

Abstract
The study of reactive oxygen species (ROS) and oxidative stress remains a very active area of biological research particularly in relation to cellular signaling and the role of ROS in disease. In the cerebral circulation, oxidative stress occurs in diverse forms of disease and with aging. Within the vessel wall, ROS produce complex structural and functional changes that have broad implications for regulation of cerebral perfusion and permeability of the blood–brain barrier. These oxidative stress-induced changes are thought to contribute to the progression of cerebrovascular disease. Here, we highlight recent findings in relation to oxidative stress in the cerebral vasculature, with an emphasis on the emerging role for NADPH oxidases as a source of ROS and the role of ROS in models of disease.
Introduction
The roles of reactive oxygen species (ROS) and oxidative stress continue to be a very active area of research in vascular biology, with broad implications in both health and disease. In general, low concentrations of ROS function as mediators and modulators of cell signaling (1, 2). By contrast, higher levels of ROS commonly contribute to vascular disease (2–4). Oxidative stress can be broadly defined as an imbalance between generation of ROS, and degradation or metabolism of ROS by the various antioxidant defense mechanisms, leading to excessive levels of ROS. The overall effects of ROS depend on local concentrations, subcellular localization, and the proximity of ROS to other target molecules.
Cerebral blood vessels are relatively more difficult to study and historically have received less attention from researchers in vascular biology. Thus, our overall understanding of the importance of ROS and oxidative stress in the cerebral circulation lags substantially behind work on vascular cells outside of the brain. Recently there has been a focus into this area of study however. This review highlights some recent findings on the role of oxidative stress in the cerebral vasculature, with an emphasis on NADPH oxidases (Nox enzymes) as a source of ROS. We will summarize evidence that ROS contribute to complex structural and functional changes within the vessel wall in the brain. Some of these changes might contribute to mechanisms of vascular protection, but many more could underlie and promote the progression of vascular disease.
What chemical species are involved?
The study and impact of redox signaling and oxidative-related mechanisms is complex in part because there are multiple, highly interactive chemical species and mechanisms involved (4, 5) (Figure 1). The ROS that are generally of most interest in the present context include the superoxide radical (O2−), hydroxyl radical (OH.) and hydrogen peroxide (H2O2). In addition, a closely related reactive nitrogen species (RNS) – peroxynitrite – also exerts important effects in the vasculature (5).
Figure 1.
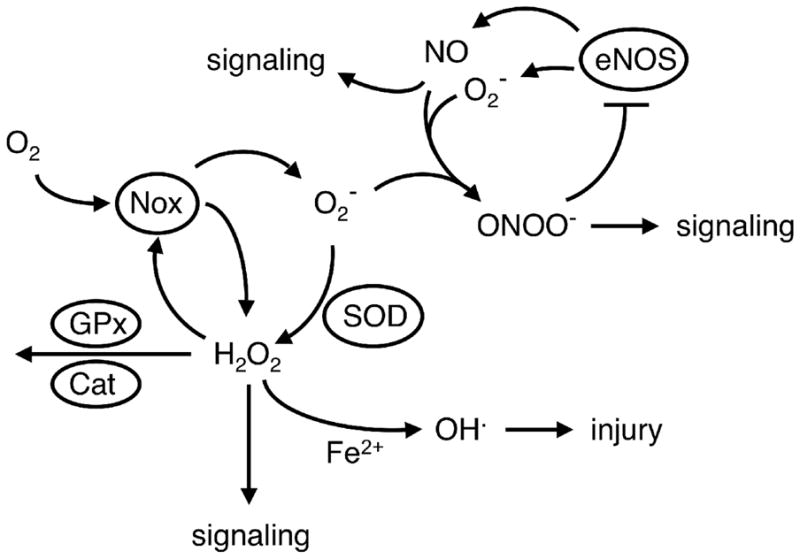
Schematic illustration of the interrelationships of various ROS and NO. NO produced by endothelial eNOS is normally a major signaling molecule. Superoxide (O2−) is produced from molecular oxygen (O2) by a variety of sources including NADPH oxidases (Nox). Once formed, superoxide can directly produce injury (not shown), can be converted by SOD into H2O2 or can react with NO to form peroxynitrite (ONOO−). The NADPH oxidase containing Nox4 can produce H2O2 directly. H2O2 is an important signaling molecule. In combination with Fe2+,. H2O2 can form the hydroxyl radical (OH·), a highly reactive ROS that causes cellular injury. H2O2 can also be degraded by glutathione peroxidase (GPx) or catalase (Cat). In addition, peroxynitrite can produce further increases in superoxide and oxidative stress by inhibiting the activity of SOD (not shown) or by oxidizing tetrahydrobiopterin, resulting in an uncoupling of eNOS where eNOS produces superoxide instead of NO (see text for further detail). Superoxide can also react with arachidonic acid and produce isoprostanes (not shown).
Superoxide can have direct effects, such as reacting with enzymes containing iron–sulphur centers (aconitase in mitochondria, for example) resulting in release of free iron and subsequent formation of the highly reactive hydroxyl radical (4, 5). In addition, the formation of multiple other biochemical species is dependent on production of superoxide, including H2O2, peroxynitrite and lipid-peroxidation products known as isoprostanes (4–7) (Figure 1). Superoxide dismutases (SOD) convert superoxide to H2O2. Nitric oxide (NO) reacts extremely efficiently with superoxide to form peroxynitrite, whereas isoprostanes are formed by the interaction of arachidonic acid and superoxide. In addition to being considered reliable markers of oxidative stress, both isoprostanes and peroxynitrite have direct effects on vascular cells (5, 6).
How common is oxidative stress in the vasculature?
All vascular cells have the potential to produce ROS. In many models of disease and aging, there is evidence that levels of ROS and/or RNS are increased and functionally important in cerebral blood vessels (Table 1). Thus, oxidative stress in the vasculature appears to be a common feature in diverse models of cerebral vascular disease and injury. As will be disussed below, oxidative stress very often negatively impacts the vessel wall. Thus, these findings have broad implications in relation to mechanisms that regulate cerebral blood flow and control permeability of the blood-–brain barrier. Importantly, many of these findings were obtained using models of major risk factors for vascular disease, stroke and cognitive decline such as hypertension and aging, indicating that the impact on brain function and viability is potentially very great.
Table 1.
Experimental models with oxidative stress in the cerebral circulation.
| Model | Phenotype | References |
|---|---|---|
| acute hypertension | ↓EDR | 8 |
| chronic hypertension | ↑ROS, ↑Nox, ↓EDR, ↓NVC hypertrophy | 9, 25, 26, 38, 60 |
| hyperhomocysteinemia | ↑ROS, ↓EDR, hypertrophy | 23, 30 |
| insulin resistance | ↑ROS, ↑Nox, ↓EDR ↓K+ channel-induced vasodilation |
13, 28 |
| hypercholesterolemia | ↑ROS ↑blood cell-vessel interactions |
69 |
| diabetes/metabolic syndrome | ↑ROS, ↑Nox, ↓EDR ↓hypoxia-induced vasodilation ↓glutamate-induced vasodilation |
14, 29, 35, 64, 86 |
| inflammation | ↑ROS, ↓EDR, | 18, 54 |
| hypoxia | ↑ROS, ↓EDR, | 87 |
| sickle cell disease | ↑blood cell-vessel interactions | 70 |
| subarachnoid hemorrhage | ↑ROS, ↑Nox, vasospasm | 39, 40, 66 |
| ischemia with reperfusion | ↓EDR, ↓vasoconstriction | 24 |
| meningitis | vasodilation | 88 |
| traumatic brain injury | ↓EDR, ↓vasoconstriction | 27 |
| Alzheimer’s disease | ↑ROS, ↓EDR, ↓NVC | 20, 21 |
| aging | ↑ROS, ↑Nox, ↓EDR, ↓NVC, | 12, 19 |
| nicotine | ↓EDR | 10 |
| alcohol | ↑ROS, ↓EDR, ↓K+ channel-induced vasodilation | 22, 36 |
| methionine synthase deficiency | ↑ROS, ↓EDR | 31 |
| SOD deficiency | ↑ROS, ↓EDR, hypertrophy ↑vasoconstriction |
11, 15, 16 |
| PPARγ interference | ↑ROS, ↓EDR, hypertrophy, | 17 |
EDR = endothelium-dependent relaxation
NVC = neurovascular coupling
ROS = reactive oxygen species in the vasculature
Nox = activity or expression of NADPH oxidase
What is the impact of oxidative stress?
Superoxide
The effects of superoxide in vascular cells are multifaceted. For example, in relation to regulation of vascular tone, superoxide can produce vasodilation or vasoconstriction (6). Superoxide may increase vascular tone by direct effects on vascular muscle or by interference with vasodilator mechanisms, including by the interaction with NO, a potent vasodilator. NO reacts with superoxide at a rate faster than the dismutation of superoxide by SOD enzymes (4, 5). Thus, the local concentration of superoxide is a key determinant of the biological half-life of NO (4, 5). The reaction of NO with superoxide results in the loss of normal NO-mediated signaling (Figure 1). As NO plays a major role in cerebral vascular biology, the loss of NO bioavailability has far-reaching implications.
The wall of larger blood vessels consists of three major components: an inner endothelium, a thicker middle layer comprised mainly of vascular muscle, and an outer adventitia which includes perivascular nerves and fibroblasts. In almost all the models shown in the Table, superoxide-mediated vascular dysfunction has been described, with many of these studies focusing on regulation of vascular tone by means of endothelium-dependent mechanisms (8–31). This same basic mechanism of dysfunction has been observed in human cerebral arteries (32).
Blood vessels are constitutively exposed to NO from two sources – endothelial and neuronal isoforms of NO synthase (eNOS and nNOS, repectively). A third source of NO is inducible NO-synthase (iNOS) which is not expressed normally but can be expressed in all vascular cells in response to a variety of stimuli (primarily proinflammatory stimuli). NO produced by eNOS is the major endothelium-derived relaxing factor in the cerebral circulation (33, 34). The impact of endothelial dysfunction in brain is particularly far reaching as eNOS-derived NO affects vascular muscle and blood elements, but also neuronal signaling and neurogenesis (34). Impairment of endothelial function and NO-signaling may contribute to cognitive decline (21).
In addition to responses that are dependent on endothelial cells, other vasodilator mechanisms are inhibited by superoxide (or other ROS). These include vascular responses to glutamate (35), activators of potassium channels (13, 28, 36), hypercapnia (23), hypoxia (29), reductions in arterial pressure (37), and neurovascular coupling (19–21, 25, 38). Vasoconstrictor responses, which may or may not be modulated by endothelium, can be impaired (24, 27) or augmented by ROS (11). Superoxide-mediated constriction of cerebral blood vessels has been described in models of vasospasm (39, 40).
The consequences of oxidative stress extend beyond the regulation of vascular tone. In both in vivo and in vitro models, superoxide or other ROS increase permeability of the blood–brain barrier (BBB) (41–43). For example, levels of superoxide, activation of matrix metalloproteinases, and increases in permeability of the BBB to large molecules are augmented in SOD-deficient mice following ischemia with reperfusion (42, 44).
In blood vessels outside the brain, many studies have shown that ROS affect vascular structure or growth (2). Angiotensin-II-induced oxidative stress, via activation of NADPH oxidase and potentially other sources of ROS (3), is thought to be a key mediator of the wall thickening known as vascular hypertrophy in some forms of hypertension (3). Similarly, hypertrophy of cerebral arterioles occurs in several forms of disease, including models of hypertension (Figure 2). Studies of SOD-deficient animals, which would be predicted to exhibit increased levels of superoxide, provide a relatively simple model to evaluate the impact of ROS on the structure of cerebral blood vessels. Genetic deficiency in the CuZn isoform of SOD produces increases in vascular superoxide, impaired NO-mediated regulation of vascular tone (due to the very efficient reaction between superoxide and NO), and hypertrophy of cerebral arterioles in the absence of increases in arterial pressure (15, 16). The effect on vascular structure is especially prominent as a deficiency in CuZn-SOD produced more vascular hypertrophy than that observed in models of hypertension, NOS deficiency or hyperhomocysteinemia (16, 17, 45–47). ROS might produce hypertrophy by inactivation of NO [which normally inhibits vascular growth (46)], or through direct activation of signaling cascades involved in growth of vascular muscle including growth factors, kinases, and transcription factors. These mechanisms have been reviewed previously (2, 3). Such structural changes may have functional consequences because vascular hypertrophy can encroach on the vessel lumen and impair maximal vasodilator capacity by increasing vascular resistance.
Figure 2.
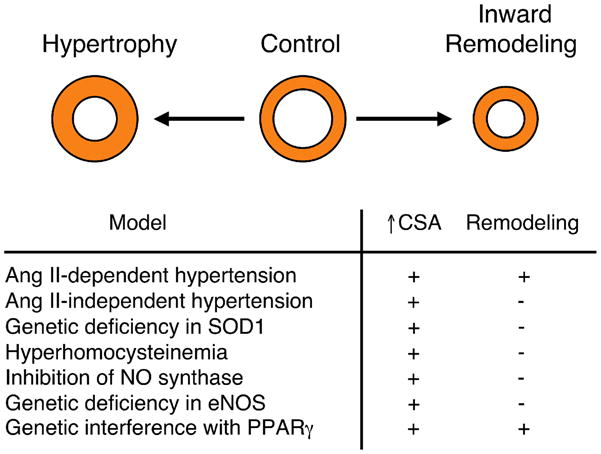
Schematic of structural changes in cerebral arterioles shown in cross-section. During disease or in response to genetic manipulation, vessels can undergo hypertrophy [increase cross-sectional area (CSA)] and/or exhibit remodeling (inward or outward changes in diameter). During angiotensin-II-dependent hypertension and in response to interference with PPARγ function (following expression of a dominant-negative variant in PPARγ), both hypertrophy and inward remodeling occur. By contrast, hypertrophy occurs in the absence of inward remodeling in models of angiotensin-II-independent hypertension (BPH-2 mice, a genetic model of hypertension thought to be renin-independent), hyperhomocysteinemia, in response to treatment with a NOS inhibitor (L-NAME), or genetic deficiency in eNOS or CuZn-SOD (SOD1). See text for additional details. Illustration based on data from Refs (16, 17, 45–47).
In addition to producing hypertrophy, angiotensin-II-dependent hypertension is associated with inward remodeling in microvessels that supply the brain (45). Inward vascular remodeling represents a rearrangement of the vessel wall resulting in a smaller lumen Figure 2). It is noteworthy that, although CuZn-SOD deficiency produced marked hypertrophy of cerebral arterioles, inward remodeling was not present in the same blood vessels (16). These findings and others (45) suggest that mechanisms that produce vascular hypertrophy differ from those that produce inward vascular remodeling. While increased superoxide can produce vascular hypertrophy (Figure 2), increases in superoxide might not be sufficient to produce inward vascular remodeling in the absence of angiotensin II, increased blood pressure, or other factors. Thus, through both direct and indirect effects, superoxide may produce diverse effects on vascular structure and regulation of vascular tone.
Hydrogen peroxide
Compared with superoxide, H2O2 is chemically more stable and diffuses much more readily across cell membranes (7). Thus, H2O2 is thought to have greater importance as a signaling molecule (7) (Figure 1). In cerebral blood vessels, H2O2 is usually a vasodilator acting directly on vascular muscle (6). H2O2 mediates vascular responses to varied stimuli, including select endothelium-dependent agonists and arachidonic acid, and seems to function in vessels as one of a family of endothelium-derived hyperpolarizing factors (4, 6, 48). H2O2 may also mediate increases in cerebral blood flow during meningitis (49).
In addition to directly altering vascular tone, H2O2 can impair vascular function, perhaps after its conversion to hydroxyl radicals (7). Endothelial dysfunction in human cerebral arteries in response to activated neutrophils is mediated by H2O2 (32). H2O2 can stimulate NADPH oxidase in vascular cells (Figure 1)and thus further increase levels of superoxide (5, 7) (Figure 1). These increases in superoxide might activate a feed-forward mechanism that further amplifies oxidative stress (7) (Figure 1).
Many studies support the concept that NO produced by eNOS affects basal tone and mediates responses to endothelium-dependent stimuli in the cerebral circulation (33, 34). By contrast, a condition known as eNOS ‘uncoupling’ may occur in which the normal flow of electrons within the enzyme is diverted such that superoxide rather than NO is produced (3, 4) A recent study suggested that eNOS is uncoupled under normal conditions in cerebral arteries and that H2O2 (produced from superoxide by SOD) is the mediator of endothelium-dependent relaxation (50). The overall importance of this latter finding and mechanism is difficult to assess, however, as results from many other studies suggest that the responses of cerebral blood vessels in several species to endothelium-dependent agonists are mediated by NO and not by ROS (see 24, 33, 51, 52 for examples). Uncoupled eNOS might contribute to oxidative stress in vascular disease (3) (Figure 1), but there has been little evidence to date showing that this form of dysfunction occurs under otherwise-normal conditions in vivo.
Peroxynitrite
The interaction of NO with superoxide results in the formation of peroxynitrite, which can affect cerebral vascular tone and alter responses to other vasoactive stimuli (6, 53). Peroxynitrite is not simply a marker of oxidative stress but on its own can produce cellular dysfunction and injury (5). For example, peroxynitrite reduces the activity of prostacyclin synthase, soluble guanylate cyclase and the mitochondrial isoform of SOD (Mn-SOD) (4, 5). Peroxynitrite activates the nuclear enzyme poly (ADP-ribose) polymerase (PARP-1) and increases expression of iNOS – additional mediators of vascular and cellular dysfunction (4, 5). Peroxynitrite also oxidizes tetrahydrobiopterin (a crucial enzyme cofactor for eNOS), thus promoting the uncoupling of eNOS, Because of its ability to inactivate Mn-SOD, oxidize tetrahydrobiopterin and uncouple NOS, peroxynitrite might further amplify oxidative stress (Figure 1). In addition to superoxide (discussed above), peroxynitrite might contribute to impairment of neurovascular coupling and endothelial function in response to angiotensin II (25) and thus may play a role in hypertension-induced effects on the vasculature. Although detrimental effects are evident (6, 25, 53), peroxynitrite may also have protective effects on the vasculature such as contributing to preconditioning-based mechanisms (54).
NADPH oxidase as a source of superoxide
NADPH oxidase is best described in phagocytes, where it comprises a membrane-bound flavocytochrome b558 [formed by p22phox and gp91phox [also referred to as Nox2)], up to three cytosolic subunits, p47phox, p67phox and p40phox, and the small G-proteins Rac1, or Rac2, and Rap1A. Once activated, the cytosolic components translocate to the plasma membrane, where they associate with the membrane-bound subunits, thus enabling electron transfer from the enzyme complex to molecular O2 and generation of superoxide (2, 3, 55). Glucose-6-phosphate dehydrogenase is a major source of the substrate NADPH within the cytosol. By contrast, although the understanding of the precise molecular makeup of the vascular NADPH oxidase is incomplete, all subunits of the phagocytic NADPH oxidase are present in vascular cells (2, 3, 56, 57). The catalytic domain of NADPH oxidase resides in the Nox2 subunit. There are several homologs of Nox2, of which Nox1, Nox4 and Nox5 appear most important for vascular cells (3) (Figure 3). Although Nox4 is expressed in all vascular cells (2), it does not require the presence of cytosolic subunits for activation (58) (Figure 3), implying unique regulatory mechanisms relative to some other Nox isoforms (59). Nox4 might also predominantly produce H2O2, rather than superoxide (59) (Figures 1 and 3). The expression of these isoforms, including their subcellular distribition, varies according to function, cell type, and tissue (2). There is much less known about the expression pattern of Nox’s in the cerebral circulation versus the systemic vasculature.
Figure 3.

Schematic representation of components of the vascular NADPH oxidases (based on Refs 2, 55–59). Nox1 is activated by homologs ofp47phox and p67phox – Nox-organizer 1 (NoxO1) and Nox-activator 1 (NoxA1). Nox2 requires recruitment ofp47phox and p67phox. Nox4 does not appear to require any cytosolic subunits, and Nox5 is activated by Ca2+. Nox1, Nox2 and Nox4 require association with p22phox to function normally. Nox4 might produce H2O2 directly (58). The subcellular localization of Nox isoforms and the site of superoxide generation varies with cell type and presumably function (2). While some isoforms are expressed in the cell membrane, additional sites of expression include the nucleus (or perinuclear) and the endoplasmic reticulum.
NADPH oxidase in the cerebral vasculature
Expression of components of NADPH oxidase has been reported in cerebral blood vessels at the level of both mRNA (Nox1, Nox2, Nox4, p22phox, p47phox and p67phox) and protein (Nox1, Nox2 and Nox4) (13, 26, 38, 60–63). At the cellular level, immunoreactivity for Nox2 was described in endothelium and adventitia of cerebral arterioles as well as in in vitro cultured endothelium from the basilar artery (where mRNA for p47phox and p67phox was also detected) (63). Nox4 mRNA expression was more abundant than expression of Nox2 or Nox1 (63). In the basilar artery, Nox1 protein levels were greater in endothelial and adventitial cells than in smooth muscle (63).
Compared to systemic blood vessels, there may be unique aspects with regard to Nox expression in the cerebral vasculature. Levels of Nox1 and Nox4 mRNA are higher than levels of Nox2 mRNA in cerebral endothelium (63), and Nox4 protein levels are greater in the basilar artery than in several extracranial blood vessels (61). Male rats express greater levels of Nox1 and Nox4 protein in the basilar artery than do female rats (62).
Changes in expression of various NADPH oxidase subunits in the brain vasculature have been described in several disease models. These changes include increases in subunit expression with hypertension (60), insulin resistance (13), diabetes (14, 64) and aging (12), as well as in response to ischemia (65), subarachnoid hemorrhage (66), salt-loading (67) and isoprostanes (67). Thus, changes in expression of components of NADPH oxidase occur under a variety of disease conditions and would be expected to contribute to increased superoxide levels.
How functionally important are NADPH oxidases? Many of the early studies examining the role of NADPH oxidase in the cerebral circulation used the pharmacological inhibitors apocynin and diphenyleneiodonium (DPI) (see Refs 10, 12, 14, 22, 41, 60, 62, 64, 66, 67 for examples). However, neither of the compounds is selective for NADPH oxidase. DPI affects other flavin-containing enzymes (some of which are potential sources of ROS) (58). Apocynin is thought to inhibit NADPH oxidase assembly (58), but might have other antioxidant effects and is reportedly not a specific inhibitor of NADPH oxidase in vascular cells (68). Thus, although the use of these compounds has frequently implicated NADPH oxidase as a key source of superoxide, these studies also have potential limitations. Nox2-deficient mice (19–21, 25, 26, 38, 41, 69, 70) and a gp91ds-tat peptide (19–21, 26, 38) that inhibits the enzyme complex (58) have been used as more-selective approaches to define the role of NADPH oxidase. The gp91ds-tat peptide affects the interaction of Nox2 with p47phox and thus inhibits assembly and activation of the oxidase complex (58). Based on sequence homology and site of action, this peptide inhibitor might also affect Nox1, but it is unlikely to affect Nox4 or Nox5 (58). Although not widely used to date, results obtained with the gp91ds-tat peptide in cerebral circulation have generally been similar to those seen in the Nox2-deficient mouse (19–21, 26, 38). Overall, this work suggests that Nox2 is a key catalytic subunit in relation to superoxide-mediated vascular dysfunction in brain in models of hypertension, Alzheimer’s disease, sickle cell disease, hypercholesterolemia and aging (19–21, 26, 38, 69, 70). Preliminary studies suggest that hypertrophy of cerebral arterioles during hypertension is also dependent on expression of Nox2 (71).
It is noteworthy that, although Nox2 might be expressed at lower levels than either Nox1 or Nox4 in vascular cells in the cerebral circulation (63), mice that are deficient in expression of Nox2 are protected from diverse stimuli that produce vascular dysfunction (see above). For example, angiotensin II does not produce endothelial dysfunction in carotid artery or cerebral blood vessels in Nox2-deficient mice (26, 38, 72, and S.C. and F.M.F., unpublished). To date, we are unaware of studies testing the functional importance of Nox1, Nox4 or Nox5 in the cerebral circulation. Considering how commonly such experimental studies are performed using rodents, it is important to recognize that while Nox5 is expressed in humans, rats and mice lack the gene for Nox5 (73). The observations that Nox4 protein expression is relatively high in cerebral arteries (61), including in males (62) and during hypertension (60), suggest that this isoform might be especially important in this vascular bed. Considering the relative higher levels of expression of Nox1 and Nox4 in cerebral vascular cells noted above (63), it will be important to define the importance of these Nox subunits in relation to vascular function and vascular growth in brain.
NADPH oxidase versus other sources of ROS
Although there has been increasing focus on defining the role of NADPH oxidases in cerebral circulation, there is still little known regarding the relative importance of this enzyme complex. While the discussion here has dealt mainly with NADPH oxidase, there are multiple potential sources of superoxide, including mitochondria, cyclooxygenases (COX), uncoupled NOS, among others (2–4). Mitochondria generate superoxide as a side product of oxidative phosphorylation and might be a particularly important source of superoxide in brain vasculature as the content of this organelle in cerebral endothelium is much greater than in other cells (74). COX has been shown repeatedly to be an important source of superoxide in brain and cerebral circulation. For example, although arachidonic-acid-induced superoxide production in aortic vascular muscle is dependent on NADPH oxidase (75), increases in superoxide and changes in tone in cerebral vessels in response to arachidonic acid are dependent on the activity of COX (48, 76). A major unanswered question is in defining what the relative importance of NADPH oxidase, mitochondria, COX, and other enzymes are as sources of ROS in the cerebral circulation. To what extent do these mechanisms interact? There are examples in cerebral blood vessels where both NADPH oxidase and COX are implicated as key mediators of dysfunction in the same model (64, 77). Because NADPH-oxidase-derived ROS might regulate expression of COX (78), and COX activity might be a determinant of NADPH oxidase expression (79), these underlying mechanisms might be more complex than was initially appreciated.
Vascular protection
Considering the major role for ROS and the emerging role for NADPH oxidase in cerebral vascular disease, it is logical to predict that interventions that inhibit expression or activity of the vascular NADPH oxidase might be beneficial for cerebrovascular disease. Effective and selective targeting of ROS sources such as NADPH oxidase might help in delaying or halting the progression of vascular disease in the brain. At present, few specific inhibitors of NADPH oxidase are available, and agents that do have some selectivity, such as the gp91ds-tat peptide (58), might be difficult to deliver chronically to desired sites of action in the brain vasculature. Because NADPH oxidase is expressed in many cells types and is a key player in the immune system, inhibitors that specifically target vascular cells should be particularly useful but also much more difficult to develop. Because angiotensin II is a prominent activator of NADPH oxidase (2, 3), drugs that affect the renin–angiotensin system can be effective regulators of NADPH oxidases. Inhibitors of angiotensin converting enzyme (ACE, which converts angiotensin I into the more active angiotensin II form) and inhibitors of angiotensin II receptors (AT1 receptors) have beneficial effects in cerebral blood vessels in patients with cerebral vascular disease and in various experimental models (see [80] for an example), but it is not known to what extent these changes are mediated through effects on NADPH oxidase.
There are agents that, while designed and utilized primarily for other purposes, might affect NADPH oxidase through pleiotropic effects. Examples of this are the ‘statin’ inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMDH) and activators of the transcription factor peroxisome proliferator activated receptor-γ (PPARγ/PPARG). Taking each of these in turn, in cerebral blood vessels, statin treatment reduced basal superoxide, activity of NADPH oxidase and improved vascular function in a model of insulin resistance (13). Furthermore, a statin protected the BBB from glutamate-induced oxidative stress and disruption that appeared to be mediated by NADPH oxidase (81).
Regarding the second class of agents, activation of PPARγ decreases expression of NADPH oxidase and AT1 receptors in vascular cells outside the brain (82, 83). However, in cerebral blood vessels, interference with PPARγ signaling (through genetic expression of a dominant-negative variant of PPARγ) increases superoxide, impairs endothelial function and causes vascular hypertrophy and inward remodeling (Table) (17). Thus, it might be anticipated that activation of PPARγ with synthetic agonists would have beneficial vascular effects. Indeed, PPARγ agonists reduce superoxide and improve endothelial function in carotid artery in hypertensive mice and rats (84, 85). These agents also reduce superoxide and activity of NADPH oxidase in brain and prevent part of the vascular remodeling that occurs during hypertension (85).
Concluding remarks
In summary, ROS play a major and diverse role in vascular biology, including vascular disease in brain. There are multiple sources of ROS, and an important role for NADPH oxidase is emerging. Some of the most important areas for future investigations concern evaluating the functional importance of NADPH oxidases relative to other sources of ROS and identifying the potential interactions between these mechanisms. Further development of therapeutic agents and delivery systems which target vascular NADPH oxidase may have beneficial effects in relation to cerebral vascular disease.
Acknowledgments
Work from our laboratory cited in this review was supported by National Institutes of Health grants HL-38901, NS-24621, HL-62984 and a Bugher Award from the American Heart Association (0575092N). S.C. was supported by a CJ Martin Fellowship from the National Health and Medical Research Council of Australia (359282) and a postdoctoral fellowship from the American Heart Association (0725643Z).
References
- 1.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 2.Lyle AN, Griendling KK. Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology. 2006;21:269–280. doi: 10.1152/physiol.00004.2006. [DOI] [PubMed] [Google Scholar]
- 3.Lee MY, Griendling KK. Redox signaling, vascular function, and hypertension. Antioxid Redox Signaling. 2008;10:1045–1059. doi: 10.1089/ars.2007.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faraci FM, Didion SP. Vascular protection: Superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24:1367–1373. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- 5.Pacher P, et al. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faraci FM. Reactive oxygen species: Influence on cerebral vascular tone. J Appl Physiol. 2006;100:739–743. doi: 10.1152/japplphysiol.01044.2005. [DOI] [PubMed] [Google Scholar]
- 7.Faraci FM. Hydrogen peroxide: Watery fuel for change in vascular biology. Arterioscler Thromb Vasc Biol. 2006;26:1931–1933. doi: 10.1161/01.ATV.0000238355.56172.b3. [DOI] [PubMed] [Google Scholar]
- 8.Wei EP, et al. Superoxide generation and reversal of acetylcholine-induced cerebral arteriolar dilation after acute hypertension. Circ Res. 1985;57:781–787. doi: 10.1161/01.res.57.5.781. [DOI] [PubMed] [Google Scholar]
- 9.Faraci FM, et al. Cerebral vascular effects of angiotensin II: New insights from genetic models. J Cerebral Blood Flow Metabol. 2006;26:449–455. doi: 10.1038/sj.jcbfm.9600204. [DOI] [PubMed] [Google Scholar]
- 10.Fang Q, et al. Inhibition of NADPH oxidase improves impaired reactivity of pial arterioles during chronic exposure to nicotine. J Appl Physiol. 2006;100:631–636. doi: 10.1152/japplphysiol.00975.2005. [DOI] [PubMed] [Google Scholar]
- 11.Faraci FM, et al. Selective cerebral vascular dysfunction in Mn-SOD-deficient mice. J Appl Physiol. 2006;100:2089–2093. doi: 10.1152/japplphysiol.00939.2005. [DOI] [PubMed] [Google Scholar]
- 12.Mayhan WG, et al. Age-related alterations in reactivity of cerebral arterioles: Role of oxidative stress. Microcirculation. 2008;15:225–236. doi: 10.1080/10739680701641421. [DOI] [PubMed] [Google Scholar]
- 13.Erdos B, et al. Rosuvastatin improves cerebrovascular function in Zucker obese rats by inhibiting NAD(P)H oxidase-dependent superoxide production. Am J Physiol. 2006;290:H1264–H1270. doi: 10.1152/ajpheart.00804.2005. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto T, et al. Vascular NAD(P)H oxidase mediates endothelial dysfunction in basilar arteries from Otsuka Long-Evans Tokushimia Fatty (OLETF) rats. Atherosclerosis. 2007;192:15–24. doi: 10.1016/j.atherosclerosis.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 15.Didion SP, et al. Increased superoxide and vascular dysfunction in CuZnSOD-deficient mice. Circ Res. 2001;91:938–944. doi: 10.1161/01.res.0000043280.65241.04. [DOI] [PubMed] [Google Scholar]
- 16.Baumbach GL, et al. Hypertrophy of cerebral arterioles in mice deficient in expression of the gene for CuZn superoxide dismutase. Stroke. 2006;37:1850–1855. doi: 10.1161/01.STR.0000227236.84546.5a. [DOI] [PubMed] [Google Scholar]
- 17.Beyer AM, et al. Interference with PPARγ signaling causes cerebral vascular dysfunction, hypertrophy, and remodeling. Hypertension. 2008;51:867–871. doi: 10.1161/HYPERTENSIONAHA.107.103648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hernanz R, et al. Hypertension alters role of iNOS, COX-2, and oxidative stress in bradykinin impairment after LPS in rat cerebral arteries. Am J Physiol. 2004;287:H225–H234. doi: 10.1152/ajpheart.00548.2003. [DOI] [PubMed] [Google Scholar]
- 19.Park L, et al. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cerebral Blood Flow Metabol. 2007;27:1908–1918. doi: 10.1038/sj.jcbfm.9600491. [DOI] [PubMed] [Google Scholar]
- 20.Park L, et al. NADPH oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid ß peptide. J Neurosci. 2005;25:1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park L, et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun H, et al. Role of NAD(P)H oxidase in alcohol-induced impairment of endothelial nitric oxide synthase-dependent dilation of cerebral arterioles. Stroke. 2006;37:495–500. doi: 10.1161/01.STR.0000199033.06678.c3. [DOI] [PubMed] [Google Scholar]
- 23.Zhang F, et al. Superoxide-dependent cerebrovascular effects of homocysteine. Am J Physiol. 1998;274:R1704–R1711. doi: 10.1152/ajpregu.1998.274.6.R1704. [DOI] [PubMed] [Google Scholar]
- 24.Nelson CW, et al. Oxygen radicals in cerebral ischemia. Am J Physiol. 1992;263:H1356–H1362. doi: 10.1152/ajpheart.1992.263.5.H1356. [DOI] [PubMed] [Google Scholar]
- 25.Girouard H, et al. Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler Thromb Vasc Biol. 2007;27:303–309. doi: 10.1161/01.ATV.0000253885.41509.25. [DOI] [PubMed] [Google Scholar]
- 26.Girouard H, et al. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through Nox-2-derived radicals. Arterioscler Thromb Vasc Biol. 2006;26:826–832. doi: 10.1161/01.ATV.0000205849.22807.6e. [DOI] [PubMed] [Google Scholar]
- 27.Kontos HA, Wei EP. Endothelium-dependent responses after experimental brain injury. J Neurotrauma. 1992;9:349–354. doi: 10.1089/neu.1992.9.349. [DOI] [PubMed] [Google Scholar]
- 28.Erdos B, et al. Cerebrovascular dysfunction in Zucker obese rats is mediated by oxidative stress and protein kinase C. Diabetes. 2004;53:1352–1359. doi: 10.2337/diabetes.53.5.1352. [DOI] [PubMed] [Google Scholar]
- 29.Phillips SA, et al. Oxidant stress and constrictor reactivity impair cerebral artery dilation in obese Zucker rats. Am J Physiol. 2005;288:R522–R530. doi: 10.1152/ajpregu.00655.2004. [DOI] [PubMed] [Google Scholar]
- 30.Dayal S, et al. Cerebral vascular dysfunction mediated by superoxide in hyperhomocysteinemic mice. Stroke. 2004;35:1957–1962. doi: 10.1161/01.STR.0000131749.81508.18. [DOI] [PubMed] [Google Scholar]
- 31.Dayal S, et al. Cerebral vascular dysfunction in methionine synthase-deficient mice. Circulation. 2005;112:737–744. doi: 10.1161/CIRCULATIONAHA.104.529248. [DOI] [PubMed] [Google Scholar]
- 32.Akopov SE, et al. The endothelium-dependent relaxation of human middle cerebral artery: Effects of activated neutrophils. Experientia. 1992;48:34–36. doi: 10.1007/BF01923601. [DOI] [PubMed] [Google Scholar]
- 33.Faraci FM, Heistad DD. Regulation of the cerebral circulation: Role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97. doi: 10.1152/physrev.1998.78.1.53. [DOI] [PubMed] [Google Scholar]
- 34.Faraci FM. Protecting the brain with eNOS: Run for your life. Circ Res. 2006;29:1029–1030. doi: 10.1161/01.RES.0000250961.47984.80. [DOI] [PubMed] [Google Scholar]
- 35.Arrick DM, et al. nNOS-dependent reactivity of cerebral arterioles in type 1 diabetes. Brain Res. 2007;1184:365–371. doi: 10.1016/j.brainres.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun H, et al. Influence of chronic alcohol consumption on inward rectifier potassium channels in cerebral arterioles. Microvasc Res. 2008;75:367–372. doi: 10.1016/j.mvr.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim CD, et al. Gene transfer of Cu/Zn SOD to cerebral vessels prevents FPI-induced CBF autoregulatory dysfunction. Am J Physiol. 2002;282:H1836–H1842. doi: 10.1152/ajpheart.00590.2001. [DOI] [PubMed] [Google Scholar]
- 38.Kazama K, et al. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res. 2004;95:1019–1026. doi: 10.1161/01.RES.0000148637.85595.c5. [DOI] [PubMed] [Google Scholar]
- 39.Kamii H, et al. Amelioration of vasospasm after subarachnoid hemorrhage in transgenic mice overexpressing CuZn-superoxide dismutase. Stroke. 1999;30:867–871. doi: 10.1161/01.str.30.4.867. [DOI] [PubMed] [Google Scholar]
- 40.Kim DE. Vascular NAD(P)H oxidase triggers delayed cerebral vasospasm after subarachnoid hemorrhage in rats. Stroke. 2002;33:2687–2691. doi: 10.1161/01.str.0000033071.99143.9e. [DOI] [PubMed] [Google Scholar]
- 41.Kahles T, et al. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 42.Kondo T, et al. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci. 1997;17:4180–4189. doi: 10.1523/JNEUROSCI.17-11-04180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kamada H, et al. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maier CM, et al. Evaluating therapeutic targets for reperfusion-related brain hemorrhage. Ann Neurol. 2006;59:929–938. doi: 10.1002/ana.20850. [DOI] [PubMed] [Google Scholar]
- 45.Baumbach GL, et al. Cerebral arteriolar structure in mice overexpressing human renin and angiotensinogen. Hypertension. 2003;41:50–55. doi: 10.1161/01.hyp.0000042427.05390.5c. [DOI] [PubMed] [Google Scholar]
- 46.Baumbach GL, et al. Structure of cerebral arterioles in mice deficient in expression of the gene for endothelial NO synthase. Circulation Res. 2004;95:822–829. doi: 10.1161/01.RES.0000146279.11923.14. [DOI] [PubMed] [Google Scholar]
- 47.Baumbach GL, et al. Structure of cerebral arterioles in cystathionine ß-synthase-deficient mice. Circulation Res. 2002;91:931–937. doi: 10.1161/01.res.0000041408.64867.1d. [DOI] [PubMed] [Google Scholar]
- 48.Sobey CG, et al. Potassium channels mediate dilatation of cerebral arterioles in response to arachidonate. Am J Physiol. 1998;275:H1606–H1612. doi: 10.1152/ajpheart.1998.275.5.H1606. [DOI] [PubMed] [Google Scholar]
- 49.Hoffmann OM, et al. Bacterial hydrogen peroxide contributes to cerebral hyperemia during early stages of experimental pneumococcal meningitis. J Cereb Blood Flow Metabol. 2007;27:1792–1797. doi: 10.1038/sj.jcbfm.9600474. [DOI] [PubMed] [Google Scholar]
- 50.Drouin A, et al. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. 2007;73:73–81. doi: 10.1016/j.cardiores.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 51.Katusic ZS, et al. Similar responsiveness of smooth muscle of the canine basilar artery to EDRF and nitric oxide. Am J Physiol. 1989;257:H1235–H1239. doi: 10.1152/ajpheart.1989.257.4.H1235. [DOI] [PubMed] [Google Scholar]
- 52.Kontos HA, et al. In vivo bioassay of endothelium-derived relaxing factor. Am J Physiol. 1988;255:H1259–H1262. doi: 10.1152/ajpheart.1988.255.5.H1259. [DOI] [PubMed] [Google Scholar]
- 53.Maneen MJ, et al. Peroxynitrite diminishes myogenic activity and is associated with decreased vascular smooth muscle F-actin in rat posterior cerebral arteries. Stroke. 2006;37:894–9. doi: 10.1161/01.STR.0000204043.18592.0d. [DOI] [PubMed] [Google Scholar]
- 54.Kunz A, et al. Neurovascular protection by ischemic tolerance: Role of nitric oxide and reactive oxygen species. J Neurosci. 2007;27:7083–7093. doi: 10.1523/JNEUROSCI.1645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dworakowski R, et al. NADPH oxidase-derived reactive oxygen species in the regulation of endothelial phenotype. Pharmacol Rep. 2008;60:21–28. [PubMed] [Google Scholar]
- 56.Touyz RM. Reactive oxygen species and angiotensin II signaling in vascular cells - implications in cardiovascular disease. Braz J Med Biol Res. 2004;37:1263–1273. doi: 10.1590/s0100-879x2004000800018. [DOI] [PubMed] [Google Scholar]
- 57.Ushio-Fukai M, Alexander RW. Reactive oxygen species as mediators of angiogenesis signaling: Role of NAD(P)H oxidase. Mol Cell Biochem. 2004;264:85–97. doi: 10.1023/b:mcbi.0000044378.09409.b5. [DOI] [PubMed] [Google Scholar]
- 58.Williams HC, Griendling KK. NADPH oxidase inhibitors: New antihypertensive agents? J Cardiovasc Pharmacol. 2007;50:9–16. doi: 10.1097/FJC.0b013e318063e820. [DOI] [PubMed] [Google Scholar]
- 59.Martyn KD, et al. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 60.Paravicini TM, et al. Increased NADPH-oxidase activity and Nox4 expression during chronic hypertension is associated with enhanced cerebral vasodilatation to NADPH in vivo. Stroke. 2004;35:584–589. doi: 10.1161/01.STR.0000112974.37028.58. [DOI] [PubMed] [Google Scholar]
- 61.Miller AA, et al. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res. 2005;97:1055–1062. doi: 10.1161/01.RES.0000189301.10217.87. [DOI] [PubMed] [Google Scholar]
- 62.Miller AA, et al. Effect of gender on NADPH-oxidase activity, expression, and function in the cerebral circulation: Role of estrogen. Stroke. 2007;38:2142–2149. doi: 10.1161/STROKEAHA.106.477406. [DOI] [PubMed] [Google Scholar]
- 63.Ago T, et al. NAD(P)H oxidases in rat basilar arterial endothelial cells. Stroke. 2005;36:1040–1046. doi: 10.1161/01.STR.0000163111.05825.0b. [DOI] [PubMed] [Google Scholar]
- 64.Mayhan WG, et al. Inhibition of NAD(P)H oxidase alleviates impaired NOS-dependent responses of pial arterioles in type 1 diabetes mellitus. Microcirculation. 2006;13:567–575. doi: 10.1080/10739680600885194. [DOI] [PubMed] [Google Scholar]
- 65.Vallet P, et al. Neuronal expression of the NADPH oxidase Nox4, and its regulation in mouse experimental brain ischemia. Neurosci. 2005;132:233–238. doi: 10.1016/j.neuroscience.2004.12.038. [DOI] [PubMed] [Google Scholar]
- 66.Shin HK, et al. Impairment of autoregulatory vasodilation by NAD(P)H oxidase-dependent superoxide generation during acute stage of subarachnoid hemorrhage in rat pial artery. J Cerebral Blood Flow Metabol. 2002;22:869–877. doi: 10.1097/00004647-200207000-00012. [DOI] [PubMed] [Google Scholar]
- 67.Ishizuka T, et al. Involvement of thromboxane A2 receptor in the cerebrovascular damage of salt-loaded, stroke-prone rats. J Hypertension. 2007;25:861–870. doi: 10.1097/HJH.0b013e3280464dc8. [DOI] [PubMed] [Google Scholar]
- 68.Heumuller S, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 69.Ishikawa M, et al. Cerebral microvascular responses to hypercholesterolemia. Roles of NADPH oxidase and P-selectin. Circ Res. 2004;94:239–244. doi: 10.1161/01.RES.0000111524.05779.60. [DOI] [PubMed] [Google Scholar]
- 70.Wood KC, et al. Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. 2005;19:989–991. doi: 10.1096/fj.04-3218fje. [DOI] [PubMed] [Google Scholar]
- 71.Chan S-L, Baumbach GL. Deficiency of Nox-2 prevents cerebral vascular hypertrophy induced by hypertension but not nitric oxide inhibition (Abstract) FASEB J. 2007;21:960.14. [Google Scholar]
- 72.Schrader LI, et al. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. 2007;27:2576–2581. doi: 10.1161/ATVBAHA.107.153080. [DOI] [PubMed] [Google Scholar]
- 73.Kawahara T, et al. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evolutionary Biol. 2007;7:109–832. doi: 10.1186/1471-2148-7-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oldendorf WH, et al. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol. 1977;1:409–417. doi: 10.1002/ana.410010502. [DOI] [PubMed] [Google Scholar]
- 75.Zafari AM, et al. Arachidonic acid metabolites mediate angiotensin II-induced NADH/NADPH oxidase activity and hypertrophy in vascular smooth muscle cells. Antioxid Redox Signaling. 1999;1:167–179. doi: 10.1089/ars.1999.1.2-167. [DOI] [PubMed] [Google Scholar]
- 76.Didion SP, et al. Superoxide levels and function of cerebral blood vessels after inhibition of CuZn-SOD. Am J Physiol. 2001;281:H1697–H1703. doi: 10.1152/ajpheart.2001.281.4.H1697. [DOI] [PubMed] [Google Scholar]
- 77.Mayhan WG, et al. Mechanism of impaired responses of cerebral arterioles during diabetes mellitus. Am J Physiol. 1991;260:H319–326. doi: 10.1152/ajpheart.1991.260.2.H319. [DOI] [PubMed] [Google Scholar]
- 78.Shi Y, Vanhoutte PM. Oxidative stress and COX cause hyper-responsiveness in vascular smooth muscle of the femoral artery from diabetic rats. Br J Pharmacol. 2008;154:639–651. doi: 10.1038/bjp.2008.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Choi SH, et al. Genetic deletion or pharmacological inhibition of cycloxygenase-1 attenuate lipopolysaccharide-induced inflammatory response and brain injury. FASEB J. 2008;22:1491–1501. doi: 10.1096/fj.07-9411com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Arrick DM, et al. Losartan improves impaired nitric oxide synthase-dependent dilatation of cerebral arterioles in type 1 diabetic rats. Brain Res. 2008;1209:128–135. doi: 10.1016/j.brainres.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuhlmann CRW, et al. Fluvastatin prevents glutamate-induced blood-brain-barrier disruption in vitro. Life Sci. 2008;82:1281–1287. doi: 10.1016/j.lfs.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 82.Sugawara A, et al. Transcriptional suppression of type 1 angiotensin II receptor gene expression by peroxisome proliferator-activated receptor-gamma in vascular smooth muscle cells. Endocrinology. 2001;142:3125–34. doi: 10.1210/endo.142.7.8272. [DOI] [PubMed] [Google Scholar]
- 83.Inoue I, et al. The ligands/activators for peroxisome proliferator-activated receptor α (PPARα) and PPARγ increase Cu2+, Zn2+-superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism. 2001;50:3–11. doi: 10.1053/meta.2001.19415. [DOI] [PubMed] [Google Scholar]
- 84.Ryan MJ, et al. The PPARγ agonist rosiglitazone improves vascular function and lowers blood pressure in hypertensive transgenic mice. Hypertension. 2004;43:661–666. doi: 10.1161/01.HYP.0000116303.71408.c2. [DOI] [PubMed] [Google Scholar]
- 85.Nakamura T, et al. Pioglitazone exerts protective effects against stroke in stroke-prone spontaneously hypertensive rats, independently of blood pressure. Stroke. 2007;38:3016–3022. doi: 10.1161/STROKEAHA.107.486522. [DOI] [PubMed] [Google Scholar]
- 86.Didion SP, et al. Cerebral vascular dysfunction in TallyHo mice: A new model of type II diabetes. Am J Physiol. 2007;292:H1579–H11583. doi: 10.1152/ajpheart.00939.2006. [DOI] [PubMed] [Google Scholar]
- 87.Xie H, et al. NF-κB activation plays a role in superoxide-mediated cerebral endothelial dysfunction after hypoxia/reoxygenation. Stroke. 2005;36:1047–1052. doi: 10.1161/01.STR.0000157664.34308.cc. [DOI] [PubMed] [Google Scholar]
- 88.McKnight AA, et al. Oxygen free radicals and the cerebral arteriolar response to group B streptococci. Ped Res. 1992;31:640–644. doi: 10.1203/00006450-199206000-00020. [DOI] [PubMed] [Google Scholar]