

Abstract
Enteric pathogens often export toxins that elicit diarrhea as a part of the etiology of disease, including toxins that affect cytoskeletal structure. Recently, we discovered that the intestinal pathogen Vibrio cholerae elicits rounding of epithelial cells that is dependent upon a gene we designated rtxA. Here we investigate the association of rtxA with the cell-rounding effect. We find that V.cholerae exports a large toxin, RTX (repeats-in-toxin) toxin, to culture supernatant fluids and that this toxin is responsible for cell rounding. Furthermore, we find that cell rounding is not due to necrosis, suggesting that RTX toxin is not a typical member of the RTX family of pore-forming toxins. Rather, RTX toxin causes depolymerization of actin stress fibers and covalent cross-linking of cellular actin into dimers, trimers and higher multimers. This RTX toxin-specific cross-linking occurs in cells previously rounded with cytochalasin D, indicating that G-actin is the toxin target. Although several models explain our observations, our simultaneous detection of actin cross-linking and depolymerization points toward a novel mechanism of action for RTX toxin, distinguishing it from all other known toxins.
Keywords: actin/cytochalasin D/rtxA/RTX toxin/Vibrio cholerae
Introduction
The disease cholera is most notorious for the six pandemics that swept Europe and the Americas during the 19th and early 20th centuries. Then, in 1961, cholera re-emerged on a pandemic scale. In 1998, nearly 300 000 cases of cholera and >10 000 deaths in 74 countries were reported to the World Health Organization (WHO), nearly a 100% increase over the previous year (WHO, 1999).
The bacterium that causes cholera is the Gram-negative pathogen Vibrio cholerae. After consumption of contaminated water or food by the host, V.cholerae colonizes the upper intestinal tract and begins to secrete the major virulence factor, the cholera toxin (CT). The action of CT causes extensive fluid loss from intestinal cells producing massive diarrhea such that patients eventually die from dehydration (Kaper et al., 1995). Although CT is clearly the most important factor in the disease cholera, CT-deficient strains of V.cholerae still elicit mild to severe diarrhea and other reactogenic symptoms in human volunteers, indicating that other toxins are likely to contribute to the pathogenesis of the disease (Kaper et al., 1984; Tacket et al., 1993; Taylor et al., 1994; Coster et al., 1995).
CT is composed of two subunits encoded by the genes ctxA and ctxB, which are located on a transmissible filamentous phage (CTXΦ) (Waldor and Mekalanos, 1996). Sequence analysis of the regions neighboring the insertion site of the CTXΦ on the V.cholerae chromosome indicated the presence of a very large open reading frame (ORF) 693 bp downstream from the CTXΦ insertion site, but oriented in the reverse orientation to that of the ctx genes (see Figure 1A) (Lin et al., 1999). At 13 635 bp, this gene, rtxA, is the largest ORF on the V.cholerae genome (Heidelberg et al., 2000).
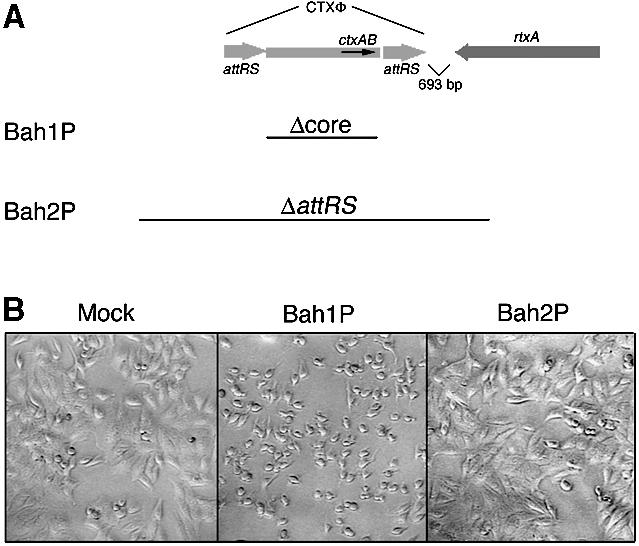
Fig. 1. Vibrio cholerae induces rounding of HEp-2 cells dependent upon rtxA. (A) Schematic representation of the CTXΦ/rtxA locus of the large chromosome of V.cholerae. Only genes relevant to this study are designated and the diagram is not drawn to scale. Bars indicate regions deleted from Bah1P and Bah2P. (B) Cell rounding after 75 min incubation of HEp-2 cells with PBS-washed bacteria. An equal volume of PBS was used as the mock infection control.
Analysis of the predicted amino acid sequence indicated that rtxA encodes a putative toxin of 4545 amino acids or 484 kDa, making it the largest single polypeptide toxin ever described. This toxin is a possible member of the RTX (repeats-in-toxin) family of pore-forming toxins, based on the presence of consensus Ca2+-binding repeat sequences at the C-terminus of the protein and the linkage of the gene with a putative toxin activation gene and type I secretion apparatus (Coote, 1992; Lin et al., 1999).
Disruption of the toxin gene had no effect on the ability of V.cholerae to lyse red blood cells (Lin et al., 1999), a characteristic previously associated with a vacuolating toxin encoded by the unlinked hlyA gene (Alm et al., 1988; Rader and Murphy, 1988; Coelho et al., 2000; Mitra et al., 2000). Mutants in the rtxA gene were, however, defective in a different phenotype: the ability of El Tor and O139 V.cholerae strains to elicit rapid rounding of tissue culture cells (Lin et al., 1999). This activity was absent from certain classes of vaccine strains as well as classical biotype strains, and these defective strains were subsequently shown to have deletions in the rtx locus (Lin et al., 1999).
Here we characterize further the association of the gene rtxA with the ability of V.cholerae to induce cell rounding. We find that the RTX toxin does not disrupt membrane integrity and thus is not likely to be a pore-forming toxin. Rather, cell rounding results from depolymerization of the actin stress fibers dependent upon the RTX toxin. Furthermore, we find that coincident with F-actin disassembly, the cellular actin is covalently cross-linked, representing a novel mechanism for a toxin to utilize to achieve actin depolymerization.
Results
Characterization of cell rounding induced by V.cholerae Bah1P
The mutant strains constructed for these experiments, Bah1P and Bah2P, are derivatives of O1 El Tor E7946, a strain previously shown to induce cell rounding (Lin et al., 1999). The ‘core’ deletions of the CTXΦ genome present in both Bah1P and Bah2P remove the genes for CT and other factors reported to induce cellular responses (Figure 1A) (Taylor et al., 1994). A second chromosomal deletion within the gene hapA eliminates hemagglutinin/protease (HA/P), a protease that releases adherent tissue culture cells (Wu et al., 1996; Mel et al., 2000). The larger CTXΦ genome deletion in Bah2P also removes the 3′ end of the gene rtxA, disrupting the function of the RTX toxin (Figure 1A) (Lin et al., 1999).
In an assay for cell rounding, Bah1P, but not the rtxA mutant Bah2P, induced rounding of human laryngeal cells (HEp-2) (Figure 1B). Similar rounding after addition of Bah1P was observed for all target cell lines that were tested, including HEp-2 cells, A549 lung epithelial carcinoma cells, Henle 407 intestinal epithelial cells, L6 rat fibroblastoma cells, Chinese hamster ovary cells, Raw264.7 and J774 macrophages, and PtK2 kidney epithelial cells. In each case, cell rounding was dependent upon rtxA since the mutant strain Bah2P did not elicit cell rounding (data not shown).
Addition of chloramphenicol, an inhibitor of bacterial protein synthesis, to the tissue culture medium inhibited cell rounding, indicating that the RTX toxin produced by bacteria after exposure to cells is the result of de novo synthesis. In contrast, addition of cycloheximide, an inhibitor of eukaryotic protein synthesis, did not inhibit cell rounding, demonstrating that synthesis of new host proteins is not essential for this process (data not shown).
Taken together, these data demonstrate that after addition of washed bacteria to tissue culture cells, the RTX toxin is produced from the gene rtxA, leading to rounding of a broad range of cell types.
Membrane integrity is intact in RTX-exposed cells
We first investigated whether the RTX toxin causes cell death through pore formation. If the RTX toxin is a pore-forming toxin, as predicted by its association with the RTX family, then membrane integrity should be compromised after infection, leading to necrosis and leakage of cytoplasmic contents. However, in standard assays for cell lysis, HEp-2 cells rounded by co-incubation with Bah1P did not release either lactate dehydrogenase or sodium chromate51 into the culture medium at levels above mock infection (Figure 2A and B). In addition, cells excluded the fluorophore DEAD-RED (Figure 2C) as well as other membrane-impermeant dyes including trypan blue and propidium iodide (data not shown). These results indicate that cell membrane integrity remains intact in cells that are rounded by co-incubation with Bah1P, and thus pore formation and necrosis cannot account for the observed cell rounding.

Fig. 2. Rounded cells have not undergone necrosis. Lactate dehydrogenase (LDH) (A) and Cr51 (B) release to the medium were measured after 3 h incubation of HEp-2 cells with bacteria. The percentage release was determined by the activity in the sample divided by the activity from cells lysed with Triton X-100. Values shown are the mean of triplicates in a single assay with the standard deviation. (C and D) HEp-2 cells incubated with Bah1P (C) or Bah2P (D) for 2 h were stained with SYTO10 (membrane permeant, green) and DEAD-RED (membrane impermeant, red) to assay membrane integrity. Photographs are representative of the field of cells. A count of >100 cells showed no significant difference in the ratio of red/green cells in Bah1P- and Bah2P-inoculated cells. Mock-infected control cells appeared similar to Bah2P shown in (D).
When bacteria were washed away 1 h post-inoculation and the cells were subsequently maintained in the presence of gentamicin, the cells remained adherent to the surface and were biochemically active 24 h later as determined by reaction with 10% Alamar blue dye (data not shown). Taken together, these data demonstrate that V.cholerae expressing the RTX toxin does not cause rapid cell death.
Depolymerization of actin stress fibers depends on the RTX toxin
A common toxic mechanism that leads to rounding of a broad range of cell types without directly affecting cell viability is alteration of the polymerization state of the actin cytoskeleton (Steele-Mortimer et al., 2000). To test whether the RTX toxin affects the state of actin polymerization, HEp-2 cells at various stages of rounding were fixed and stained with fluorescently labeled phalloidin to visualize polymerized F-actin (Figure 3). The actin stress fibers in HEp-2 cells exposed to Bah1P are completely depolymerized. At 1 h, the actin within the rounding cells is found in dense phalloidin-staining aggregates (Figure 3B) that were also observed in cells treated with cytochalasin D, a drug that inhibits formation of actin filaments (Figure 3D). After 2 h, the RTX-treated cells ceased to bind phalloidin, indicating a total absence of polymerized F-actin (Figure 3F and G). Depolymer ization of stress fibers is not observed in cells exposed to the rtxA mutant Bah2P (Figure 3C and H), demonstrating that disassembly of actin requires the RTX toxin.

Fig. 3. Depolymerization of actin stress fibers depends on rtxA. A total of 105 HEp-2 cells were inoculated with PBS (A and E), 107 Bah1P (B, F and G), 107 Bah2P (C and H) or 2.5 µM cytochalasin D (D, I and J). Inoculated cells were incubated at 37°C in 5% CO2 for either 1 (A–D) or 2 h (E–J), after which cells were stained with fluorescent phalloidin (green). (G and J) Cells counterstained with propidium iodide (red) to facilitate location of cells in (F) and (I), respectively.
Two other putative toxins of V.cholerae, zonal occludens toxin (ZOT) and the protease HA/P, have also been associated with rearrangements of F-actin (Fasano et al., 1995; Wu et al., 1996). Since the RTX+ strain Bah1P used in our studies is deleted for both the zot and hapA genes, the cell biological effects we observe cannot be due to these other putative toxins. Thus, we conclude that the depolymerization of actin fibers is due to RTX toxin.
Rho in RTX-exposed cell extracts is ADP-ribosylated by exoenzyme C3
The amino acid stretch from amino acid 3376 to 3625 of the RTX toxin was found to have sequence similarity (22% identity, 40% similarity) to a region of Clostridium difficile toxin A (CdA) (Figure 4A), another very large toxin that causes depolymerization of actin (Fiorentini and Thelestam, 1991; Boquet et al., 1998). CdA is known to transfer a UDP-glucose molecule to Thr37 of Rho and Thr35 of Rac and Cdc42, rendering these GTPases biologically inactive (Boquet et al., 1998). The modification protects Rho in cell extracts from in vitro ADP-ribosylation by Clostridium botulinum exoenzyme C3, a toxin that specifically targets RhoA, B and C (Dillon et al., 1995; Just et al., 1995). Although the region of amino acid similarity between RtxA and CdA lies outside the catalytic domain of CdA (Faust et al., 1998), RTX-exposed cells were tested for a CdA-like modification of Rho.

Fig. 4. The RTX toxin does not modify the Rho GTPase similarly to C.difficile toxin A (CdA). (A) A PSI-BLAST search (Altschul et al., 1997) comparing RtxA with the DDBJ/EMBL/GenBank database detected a region of limited sequence similarity to CdA as shown (score = 46.9, E = 0.002). (B) To check for a CdA-like modification of Rho in Bah1P-infected cells, Rho protein in 50 µg of cell extract was ADP-ribosylated with exoenzyme C3 in vitro. Prior modification of Rho by CdA protects cells from further modification by C3 as shown in lane 8.
Rho proteins in cell extracts from cells co-incubated with Bah1P or Bah2P were not protected from in vitro ADP-ribosylation by exoenzyme C3 (Figure 4). In contrast, control cells exposed to purified CdA toxin did show protection from exoenzyme C3. These data demonstrate that a CdA-like modification of Rho is not responsible for actin depolymerization. In addition, a shift in electrophoretic mobility of Rho was not detected in Bah1P-exposed cell extracts, suggesting that a different modification of Rho also did not occur, although these data do not rule out other effects on signaling through the Rho family GTPases.
Actin is modified in RTX-exposed cells
Surprisingly, in the previous experiment, it was observed that a 42 kDa band was absent from Coomassie Blue-stained SDS–polyacrylamide gels of cell extracts from RTX-exposed cells as compared with control extracts (data not shown). This observation suggested that the target of the toxin may be actin itself.
Western blot analysis of cell extracts using anti-actin antibody showed that virtually all of the monomeric actin in Bah1P-exposed HEp-2 cells was absent and replaced by larger immunologically cross-reactive species (Figure 5). In cells rounded by exposure to either cytochalasin D or CdA, the size of actin was not altered, demonstrating that this effect is not a general consequence of F-actin disassembly (Figure 5). Furthermore, cells exposed to Bah2P did not show the alteration in the size of actin, demonstrating that the effect is dependent upon rtxA. These data indicate that the actin has been altered directly in RTX toxin-exposed cells.

Fig. 5. In vivo cross-linking of actin depends on rtxA. Western blot detection of (A) actin or (B) tubulin in cell extracts prepared from HEp-2 cells after co-incubation with PBS (mock), PBS-washed bacterial cultures (as indicated), 2.5 µM cytochalasin D (CytD) or 5 nM C.difficile toxin A (CdA) for the time intervals indicated at the top.
Cellular actin is cross-linked covalently in RTX-exposed cells
Closer examination of the slowly migrating actin-related species showed that they varied by 42 kDa intervals, suggesting that G-actin monomers (42 kDa) were being cross-linked stably in toxin-treated cells (Figure 5). This RTX toxin-induced ‘actin laddering’ on SDS–polyacryl amide gels was observed even when RTX toxin-treated HEp-2 cells were boiled in SDS–PAGE buffer for up to 10 min in the presence of 0.2 M dithiothreitol (DTT), 5% β-mercaptoethanol, 6 M urea or 1% Triton X-100, indicating that the cellular actin is cross-linked by a non-sulfhydryl bond (data not shown). The stability of the toxin-induced actin cross-linkage strongly suggests that these bonds are covalent.
To test this hypothesis, all the Triton-soluble actin of Bah1P-exposed HEp-2 cell extracts was purified over a DNase I affinity column. The 84 kDa ‘dimer’ form was stable even after elution from the DNase I column in 50% formamide, further indicative of a covalent linkage (Figure 6). This form was purified further by excision from a 7.5% SDS–polyacrylamide preparative gel, digested to completion with trypsin and the resulting peptide fragments separated by HPLC. The amino acid sequences of 40 individual digestion products were determined. Thirty-nine of the peptide sequences, representing 20 independent sequences, could be aligned directly with the amino acid sequence of human β-actin (DDBJ/EMBL/GenBank accession No. NP 001092). Interestingly, the sole non-actin peptide was probably derived from calnexin (DDBJ/EMBL/GenBank accession No. A46673) (Figure 6). Calnexin, a molecular chaperone that binds aggregated proteins, migrates on SDS–PAGE with an apparent mol. wt of 88 kDa, suggesting that it may co-purify with the 84 kDa cross-linked actin (Bergeron et al., 1994). However, the absence of another 42 kDa protein at a proportion equal to the actin species demonstrates that the 84 kDa form is a homodimer. These data confirm that exposure of HEp-2 cells to V.cholerae expressing RTX toxin results in the covalent cross-linking of cellular actin monomers to themselves.

Fig. 6. The 84 kDa actin species is a dimer of actin. The Coomassie Blue-stained SDS–polyacrylamide gel on the left shows actin species following elution from a DNase I affinity column in G-buffer containing 50% formamide. The sequences of 21 independent peptides derived from trypsin fragments of dimer form are shown on the right alongside the corresponding protein that the peptide matches most closely.
The RTX toxin is exported to culture supernatant fluids
In our previous report, we stated that the RTX toxin activity was not present in culture supernatant fluids (Lin et al., 1999). However, given that rtxA is found adjacent to genes for a type I secretion apparatus, it is likely that the toxin is exported. To assess this problem more carefully, we switched to the strain KFV43. KFV43 is a derivative of the El Tor strain N16961 with a deletion in hapA (Fullner and Mekalanos, 1999). In a survey of wild-type El Tor strains, it was noted that both cell rounding and cross-linking after co-incubation of HEp-2 cells with the strain KFV43 tended to progress more rapidly under identical infection protocols than observed for Bah1P or its wild-type parent E7946P (data not shown). This observation suggested that KFV43 is an overproducer of RTX toxin compared with Bah1P and that this strain might serve as a better source for detection of RTX toxin in culture supernatant fluids (data not shown).
Indeed, when sterile culture supernatant fluids from logarithmic phase cultures of KFV43 were mixed 1:1 with tissue culture media and added to cells, cell rounding was observed (data not shown). No activity was detected if the cultures were grown to stationary phase. Cell rounding activity in these supernatant fluids could be concentrated 300-fold by ultrafiltration using a 100 kDa cut-off cartridge and 300 kDa cut-off membrane (data not shown). For comparison, concentrated supernatant fluids from CW128, an isogenic rtxA mutant of KFV43, were also prepared by an identical protocol.
These concentrated preparations were examined for the presence of the RTX toxin by western blotting using an antibody raised against a 138 kDa subfragment of RtxA that had been purified from Escherichia coli as a His6-tagged protein. Numerous bands were detected by the RtxA-specific antibody that were not detected in either the rtxA mutant preparation or by the pre-immune serum. Several large bands consistent with the predicted full-length size of RtxA were detected, but the majority of the protein appears as breakdown products of ∼115, 100 and 65 kDa (Figure 7).

Fig. 7. Detection of RtxA in concentrated culture supernatant fluids by western blotting. A 2.6 µg aliquot of concentrated culture supernatant fluids from KFV43 (RTX+) and CW128 (RTX–) was loaded in each lane. RtxA was detected using pre-immune rabbit serum (left) or serum raised against RtxA138 (right). Arrows on the right denote the location of RtxA-specific bands. Those marked with an asterisk denote the three major breakdown products.
When added to HEp-2 cells, these preparations induce both cell rounding and cross-linking of actin (Figure 8). The activities in these preparations were neutralized by pre-incubation with the RtxA-specific antibody, but not by pre-immune sera or a CT-specific antibody (Figure 8). Taken together, these data demonstrate that the RTX protein is exported to culture supernatants during logarithmic growth and that the RtxA protein (or one of its major breakdown products) represents the active RTX toxin moiety responsible for both cell rounding and actin cross-linking.
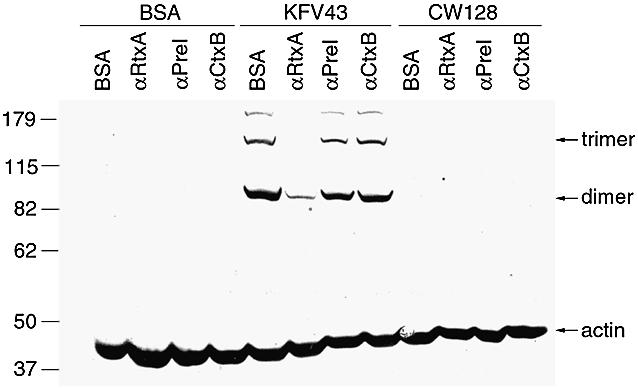
Fig. 8. Antibody neutralization of RtxA inhibits actin cross-linking. A 30 µg aliquot of IgG1 from the indicated rabbit sera was added to 4 µg of concentrated supernatant preparations of KFV43 (RTX+) or CW128 (RTX–). Bovine serum albumin (BSA) was substituted for either antibody or supernatant preparation in equal concentration for control samples. The mixtures were incubated on ice for 30 min and then were added to 5 × 105 HEp-2 adherent cells covered with RPMI 1640 medium without additions. Cells were incubated at 37°C in 5% CO2 for 75 min prior to western blot detection of actin.
G-actin is a substrate for actin cross-linking
The RTX toxin could either act on the monomeric G-actin form, leading to depolymerization, or it could break F-actin chains directly. To test whether G-actin can serve as a substrate for the RTX toxin, the actin in HEp-2 cells was depolymerized with cytochalasin D, similarly to that shown in Figure 3D, prior to addition of concentrated culture supernatant fluids. Although the cross-linking activity was reduced by ∼50%, cells rounded with cytochalasin D do show cross-linking of actin (Figure 9). This result indicates that G-actin is a substrate for the RTX toxin activity.
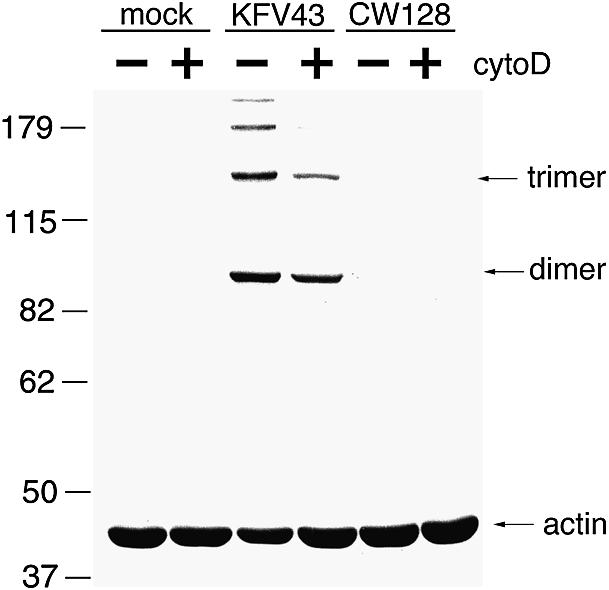
Fig. 9. Cytochalasin D does not block actin cross-linking. Cells were incubated with 2.5 µM cytochalasin D for 1 h, at which time cells were visually rounded. A 4 µg aliquot of concentrated supernatant preparations of KFV43 (RTX+) or CW128 (RTX–) was then added. Cells were incubated at 37°C in 5% CO2 for 2 h prior to western blot detection of actin.
Discussion
In this report, we have presented both biochemical and cell biological evidence for the depolymerization of F-actin stress fibers in a manner that produces a novel by-product: a highly cross-linked complex of actin dimers, trimers and higher multimers. We have shown that addition of live bacteria to cells initiates this process and that both actin depolymerization and actin cross-linking depend on the presence of the gene rtxA. Furthermore, we have shown that toxin exported to the supernatant fluids also has these activities and that this toxin can be neutralized with an RtxA-specific antibody.
Therefore, the mechanism by which the gene rtxA is linked to cell rounding is through production of the secreted RTX toxin, which then interacts with cells, leading to depolymerization of stress fibers and cross-linking of actin. Preliminary peptide mapping of the cross-linked products suggests that the cross-link may occur between two peptide fragments in the N-terminal portion of actin protein. However, extensive chemical characterization will be necessary before the exact nature of the cross-link is known. Indeed, our current mapping strategy has not excluded the possibility that more than one chemical cross-link can be formed. Thus, the actin in the higher cross-linked forms may be a ‘web’ instead of a chain.
Numerous toxins and type III secreted effector proteins have now been reported that mediate depolymerization of actin through action on the Rho family GTPases, by either direct modification or indirect action through their roles as GTP exchange factors (GEFs) and GTP-activating proteins (GAPs) (Lerm et al., 2000; Steele-Mortimer et al., 2000). In our system, cross-linking of actin was not observed when cells were rounded by CdA, a toxin that inactivates Rho, Rac and Cdc42 by the addition of UDP-glucose (Boquet et al., 1998). Thus, down-regulation of the Rho family GTPase alone does not lead to cross-linking of actin. This result indicates that the RTX toxin is unlikely to act through the Rho GTPase signaling pathway.
To explain how the RTX toxin causes both actin depolymerization and cross-linking, we favor a model more closely related to those proposed for the action of the clostridial binary toxins, including C.spiroforme toxin, C.botulinum C2, C.perfringens iota toxin and an ADP-ribosyltransferase from C.difficile. These toxins all ADP-ribosylate G-actin at Arg177 (Boquet et al., 1998). The modified G-actin is unable to undergo polymerization and therefore acts as a capping protein at the barbed end of the actin filament. The unmodified pointed end continues to depolymerize, leading to a disorganization of the cytoskeleton and rounding of the target cells (Boquet et al., 1998; Popoff, 1998). By analogy, cross-linking of actin into dimers and higher order cross-linked products would also deplete the pool of G-actin available for polymerization at the barbed end of actin filaments. A shift in the equilibrium between G- and F-actin would occur, ultimately resulting in F-actin disassembly (Figure 10). In concordance with this proposed model, we have found that rounding of cells with cytochalasin D prior to addition of the RTX toxin does not disrupt cross-linking of actin, indicating that the toxin does target monomeric G-actin. Thus, we suggest that G-actin is cross-linked directly by the RTX toxin, removing it from the actin pool, and that the stress fibers then depolymerize as the dynamic equilibrium shifts.
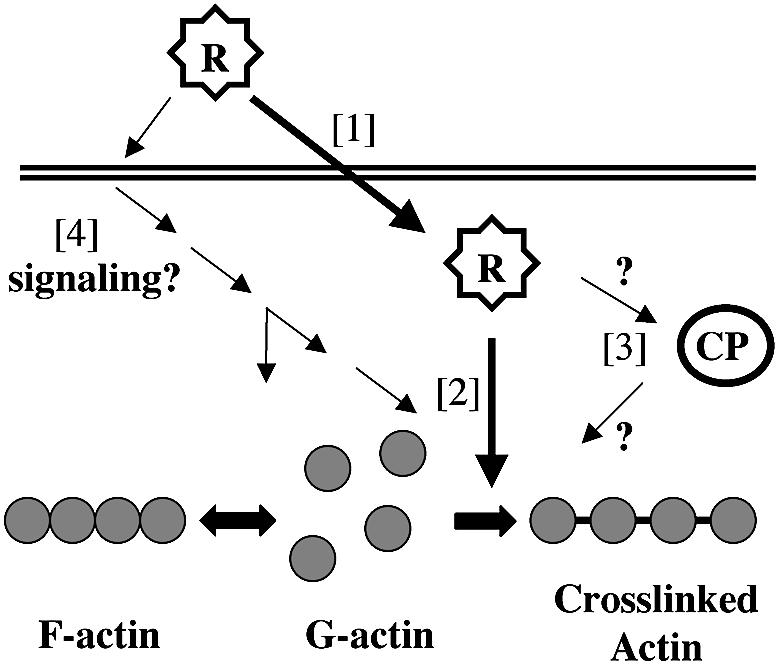
Fig. 10. Proposed models for the action of the RTX toxin. The secreted toxin (R) could enter the cells [1] gaining direct access to the cytoplasmic G-actin [2]. The RTX toxin could thus be the enzyme that then covalently cross-links G-actin, disrupting the equilibrium between F- and G-actin, leading to the depolymerization of stress fibers. Alternatively, RTX could activate an endogenous cross-linking protein (CP), which then carries out the cross-linking reaction [3]. Finally, RTX may function either from within or from outside the cell to activate signal pathways to activate endogenous proteins for both depolymerization and cross-linking [4].
This simple model, however, is not the sole viable model. We have been unable to detect in vitro cross-linking activity when concentrated supernatant fluid containing RTX toxin is incubated with either purified actin or HEp-2 cell lysates in the presence of ATP, GTP and/or divalent cations. Our inability to detect this activity may simply indicate that the reaction requires unknown cofactors or intracellular modification of the full-length toxin. However, this experiment may also predict that the RTX toxin works by an indirect mechanism.
As an alternative model, the RTX toxin could stimulate activity of an endogenous cross-linking protein (Figure 10). An attractive cellular target within this model are the transglutaminases (TGases), a family of enzymes that catalyze a Ca2+-dependent acyl transfer reaction between a γ-carboxyamide group of glutamine and ε-amino group of lysine or other primary amines (Chen and Mehta, 1999). Such enzymes have been associated with cross-linking of proteins involved in cell growth and differentiation, wound healing, receptor-mediated endocytosis, apoptosis and the formation of plaques in Alzheimer’s patients (Murthy et al., 1998; Chen and Mehta, 1999).
Both in vivo and in vitro studies have demonstrated that actin can serve as a substrate for TGases. For example, bacterial TGase has been used in vitro to introduce photo-cross-linking reagents at glutamine residues of actin for the study of actin structure (Hegyi et al., 1998; Kim et al., 1998).
Furthermore, it has been shown that cytoplasmic actin can be a substrate for TGase in vivo. The lysine derivative DALP (3-[Nα[Nε-[2′,4′-dinitrophenyl]-amino-n-hexanoyl]- l-lysylamido]-propane-1-ol) is a membrane-permeant amine substrate for tissue TGase. In leukemia-derived human cell lines HL-60 and U937 that were induced to undergo apoptosis, cytoplasmic actin was labeled with DALP, indicating that actin is a target for TGase in these cell lines. Although the majority of DALP label attached to the 43 kDa form of actin, other higher species that reacted with an actin-specific antibody were also labeled, although these were not identified as multimers of actin (Nemes et al., 1997). Thus, it is conceivable that the RTX toxin acts via a similar TGase in cells to cross-link actin without inducing the cells to undergo apoptosis.
Other models for the activity of the RTX toxin can be formulated if it is presumed that F-actin depolymerization and cross-linking are not interconnected activities (Figure 10). For example, the RTX toxin could work indirectly to alter the intracellular environment or signal transduction pathways such that many processes are affected concurrently. A modulation of a signal molecule could affect the activity of the Rho GTPase signaling pathway leading to actin depolymerization while simultaneously activating the TGase or other cross-linking protein. Thus, cross-linking would not be the direct cause of the depolymerization, but rather a concurrent response to a common signal.
Similarly, cross-linking and actin depolymerization activities could be separate activities mediated by different domains of the 484 kDa toxin protein. Both Salmonella typhimurium SptP and Pseudomonas aeruginosa ExoS function as enzymes as well as GAP proteins (Kaniga et al., 1996; Ganesan et al., 1998; Fu and Galán, 1999; Goehring et al., 1999; Pederson et al., 1999). In each case, these two activities are associated with different domains of the toxin. Validation of a dual activity model for the RTX toxin would require demonstration that actin cross-linking and depolymerization of actin are separable activities. As yet, we have not observed rounding in the absence of cross-linking or the reverse.
Regardless of the mechanism, it is clear that the RTX toxin has a dramatic effect on target cells in vitro. However, it remains to be shown whether the RTX toxin is an important factor in V.cholerae pathogenesis. Deletion of the ctxA and ctxB genes that encode CT created strains that are still pathogenic in human volunteer studies, albeit with milder symptoms. In some volunteer studies, a second toxin gene, including genes encoding HlyA (Tacket et al., 1993), HA/P (Benítez et al., 1999) or RTX toxin (Taylor et al., 1994), have also been eliminated prior to toxin administration to human volunteers. For each of these strains, mild symptoms still persisted. Thus, none of these toxins represent ‘the reactogenic factor’, but rather reactogenicity may reflect a combined contribution of each of these factors. Indeed, a strain deleted of all potential reactogenic factors has never been tested in human volunteers. Our demonstration of the dramatic cellular consequence of RTX exposure, coupled with recent evidence that both HA/P (Wu et al., 1996, 1998, 2000; Mel et al., 2000) and hemolysin (Coelho et al., 2000; Mitra et al., 2000) also have devastating cellular effects, suggests that such studies may be warranted. Such a multiple toxin mutant may lead finally to the production of a safe, live, oral vaccine that could be implemented worldwide to control the incidence of cholera.
Materials and methods
Bacterial strains
Bah1 and Bah2 are vaccine derivatives of the V.cholerae O1 Ogawa strain E7946 (Taylor et al., 1994). hapA was deleted from these strains to create Bah1P, Bah2P and E7946P, respectively, using the sacB-based counterselectable plasmid pHapSal1 as previously described (Finkelstein et al., 1992; Fullner and Mekalanos, 1999). KFV43 is a ΔhapA derivative of the V.cholerae O1 Inaba strain N16961 (Fullner and Mekalanos, 1999). CW128 is a derivative of KFV43 with a 5.5 kb internal deletion in the rtxA gene that inactivates the activity of the RTX toxin (C.Walchle and J.J.Mekalanos, unpublished).
Cell rounding and actin depolymerization
Bacterial cultures, grown for 16 h in LB broth at 30°C and washed with phosphate-buffered saline (PBS), were added to adherent cells (m.o.i. ∼200) covered with fresh RPMI 1640 medium containing 10 mM HEPES buffer, 2 mM glutamine and 10% fetal calf serum. For some experiments, 5 µg/ml chloramphenicol or 200 µg/ml cycloheximide was added 60 min prior to the addition of bacteria. Cell rounding was monitored with a Nikon Diaphot 200 inverted microscope equipped with a computer interface. For visualization of actin, cells grown on glass coverslips were stained with Molecular Probes Alexa-488-labeled phalloidin according to the manufacturer’s instructions. Cells were counterstained with propidium iodide to facilitate location of cells in the field. Coverslips mounted with ProLong Antifade mounting solution (Molecular Probes, Inc.) were viewed with a Nikon Eclipse TE300 fluorescence microscope.
Cell lysis assays
For all cell lysis assays, cells were co-incubated with PBS-washed bacteria at an m.o.i. of ∼200. Assay for release of lactate dehydrogenase to the medium was carried out using the Promega CytoTox96 Non-radioactive Cytotoxicity kit according to the manufacturer’s instructions using HEp-2 cells seeded into 96-well dishes in phenol red-free medium. For measurement of Cr51 release, non-adhered HEp-2 cells were mixed with sodium chromate51 and incubated at 37°C for 1 h. Cells loaded with Cr51 were washed three times in medium and then were seeded into a 96-well dish. After 3 h, the cells had adhered to the surface. After cells were exposed to bacteria, Cr51 released to the medium was measure in a Wallac 1470 Wizard Automatic Gamma Counter. Membrane integrity analysis using the fluorophores SYTO10 and DEAD-RED was carried out using reagents in the Molecular Probes Live/Dead Reduced Biohazard Viability/Cytotoxicity kit according to the manufacturer’s instructions.
ADP-ribosylation of cell extracts with exoenzyme C3
Labeling of cell extracts by C.botulinum exoenzyme C3 (List Laboratories) was carried out as described by Dillon et al. (1995). Subconfluent HEp-2 cells seeded into 100 mm tissue culture dishes were co-incubated with bacteria at an m.o.i. of ∼200 or 1 nM CdA (kindly provided by I.Castagliuolo and J.Lamont). Cell rounding in response to bacteria was allowed to proceed for 80 min while CdA-exposed cells were processed after 6 h when cells appeared similar to cells rounded by Bah1P. Cells were washed in PBS, recovered by scraping and pelleted at 1000 g. Cells were resuspended in extract buffer [1% Triton X-100, 20 mM Tris pH 7.5, 1 mM MgCl2, 125 mM NaCl, 1 mM EGTA, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 µg/ml pepstatin A, 0.5 µg/ml leupeptin] and were frozen at –20°C. A 50 µg aliquot of protein extract was added to the reaction buffer (10 µM cold NAD, 250 nM [32P]NAD, 10 mM HEPES pH 8.0, 2 mM MgCl2, 1 mM DTT, 200 µM GTP, 10 mM thymidine, 15 mM isoniazid, 80 nM exoenzyme C3) and the reactions were incubated at 30°C for 1 h. Reactions were stopped by boiling in SDS–PAGE sample buffer for 5 min. Samples were loaded onto a 15% SDS–polyacrylamide gel, electrophoresed, and ADP-ribosylated proteins were visualized by exposing the gel to the Bio-Rad Model GS505 PhosphorImager.
Purification of actin and peptide sequencing
Purification of actin was based on the protocol of Goode et al. (1999). A total of 108 HEp-2 cells seeded into 500 cm2 tissue culture plates were exposed to Bah1Δhap (m.o.i. ∼200) for 65 min. The cells were washed once in PBS, collected by scraping and pelleted by centrifugation at 1000 g. Pelleted cells were lysed in 5 ml of G-buffer (10 mM Tris pH 7.5, 0.2 mM CaCl2, 0.2 mM ATP, 0.2 mM DTT) containing 1% Triton X-100, and the Triton-insoluble fraction was removed by centrifugation at 26 000 g. A 1 ml aliquot of the soluble fraction was loaded onto a 2 ml DNase I–Affigel 10 column. The column was washed with two column volumes of G-buffer, two column volumes of G-buffer with 0.2 M NH4Cl and two column volumes of G-buffer with 10% deionized formamide, and was eluted with G-buffer containing 50% formamide. Pooled eluants from five column runs were dialyzed into G-buffer, concentrated with a Centricon-30 microconcentrator, boiled in SDS–PAGE buffer and then loaded onto a preparative 7.5% SDS–polyacrylamide gel. Coomassie Blue-stained proteins corresponding to the 42 and 84 kDa forms of actin were excised, rinsed in 50% acetonitrile and frozen at –20°C for HPLC analysis. Both forms were digested with trypsin and the sequence of the peptide fragments was determined at the Harvard Microchemistry Facility by microcapillary reverse-phase HPLC non-electrospray tandem mass spectrometry (µLC/MS/MS) on a Finnigan LCQ quadrupole ion trap mass spectrometer (LeRoy et al., 1998).
Concentrated supernatant preparations
Supernatants from four 500 ml flasks of KFV43 or CW128 culture grown in LB broth at 37°C to log phase (A600 = 0.3–0.5) were collected by centrifugation at 13 000 g. Pooled supernatants were filtered over a 0.22 µm Corning cellulose acetate filter unit and concentrated to 150 ml across an Amicon spiral-wound ultrafiltration cartridge with a 100 kDa cut-off. The retentate was supplemented in three changes with 1 l of TE (20 mM Tris pH 8.0, 1 mM EDTA, 0.2 mM PMSF). The supernatants were concentrated further in a 50 ml Amicon Spin-cell apparatus fitted with a 300 kDa cut-off filter with one supplementation of 50 ml of TE buffer to a final volume of 6.3 ml (300-fold total concentration). Glycerol was added to 10% (v/v) and preparations were frozen and stored at –80°C in aliquots.
Western blot for actin laddering
A total of 5 × 105 toxin- or mock-treated HEp-2 cells were washed once with PBS, released in PBS with 0.1 mM EDTA, and recovered in RPMI 1640 medium by scraping. Cells were collected by centrifugation at 8000 g and resuspended in SDS–PAGE buffer containing 0.2 M DTT. These cell lysates were heated in boiling water for 5 min unless otherwise stated, electrophoresed in 8% SDS–polyacrylamide gels and then transferred to nitrocellulose. Antibody detection with 1:2000 polyclonal anti-actin (Sigma) or monoclonal anti-tubulin (Sigma) antibodies was performed using 1:2000 anti-rabbit IgG–alkaline phosphatase (AP) conjugate as a secondary antibody (Boehringer Mannheim) and as a substrate for colorimetric development with 5-bromo 4-chloro indolyl phosphate p-toluidine (Bachem) and nitroblue tetrazolium (Sigma) (Ausubel et al., 1990).
Production of RtxA138- and RTX-specific antibody
pWL126 contains a 4 kb EcoRI–HindIII subfragment of the rtxA gene in pET22b (Novagen). A 138 kDa His-tagged product, designated RtxA138, was purified from E.coli BL21(DE3)(pLysS) cells in the presence of 6 M urea on a nickel column by protocols recommended by the manufacturer (Novagen). A New England White rabbit was immunized with RtxA138 for the production of a polyclonal antiserum. For secondary boosts, the antigen was purified further by gel filtration chromatography or by electroelution. For antibody neutralization assays, the IgG1 fraction was isolated by passage of serum over a protein A–agarose column (Harlow and Lane, 1988).
Western blot for detection of RTX toxin
Concentrated culture supernatant fluids from KFV43 (RTX+) and CW128 (RTX–) were loaded in each lane of a 4–15% gradient SDS–polyacryl amide gel (Bio-Rad). Gels were transferred in a Bio-Rad Mini Trans-Blot transfer cell at 350 mA for 2 h at 4°C. RTX toxin was detected using a 1:1000 dilution of serum and the Amersham ECL for western blotting kit.
Acknowledgments
Acknowledgements
We thank J.Lamont and I.Castagliuolo for providing purified toxin A, W.Lin for constructing plasmid pWL126, Cynthia Walchle for providing CW128, and the Harvard Microchemistry Facility for the peptide sequence analysis. T.Mitchison, E.Rubin, B.Guo, D.Higgins and J.Mogridge are thanked for their helpful suggestions. This work was supported by NIH grant AI-18045 to J.J.M. K.J.F. was supported by an NRSA postdoctoral fellowship AI-10395.
References
- Alm R.A., Stroeher,U.H. and Manning,P.A. (1988) Extracellular proteins of Vibrio cholerae: nucleotide sequence of the structural gene (hlyA) for the haemolysin of the haemolytic El Tor strain O17 and characterization of the hlyA mutation in the non-haemolytic classical strain 569B. Mol. Microbiol., 2, 481–488. [DOI] [PubMed] [Google Scholar]
- Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1990) Current Protocols in Molecular Biology. Greene Publishing Associates and Wiley-Interscience, New York, NY. [Google Scholar]
- Benítez J.A. et al. (1999) Preliminary assessment of the safety and immunogenicity of a new CTXΦ-negative, hemagglutinin/protease-defective El Tor strain as a cholera vaccine candidate. Infect. Immun., 67, 539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron J.J.M., Brenner,M.B., Thomas,D.Y. and Williams,D.B. (1994) Calnexin: a membrane-bound chaperone of the endoplasmic reticulum. Trends Biochem. Sci., 19, 124–128. [DOI] [PubMed] [Google Scholar]
- Boquet P., Munro,P., Fiorentini,C. and Just,I. (1998) Toxins from anaerobic bacteria: specificity and molecular mechanisms of action. Curr. Opin. Microbiol., 1, 66–74. [DOI] [PubMed] [Google Scholar]
- Chen J.S.K. and Mehta,K. (1999) Tissue transglutaminases: an enzyme with a split personality. Int. J. Biochem. Cell Biol., 31, 817–836. [DOI] [PubMed] [Google Scholar]
- Coelho A., Andrade,J.R.C., Vicente,A.C.P. and DiRita,V.J. (2000) Cytotoxic cell vacuolating activity from Vibrio cholerae hemolysin. Infect. Immun., 68, 1700–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coote J.G. (1992) Structural and functional relationships among the RTX toxin determinants of Gram-negative bacteria. FEMS Microbiol. Rev., 8, 137–161. [DOI] [PubMed] [Google Scholar]
- Coster T.S. et al. (1995) Safety, immunogenicity and efficacy of live attenuated Vibrio cholerae O139 vaccine prototype. Lancet, 345, 949–952. [DOI] [PubMed] [Google Scholar]
- Dillon S.T., Rubin,E.J., Yakubovich,M., Pothoulakis,C., LaMont,J.T., Feig,L.A. and Gilbert,R.J. (1995) Involvement of Ras-related Rho proteins in the mechanism of action of Clostridium difficile toxin A and toxin B. Infect. Immun., 63, 1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasano A. et al. (1995) Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J. Clin. Invest., 96, 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust C., Ye,B. and Song,K.-P. (1998) The enzymatic domain of Clostridium difficile toxin A is located within the N-terminal domain. Biochem. Biophys. Res. Commun., 251, 100–105. [DOI] [PubMed] [Google Scholar]
- Finkelstein R.A., Boesman-Finkelstein,M., Chang,Y. and Häse,C.C. (1992) Vibrio cholerae hemagglutinin/protease, colonial variation, virulence and detachment. Infect. Immun., 60, 472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentini C. and Thelestam,M. (1991) Clostridium difficile toxin A and its effects on cells. Toxicon, 29, 543–567. [DOI] [PubMed] [Google Scholar]
- Fu Y. and Galán,J.E. (1999) A Salmonella protein antagonizes Rac-1 and Cdc42 to mediate host-cell recovery after bacterial invasion. Nature, 401, 293–297. [DOI] [PubMed] [Google Scholar]
- Fullner K.J. and Mekalanos,J.J. (1999) Genetic characterization of a new type IV pilus gene cluster found in both classical and El Tor biotypes of Vibrio cholerae. Infect. Immun., 67, 1393–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan A.K., Frank,D.W., Misra,R.P., Schmidt,G. and Barbieri,J.T. (1998) Pseudomonas aeruginosa exoenzyme S ADP-ribosylates Ras at multiple sites. J. Biol. Chem., 273, 7332–7337. [DOI] [PubMed] [Google Scholar]
- Goehring U.-M., Schmidt,G., Pederson,K.J., Aktories,K. and Barbieri,J.T. (1999) The N-terminal domain of Pseudomonas aeruginosa exoenzyme S is a GTPase-activating protein for Rho GTPases. J. Biol. Chem., 274, 36369–36372. [DOI] [PubMed] [Google Scholar]
- Goode B.L., Wong,J.J., Butty,A.-C., Peter,M., McCormack,A., Yates,J.R., Drubin,D.G. and Barnes,G. (1999) Coronin promotes the rapid assembly and cross-linking of actin filaments and may link the actin and microtubule cytoskeletons in yeast. J. Cell Biol., 144, 83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E. and Lane,D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Hegyi G., Mak,M., Kim,E., Elzinga,M., Muhlrad,A. and Reisler,E. (1998) Intrastrand cross-linked actin between Gln-41 and Cys-374. I. Mapping of sites cross-linked in F-actin by N-(4-azido- 2-nitrophenyl) putrescine. Biochemistry, 37, 17784–17792. [DOI] [PubMed] [Google Scholar]
- Heidelberg J.F. et al. (2000) DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature, 406, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just I., Selzer,J., von Eichel-Streiber,C. and Aktories,K. (1995) The low molecular mass GTP-binding protein Rho is affected by toxin A from Clostridium difficile. J. Clin. Invest., 95, 1026–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniga K., Uralili,J., Bliska,J.B. and Galandrini,R. (1996) A secreted protein tyrosine phosphatase with modular effector domains in the bacterial pathogen Salmonella typhimurium. Mol. Microbiol., 21, 633–641. [DOI] [PubMed] [Google Scholar]
- Kaper J.B., Lockman,H., Baldini,M.M. and Levine,M.M. (1984) Recombinant nontoxinogenic Vibrio cholerae strains as attenuated cholera vaccine candidates. Nature, 308, 655–658. [DOI] [PubMed] [Google Scholar]
- Kaper J.B., Morris,J.G.J. and Levine,M.M. (1995) Cholera. Clin. Microbiol. Rev., 8, 48–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E., Phillips,M., Hegyi,G., Muhlrad,A. and Reisler,E. (1998) Intrastrand cross-linked actin between Gln-41 and Cys-374. II. Properties of cross-linked oligomers. Biochemistry, 37, 17793–17800. [DOI] [PubMed] [Google Scholar]
- Lerm M., Schmidt,G. and Aktories,K. (2000) Bacterial protein toxins targeting Rho GTPases. FEMS Microbiol. Lett., 188, 1–6. [DOI] [PubMed] [Google Scholar]
- LeRoy G., Orphanides,G., Lane,W.S. and Reinberg,D. (1998) Requirement of RSF and FACT for transcription of chromatin templates in vitro. Science, 282, 1900–1904. [DOI] [PubMed] [Google Scholar]
- Lin W., Fullner,K.J., Clayton,R., Sexton,J.A., Rogers,M.B., Calia,K.E., Calderwood,S.B., Fraser,C. and Mekalanos,J.J. (1999) Identification of a Vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc. Natl Acad. Sci. USA, 96, 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mel S.F., Fullner,K.J., Wimer-Mackin,S., Lencer,W.I. and Mekalanos,J.J. (2000) Association of protease activity in Vibrio cholerae vaccine strains with decreases in transcellular epithelial resistance of polarized T84 intestinal cells. Infect. Immun., in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra R., Figueroa,P., Mukhopadhyay,A.K., Shimada,T., Takeda,Y., Berg,D.E. and Nair,G.B. (2000) Cell vacuolation, a manifestation of the El Tor hemolysin of Vibrio cholerae. Infect. Immun., 68, 1928–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy S.N.P., Wilson,J.H., Lukas,T.J., Kuret,J. and Lorand,L. (1998) Cross-linking sites of the human Tau protein, probed by reactions with human transglutaminase. J. Neurochem., 71, 2607–2614. [DOI] [PubMed] [Google Scholar]
- Nemes Z.J., Adány,R., Balázs,M., Boross,P. and Fésüs,L. (1997) Identification of cytoplasmic actin as an abundant glutaminyl substrate for tissue transglutaminase in HL-60 and U937 cells undergoing apoptosis. J. Biol. Chem., 272, 20577–20583. [DOI] [PubMed] [Google Scholar]
- Pederson K.J., Vallis,A.J., Aktories,K., Frank,D.W. and Barbieri,J.T. (1999) The amino-terminal domain of Pseudomonas aeruginosa ExoS disrupts actin filaments via small-molecular weight GTP-binding proteins. Mol. Microbiol., 32, 393–401. [DOI] [PubMed] [Google Scholar]
- Popoff M.R. (1998) Interactions between bacterial toxins and intestinal cells. Toxicon, 36, 665–685. [DOI] [PubMed] [Google Scholar]
- Rader A.E. and Murphy,J.R. (1988) Nucleotide sequences and comparison of the hemolysin determinants of Vibrio cholerae El Tor RV79(Hly+) and RV79(Hly–) and classical 569B(Hly–). Infect. Immun., 56, 1414–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele-Mortimer O., Knodler,L.A. and Finlay,B.B. (2000) Poisons, ruffles and rockets: bacterial pathogens and the host cell cytoskeleton. Traffic, 1, 107–118. [DOI] [PubMed] [Google Scholar]
- Tacket C.O., Losonsky,G., Nataro,J.P., Cryz,S.J., Edelman,R., Fasano,A., Michalski,J., Kaper,J.B. and Levine,M.M. (1993) Safety and immunogenicity of live oral cholera vaccine candidate CVD110, a ΔctxA Δzot, Δace derivative of El Tor Ogawa Vibrio cholerae. J. Infect. Dis., 168, 1536–1540. [DOI] [PubMed] [Google Scholar]
- Taylor D.N. et al. (1994) Development of a live, oral, attenuated vaccine against El Tor cholera. J. Infect. Dis., 170, 1518–1523. [DOI] [PubMed] [Google Scholar]
- Waldor M.K. and Mekalanos,J.J. (1996) Lysogenic conversion by a filamentous phage encoding cholera toxin. Science, 272, 1910–1914. [DOI] [PubMed] [Google Scholar]
- WHO (1999) Cholera, 1998. Wkly Epidemiol. Rec., 74, 257–259. [PubMed] [Google Scholar]
- Wu Z., Milton,D., Nybom,P., Sjö,A. and Magnusson,K.-E. (1996) Vibrio cholerae hemagglutinin/protease (HA/protease) causes morphological changes in cultured epithelial cells and perturbs their paracellular barrier function. Microb. Pathog., 21, 111–123. [DOI] [PubMed] [Google Scholar]
- Wu Z., Nybom,P., Sudqvist,T. and Magnusson,K.-E. (1998) Endogenous nitric oxide in MDCK-I cells modulates the Vibrio cholerae haemagglutinin/protease (HA/P)-mediated cytoxicity. Microb. Pathog., 24, 321–326. [DOI] [PubMed] [Google Scholar]
- Wu Z., Nybom,P. and Magnusson,K.-E. (2000) Distinct effects of the Vibrio cholerae haemagglutinin/protease on the structure and localization of the tight junction-associated proteins occludin and ZO-1. Cell. Microbiol., 2, 11–18. [DOI] [PubMed] [Google Scholar]