

Abstract
Heterotrimeric guanine nucleotide-binding proteins (G proteins) composed of three subunits α, β, γ mediate activation of multiple intracellular signaling cascades initiated by G protein-coupled receptors (GPCRs). Previously our laboratory identified small molecules that bind to Gβγ and interfere with or enhance binding of select effectors with Gβγ. To understand the molecular mechanisms of selectivity and assess binding of compounds to Gβγ, we used biophysical and biochemical approaches to directly monitor small molecule binding to Gβγ. Surface plasmon resonance (SPR) analysis indicated that multiple compounds bound directly to Gβγ with affinities in the high nanomolar to low micromolar range but with surprisingly slow on and off rate kinetics. While the koff was slow for most of the compounds in physiological buffers, they could be removed from Gβγ with mild chaotropic salts or mildly dissociating collision energy in a mass-spectrometer indicating that compound-Gβγ interactions were non-covalent. Finally, at concentrations used to observe maximal biological effects the stoichiometry of binding was 1:1. The results from this study show that small molecule modulation of Gβγ-effector interactions is by specific direct non-covalent and reversible binding of small molecules to Gβγ. This is highly relevant to development of Gβγ targeting as a therapeutic approach since reversible, direct binding is a prerequisite for drug development and important for specificity.
Keywords: G protein, Surface Plasmon Resonance, Protein-ligand interactions, non-covalent binding
1. Introduction
A cell’s ability to detect and accurately respond to its microenvironment is of paramount importance in maintaining basic biological processes such as tissue homeostasis, development and immunity. To do so, cells employ many different cellular response mechanisms to relay external sensory stimuli to internal effectors. A major such pathway, involves guanine nucleotide-binding proteins (G proteins)1 that are involved in second-messenger cascades triggered by G-protein-coupled receptors (GPCR’s). Typically, G protein signaling pathways are composed of a G protein-coupled seven transmembrane receptor/s (GPCR), heterotrimeric G proteins, and intracellular effectors [12]. Heterotrimeric G-proteins are composed of Gα subunit and Gβγ subunits. Gβγ subunits interact with many different effector proteins to regulate cellular physiology. These include adenylate cyclase (AC), G protein coupled receptor kinase 2 (GRK2) [30], phospholipase Cβ1, β2 and β3 isoforms [4,29,39], inward rectifying potassium channels (GIRK) [24,27], phosphoinositide 3 kinase γ (PI3Kγ) [41,42], and PI3Kβ [13] isoforms, and N-type calcium channels [15].
Various studies suggest that pharmacological targeting of Gβγ could be of therapeutic value. Blocking Gβγ signaling with a c-terminal Gβγ-binding fragment of G protein-coupled receptor kinase-2 (GRK2ct) inhibits development of heart failure in various animal models [20,32]. Gβγ-dependent regulation of PI3Kγ plays a central role in leukocyte migration in response to chemoattractants [14,23] suggesting that blocking this interaction could be used to inhibit neutrophil dependent inflammation. Roles for Gβγ-dependent signaling in μ-opioid potency [44], drug addiction [45], cancer metastasis and prostate cancer [3] have all been suggested. While Gβγ subunits are ubiquitous, their participation in activation particular downstream signaling processes is more restricted [38]. For example, while Gβγ-dependent regulation of PLCβ is prevalent in immune cells and some other cell types, Gαq-dependent PLCβ is the dominant G protein dependent PLC pathway in most cell types. Thus pharmacological specificity could come from inhibiting particular Gβγ-target couplings thereby restricting the pharmacological effects to specific tissues and pathways.
To identify small molecule compounds that could bind to Gβγ and inhibit interactions with downstream effectors we screened the NCI diversity set library using a peptide competition approach [2]. Subsequently we and others [16,19,46] have shown that small molecules identified in this screen selectively modulate Gβγ-effector interactions in vitro and in cells. Additionally one class of molecules, M119/gallein, is efficacious in various in vivo models including, mouse models of opioid-dependent antinociception [2,25], inflammation [22], and cardiac hypertrophy [5].
Here we describe characterization of Gβγ-small molecule interactions using direct biophysical methods to determine the binding mechanisms for various compound classes. Prior to this study our understanding of small molecule-Gβγ interactions primarily arose from determinations of IC50 values using indirect phage-displayed peptide competition ELISA assays [2]. We were interested in identifying molecules that directly bound to Gβγ non-covalently by a single site 1:1 binding mechanism. This is critical for development of these molecules as a pharmacological tool because reactive molecules tend to be non-specific and single site binding implies a specific binding mode relevant to the compound’s mechanism of action. Several non-specific mechanisms for inhibition of proteins by small molecules are possible. One is covalent protein modification through reactions of compounds with nucleophiles such as SH or NH moieties in the protein. Another possibility is promiscuous-aggregation based binding of small molecule aggregates to protein surfaces leading to a general non-specific inhibition of protein function [6]. In the course of this work we found two mechanisms for inhibition by two general groups of compounds; 1) reversible covalent reactions with cysteine residues in the protein interaction surface ([8] and Dessal and Smrcka unpublished data), and in the work described here, 2) reversible non-covalent binding of compounds to Gβγ. Understanding these mechanisms is critical to determining whether use of these compounds can be leads for a viable therapeutic strategy.
2. Materials and Methods
2.1 Small molecules
Compounds for this study were obtained from the National Cancer Institute repository except when indicated otherwise. Compound abbreviations are as follows: M119 (NSC119910) 2-(3,4,5-trihydroxy-6-oxoxanthen-9-yl)cyclohexane-1-carboxylic acid, M119B (NSC119892) 2-(3-hydroxy-6-oxoxanthen-9-yl)cyclohexane-1-carboxylic acid, 372 (NSC115372) 1-amino-4-(3-aminoanilino)-9,10-dioxoanthracene-2-sulfonic acid, and M201 (NSC201400) 7-amino-1,2,3,10-tetramethoxy-6,7-dihydro-5H-benzo[a]heptalen-9-one. Gallein, 3′, 4′, 5′, 6′-tetrahydroxyspiro[2-benzofuran-3, 9′-xanthene]-1-one was purchased from Acros Organics, Geel, Belgium or TCI America, Portland Oregon. All compounds were prepared as 50mM stocks in DMSO with care taken to subject them to minimum freeze thaw cycles.
2.2 Purification of biotinylated Gβγ
Biotinylated Gβ1γ2 subunits were expressed in 1L of Hi5 cells (at a density of 1.5×106cells/mL) triply infected with 2nd pass amplified baculoviruses encoding 6his-αi (4mL), Gγ2 (10mL) and N-terminally biotin acceptor tagged-Gβ1 (10mL) and biotin-protein ligase, BirA, in a bicistronic baculovirus expression vector. Proteins were purified as described by Gilman and colleagues [21] and modified by [7]. Gβγ protein produced by this method was confirmed to be biotinylated at the N terminus of the Gβ subunit by Western blotting with neutravidin linked HRP and pulldown by streptavidin agarose.
2.3 Surface Plasmon Resonance
Direct binding of small molecules was assessed using a Reichert SR7000 Surface Plasmon Resonance dual channel instrument equipped with a separate auto sampler and syringe pump (Reichert, Depew, NY). The basis for surface preparation was adapted from Wu et, al [43] and Rebois et al [31] with minor modifications. Sensor chips with mixed self assembled monolayers composed of 90% monothiol alkane PEG3K-OH and 10% monothiol alkane PEG6K-COOH (part number 13206061, Reichert, Depew, NY) were mounted on the instrument using immersion oil. The flow cell block was then mounted, effectively separating the sensor chip into two identical flow cells after which running buffer (degassed double distilled water) was allowed to flow over both flow cells using the syringe pump. The two flow cells were connected in series by a short length of tubing and the surface was primed by injecting an alkaline salt solution (2M NaCl and 10mM NaOH). Free carboxyl groups on the surface were modified by injecting a mixture of 0.1 M 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride and 0.05 M N-hydroxysuccinimide at a flow rate of 5 μl/min to generate a reactive succinimide ester surface. Neutravidin (100 mg/ml; prepared in 20 mM sodium acetate buffer, pH 5.5) was coupled to the sensor via free amine coupling to the immobilized succinimide, followed by quenching the remaining activated succinimide esters with 1 M ethanolamine, pH 8.5. One flow cell was then uncoupled from the buffer flow (henceforth called the reference cell) and the running buffer was changed to Gβγ binding buffer (50mM HEPES, pH 7.6, 1 mM EDTA, 100 mM NaCl, 0.1% C12E10, and 1 mM dithiothreitol). Biotinylated Gβ1γ2 (bGβ1γ2) was bound to the flow cell (henceforth called the Gβγ cell) to achieve between 2000 – 10000 micro-refractive index units (determined by experiment). After washing away free Gβγ the reference cell was reconnected to the main buffer flow. Surface preparation is completed by injection of 1mM free biotin to block all unbound neutravidin binding sites on both the Gβγ and reference cells.
2.4 Direct binding of ligands and data analysis
Direct binding of ligands (small molecules/peptides) was done at 25°C. In the case of small molecules, the running buffer was replaced by Gβγ binding buffer + 1.2% Dimethyl Sulfoxide (DMSO). The Gβγ binding buffer contained DTT to inhibit redox reactions of compounds with free cysteines on Gβγ and 0.1% C12E10 to disrupt small molecule aggregates [9]. Thus any interactions observed are not likely dependent on redox chemistry or nonspecific promiscuous aggregate binding. Temperature compensation and buffer calibration was done prior to any/all data collection using SPR direct access software. A series of dilutions of compounds were prepared in binding buffer and injected for 7 min followed by a dissociation phase of 10 min at a flow rate of 50 μL/min. To reference for drift, two blank injections were performed between compound injections and used to apply corrections. The sensorgrams were corrected for nonspecific background by running the same experiment in series over the identical reference cell. Actual subtractions (for drift and non-specific background binding) were done using Reichert SPR v4.5 software (Reichert, Depew, NY). Bulk shift effects and refractive index changes due to differences in DMSO concentration between running buffer and solvent concentrations were corrected using Scrubber 2.0 (Biologic) software. Best fit kinetic parameters were determined using either a kinetic titration model, which accounts for incomplete dissociation between injections [17] or the standard surface regeneration model [33]. Data analysis was carried out using either Scrubber 2.0 (Biologic) or ClampXP version 3.5 and plotted using Graphpad 5.
2.5 Mass spectrometric analysis
To assess the reversibility and stoichiometry of compound binding to Gβγ 5μM Gβ1γ2C68S protein in 20mM Ammonium Acetate pH 7.0 buffer, was mixed with 100μM gallein and incubated for 30 minutes at room temperature. Gβγ or Gβγ-gallein complexes were separately and directly analyzed using a Waters “Synapt” nano ESI QToF Mass Spectrometer specifically designed for analysis of intact non-covalent protein complexes [36].
2.6 Gallein-Gβγ stoichiometry determination using [3H]-gallein
Commercially available gallein (Acros Organics, Geel, Belgium) at 99% purity, was labeled with [3H] by tritium exchange (American Radiolabeled Chemicals, USA). The resulting radiolabeled product was purified and analyzed by C18 HPLC chromatography with absorbance detection, relative to a purified gallein standard to determine purity and specific activity. The final radiolabeled product was 99% pure with a specific activity of 12 Ci/mmol. Gβ1γ2 was mixed with [3H]-gallein diluted with unlabeled gallein at various specific activities to give final concentrations of 1μM Gβγ and the indicated concentrations of gallein in a total reaction volume of 60μL and incubated for 1 hour at room temperature in Gβγ binding buffer. The Gβγ-[3H]-gallein complex was separated from unbound [3H]-gallein using a Zeba™ microspin desalting columns (Thermo scientific) and measured by liquid scintillation counting. To calculate binding stoichiometry Gβγ concentrations were corrected for loss of Gβγ in the column that ranged from 25 to 50%.
3. Results
3.1 Surface Plasmon Resonance based detection of small molecule binding to Gβγ subunits
A variety of biophysical techniques are possible for direct measurement of small molecule ligand, protein interactions. We chose a surface plasmon resonance (SPR) based method for monitoring direct small molecule binding to Gβγ. A major advantage of SPR over isothermal titration calorimetry, for example, is that only small amounts of unlabeled protein are required [26] and it provides kinetic data (kon and koff values) for protein-ligand interactions [34]. A potential difficulty is that the method is ideally suited for monitoring large protein binding interactions because the sensitivity of the method is directly proportional to the mass of the binding ligand. However various investigators have successfully employed SPR to measure small molecule binding to proteins by increasing the surface density of the bound target [18,28].
Gβ1γ2 subunits with site specific biotin modification at the amino terminus of the Gβ subunit were prepared using in vivo biotinylation of a biotin acceptor tag fused to the N-terminus of Gβ1(bGβ1) and coexpression of biotin-ligase in insect cells. Neutravidin was directly coupled to the SPR chip via primary amine coupling followed by binding of bGβ1γ2 as described in methods. We standardized the Neutravidin surface density to 4000–5000μRIU which could yield a maximum of 12000μRIU Gβγ subunit immobilization (or less when appropriate). High immobilization densities are required for detection of binding of low molecular weight ligands. To determine if the Gβγ hot spot was accessible for small molecule binding we tested a hot spot-binding peptide, SIGK, for binding to immobilized Gβ1γ2 (Figure 1, A and B). The peptide bound to Gβγ with an affinity of 3.9±0.5μM in close agreement with IC50 values obtained from competition ELISA experiments [35]. Control peptide SIGK(L9A) which was non-binding in the ELISA experiments [35] did not elicit a response associated with binding to Gβγ in the SPR assay (data not shown).
Figure 1.

A) Binding of SIGK peptide (black, 5 different concentrations) to a surface of immobilized Gβ1γ2. The experiment was carried out at room temperature with a flow rate of 50 μL/min in Gβγ-binding buffer (50mM HEPES, pH 7.6, 1 mM EDTA, 100 mM NaCl, 0.1% C12E10, and 1 mM dithiothreitol). Each concentration was injected for 5 minutes followed by a dissociation phase of 10 minutes. No surface regeneration was needed. Each concentration was injected in triplicate. Data was corrected for non specific background binding (by subtraction of sensorgrams from the reference cell) and instrument drift (using blank buffer injections) with subsequent removal of bulk shifts of refractive index using scrubber 2 (biologic) software. Data (black dots) were fit (red lines) to a simple bimolecular model. Kinetic parameters from this experiment were, kon = 3.6×103 M−1s−1, koff = 1.7×10−2 s−1, with a derived Kd (koff/kon) = 4.6μM. The average Kd from 3 independent experiments was 3.9±0.5μM. B) Plot of Rmax data vs. peptide concentration from the data in A. Each data point is ± SEM from 3 separate experiments.
Initial experiments with small molecule binding to Gβγ were carried out with M119 (Figure 2), owing to the abundance of in vitro and in vivo data demonstrating that M119 and the related compound gallein inhibit Gβγ-dependent signaling processes [2,5,22]. Figure 2A and B show representative association phases for two concentrations of M119, fit to a one phase exponential association equation. Data between 300 nM and 10 μM fit well with this equation but fit results for data from 30 and 100μM concentrations of M119 were poor. Since 10 μM M119 is saturating for most of its biological effects, all SPR experiments were performed at less than or equal to 10 μM. Surprisingly, M119 bound very slowly, atypical of most common small molecule protein interactions where binding is often diffusion limited [28,40]. As a first approximation the data was analyzed using an equilibrium binding analysis [11]. Since the binding did not reach saturation during each binding interaction due to the slow on rate, Rmax(obs) values were estimated from the one phase exponential association equation for each concentration. Rmax(obs) values were plotted as a function of concentration and fit using linear least squares fitting to a 1:1 Langmuir isotherm (Figure 2C, Table 1). The Ka obtained from this single data set was equal to 2.16×106 M−1 giving a Kd of 460 nM.
Figure 2.

SPR analysis of M119 direct binding to Gβ1γ2 analyzed by either equilibrium estimation or kinetic titration. M119 was injected at various concentrations (from 300 nM to 10μM) for 5 minutes each followed by 7 min dissociation phases. A and B) Representative data from individual association phases for two different concentrations of M119 (black dots) were fit (red line) separately (after removal of bulk solvent effects) using linear least squares fitting to a one phase exponential association equation to derive R max, R=Rmax(obs)*(1−exp(−K*X)), where R = response (in μRIU), Rmax(obs) = maximal response, K = observed on rate, X = time (in seconds). C) The derived Rmax(obs) values from 4 different concentrations as in A and B were plotted as a function of concentration and fit using linear least squares to a 1:1 Langmuir isotherm (Rmax(obs)= Rmax * ((Ka * X)/(Ka * (X +1)) where Rmax = Maximum binding of M119, Ka = Association constant for M119, X = concentration of M119. Ka obtained = 2.16×106 M−1. D) Alternatively, the entire continuous trace is shown with concentrations of M119 as indicated, and was globally fit to the data using a kinetic titration model in ClampXP software. Fits are shown in red with the inset showing the deviations (residuals) from the actual data. Kinetic parameters estimated by both approaches are reported in Table 1. These data are from a single experiment. Average data from multiple experiments are shown in Table 2.
Table 1.
Binding parameters from two different analyses of M119 binding to Gβγ. Data are from a single experiment.
| Equilibrium titration | Kinetic titration | |
|---|---|---|
| kon (M−1s−1) | 1027±8 | |
| koff (s−1) | 1.2±0.5×10−4 | |
| Kd (M) | 4.6×10−7 | 1.2×10−7 |
To analyze the data more rigorously, kinetic data was collected for both association and dissociation phases at various concentrations of M119 (Figure 2D). Surprisingly, dissociation of M119 was also quite slow (Figure 2D) and did not allow return to baseline between each injection. To account for incomplete dissociation between injections we used a kinetic titration approach as described by Myszka and colleagues [17] to obtain binding data. Global fitting of the single data set in figure 2D to the parameters in the kinetic titration model yielded a Kd of 120 nM (Table 1) similar to the Kd estimated from equilibrium analysis in Figure 2C. The average Kd for M119 binding to Gβγ from multiple kinetic titration experiments is 276±70 nM and is shown in Table 2. The binding rates and constants obtained from kinetic titration analysis were independent of flow rate over a wide range (25 – 100μL/min) indicating a lack of mass transfer effects.
Table 2.
Parameters for binding of various compounds to Gβγ determined by kinetic titration
| Kd (μM) | kon (M−1s−1) | koff (×10−4s−1) | |
|---|---|---|---|
| M119 (N=5) | 0.275±70 | 830±200 | 2.0±0.8 |
| Gallein (N=5) | 0.407±0.24 | 870±112 | 3.5±0.45 |
| M201 (N=2) | 67.1±27 | 91.5±13 | 60±20 |
| 372 (N=4) | 2±0.3 | 390±93 | 8.6±2.4 |
Overall these data demonstrate that M119 binds directly and reversibly to Gβγ, but surprisingly the kon and koff were quite slow. The apparent Kd is submicromolar and the data are best fit with a 1:1 binding model. Since the experiments are done in the presence of DTT, EDTA and 0.1% non-ionic detergent, it suggests that M119 is not utilizing a redox-based, metal ion- dependent, or promiscuous-aggregation based mechanism for binding to Gβγ.
3.2 SPR direct binding analysis of other small molecules to Gβγ
Well over 20 structurally distinct Gβγ modulating small molecules were identified in our initial screen. Some of these compounds have since been shown to act by a redox based mechanism. Here we selected 3 representative compounds with distinct structural moieties and biological functions, that did not bind to Gβγ using redox chemistry [8]. These were gallein, M201 (NSC201400) and 372 (NSC115372) (Fig 3A). Biological activities have been previously reported for gallein and M201 [2].
Figure 3.

SPR analysis of other small molecule binders to Gβγ. A) Structures of small molecules tested. Representative SPR traces for B) gallein C) M201 and D) 372 are shown. For all data sets, sequentially increasing concentrations of compound were injected for 7 minutes and allowed to dissociate for 10 minutes. Data is shown as black dots while the global fits are shown as a red line. The inset represents deviations (residuals) compared to the average fitted kinetic parameters. Kinetic and equilibrium binding constants from this analysis are in Table 2.
Kinetic titration experiments and analysis was carried out for each individual compound over separate Gβγ immobilized surfaces. Non-specific/multiple binding sites were observed above 30 μM gallein and 20 μM 372. Since 10 μM gallein was a maximally effective concentration in biological assays; all SPR assays were carried out to a maximum of 10 μM for gallein and 15 μM for 372. All 3 compounds bound to immobilized Gβγ with slow association and dissociation kinetics consistent with those observed for M119 (Fig 3, B, C and D; Table 2). The koff for both gallein and 372 were 10 fold slower than that of M201 resulting in incomplete dissociation of both these compounds in the time course of this experiment compared with M201 which showed full dissociation to baseline levels. To examine the on and off rates for gallein in more detail we conducted an extended binding and dissociation time course for one concentration of gallein (supplemental figure 1). These results are consistent with slow association of gallein with Gβγ followed by a slow, but fully reversible dissociation of gallein from Gβγ. These data indicate that all of these compounds bind directly to Gβγ but that the binding and dissociation kinetics are surprisingly slow for multiple compounds.
3.3 Reversibility of binding of gallein and other compounds to Gβγ
Since gallein, M119 and 372 dissociation from Gβγ was slowly reversible, we tested if these molecules bound by a covalent mechanism. As one approach, we searched for ionic solution conditions that would rapidly and completely strip these compounds from the Gβγ surface. We tested multiple different solution conditions and identified a cocktail of low concentration chaotropic agents (610 mM MgCl2, 205 mM Urea and 610 mM Guanidine-HCl) that completely (>85%) and rapidly removed all 3 classes of Gβγ hot spot-binding small molecules from Gβγ and allowed for complete rebinding of all of the molecules. Figure 4A shows an example experiment demonstrating gallein binding, restoration of the baseline during regeneration, and rebinding gallein after regeneration.
Figure 4.

Mild ionic stripping reverses gallein binding to Gβγ. A) Gallein was injected over the Gβγ surface at the indicated lines with an 3, 30 second pulses of regeneration buffer containing 610 mM MgCl2, 205 mM Urea and 610 mM Guanidine-HCl. Data shown is raw data from the channel containing immobilized protein without reference subtractions, drift or bulk shift corrections. B) Binding of gallein was conducted at the indicated concentrations for 7 minutes followed by a dissociation phase of 10 minutes. Between each concentration surface regeneration was carried 3, 30s pulses of regeneration cocktail as in A. Each concentration was repeated in triplicate (data shown as black dots). Response versus time plots of for all concentrations of gallein binding to Gβγ (black dots) globally fit (red lines) to a simple bimolecular model in scrubber 2. Kinetic and equilibrium constants from this analysis are kon = 981 M−1s−1, koff = 3.19×10−4 s−1, derived KD (koff/kon) = 325nM. Average values for gallein and other compounds from multiple experiments are reported in Table 3.
To demonstrate that the stripping treatment completely reversed binding of the small molecules yet left Gβγ intact, we used the chaotropic cocktail described above in a SPR regeneration protocol to determine binding constants for gallein and 372 [33]. Surface regeneration was carried out between each association, dissociation cycle by giving 3, 30s pulses of regeneration cocktail. Representative data is shown for gallein and fits are given in Figure 4B. Kinetic and equilibrium constants determined by this approach (Table 3) were very similar to those determined in experiments in figures 3B and 3D and reported in Table 2 that did not involve regeneration. This provides strong support for the accuracy of the kinetic and equilibrium binding data in Tables 1 and 2 and indicates that regeneration to remove compound does not damage the Gβγ subunit complex. Compounds covalently associated with Gβγ would be unlikely to be removed by this relatively mild treatment, indicating that binding of these compounds to Gβγ does not involve covalent modification of Gβγ subunits.
Table 3.
Parameters for binding to Gβγ determined with a surface Regeneration protocol.
| Kd (μM) | kon (M−1s−1) | koff (×10−4s−1) | |
|---|---|---|---|
| Gallein (N=2) | 0.35±0.03 | 926±55 | 3.2±0.27 |
| 372 (N=3) | 2.7±0.45 | 267±137 | 8.2±5.1 |
3.4 Non-covalent mass spectrometry to assess binding stoichiometry and reversibility
As an alternate approach to assessing binding stoichiometry and reversibility we used a low energy electrospray TOF mass spectrometry approach that measures non-covalently associated protein complexes [36] to analyze Gβ1His6γ2C68S small molecule complexes. The electrospray ionization conditions for introducing the protein complex into the gas phase for MS are sufficiently mild that non-covalent complexes are observable.
Gβγ-gallein complexes were formed by addition of 100 μM gallein to 5 μM Gβ1His6γ2C68S. The Gγ2C68S mutation prevents lipid modification at Gγ2C68 obviating the requirement for detergent in the analysis of Gβγ. Figure 5A shows a representative +12 ion of Gβ1His6γ2C68S subunits while Figure 5B shows Gβ1γ2(C68S) with bound gallein. In both cases the major peak at 3837.8 (46044 Da) corresponds to the Gβ1His6γ2C68S heterodimer with Gβ1 N-terminally processed and acetylated, and unprocessed His6γ2C68S. Peak heterogeneity is due primarily to the presence of various incomplete N-terminal processed forms of Gγ (N terminus cleaved unacetylated and uncleaved unacetylated) and NH3+ adducts.
Figure 5.

Mass spectrometry of intact Gβ1His6γ2 C68S protein with bound Gallein. Gβ1γ2 (C68S) (5μM) in 20mM Ammonium Acetate pH 7.0 buffer was analyzed in a Waters “Synapt” nano ESI QToF Mass Spectrometer. Multiple charged species from +11 to +13 ions were observed but only data for +12 charged species is shown for simplicity. A) Mass spectrum for Gβγ alone. B) Gβγ bound to gallein. The major peak in both panels of 3837.8 (46044 Da) corresponds to the Gβγ heterodimer containing Gβ after initiator Met cleavage and N-terminal acetylation and His6γ2C68S without N-terminal Met cleavage.
Figure 5B shows a single peak at 3837.87 for Gβγ without bound gallein, one major peak corresponding to exactly 1 molecule of bound gallein (peak 3868.29) and two minor peaks with exactly 2 (peak 3898.42) and 3 (peak 3930.79) Gβγ bound gallein molecules. Since Gβγ-gallein interactions are non-covalent, even though the Gβγ-complex was formed at saturating gallein concentration, much of the bound gallein is likely dissociated during the initial electrospray ionization. Nevertheless, the relative abundance of the remaining peaks likely represents the relative stability of those complexes in the gas phase. The peak corresponding to 1 molecule of bound gallein is the predominant gallein bound form. No species representing a Gβγ-bound aggregate of gallein was observable. Thus if any aggregate binding does occur in solution it is likely relatively weak and unobservable by this method. The minor peaks corresponding to 2 and 3 molecules of bound gallein likely represent less energetically favorable Gβγ-gallein complexes and thus are less predominant in the spectrum. Since these complexes were formed at 100 μM gallein (100 times the Kd for gallein-Gβγ interactions), the presence of weaker binders is not unexpected. This data indicates that stoichiometries as high as 3:1 gallein: βγ are possible at high concentration (ie > 30 μM gallein) but the most energetically stable form is the 1:1 complex that is likely to be found at concentrations 10 μM or less which is the biologically relevant concentration of gallein.
To provide additional evidence that gallein binding is reversible, we increased the collision cell voltage to disrupt non-covalent interactions but leave covalent interactions intact. In Figure 6A is the spectrum at low collision energy showing three multiply charged Gβγ-gallein ions (+13, +12 and +11) with peaks corresponding to bound gallein. At higher collision energy (Figure 6B) the peaks corresponding to different Gβγ-bound species of gallein are absent while peaks corresponding to +11, +12 and +13 Gβγ ions remained. Additional peaks corresponding to free β subunits were also observed (4144.39, 3730.16). Thus at collision energies that maintain some non-covalent Gβ-Gγ interactions, gallein binding is fully reversed. These data are in stark contrast with what we reported for covalent modifiers of Gβγ subunits where incubation with compound completely depletes the free Gβγ pool and only species corresponding to covalently modified β subunits are observed at higher collision energies [8]. These data strongly support the notion that the most energetically favorable binding of gallein to Gβγ is in a 1:1 stoichiometry, and that the binding is non-covalent and fully reversible.
Figure 6.

Collision induced dissociation reverses gallein binding to intact Gβγ. A) Mass spectrum of Gβγ -gallein complex measured at low collision cell voltage. B) Mass spectrum of Gβγ-gallein complex measured at high collision cell voltage.
Radioactive ligand binding to assess binding stoichiometry
As another approach to assess gallein:Gβγ binding stoichiometry we carried out a simple binding experiment using 3H labeled gallein as a tracer and a spin-through size exclusion column to separate bound from free gallein. In the absence of Gβγ there is a small leak of gallein in the Gβγ fraction (Fig 7A column 1). In the presence of Gβγ the binding in this fraction is significantly increased in a manner that is eliminated by inclusion of excess of unlabeled “cold” gallein (Fig. 7A columns 2 and 3). Thus, this assay detects [3H]-gallein specifically bound to Gβγ. Several gallein concentrations were tested by this method and molar binding stoichiometries were calculated. The amount of binding increased with increasing concentrations to a maximum of 1.6 moles of gallein to 1 mole of Gβγ. This 1.6:1 stoichiometry only occurred at concentrations of 100 μM gallein or greater and at 30 μM or less the binding stoichiometry was 1:1 or less. As stated earlier, gallein is saturating in most biological assays at 10 μM so the relevant binding stoichiometry that achieves complete inhibition of Gβγ functions is close to 1:1.
Figure 7.
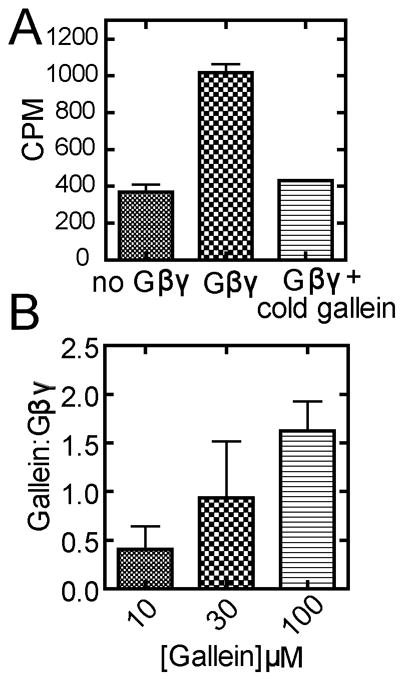
[3H]-Gallein binding to Gβγ. A) 1 μM Gβ1γ2 was mixed with 10 μM gallein (containing [3H]-gallein, 24,000 cpm) followed by incubation and gel filtration to remove unbound gallein as described in methods. Background [3H]-gallein eluting from the column in the absence of Gβγ was determined by loading [3H]-gallein without βγ added and was less that 2% of the total cpm loaded onto the column. Excess unlabeled “cold” gallein (1mM) in the presence of [3H]-gallein (24,000 cpm) was used to determine non-specific binding. Data shown is representative of 3 separate experiments. B) Gβγ was incubated with various concentrations of [3H]-gallein and the binding stoichiometry determined after subtracting background leak as determined in A. 10 and 30 μM concentrations were not statistically different from each other but 100 μM was statistically different from 10 μM p<0.05. Data are the combined results from 3 separate experiments performed in duplicate.
4. Discussion
We previously identified multiple small molecules that bind to G protein βγ subunits and inhibit Gβγ subunit functions. One class of these molecules in particular, gallein/M119, has been extensively tested by us and others in various in vitro and in vivo experiments that suggest specificity and efficacy of these molecules [1,2,5,10,19,22,25]. Despite this extensive data, detailed analysis of the biophysical mechanism for action of these compounds has not been reported. This is important because there are potential artifactual and non-specific mechanisms for small molecule inhibition of protein function and for this to be considered as a viable approach to therapeutics, non-specific reactivity or binding mechanisms have to be avoided. These non-specific mechanisms include reversible and irreversible covalent modification of the protein target by the small molecule, small molecule aggregation resulting in non-specific coating and inhibition of the protein target, redox modification of the protein as well as others. In fact we have reported that some small molecules reversibly modify Gβγ by formation of disulfides or mixed disulfides with a specific cysteine residue in the Gβγ hot spot [8]. Our previous data indicating selectivity of gallein and other small molecules for inhibition of different G protein effectors suggests the binding of gallein to Gβγ is not via these non-specific mechanisms [2]. Nevertheless, to further consider small molecule inhibition of Gβγ as a viable direction for drug development it is important that these issues be resolved with direct methods.
In this report we use several biophysical methods to show that gallein/M119 and two other classes of molecules bind directly to Gβγ. A surprising result from direct binding analysis of the different classes of Gβγ hot spot-binding small molecules was the remarkably slow kinetics of binding and dissociation of these compounds compared to the much faster kinetics of Gβγ hot spot-binding peptides (i.e. SIGK. Fig. 1.). The reason for this slow binding is not understood. One possibility is that the compounds bind to a single conformation explored by the Gβγ binding surface. It is widely thought that Gβγ does not undergo major conformational changes but recent NMR data suggests that the Gβγ hot spot is highly dynamic [37]. If the binding surface is exploring multiple conformations it could be that a specific minor conformation is required for small molecule binding. If adopting such a conformation were rate limiting for complex formation this could underlie a slow on-rate constant.
The slow dissociation of gallein and 372 from Gβγ in the SPR experiments suggested possible covalent interactions. The observation of binding in the presence of reducing agent and EDTA suggested that the molecules were not binding in a redox or metal ion-dependent manner. Analysis by intact protein mass spectrometry demonstrated that gallein could be completely dissociated from Gβγ at collision energies that maintain some intact Gβγ heterotrimer, and gallein and 372 could be completely stripped from Gβγ with a mixture of low concentration chaotropic agents. This strongly suggests that gallein and M119 bind in a reversible non-covalent manner.
The SPR binding data agree very closely with a simple bimolecular binding interaction model, and the major binding mode assessed by non-covalent mass spectrometry and [3H]-gallein binding at relevant concentrations was close to 1:1. Potential formation of ordered aggregates was tested by Dynamic Light Scattering analysis (DLS). None of the small molecules tested showed aggregate formation at levels measurable by DLS (data not shown). These compounds also do not appear as promiscuous hits for aggregators in commonly carried out screening experiments. Additionally the experiments were done in detergent conditions which have been shown to disrupt aggregates [9].
5. Conclusion
The key goal of this study was to establish that the various small molecules of interest directly interact with Gβγ in a reversible, specific and stoichiometric manner. The data presented strongly support this conclusion and along with recent studies in animal models suggest [5,25] that use of molecules such as those shown here represent viable starting points for development of lead molecules for therapeutics.
Supplementary Material
Highlights.
An SPR method is presented for examining small molecule binding to G protein βγ subunits.
SPR analysis demonstrates direct reversible binding of small molecules to Gβγ.
Rate constants for small molecule binding and dissociation are surprisingly slow.
mass spectrometry demonstrates a predominantly 1 :1, non-covalent binding mode for gallein
Radioligand binding supports a 1:1 binding mode of gallein to Gβγ.
Acknowledgments
This work was supported by the National Institutes of Health Institute of General Medicine [Grant 5R01GM081772]. The authors would like to thank Thomas Ryan, Chief Scientist Reichert Inc (Depew, NY) for his help in the preparation of Figure 2 and also guidance in carrying out SPR experiments in this text.
Footnotes
Non standard abbreviations: GPCR, G protein-coupled receptor; SIGK, peptide with sequence SIGKAFKILGYPDYD; Gβγ, Heterotrimeric G protein βγ subunits; DTT, Dithiothreitol; SPR, Surface Plasmon Resonance; ELISA, Enzyme Linked Immunosorbent Assay; PLC, Phospholipase C; GRK2, G protein-coupled receptor kinase 2; TBS, Tris-buffered Saline
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bianchi E, Norcini M, Smrcka A, Ghelardini C. Supraspinal Gβγ-dependent stimulation of PLCβ originating from G inhibitory protein-μ opioid receptor-coupling is necessary for morphine induced acute hyperalgesia. J Neurochem. 2009;111:171–180. doi: 10.1111/j.1471-4159.2009.06308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonacci TM, Mathews JL, Yuan C, Lehmann DM, Malik S, Wu D, Font JL, Bidlack JM, Smrcka AV. Differential Targeting of Gβγ-Subunit Signaling with Small Molecules. Science. 2006;312:443–446. doi: 10.1126/science.1120378. [DOI] [PubMed] [Google Scholar]
- 3.Bookout AL, Finney AE, Guo R, Peppel K, Koch WJ, Daaka Y. Targeting Gβγ Signaling to Inhibit Prostate Tumor Formation and Growth. J Biol Chem. 2003;278:37569–37573. doi: 10.1074/jbc.M306276200. [DOI] [PubMed] [Google Scholar]
- 4.Camps M, Hou C, Sidiropoulos D, Stock JB, Jakobs KH, Gierschik P. Stimulation of phospholipase C by G-protein βγ-subunits. Eur J Biochem. 1992;206:821–831. doi: 10.1111/j.1432-1033.1992.tb16990.x. [DOI] [PubMed] [Google Scholar]
- 5.Casey LM, Pistner AR, Belmonte SL, Migdalovich D, Stolpnik O, Nwakanma FE, Vorobiof G, Dunaevsky O, Matavel A, Lopes CM, Smrcka AV, Blaxall BC. Small molecule disruption of G βγ signaling inhibits the progression of heart failure. Circ Res. 2010;107:532–539. doi: 10.1161/CIRCRESAHA.110.217075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coan KED, Shoichet BK. Stoichiometry and Physical Chemistry of Promiscuous Aggregate-Based Inhibitors. J Am Chem Soc. 2008;130:9606–9612. doi: 10.1021/ja802977h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis TL, Bonacci TM, Sprang SR, Smrcka AV. Structural Definition of a Preferred Protein Interaction Site in the G protein β1γ2 heterodimer. Biochem. 2005;44:10593–10604. doi: 10.1021/bi050655i. [DOI] [PubMed] [Google Scholar]
- 8.Dessal AL, Prades R, Giralt E, Smrcka AV. Rational Design of a Selective Covalent Modifier of G Protein βγ Subunits. Mol Pharmacol. 2011;79:24–33. doi: 10.1124/mol.110.068155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nat Protoc. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Marcos M, Ghosh P, Farquhar MG. GIV is a nonreceptor GEF for Gαi with a unique motif that regulates Akt signaling. Proc Natl Acad Sci U S A. 2009;106:3178–3183. doi: 10.1073/pnas.0900294106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gesellchen F, Zimmermann B, Herberg FW. Direct optical detection of protein-ligand interactions. Methods Mol Biol. 2005;305 doi: 10.1385/1-59259-912-5:017. [DOI] [PubMed] [Google Scholar]
- 12.Gilman AG. G proteins: transducers of receptor-generated signals. Ann Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 13.Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, Meek S, Smith AJH, Okkenhaug K, Vanhaesebroeck B. The p110β isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110γ. Proc Natl Acad Sci U S A. 2008;105:8292–8297. doi: 10.1073/pnas.0707761105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirsch E, Katanaev VL, Garlanda C, Azzolino O, Pirola L, Silengo L, Sozzani S, Mantovani A, Altruda F, Wymann MP. Central Role for G Protein-Coupled Phosphoinositide 3-Kinase γ in Inflammation. Science. 2000;287:1049–1053. doi: 10.1126/science.287.5455.1049. [DOI] [PubMed] [Google Scholar]
- 15.Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Adv Second Messenger Phosphoprotein Res. 1999;33:131–151. doi: 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- 16.Irannejad R, Wedegaertner PB. Regulation of constitutive cargo transport from the trans-Golgi network to plasma membrane by Golgi-localized G protein βγ subunits. J Biol Chem. 2010;285:32393–32404. doi: 10.1074/jbc.M110.154963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karlsson R, Katsamba PS, Nordin H, Pol E, Myszka DG. Analyzing a kinetic titration series using affinity biosensors. Anal Biochem. 2006;349:136–147. doi: 10.1016/j.ab.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 18.Karlsson R, Kullman-Magnusson M, Hamalainen MD, Remaeus A, Andersson K, Borg P, Gyzander E, Deinum J. Biosensor analysis of drug-target interactions: direct and competitive binding assays for investigation of interactions between thrombin and thrombin inhibitors. Anal Biochem. 2000;278:1–13. doi: 10.1006/abio.1999.4406. [DOI] [PubMed] [Google Scholar]
- 19.Kirui JK, Xie Y, Wolff DW, Jiang H, Abel PW, Tu Y. Gβγ signaling promotes breast cancer cell migration and invasion. J Pharmacol Exp Ther. 2010;333 doi: 10.1124/jpet.109.164814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koch WJ, Rockman HA, Samama P, Hamilton R, Bond RA, Milano CA, Lefkowitz RJ. Cardiac function in mice overexpressing the β-adrenergic receptor kinase or a βARK inhibitor. Science. 1995;268:1350–1353. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- 21.Kozasa T, Gilman AG. Purification of recombinant G proteins from Sf9 cells by hexahistidine tagging of associated subunits. Characterization of α12 and inhibition of adenylyl cyclase by αz. J Biol Chem. 1995;270:1734–1741. doi: 10.1074/jbc.270.4.1734. [DOI] [PubMed] [Google Scholar]
- 22.Lehmann DM, Seneviratne AMPB, Smrcka AV. Small Molecule Disruption of G Protein βγ Subunit Signaling Inhibits Neutrophil Chemotaxis and Inflammation. Mol Pharmacol. 2008;73:410–418. doi: 10.1124/mol.107.041780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLCβ-2 and -3 and PI3Kγ in Chemoattractant-Mediated Signal Transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- 24.Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. The βγ subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- 25.Mathews JL, Smrcka AV, Bidlack JM. A Novel Gβγ-Subunit Inhibitor Selectively Modulates μ-Opioid-Dependent Antinociception and Attenuates Acute Morphine-Induced Antinociceptive Tolerance and Dependence. J Neurosci. 2008;28:12183–12189. doi: 10.1523/JNEUROSCI.2326-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mullett WM, Lai EP, Yeung JM. Surface plasmon resonance-based immunoassays. Methods. 2000;22 doi: 10.1006/meth.2000.1039. [DOI] [PubMed] [Google Scholar]
- 27.Nakajima Y, Nakajima S, Kozasa T. Activation of G protein-coupled inward rectifier K+ channels in brain neurons requires association of G protein βγ subunits with cell membrane. FEBS Lett. 1996;390:217–220. doi: 10.1016/0014-5793(96)00661-8. [DOI] [PubMed] [Google Scholar]
- 28.Papalia GA, Leavitt S, Bynum MA, Katsamba PS, Wilton R, Qiu H, Steukers M, Wang S, Bindu L, Phogat S, Giannetti AM, Ryan TE, Pudlak VA, Matusiewicz K, Michelson KM, Nowakowski A, Pham-Baginski A, Brooks J, Tieman BC, Bruce BD, Vaughn M, Baksh M, Cho YH, Wit MD, Smets A, Vandersmissen J, Michiels L, Myszka DG. Comparative analysis of 10 small molecules binding to carbonic anhydrase II by different investigators using Biacore technology. Anal Biochem. 2006;359 doi: 10.1016/j.ab.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 29.Park D, Jhon D-Y, Lee C-W, Lee K-H, Goo Rhee S. Activation of phospholipase C isozymes by G protein βγ subunits. J Biol Chem. 1993;268:4573–4576. [PubMed] [Google Scholar]
- 30.Pitcher JA, Inglese J, Higgins JB, Arriza JL, Casey PJ, Kim C, Benovic JL, Kwatra MM, Caron MG, Lefkowitz RJ. Role of βγ subunits of G proteins in targeting the β-adrenergic receptor kinase to membrane-bound receptors. Science. 1992;257:1264–1267. doi: 10.1126/science.1325672. [DOI] [PubMed] [Google Scholar]
- 31.Rebois RV, Schuck P, Northup JK. Elucidating kinetic and thermodynamic constants for interaction of G protein subunits and receptors by surface plasmon resonance spectroscopy. Methods Enzymol. 2002;344:15–42. doi: 10.1016/s0076-6879(02)44703-9. [DOI] [PubMed] [Google Scholar]
- 32.Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross J, Jr, Lefkowitz RJ, Koch WJ. Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci U S A. 1998;95:7000–7005. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roden LD, Myszka DG. Global analysis of a macromolecular interaction measured on BIAcore. Biochem Biophys Res Commun. 1996;225:1073–1077. doi: 10.1006/bbrc.1996.1297. [DOI] [PubMed] [Google Scholar]
- 34.Schuck P. Reliable determination of binding affinity and kinetics using surface plasmon resonance biosensors. Curr Opin Biotechnol. 1997;8 doi: 10.1016/s0958-1669(97)80074-2. [DOI] [PubMed] [Google Scholar]
- 35.Scott JK, Huang SF, Gangadhar BP, Samoriski GM, Clapp P, Gross RA, Taussig R, Smrcka AV. Evidence that a protein-protein interaction ‘hot spot’ on heterotrimeric G protein βγ subunits is used for recognition of a subclass of effectors. EMBO J. 2001;20:767–776. doi: 10.1093/emboj/20.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharon M, Robinson CV. The role of mass spectrometry in structure elucidation of dynamic protein complexes. Annu Rev Biochem. 2007;76:167–193. doi: 10.1146/annurev.biochem.76.061005.090816. [DOI] [PubMed] [Google Scholar]
- 37.Smrcka AV, Kichik N, Tarrago T, Burroughs M, Park MS, Itoga NK, Stern HA, Willardson BM, Giralt E. NMR analysis of G-protein βγ subunit complexes reveals a dynamic G(α)-Gβγ subunit interface and multiple protein recognition modes. Proc Natl Acad Sci U S A. 2010;107:639–644. doi: 10.1073/pnas.0909503107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smrcka AV, Lehmann DM, Dessal AL. G protein betagamma subunits as targets for small molecule therapeutic development. Comb Chem High Throughput Screen. 2008;11:382–395. doi: 10.2174/138620708784534761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smrcka AV, Sternweis PC. Regulation of purified subtypes of phosphatidylinositol specific phospholipase C β by G protein α and βγ subunits. J Biol Chem. 1993;268:9667–9674. [PubMed] [Google Scholar]
- 40.Stenlund P, Frostell-Karlsson A, Karlsson OP. Studies of small molecule interactions with protein phosphatases using biosensor technology. Anal Biochem. 2006;353 doi: 10.1016/j.ab.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 41.Stephens L, Smrcka A, Cooke FT, Jackson TR, Sternweis PC, Hawkins PT. A novel, phosphoinositide 3-kinase activity in myeloid-derived cells is activated by G-protein βγ-subunits. Cell. 1994;77:83–93. doi: 10.1016/0092-8674(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 42.Stephens LR, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, Smrcka AV, Thelen M, Cadwallader K, Tempst P, Hawkins PT. The Gβγ Sensitivity of a PI3K is Dependent upon a Tightly Associated Adaptor, p101. Cell. 1997;89:105–114. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 43.Wu L, Huang MH, Zhao JL, Yang MS. Study of MMLV RT- binding with DNA using surface plasmon resonance biosensor. Acta Biochim Biophys Sin (Shanghai) 2005;37:634–642. doi: 10.1111/j.1745-7270.2005.00088.x. [DOI] [PubMed] [Google Scholar]
- 44.Xie W, Samoriski GM, McLaughlin JP, Romoser V, Smrcka A, Hinkle PM, Bidlack JM, Gross RA, Jiang H, Wu D. Genetic alteration of phospholipase Cβ3 expression modulates behavioral and cellular responses to μ opioids. Proc Natl Acad Sci USA. 1999;96:10385–10390. doi: 10.1073/pnas.96.18.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao L, Fan P, Jiang Z, Mailliard WS, Gordon AS, Diamond I. Addicting drugs utilize a synergistic molecular mechanism in common requiring adenosine and Gi-βγ dimers. Proc Natl Acad Sci U S A. 2003;100:14379–14384. doi: 10.1073/pnas.2336093100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao T, Nalbant P, Hoshino M, Dong X, Wu D, Bokoch GM. Signaling requirements for translocation of P-Rex1, a key Rac2 exchange factor involved in chemoattractant-stimulated human neutrophil function. J Leukoc Biol. 2007;81:1127–1136. doi: 10.1189/jlb.0406251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.