a two-hybrid interaction assay we tested various parts of She3p for their binding to the C-terminal tail region of Myo4p (amino acids 923–1472, see Figure 1A). Both full-length She3p and the N-terminal half of She3p (amino acids 1–197) bind to the Myo4p tail in this assay whereas the C-terminal half of She3p (amino acids 197–426) cannot (Figure 1B). This interaction is specific since the tail of the related Myo2p myosin (amino acids 929–1574) does not bind to She3p (Figure 1B). Both the N-terminal region of She3p and the first 150 amino acids of the Myo4p tail have been predicted to form a coiled-coil structure that could serve as interaction domain (Haarer et al., 1994; Jansen et al., 1996).

Fig. 1. She3p binds to the motor Myo4p and to She2p via different domains. (A) Schematic representation of the constructs used in the in vivo and in vitro interaction assays. Numbers below boxes correspond to amino acid position. Hatched boxes in Myo4p and She3p represent predicted coiled-coil regions. (B) Two hybrid interaction analysis. She2-, Myo4 tail- and Myo2 tail-Gal4 DNA binding domain fusions (‘bait’) were coexpressed with Gal4 activation domain alone or fusions (‘prey’) to Myo4p tail, She3p, She3p N-terminus and She3p C-terminus. Interaction was indicated by growth on medium lacking histidine. Whereas She2p showed interaction with full-length She3p and the She3p C-terminus, Myo4p tail interacted with She3p and She3p N-terminus. (C) In vitro interaction of She3p with Myo4p tail and She2p. Recombinant GST fusions of She2p, Myo4p tail, a Myo4 tail lacking the predicted coiled-coil domain or GST alone were immobilized to glutathione–Sepharose beads and incubated with in vitro translated 35S-labelled She3p (double arrows). Bound She3p was eluted, and applied to an SDS–PAGE gel together with one-fifth of the in vitro translated She3p used for pulldown (‘input’). Numbers indicate positions of molecular weight markers (in kDa). The band seen at 40 kDa is a degradation product of She3p. (D) In vitro interaction of She2p with the C-terminus of She3p. In vitro translated domains of She3p were affinity purified with GST–She2p beads as described above. Only the She3p C-terminal domain bound to GST–She2p.
In parallel studies, we wanted to elucidate the role of She2p in ASH1 mRNA localization. In an attempt to identify interaction partners of She2p by a two-hybrid interaction approach we identified She3p as a putative binding partner (see Materials and methods). Out of 119 She2p-interacting clones isolated in an unbiased two-hybrid screen, 33 contained inserts spanning different portions of the SHE3 open reading frame (ORF). The smallest overlapping fragment of all inserts covered the She3p C-terminus (amino acids 213–426), which lacks the coiled-coil part of She3p (data not shown). This prompted us to test directly for an interaction of She2p with different parts of She3p and with the Myo4p tail. Both full-length She3p and its C-terminal half showed an interaction with She2p (Figure 1B). In contrast, no binding was detected with either the She3p N-terminus or the Myo4p tail. In order to confirm our data, the SHE2 and SHE3 ORFs were swapped between the GAL4 DNA-binding and activation domain vectors, which resulted in an identical activation pattern of the two-hybrid reporter genes (data not shown).
In order to verify the binding of She3p to She2p and Myo4p we tested whether the proteins associate in vitro. Recombinantly expressed glutathione S-transferase (GST) fused to She2p or Myo4p tail containing or lacking the coiled-coil region of Myo4 (GST–Myo4tail or GST–Myo4tailΔcoil, respectively) was immobilized on glutathione–Sepharose beads. Full-length She3p and its N- and C-terminal domains were produced by in vitro translation in the presence of [35S]methionine. GST–She2 as well as GST–Myo4tail beads but not beads with GST–Myo4tailΔcoil or GST alone precipitated in vitro translated She3p (Figure 1C). This demonstrates a direct interaction of the proteins. The C-terminal but not the N-terminal domain of She3p bound to GST–She2, supporting our two-hybrid assay results (Figure 1D). In contrast, unlike in the two-hybrid assay, the Myo4p tail domain did not bind significantly to either the N- or the C-terminal domain of She3p in vitro (data not shown).
To analyse the interaction of She3p and Myo4p in vivo, we generated a yeast strain that carries epitope-tagged versions of Myo4p fused to the haemagglutinin (HA) tag and She3p fused to two IgG-binding domains of protein A (Grandi et al., 1993). Cell extracts from this strain were fractionated on a 5–25% sucrose gradient. Both Myo4p– HA and She3p–ProtA comigrate on this gradient between 7.4S and 19S (Figure 2A). The comigration was independent of RNA since extensive RNase A digestion of the extract before fractionation resulted in a similar distribution of Myo4p–HA and She3p–ProtA in the gradient. RT–PCR analysis showed that no ASH1 or SIC1 RNA could be detected after RNase A treatment, indicating a successful removal of RNA (data not shown). In order to ensure that the comigration reflected an association of the two proteins, we immunoprecipitated She3p–ProtA. Precipitation of She3p–ProtA from the peak fractions (4–9) of the RNase A-treated extract resulted in coprecipitation of Myo4–HA (Figure 2B), indicating the tight RNA-independent association of these two proteins in vivo.

Fig. 2. She3p association with Myo4p in vivo is RNA independent. (A) Western blot analysis of a 5–25% sucrose gradient fractionation of a cell extract from strain RJY758 (MYO4–HA6, SHE3-TEVProtA, YEplac181-ASH1) after treatment with RNase A (upper panels) or mock treatment (lower panels). Numbers on top of the panels indicate fraction number (1 = top), numbers on the bottom indicate migration behaviour (in Svedberg units) of protein standards in a parallel gradient. (B) Immunoprecipitation of She3–ProtA by IgG–agarose beads from peak fractions of the RNase A-treated extracts reveals coprecipitation of Myo4p. Upper panel: western blot analysis using anti-HA antibody 12CA5; lower panel: western blot analysis using peroxidase–anti-peroxidase complex to detect She3–protein A fusion.
In summary, our results suggest that discrete domains of She3p bind to Myo4p and She2p, and implicate a role of She3p in linking the myosin tail to She2p.
She2p associates with ASH1 mRNA independently of She3p and Myo4p
Both Myo4p and She3p have been shown to associate with ASH1 mRNA in vivo (Münchow et al., 1999; Takizawa and Vale, 2000). Disruption of SHE2 abolishes Myo4p’s association with the mRNA. We wondered whether the same is true for association of She3p and ASH1 mRNA. Yeast strains were created that contain myc epitope-tagged versions of She3p either in the background of functional or non-functional She2p or Myo4p. Extracts were prepared from these strains and myc-tagged She3p precipitated using monoclonal anti-myc antibodies. Coprecipitated ASH1 mRNA was detected by RT–PCR (Münchow et al., 1999).
Disruption of SHE2 or MYO4 did not significantly change the level of ASH1 mRNA in the cell extract (‘Total’, Figure 3A, lanes 1–7). However, whereas ASH1 mRNA could be detected in She3p–myc precipitates from both MYO4+ and myo4Δ cell extracts (‘Pellet’, Figure 3A, lanes 5 and 7), She3p–myc did not coprecipitate ASH1 mRNA in the absence of SHE2 (Figure 3A, lane 6), suggesting an essential function of She2p for the association of both She3p and Myo4p (Münchow et al., 1999) with ASH1 mRNA. This prompted us to check for an association of She2p with ASH1 mRNA. Like She3p–myc, a myc epitope-tagged version of She2p coprecipitates ASH1 mRNA (Pellet, Figure 3A, lane 2). In contrast to She3p, She2p still coprecipitates ASH1 in the absence of Myo4p or She3p (Pellet, Figure 3A, lanes 3 and 4), indicating a MYO4/SHE3-independent association of She2p and ASH1 mRNA in vivo.
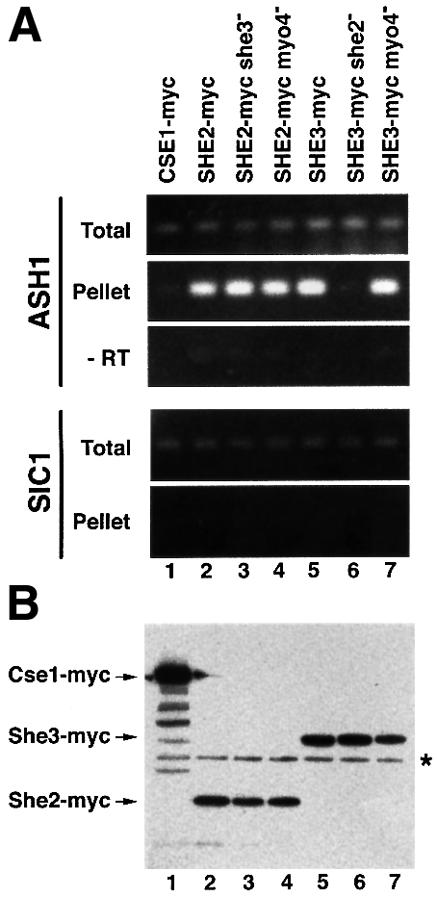
Fig. 3. Specific coprecipitation of ASH1 mRNA with myc-tagged She3p and She2p. Myc-tagged proteins from cell extracts of strains containing or lacking functional SHE genes were precipiated with anti-myc antibody 9E11 coupled to magnetic beads essentially as described in Münchow et al. (1999). Eluted material was prepared for RT–PCR or western blot analysis. (A) RT–PCR analysis. A 247 bp fragment covering part of the ASH1 3′UTR or a 316 bp fragment covering the 3′ part of SIC1 mRNA were amplified from total extract and immunoprecipitates of myc-tagged proteins. No PCR product was observed when reverse transcriptase was omitted (‘–RT’). (B) To show equal efficiency of immunoprecipitations in myc-tagged strains, identical aliquots of the immunoprecipitates were separated on 10% SDS–PAGE and myc-tagged proteins detected by western blotting using anti-myc antibody 9E10. Lane 1, Cse1p–myc; lanes 2–4, She2p–myc; lanes 5–7, She3–myc. An asterisk marks the position of the antibody heavy chain.
To demonstrate the specificity of coprecipitation we used a myc-tagged version of Cse1p, a protein involved in nuclear export (Solsbacher et al., 1998). In addition, we tested for coprecipitation of the myc-tagged proteins with a second mRNA, SIC1, which shows a similar expression profile and abundance to ASH1 (Schwob et al., 1994). Both controls demonstrate that ASH1 mRNA specifically coprecipitates with She3p–myc and She2p–myc since no ASH1 mRNA could be detected in Cse1p–myc precipitates and no significant amounts of SIC1 mRNA were detected in the She protein immunopellets.
She2p binds directly to ASH1 mRNA
Since She2p associates with ASH1 mRNA in vivo independently of She3p and Myo4p, we wondered if She2p might bind directly to ASH1 mRNA and might therefore be the ASH1 RNA-binding protein. To test this, we used a GST pulldown assay (Trifillis et al., 1999). A recombinantly produced GST–She2 fusion protein was immobilized on glutathione–Sepharose beads. In parallel, GST alone was purified and similarly immobilized. 32P-labelled ASH1 mRNA and control transcripts were generated in vitro by SP6 polymerase. Transcripts were mixed with identical amounts of immobilized GST and GST–She2 proteins in the presence of competitor tRNA and the resulting protein–RNA complexes were purified (see Materials and methods). Bound RNA was eluted and aliquots were removed for quantification or applied to denaturing agarose or polyacrylamide gels. The results of the pulldown experiments are summarized in Figure 4A. No RNA was coprecipitated with GST alone (Figure 4B and data not shown). Similar negative results were obtained when we used a fusion of GST and the RNA-binding coat protein (Peabody, 1990) from phage MS2 (data not shown).

Fig. 4. Purified GST–She2p binds ASH1 RNA in vitro through the localization elements E1–E3. (A) Overview of the results of ASH1–GST–She2p pulldown experiments. At the top of the panel ASH1 mRNA is sketched with the positions of the localization elements E1, E2A, E2B and E3 (hatched bars). Numbers below indicate nucleotide position (AUG = 1). Arrows indicate the start and end of the RNAs used in the pulldown experiments. Numbers in parentheses on the left indicate the start and end of the RNAs on the nucleotide level. The right column indicates relative recovery of input RNA on the glutathione beads. +++, recovery >50%; ++, >35%; +, >15%; –, <10%. Results are mean values of three to five independent experiments. (B) Representative denaturing polyacrylamide gel showing specific precipitation of 32P-labelled localization elements E2B, E3 and ASH1-U by GST–She2p (lanes 6–8) but not of the mutant U* or an RNA lacking localization elements (‘inter’, lanes 9 and 10). No binding of RNA was observed when beads where loaded with GST (lanes 11–15). Lanes 1–5 show input RNAs. (C) Schematic overview of predicted secondary structures of localization elements E3, ASH1-U and U*. The arrow indicates disrupted doubled-stranded RNA stem in mutant element U*.
GST–She2p was able to bind specifically to full-length ASH1 mRNA including the 5′ and 3′UTRs (Figure 4A). Furthermore, binding was also detected to shorter transcripts that contained at least one of the described RNA localization elements E1–E3 (Chartrand et al., 1999). Strongest binding (>50% of the input material precipitated) was observed to transcripts containing elements E1, E1 plus E2A/B (‘ΔE3’), or E3 plus the remaining 3′UTR (‘ΔE1ΔE2’). Each of these fragments has been shown to be able to localize a lacZ reporter RNA to the daughter cell (Chartrand et al., 1999). The shortest RNA fragment that bound to GST–She2p was a subfragment of the E3 localization element, element ASH1-U (also named U), which can also localize a reporter RNA (Gonzalez et al., 1999) (see also Figure 4C). In contrast, a mutant version of element U (‘U*’) with a disruption of the distal double-stranded stem abolished binding to She2p (Figure 4B, lane 9, and C). The disruption of this double-stranded RNA stem resulted in a loss of RNA localization in vivo (Gonzalez et al., 1999). No binding of GST–She2p was observed to RNAs lacking E elements (‘ΔE elements’, ‘inter’), ASH1 antisense RNA or RNA encoding yeast Srp54 protein (Hann and Walter, 1991) (Figure 4A and B).
Interestingly, a GST–She3p fusion protein was able to bind weakly to labelled ASH1 RNA (data not shown). Approximately 15% of the input RNA coprecipitated with GST–She3p. However, in contrast to GST–She2p, binding was not specific since no significantly stronger precipitation of ASH1 full-length RNA than of control RNAs (antisense ASH1 or SRP54 RNA) was observed in our assay (data not shown).
To demonstrate that the interaction of GST–She2p with ASH1 mRNA is authentic we used a second method to test for protein–RNA binding. Labelled RNAs covering localization elements ASH1-U, or -U* (76 nucleotides each) were mixed with increasing amounts of purified GST or GST fused to She2p or She3p and complex formation was determined by a mobility shift assay (Figure 5A). GST, GST–She3p or GST–MS2 (data not shown) failed to bind to ASH1-U whereas GST–She2p formed a complex with element ASH1-U (black arrowhead). Similar binding was observed to E3 (Supple mentary data, available at The EMBO Journal Online) as well as a weak but significant binding to the E2B element (data not shown). In contrast, no mobility shift was observed when She2p was incubated with mutant U* (Figure 5A) or another double-stranded RNA, iron-response element (IRE, Figure 5C), indicating that She2p does not recognize all stem–loop-forming RNA structures.

Fig. 5. She2p binding to ASH1-U is specific and enhanced by She3p. Probes used are indicated at the bottom of each panel, proteins at the top. (A) RNA mobility shift assay using 32P-labelled probes of ASH1-U or mutant U* and increasing concentrations of GST, GST–She2p (2.5, 5.0, 7.5, 10, 20, 40 ng/µl) or GST–She3p (10, 20, 30, 40, 60 ng/µl). The arrowhead indicates the position of free probe and the black arrow the position of an ASH1-U RNA complex with GST–She2p that cannot be detected with GST–She3p or mutant U*. (B) Mobility shift using constant GST–She2p (2.5 ng/µl) and increasing concentrations of GST or GST–She3p (10, 20, 30, 40, 60 ng/µl). Increasing the amount of She3p but not of GST enhances She2p–ASH1-U complex formation (black arrow) and leads to the formation of an RNA–protein complex with lower mobility (open arrow). (C) Mobility shift assay using the IRE as probe. No complex formation is observed.
The mobility shift assay also allowed us to address the question of binding cooperativity between She2p, She3p and ASH1-U. The presence of GST–She3p but not of GST in a binding reaction of GST–She2p and ASH1-U enhanced the complex formation of She2p and ASH1 RNA (Figure 5B). In addition, a new complex with lower mobility (open arrow) appeared, suggesting the formation of a higher order complex.
In summary, our analysis demonstrates the direct binding of ASH1 mRNA to She2p in vivo and in vitro. Binding of She2p to the mRNA depends on the presence of functional localization elements E1–E3 within the RNA and is enhanced by She3p.
She2p moves to the bud in an ASH1 RNA-dependent manner
The association of She2p and ASH1 mRNA was surprising because of previous findings that, unlike She3p and Myo4p, She2p does not colocalize with ASH1 (Jansen et al., 1996; Bertrand et al., 1998). We wondered whether the apparent lack of colocalization could be due to an inaccessibility of She2p by anti-She2p antibodies once the protein has become part of the putative localization particle. We therefore used a 9-fold myc epitope to tag She2p at the C-terminus with the rationale that a sufficient number of myc epitopes would stick out of the localization particle to detect the tagged protein by indirect immunofluorescence.
Yeast strains were constructed that express a functional myc9-tagged She2p and carry either a high-copy plasmid with ASH1 under the control of its own promoter (2µ ASH1) or of the inducible GAL1 promoter (GAL1-ASH1). Although ASH1 mRNA is normally only expressed and transported in late anaphase cells, it has the potential to localize to the bud at earlier stages of the cell cycle when expressed from a heterologous promotor (Long et al., 1997).
In a population of cells that have a higher dosage of ASH1 (2µ-ASH1), ∼40% (18/47 counted) of anaphase cells with an ASH1 signal at the bud tip also show a She2p–myc9 signal overlapping with the RNA staining (Figure 6A and B). In cells carrying the GAL1-ASH1 plasmid, She2p–myc is detected throughout the cytoplasm under non-inducing conditions (Figure 6D). Some staining is also visible at the position of the nucleus (Figure 6, compare D with DAPI staining in F), indicating that She2p might be distributed between both nucleus and cytoplasm. Upon induction of ASH1 by addition of galactose to the culture, the cells show an additional strong staining of She2p–myc at the bud tip (Figure 6G). This staining overlaps with the position of ASH1 mRNA as detected by in situ hybridization (Figure 6H). No She2p–myc staining at the bud tip was detected when cells carrying a control plasmid without the ASH1 gene were induced (Figure 6K). In addition, no accumulation of She2p–myc at the bud tip was observed when ASH1 was induced in a strain carrying a deletion of SHE3 (data not shown). In contrast to She2p, She3p–myc localizes to the bud tip independently of ASH1 induction (Figure 6N).

Fig. 6. She2p–myc9 localizes to the bud tip upon ASH1 expression. Overexpression of ASH1 mRNA from a high-copy plasmid (A–C) or from the heterologous GAL1 promoter (G–H) induces a shift of She2p localization to the bud tip (arrowheads). Localization of She3–myc6 to the bud tip is independent of ASH1 expression (N–P). A combination of indirect immunofluorescence against myc-tagged proteins and in situ hybridization against ASH1 mRNA was performed as described in Materials and methods. Immunofluorescence staining against She2p–myc9 is shown in (A), (D), (G) and (K), whereas staining in (N) reflects She3p–myc6 localization. (B), (E), (H), (L) and (O) show localization of ASH1 mRNA by in situ hybridization using CY3-labelled ASH1 antisense oligos. (C), (F), (I), (M) and (P) are composite images of differential interference contrast (DIC) and DNA staining by DAPI. Bar, 5 µm.
Our results demonstrate that She2p moves to the bud tip upon ASH1 expression and suggest She2p is transported to its destination site together with ASH1 mRNA. The data are supported by recent findings of Takizawa and Vale (2000) that an epitope-tagged version of She2p with 13 myc epitopes colocalizes with an ASH1 RNA-containing particle.
She2p binds to Myo4p–She3p in an ASH1-dependent manner
How is She2p recruited to the daughter cell tip? Both Myo4p and She3p can localize to the daughter cell tip independently of ASH1 RNA. Does She2p therefore bind in an ASH1-dependent manner to Myo4p–She3p to become localized? To test this idea, we generated strains containing She proteins with different epitopes (Myo4p– HA6, She3p–ProtA, She2p–myc3) and a plasmid that allowed galactose-dependent ASH1 RNA induction (‘+ASH1’) or the induction of an unrelated RNA (‘–ASH1’). Cell extracts were prepared after galactose induction, and She3p–ProtA precipitated with IgG– agarose beads. Coprecipitated proteins were detected by western blotting using antibodies against the HA or myc epitope (Figure 7). Whereas coprecipitation of Myo4p with She3p was independent of ASH1 induction (Figure 7, lanes 4–6), She2p was enriched in immune pellets from extracts of cells where ASH1 had been induced (Figure 7, compare lanes 5 and 6). To prove inevitably that the band detected at the position of She2–myc is not a breakdown product of She3–ProtA, we used a control strain that expresses Myo4–HA and She3p–ProtA but lacks a myc epitope (‘control’, Figure 7, lanes 1 and 4). The absence of a band at the size of She2p–myc clearly shows that the detected band in lane 6 corresponds to She2–myc. Interestingly, we also observed a very weak She2p band in precipitates from cells without overexpressed ASH1 (Figure 7, lane 5). This could reflect a weak RNA-independent She2p–She3p binding or might be due to low amounts of endogenous ASH1 mRNA in the non-induced cells.
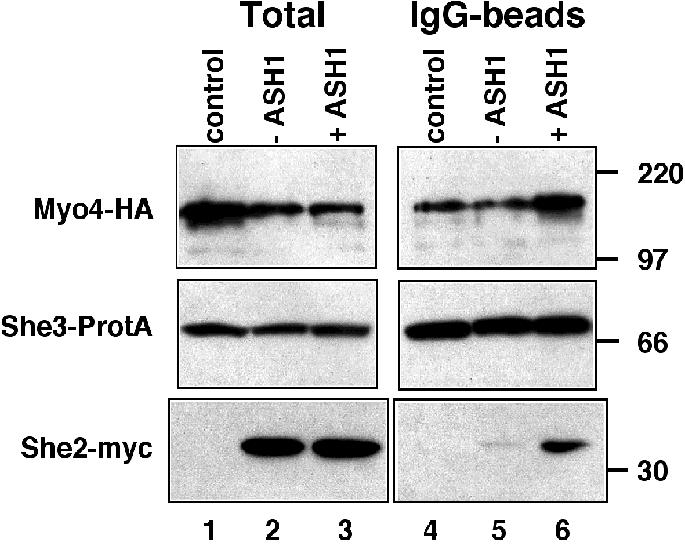
Fig. 7. Coprecipitation of She2–myc with She3–ProtA and Myo4–HA upon ASH1 RNA overexpression. Western blot analysis of cell extracts from strains RJY750 (SHE3–ProtA, MYO4–HA6, ‘control’), RJY756 (SHE3–ProtA, MYO4–HA6, SHE2–myc3, pGAL1; ‘–ASH1’) and RJY757 (SHE3–ProtA, MOY4–HA6, SHE2–myc3, pGAL1-ASH1; ‘+ASH1’), and corresponding pellets from an anti-protein A precipitation (‘IgG-beads’). The blot was cut into three pieces and probed with antibodies against HA, myc or protein A. Numbers on the right indicate the positions of size markers (in kDa).
We conclude from our data that, in vivo, ASH1 RNA reinforces the association of She2p with the Myo4p–She3p complex.
Discussion
The interaction network of Myo4p, She2p, She3p and ASH1 mRNA that we identified provides a first detailed description of how a localized mRNA is tethered to its cognate motor protein.
Tethering of localized RNA and motor protein depends on the function of two She proteins whose role in RNA localization has previously not been understood. ASH1 mRNA is bound by an RNA-binding zipcode-specific protein, She2p. A motor protein-associated linker protein, She3p, interacts with this RNA-binding protein. She3p appears to bind the motor, Myo4p, and the RNA-binding protein, She2p, via different domains.
Several experimental findings demonstrate that She2p is the crucial RNA-binding protein. In vivo, deletion of MYO4 or SHE3 does not alter She2p’s ability to coprecipitate ASH1 mRNA, suggesting a direct and motor protein-independent association of She2p with the RNA. She2p can bind ASH1 mRNA directly in vitro and this binding occurs via the defined ASH1 RNA localization elements E1, E2A/B and E3. Deletion analysis showed that transcripts with only one localization element coprecipitated with GST–She2p as efficiently as full-length RNA. This suggests that She2p can bind independently to any of the localization elements and does not have a preference for a specific element. Similar results have been obtained by an in vitro cross-linking approach using recombinant She2p and RNA localization elements (R.Long, personal communication).
How can She2p recognize all three independent ASH1 RNA localization zipcodes? The differences in sequence of the three localization elements strongly suggest a common structural feature. E1, E3 and ASH1-U have been predicted to form double-stranded stem–loop structures with several bulges (Chartrand et al., 1999; Gonzalez et al., 1999). Structure prediction of the E2B localization element also suggests the presence of a stable stem–loop structure (data not shown). Mutations within the terminal stem of E1 and E3 disrupt the localizing activity of the zipcode elements (Chartrand et al., 1999). Consistent with these data we have shown that She2p binds directly to the shortest wild-type zipcode (ASH1-U) but not to a mutant version with a partially disrupted stem (Gonzalez et al., 1999), suggesting that She2p might recognize the terminal stem–loop structure of the zipcodes.
She2p does not show any sequence homology to other proteins in the databases. In addition, She2p does not contain sequence motifs found in other classes of RNA-binding proteins (Query et al., 1989; Bycroft et al., 1995; Adinolfi et al., 1999). We propose that She2p represents a novel type of RNA-binding protein.
What is the function of the other She protein essential for ASH1 mRNA localization, She3p? The similar localization pattern of She3p and Myo4p at all stages of the cell cycle suggested a tight association of She3p and Myo4p (Jansen et al., 1996). Our results indicate that this association is direct. First, Myo4p and She3p copurify in a two-step purification in the absence of RNA. Secondly, She3p binds to the Myo4p C-terminal domain but not to that of the related Myo2p myosin in a two-hybrid interaction trap. Interaction requires the presence of She3p’s predicted coiled-coil domain (Jansen et al., 1996). Thirdly, a GST fusion of the C-terminal myosin tail binds directly to an in vitro translated She3p. This binding depends on the presence of coiled-coil domains within the Myo4p tail (amino acids 923–1075). Taken together, two-hybrid and GST pulldown data suggest an interaction of Myo4 tail and She3p through their coiled-coil domains. However, so far we have not been able to demonstrate this interaction in vitro. Coiled-coil domains within class V myosin tails have been suggested to be essential only for heavy chain dimerization (Mermall et al., 1998). Our findings might add an additional function to this domain of class V myosins.
In summary, our data demonstrate a direct interaction of She3p and the myosin motor protein Myo4p. Similar findings have recently been published by Takizawa and Vale (2000).
Whereas She3p’s N-terminal half seems to bind to the myosin motor, its C-terminus is involved in She2p binding. A direct ASH1 RNA-independent binding of She2p and She3p is suggested by our in vitro pulldown experiments. In contrast, our in vivo experiments indicate that ASH1 mRNA reinforces the She2p–She3p interaction. Overexpression of ASH1 from a strong GAL1 promoter strongly increases the quantity of She2p coprecipitating with She3p and Myo4p. Consistent with this finding, She2p localization changes upon ASH1 expression and, like ASH1 mRNA, She2p becomes concentrated at the bud tip. In addition to ASH1, this movement depended on the presence of She3p (data not shown). Our finding that She3p enhances She2p–ASH1 mRNA complex formation suggests some cooperativity of binding between She2p, She3p and ASH1 mRNA that might result in a more stable RNA–protein complex.
Our results suggest a model in which ASH1 mRNA is associating with the RNA-transporting motor protein Myo4p through the interaction with two She proteins, She2p and She3p. She2p seems to be the RNA-binding protein of the RNA–motor protein complex. She3p’s major role seems to be to form a bridge between the heavy chain of the myosin motor, Myo4p, and the She2p–ASH1 RNP, and it can therefore be considered as a specific adapter for the ASH1 RNP.
It is interesting to note that, although currently no She3p homologue has been identified, the protein shows functional analogy to the Drosophila swallow (Swa) protein involved in bicoid RNA localization (Schnorrer et al., 2000). Like Swa, She3p binds directly to a motor protein (cytoplasmic dynein or myosin, respectively) via its coiled-coil domain. Furthermore, localization of She3p and Swa to their destination sites is independent of their cognate RNA. Swa contains a putative RNA-binding motif and might therefore bind the transported RNA directly, although this has not been shown.
Although our data suggest that Myo4, She2p and She3p are part of the ASH1 mRNP that is localized to the daughter cell tip, we have no evidence that they are the only proteins of this complex. A full description of the protein content of the ASH1 mRNP must await its biochemical purification.
Materials and methods
Yeast strains and methods
All yeast strains used in this study are listed in Table I and were derived from W303a (Rothstein, 1983) with the exception of RJY786–RJY791, which have an RS453 background (Segref et al., 1997). Sequences of applied oligonucleotides are published in Supplementary Table II. Media preparation and manipulation of yeast cells were carried out essentially as described (Adams et al., 1997). Transformation was performed according to Gietz and Schiestl (1995). Gene disruption of various SHE genes with the Schizosaccharomyces pombe HIS3 marker gene was achieved using a PCR-based method described by Wach et al. (1997). In all cases of PCR applications, proofreading polymerases were used. Amplified constructs were verified by sequencing.
Table I. Yeast strains used in this study.
| Name | Relevant genotype | Source |
|---|---|---|
| L40 | Mata, hisD200, trp1-901, leu2-3,112, ade2, LYS2::(4lexAop-HIS3), URA3::(8lexAop-lacZ), GAL4 | Nonaka et al. (1995) |
| PJ69-4α | Matα, trp1-901, leu2-3,112, ura3-52, his3-200, gal4Δ; gal80Δ; GAL2-ADE2, LYS2::GAL1-HIS3, met2::GAL7-lacZ | James et al. (1996) |
| RJY375 | Mata, pep4::URA3, CSE1–myc9::HIS3, YEplac181-ASH1 | Münchow et al. (1999) |
| RJY505 | Matα, HO-ADE2, pep4::LEU2, SHE3–myc6, YEplac195-ASH1 | D.Djandji, unpublished |
| RJY574 | Matα, HO-ADE2, pep4::LEU2, SHE3–myc6, myo4::HIS3MX6, YEplac195-ASH1 | this work |
| RJY634 | Mata, pep4::URA3, SHE3–myc6, MYO4–HA6, she2::HIS3MX6, YEplac181-ASH1 | this work |
| RJY635 | Mata, SHE2myc3::HIS3MX6, YEplac181-ASH1 | this work |
| RJY636 | Mata, SHE2myc3::HIS3MX6, she3::URA3, YEplac181-ASH1 | this work |
| RJY750 | Matα, HO-ADE2, pep4::URA3, SHE3-TEVProA::HIS3MX6, MYO4–HA6 | this work |
| RJY756 | Mata, HO-ADE2, pep4::URA3, SHE3-TEVProA::HIS3MX6, MYO4–HA6, SHE2–myc3, p415-GAL1 | this work |
| RJY757 | Mata, HO-ADE2, pep4::URA3, SHE3-TEVProA::HIS3MX6, MYO4–HA6, SHE2–myc3, p415-GAL1-ASH1 | this work |
| RJY758 | Matα, HO-ADE2, pep4::URA3, SHE3-TEVProA::HIS3MX6, MYO4–HA6, YEplac181-ASH1 | this work |
| RJY785 | Mata, SHE2–myc3::HIS3MX6, myo4::URA3, YEplac181-ASH1 | this work |
| RJY786 | Mata, mex67::HIS3, SHE2myc9::klTRP1, pUN100-MEX67, YEplac195 | this work |
| RJY787 | Mata, mex67::HIS3, SHE2myc9::klTRP1, pUN100-MEX67, YEplac195-ASH1 | this work |
| RJY788 | Mata, mex67::HIS3, SHE2myc9::klTRP1, pUN100-MEX67, p416-GAL1 | this work |
| RJY789 | Mata, mex67::HIS3, SHE2myc9::klTRP1, pUN100-MEX67, p416-GAL1-ASH1 | this work |
| RJY791 | Mata, mex67::HIS3, SHE3myc9::klTRP1, pUN100-mex67-5, p416-GAL1-ASH1 | this work |
Strains RJY375–RJY785 are derivatives of W303a (also named K699) with the following full genotype: Mata, ade2-1, trp1-1, can1-100, leu2-3,112, his3-11,15, ura3, GAL, psi+. Strains RJY786–RJY791 are derived from RS453, whose genotype is: Mata, ade2, his3, leu2, trp1, ura3, his3.
Epitope tagging of MYO4, SHE2 and SHE3
Epitope tagging of SHE2 with three myc epitopes and of MYO4 with six HA epitopes has been described (Jansen et al., 1996). In order to generate versions of SHE2 with nine myc epitopes and of SHE3 with two ZZ domains of protein A we followed the ‘one-step tagging’ method described by Knop et al. (1999). Transformants were checked by Southern analysis for correct integration. All epitope-tagged proteins were fully functional.
Two-hybrid interaction analysis
Myosin tail-encoding regions of the MYO2 ORF (amino acids 929–1574) and MYO4 ORF (amino acids 923–1472) were amplified using primer pairs omyo2-BH1 and omyo2-PstI or omyo4-2hyb2 and omyo4-2hyb3. The BamHI and PstI sites, or the EcoRI and BamHI sites, respectively, generated this way were used to clone the PCR products into the bait vector pBTM116. The SHE3 ORF was amplified using primers RJO20 and RJO21, creating EcoRI or BamHI sites at the ends. SHE3-N (encoding amino acids 1–197) and SHE3-C (encoding amino acids 197–426) were amplified similarly, using primer pairs RJO20 and RJO89 or RJO21 and RJO90, respectively. Amplified and digested DNA fragments were cloned into pGAD424 (Clontech Laboratories). The SHE2 coding region was amplified using primers RJO18 and RJO19, digested with EcoRI and BamHI and cloned into the bait vector pGBDU-C1 (James et al., 1996). Yeast strain L40 (Nonaka et al., 1995) or PJ694A (James et al., 1996) was co-transformed with the resulting bait plasmids and pGAD424-SHE3 constructs. Transformants were selected on appropriate medium and subsequently tested for activation of the lexA-HIS3 (L40) or GAL1-HIS3 (PJ694A) reporter genes.
For the two-hybrid interaction screen with SHE2, PJ694A containing pGBDU-SHE2 was transformed with three GAL4 activation domain libraries carrying yeast genomic DNA fragments in different reading frames (James et al., 1996). Of ∼9 × 106 transformants, 1362 were selected in the first round because they were able to grow on medium lacking histidine. Of these clones, 119 grew on his–ade– medium and were subjected to further analysis by plasmid rescue, PCR–restriction analysis of the inserts and sequencing. Thirty-three of the clones analysed contained either full-length SHE3 or 3′ fragments of the SHE3 ORF encoding the She3p C-terminus.
In situ hybridization and immunofluorescence
For ASH1 induction, we used a centromeric plasmid containing ASH1 under the control of the strong GAL1 promoter. A fragment of ASH1 (from ASH1 ATG to nucleotide 510 of the 3′UTR) was amplified using primers RJO141 and RJO142. After digestion with BamHI and XhoI, the DNA fragment was cloned into p415-GAL1 or p416-GAL1 (Mumberg et al., 1994). The resulting plasmids and the empty vectors were transformed into yeast resulting in strains RJY788 (SHE2–myc9, p416-GAL1), RJY789 (SHE2–myc9, p416-GAL1-ASH1), RJY756 (MYO4– HA6, SHE2–myc3, SHE3-TEVProtA, p415-GAL1) and RJY757 (MYO4– HA6, SHE2–myc3, SHE3-TEVProtA, p415-GAL1-ASH1). For induction, stationary-phase cells grown overnight in 2% raffinose minimal medium lacking uracil were diluted into YEP/2% raffinose and grown for two generations at 30°C. Galactose was added to a final concentration of 3% and cells were induced for 1 h before fixation or cell lysis with glass beads. Combined in situ hybridization against ASH1 mRNA and immunofluorescence against myc-tagged proteins were performed as described (Long et al., 1997) with the modification that anti-mouse Alexa-488 antibodies were used as secondary antibodies (diluted 1:1000) and that all manipulations were performed on multiwell slides in order to minimize antisense probe and antibody consumption.
Immunoprecipitation of epitope-tagged proteins
Immunoprecipitation of myc-tagged proteins and subsequent detection of ASH1 or SIC1 mRNAs by RT–PCR were performed essentially as described (Münchow et al., 1999). Two changes have been introduced to the protocol. The new breakage buffer A contained 50 mM HEPES–KOH pH 7.3, 20 mM potassium acetate, 2 mM EDTA, 0.1% Triton X-100, 5% glycerol and a protease inhibitor cocktail (Roche). The anti-mouse IgG2a magnetic beads (Dynal) were blocked three times for 10 min in breakage buffer containing tRNA (0.1 mg/ml). Three independent RT–PCRs were performed from each immune pellet in order to minimize RT–PCR-dependent amplification variation.
Immunoprecipitation of She3–ProtA and detection of coprecipitated proteins were performed as follows. Logarithmically growing cells (3 × 108) of strains RJY750, RJY756 and RJY757 (induced by addition of 3% galactose) were disrupted with glass beads in 300 µl of breakage buffer containing a protease inhibitor cocktail. Extracts were cleared by centrifugation (2 min at 5000 g). After addition of IgG–agarose beads (Sigma), extracts were incubated for 1 h at 4°C. Beads were pelleted and washed with breakage buffer containing 50 mM potassium acetate. Bound proteins were eluted with hot Laemmli buffer. Equivalent amounts of the cell extract (total) and the eluted proteins (pellet) were separated by PAGE and blotted, and epitope-tagged proteins were detected using monoclonal antibodies 12CA5 (anti-HA), 9E10 (anti-myc) or peroxidase–anti-peroxidase conjugate (PAP; Sigma).
Cell extract fractionation by sucrose gradient centrifugation
Logarithmically growing cells (2 × 109) of strain RJ758 (SHE3-TEVProtA, MYO4–HA6, YEplac181-ASH1) were pelleted and the pellet resuspended in an equal volume of 2× breakage buffer B (100 mM HEPES–KOH pH 7.3, 100 mM potassium acetate, 10 mM magnesium acetate, 0.2% Triton X-100, 10% sucrose) including protease inhibitors. After cell lysis with glass beads, extracts were clarified by centrifugation (10 min at 1000 g). Mg-ATP was added to a final concentration of 5 mM and extracts were treated at 4°C for 10 min either with 10 µg of RNase A or with 400 U of RNase inhibitor (Promega). Extracts were loaded onto a 5–25% sucrose gradient (in 1× breakage buffer B without Triton X-100) and centrifuged at 4°C for 16 h at 100 000 g. Fractions (500 µl) were collected from the top and aliquots separated on 8% PAGE. After blotting, the membrane was cut into two halves and probed for the presence of either She3p–ProtA or Myo4p–HA6.
GST pulldowns and electrophoretic mobility shift assays
For expression of recombinant MS2, She2p, She3p and Myo4p tail, we generated fusions to GST in pGEX4T2 (Pharmacia). The ORF of MS2 coat protein (Peabody, 1990) was amplified from pG14-MS2-GFP (Bertrand et al., 1998) using primers that added BamHI and XhoI sites at the 5′ or 3′ terminus (RJO261 and RJO262). The SHE2 ORF was cloned as a BamHI–EcoRI fragment in-frame with GST. Restriction sites were generated by PCR with primers RJO232 and RJO233. Similarly, DNA fragments carrying the SHE3 ORF or MYO4 coding for amino acids 923–1471 (‘Myo4p tail’) or amino acids 1075–1471 (‘Myo4p tailΔcoil’) with new BamHI and SalI sites were created by PCR and cloned in-frame with GST. Expression and purification procedures of all fusion proteins are available as Supplementary data.
For in vitro translation of She3p, the SHE3 ORF was amplified by PCR using oligonucleotides RJO230 and RJO231. SHE3-N and SHE3-C were generated using primer pairs RJO230 and RJO271 or RJO231 and RJO272. The PCR products were cloned as a BamHI–EcoRI fragment into pGEM-3Z. Coupled in vitro transcription–translation in the presence of 35S-labelled methionine was performed with the TNT Quick system (Promega) according to the manufacturer’s suggestions. An aliquot of the translation extract (1/10) was incubated for 60 min at room temperature with immobilized GST fusion proteins in 1 ml of binding buffer (50 mM NaCl, 20 mM Tris–HCl pH 8.0, 0.5% NP-40, 0.1 mg/ml insulin) and washed four times with binding buffer lacking insulin. Bound proteins were eluted with hot Laemmli buffer, separated by 12% SDS–PAGE, and labelled proteins detected by autoradiography.
For pulldown experiments with in vitro transcribed RNAs and immobilized GST fusion proteins, we followed the protocol of Trifillis et al. (1999). Full-length SRP54 (a gift from M.Pool) and ASH1 genes were cloned into pGEM-3Z and desired subfragments of ASH1 were created by successive deletion of ASH1 using convenient restriction sites. A detailed cloning strategy of the diverse fragments is published as Supplementary data. [32P]CTP-labelled in vitro transcripts were generated using Promega’s Riboprobe SP6 transcription system. Labelled RNA (106 c.p.m.) was added to 1 µg of immobilized GST fusion protein in 400 µl of RNA-binding buffer (RBB: 10 mM Tris–HCl pH 7.5, 150 mM KCl, 1.5 mM MgCl2) containing 10 U of RNasin (Promega). After binding at 4°C for 1 h, beads were pelleted, washed with RBB containing 1 mg/ml heparin followed by four washes with RBB with 0.1% Triton X-100. RNA was extracted from the beads and resuspended in 50 µl of TE. One microlitre was used for quantitation using Cerenkov counting and 5 µl were loaded onto denaturing polyacrylamide or agarose gels.
For electrophoretic mobility shift analysis, we followed the protocol of Black et al. (1998). Increasing amounts of recombinant proteins were mixed with in vitro transcribed and labelled RNAs. Assembled complexes were separated from unbound probe by electrophoresis for 5 h at 170 V through a 5% polyacrylamide gel in 0.5× TBE.
Supplementary data
Supplementary data to this paper are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank U.von Ahsen, P.Chartrand, E.Hurt, P.James, M.Pool, E.Schiebel and W.Zachariae for plasmids and strains, D.Görlich, F.Weismann and G.Merdes for help with recombinant protein expression, R.Long and R.Singer for communicating results prior to publication. We also thank M.Kiebler and A.Jaedicke for comments on the manuscript. R.-P.J. is a recipient of a grant from the DFG (JA696/2).
References
- Adams A., Gottschling,D.E., Kaiser,C.A. and Stearns,T. (1997) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Adinolfi S., Bagni,C., Castiglione Morelli,M.A., Fraternali,F., Musco,G. and Pastore,A. (1999) Novel RNA-binding motif: The KH module. Biopolymers, 51, 153–164. [DOI] [PubMed] [Google Scholar]
- Bashirulla A., Cooperstock,R.L. and Lipshitz,H.D. (1998) RNA localization in development. Annu. Rev. Biochem., 67, 335–394. [DOI] [PubMed] [Google Scholar]
- Bassell G.J., Oleynikov,Y. and Singer,R.H. (1999) The travels of mRNAs through all cells large and small. FASEB J., 13, 447–454. [DOI] [PubMed] [Google Scholar]
- Bertrand E., Chartrand,P., Schaefer,M., Shenoy,S.M., Singer,R.H. and Long,R.M. (1998) Localization of ASH1 mRNA particles in living yeast. Mol. Cell, 2, 437–445. [DOI] [PubMed] [Google Scholar]
- Black D.L., Chan,R., Min,H., Wang,J. and Bell,L. (1998) The electrophoretic mobility shift assay for RNA binding proteins. In Smith,C.W.J. (ed.), RNA–Protein Interactions. Oxford University Press, Oxford, UK, pp. 109–136. [Google Scholar]
- Bobola N., Jansen,R.-P., Shin,T.H. and Nasmyth,K. (1996) Asymmetric accumulation of Ash1p in postanaphase nuclei depends on a myosin and restricts yeast mating-type switching to mother cells. Cell, 84, 699–709. [DOI] [PubMed] [Google Scholar]
- Bycroft M., Grünert,S., Murzin,A.G., Proctor,M. and St Johnston,D. (1995) NMR solution structure of a dsRNA binding domain from Drosophila staufen protein reveals homology to the N-terminal domain of ribosomal protein S5. EMBO J., 14, 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capri M., Santoni,M.J., Thomas-Delaage,M. and Ait-Ahmed,O. (1997) Implication of a 5′ coding sequence in targeting maternal mRNA to the Drosophila oocyte. Mech. Dev., 68, 91–100. [DOI] [PubMed] [Google Scholar]
- Carson J.H., Worboys,K., Ainger,K. and Barbarese,E. (1997) Translo cation of myelin basic protein mRNA in oligodendrocytes requires microtubules and kinesin. Cell Motil. Cytoskel., 38, 318–328. [DOI] [PubMed] [Google Scholar]
- Chartrand P., Meng,X.-H., Singer,R.H. and Long,R.M. (1999) Structural elements required for the localization of ASH1 mRNA and of a green fluorescent protein reporter particle in vivo. Curr. Biol., 9, 333–336. [DOI] [PubMed] [Google Scholar]
- Cosma M.P., Tanaka,T. and Nasmyth,K. (1999) Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle- and developmentally regulated promoter. Cell, 97, 299–311. [DOI] [PubMed] [Google Scholar]
- Evangelista M., Blundell,K., Longtine,M.S., Chow,C.J., Adames,N., Pringle,J.R., Peter,M. and Boone,C. (1997) Bni1p, a yeast formin linking Cdc42p and the actin cytoskeleton during polarized morphogenesis. Science, 276, 118–122. [DOI] [PubMed] [Google Scholar]
- Gietz R.D. and Schiestl,R.H. (1995) Transforming yeast with DNA. Methods Mol. Cell. Biol., 5, 255–269. [Google Scholar]
- Gonzalez I., Buonomo,S.B.C., Nasmyth,K. and von Ahsen,U. (1999) ASH1 mRNA localization in yeast involves multiple secondary structural elements and Ash1 protein translation. Curr. Biol., 9, 337–340. [DOI] [PubMed] [Google Scholar]
- Grandi P., Doye,V. and Hurt,E.C. (1993) Purification of NSP1 reveals complex formation with ‘GLFG’ nucleoporins and a novel nuclear pore protein NIC96. EMBO J., 12, 3061–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarer B.K., Petzold,A., Lillie,S.H. and Brown,S.S. (1994) Identification of MYO4, a second class V myosin gene in yeast. J. Cell Sci., 107, 1055–1064. [DOI] [PubMed] [Google Scholar]
- Hann B.C. and Walter,P. (1991) The signal recognition particle in S.cerevisiae. Cell, 67, 131–144. [DOI] [PubMed] [Google Scholar]
- James P., Halladay,J. and Craig,E.A. (1996) Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics, 144, 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R.-P., Dowzer,C., Michaelis,C., Galova,M. and Nasmyth,K. (1996) Mother cell-specific HO expression in budding yeast depends on the unconventional myosin Myo4p and other cytoplasmic proteins. Cell, 84, 687–697. [DOI] [PubMed] [Google Scholar]
- Kiebler M.A. and DesGroseillers,L. (2000) Molecular insights into mRNA transport and local translation in the mammalian nervous system. Neuron, 25, 19–28. [DOI] [PubMed] [Google Scholar]
- Knop M., Siegers,K., Pereira,G., Zachariae,W., Winsor,B., Nasmyth,K. and Schiebel,E. (1999) Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast, 15, 963–972. [DOI] [PubMed] [Google Scholar]
- Lasko P. (1999) RNA sorting in Drosophila. FASEB J., 13, 421–433. [DOI] [PubMed] [Google Scholar]
- Long R.M., Singer,R.H., Meng,X., Gonzalez,I., Nasmyth,K. and Jansen,R.-P. (1997) Mating type switching in yeast controlled by asymmetric localization of ASH1 mRNA. Science, 277, 383–387. [DOI] [PubMed] [Google Scholar]
- Mermall V., Post,P.L. and Mooseker,M.S. (1998) Unconventional myosins in cell movement, membrane traffic and signal transduction. Science, 279, 527–533. [DOI] [PubMed] [Google Scholar]
- Mowry K.L. (1999) RNA sorting in Xenopus oocytes and embryos. FASEB J., 13, 435–445. [DOI] [PubMed] [Google Scholar]
- Mumberg D., Müller,R. and Funk,M. (1994) Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res., 22, 5767–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münchow S., Sauter,C. and Jansen,R.-P. (1999) Association of the class V myosin Myo4p with a localised messenger RNA in budding yeast depends on She proteins. J. Cell Sci., 112, 1511–1518. [DOI] [PubMed] [Google Scholar]
- Nonaka H., Tanaka,K., Hirano,H., Fujiwara,T., Kohno,H., Umikawa,M. and Mino,A. (1995) A downstream target of RHO1 small GTP-binding protein is PKC1, a homolog of protein kinase C, which leads to activation of the MAP kinase cascade in Saccharomyces cerevisiae. EMBO J., 14, 5931–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peabody D.S. (1990) Translational repression by bacteriophage MS2 coat protein expressed from a plasmid. J. Biol. Chem., 265, 5684–5689. [PubMed] [Google Scholar]
- Query C.C., Bentley,R.C. and Keene,J.D. (1989) A common RNA recognition motif identified within a defined U1 RNA binding domain of the 70k U1 snRNP protein. Cell, 57, 89–101. [DOI] [PubMed] [Google Scholar]
- Rothstein R.J. (1983) One-step gene disruption in yeast. Methods Enzymol., 101, 202–211. [DOI] [PubMed] [Google Scholar]
- Schnapp B.J. (1999) RNA localization: a glimpse of the machinery. Curr. Biol., 9, R725–R727. [DOI] [PubMed] [Google Scholar]
- Schnorrer F., Bohmann,K. and Nuesslein-Volhard,C. (2000) The molecular motor dynein is involved in targeting Swallow and bicoid RNA to the anterior pole of Drosophila oocytes. Nature Cell Biol., 2, 185–190. [DOI] [PubMed] [Google Scholar]
- Schwob E., Böhm,T., Mendenhall,M.D. and Nasmyth,K. (1994) The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S.cerevisiae. Cell, 79, 233–244. [DOI] [PubMed] [Google Scholar]
- Segref A., Sharma,K., Doye,V., Hellwig,A., Huber,J., Lührmann,R. and Hurt,E. (1997) Mex67p, a novel factor for nuclear mRNA export, binds to both poly(A)+ RNA and nuclear pores. EMBO J., 16, 3256–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer R.H. (1993) RNA zipcodes for cytoplasmic adresses. Curr. Biol., 3, 719–721. [DOI] [PubMed] [Google Scholar]
- Solsbacher J., Maurer,P., Bischoff,F.R. and Schlenstedt,G. (1998) Cse1p is involved in export of yeast importin α from the nucleus. Mol. Cell. Biol., 18, 6805–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa P.A. and Vale,R.D. (2000) The myosin motor, Myo4p, binds Ash1 mRNA via the adapter protein, She3p. Proc. Natl Acad. Sci. USA, 97, 5273–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa P.A., Sil,A., Swedlow,J.R., Herskowitz,I. and Vale,R.D. (1997) Actin-dependent localization of an RNA encoding a cell-fate determinant in yeast. Nature, 389, 90–93. [DOI] [PubMed] [Google Scholar]
- Thio G.L., Ray,R.P., Barcelo,G. and Schupbach,T. (2000) Localization of gurken RNA in Drosophila oogenesis requires elements in the 5′ and 3′ regions of the transcript. Dev. Biol., 221, 435–446. [DOI] [PubMed] [Google Scholar]
- Trifillis P., Day,N. and Kiledjian,M. (1999) Finding the right RNA: identification of cellular mRNA substrates for RNA-binding proteins. RNA, 5, 1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A., Brachat,A., Alberti-Segui,C., Rebischung,C. and Phillippsen,P. (1997) Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast, 13, 1065–1075. [DOI] [PubMed] [Google Scholar]
- Wendland B., McCaffery,J.M., Xiao,Q. and Emr,S.D. (1996) A novel fluorescence-activated cell sorter-based screen for yeast endocytosis mutants identifies a yeast homologue of mammalian eps15. J. Cell Biol., 135, 1485–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]