

Abstract
Background
Acute allograft rejection requires a multifaceted immune response involving trafficking of immune cells into the transplant and expression of effector cell functions leading to graft destruction. The chemokine receptor CXCR3 and its ligands, CXCL9, CXCL10 and CXCL11, constitute an important pathway for effector cell recruitment post-transplant. However, analysis of CXCR3 expression and function has been hampered by a general lack of availability of a neutralizing anti-CXCR3 monoclonal antibody (mAb) for use in experimental models.
Methods
We report the generation, characterization and use of CXCR3-173, a new hamster mAb specific for mouse CXCR3 that recognizes CXCR3 on cells from wild-type but not CXCR3-/- mice.
Results
Using CXCR3-173 mAb, we demonstrate CXCR3 expression on primary memory phenotype CD4+ and CD8+ T cells, naturally occurring CD4+CD25+ Foxp3+ regulatory T cells, natural killer T (NKT) cells, and ~25% of NK cells. CXCR3-173 blocked chemotaxis in vitro in response to CXCL10 or CXCL11 but not CXCL9. When injected into mice, this mAb significantly prolonged both cardiac and islet allograft survival. When combined with a subtherapeutic regimen of rapamycin, CXCR3-173 mAb induced long-term (>100 d) survival of cardiac and islet allografts. The in vivo effects of CXCR3-173 mAb were not associated with effector lymphocyte depletion.
Conclusion
These data highlight the utility of CXCR3-173 mAb in developing immunotherapeutic approaches to inhibit transplant rejection and potentially other immune-mediated diseases in murine models.
Keywords: transplant rejection, chemokine receptor, immunotherapy
INTRODUCTION
CXCR3 (CD183) is a seven transmembrane spanning, G-protein coupled chemokine receptor important in CD4+ T cell responses to allografts (1, 2) host responses to infection (3), and NK cell-dependent priming of CD4+ T cells in lymph nodes (4). CXCR3 has three ligands, CXCL9 (Mig), CXCL10 (IP-10) and CXCL11 (ITAC) that are induced by IFN-γ, IFN-α/β or other pro-inflammatory cytokines (e.g. TNF-α). CXCR3 has been shown to mediate the major effects of CXCL9, CXCL10 and CXCL11. While initial in vitro studies suggested that the three CXCR3 ligands display significant redundancy in the biologic responses they induce, subsequent work showed that the interactions between CXCR3 and its respective ligands result in distinct biologic functions due to subtle differences in ligand binding, as well as the timing and cellular source of ligand production (1, 3, 5, 6).
Most recently, an understanding of the physiologic functions of the receptor for CXCL9, CXCL10 and CXCL11 has been complicated by two additional findings. An alternatively spliced form of CXCR3, designated CXCR3-B, was found on human endothelium and was proposed to mediate the angiostatic actions of CXCR3 ligands (7). However, no evidence for the CXCR3-B transcript has been found in mice suggesting that this isoform of the receptor arises in a species restricted manner ((8) and R.U. unpublished). In addition, the CXCR3 ligand CXCL11 has been shown to interact with CXCR7/RDC1 and genetic deletion of CXCR7 results in defects in cardiac and vascular development resulting in 95% death of neonates within the first 24 hours of birth (9, 10). Moreover, no hematopoietic defects in CXCR7-/- mice were observed.
A critical role for CXCR3 in regulating host alloresponses was established as a result of the markedly delayed tempo of rejection in fully MHC-mismatched cardiac allograft recipients that were genetically deficient in CXCR3 (2, 11). However, evidence that additional chemokine receptors and their ligands contribute to the trafficking of effector lymphocytes into allografts (12) highlights the need for further assessment of the role of CXCR3 in promoting allograft rejection. Thus, although CXCR3-/- mice display deficient allograft rejection responses, it remains unclear whether this reflects a requirement for CXCR3 during the rejection response per se or an essential role for CXCR3 during the development of the cells that mediate rejection. Second, the identity of CXCR3+ cells that participate in allograft rejection in mouse models is hampered by the lack of reagents to track and mark these cells. Specifically, none of the currently available mouse CXCR3 antibody reagents affect CXCR3 function on naturally occurring cells and only one available mAb is capable of detecting murine CXCR3 on intact cells. Finally, the CXCR3-/- mouse is unsuited to test immune therapies that transiently block CXCR3 function.
To analyze CXCR3 expression and function in mice, we generated and characterized mAbs to mouse CXCR3. We then used one of these mAbs (CXCR3-173) to study CXCR3 expression on mouse splenocyte populations and, in doing so, identified a novel expression pattern of CXCR3 on mouse NK cells, Foxp3+ regulatory T cells (Tregs) and memory phenotype CD8+ T cells. We also found that CXCR3-173 mAb administration prolonged cardiac allograft survival without depleting the critical CD4+ effector T cell population. A similar increased survival of islet allografts with CXCR3-173 mAb treatment was observed. Finally, combining CXCR3-173 mAb treatment with low-dose rapamycin resulted in long-term survival of both cardiac and islet allografts. Thus, using this novel mAb, we have established a critical role for CXCR3 in allograft rejection and identified several potential cellular effectors of the CXCR3-dependent allograft rejection response. We propose that this antibody will be of use in defining the temporal requirements of CXCR3 during allograft rejection and other disease models, and will thereby serve as a valuable tool in assessing the preclinical efficacy of CXCR3 antagonists.
MATERIALS AND METHODS
Animals
C57BL/6 (H2b) and BALB/c (H2d) mice were obtained from the National Cancer Institute (Bethesda, MD), CXCR3-/- mice have been described (2), and Foxp3gfp mice were a generous gift from A.Y. Rudensky (University of Washington, Seattle, WA) (13). Armenian hamsters were from Cytogen Research and Development (Cambridge, MA). Animal studies were performed under approved protocols of the Animal Studies Committees of Washington University and the Children's Hospital of Philadelphia.
Cell Lines
A pSRαneo/mouse CXCR3 plasmid was used to transfect BaF3 cells. A single neomycin resistant clone with high CXCR3 expression (designated BaF3/mCXCR3) was utilized.
Antibodies
We purchased antibodies to CD11b (M1/70), CD3 (145-2C11), NK1.1 (PK136), CD4 (RM4-5), CD8 (Ly-2, 53-6.7), CD44 (Pgp-1, Ly24) and isotype control hamster IgG1 (anti-TNP) from BD Biosciences (San Diego, CA). CXCR3-173 mAb was conjugated with allophycocyanin (APC) as described (M. Roederer, Conjugation of monoclonal antibodies http://www.drmr.com/abcon/). Cells were studied using a FACSCalibur (BD BioSciences) and data analyzed with FlowJo software (Tree Star, Ashland, OR).
Hamster mAb Production
Hamsters were immunized with a multiple antigen peptide (MAPS8) encompassing amino acids 1-37 of mouse CXCR3 in complete Freund's adjuvant, and boosted with peptide in incomplete Freund's adjuvant (14). Hamsters showing ELISA seropositivity for CXCR3 peptide were boosted and hybridomas generated as described (14). Hybridoma supernatants were screened by FACS for CXCR3-specific staining and 3 positive cell lines were identified. One of these, CXCR3-173 was selected for further characterization and repeatedly cloned by limiting dilution, purified from hybridoma supernatants by Protein A affinity chromatography, and were tested for staining, CXCR3 blockade in vitro, and endotoxin levels (Cambrex BioSciences, Walkersville, MD); PIP Armenian hamster mAb (anti-GST) served as a control.
Ligand Binding
Human 125I-CXCL10 (Perkin Elmer, Boston, MA) and 125I-CXCL11 (Amersham, Arlington Heights, IL) were used for binding assays as described (15), and assays were repeated at least twice. Briefly, 2×105 BaF3/mCXCR3 cells were incubated in binding buffer (50 mM Hepes, pH 7.4, 1 mM CaCl2, 5 mM MgCl2, 0.5% BSA) with control or CXCR3-173 mAb (1 hr, 4 °C). Subsequently, 0.05 nM of labeled chemokine was added and incubation continued (1 hr, 4 °C). Non-specific binding was determined by adding excess unlabeled chemokine. Cell-receptor bound and free fractions were separated by spinning the cell suspension through a 6:4 mixture of dibutylpthalate:dioctylpthalate oil (14).
FACS-Based Chemokine Binding Inhibition Assay
BaF3/mCXCR3 cells were incubated in binding buffer (1 hr, 4 °C) with increasing concentrations of specified mouse CXCR3 ligands (PeproTech, Rocky Hill, NJ), followed by staining of cells with APC-conjugated CXCR3-173 mAb. To prevent receptor internalization, cells were kept at 4 °C throughout the experiment. The mean channel shift (MCS) was calculated and the percent decrease in staining determined relative to cells stained without chemokine.
Chemotaxis Assays
Assays were performed in triplicate in 96-well ChemoTx plates with a 5 μM pore size (NeuroProbe, Gaithersburg, MD) using ConA (1 mg/ml) stimulated C57BL/6 splenocytes. Activated cells were preincubated with control or CXCR3-173 antibody for 1 hr at 4 °C and at 37 °C for 1 hr with mouse CXCL9 (200 ng/ml), mouse CXCL10 (25 ng/ml) or mouse CXCL11 (25 ng/ml). Chemokines were titrated and concentrations used were in the linear range of a dose response analysis (data not shown). CyQuant GR dye (Invitrogen, Carlsbad, CA) was used to calculate the number of migrated cells based on a standard curve.
Pharmacokinetics and Immunogenicity of CXCR3-173
CXCR3-173 half-life was measured as described by Sheehan et al. (16). Groups of mice were injected with 250 μg of CXCR3-173 mAb and serum was harvested over a 10-day period. Clearance of CXCR3-173 mAb was measured by quantitating serum mAb levels by ELISA. Immunogenicity was tested by serially injecting mice with CXCR3-173 mAb and measuring accumulation of mAb over a one month period (16).
Allografts
Transplantation of fully MHC-mismatched cardiac allografts (BALB/c->C57BL/6) was performed as described (2), using 6 allografts/group. For mAb treatments, allografted mice were treated intraperitoneally with 500 μg PIP (control Armenian hamster IgG1) or 500, 200 or 100 μg CXCR3-173 mAb every other day for two weeks. We also tested low dose rapamycin (0.1 mg/kg/day, daily for two weeks, Alexis Biochemicals) alone or in conjunction with CXCR3-173 mAb at both the 500 μg and 200 μg regimens. Rejection was monitored by daily palpation of grafts and confirmed by histology. Islet allografts were performed as described (17). Primary function was defined as a reduction of blood glucose to <200 mg/ml post-transplant, and rejection as a rise in blood glucose to >300 mg/ml.
Pathology
Allografts were harvested at 7 days post-transplant or at rejection, and portions were formalin-fixed for light microscopy and snap-frozen for immunopathology. Paraffin sections were stained using hematoxylin and eosin, and cryostat sections were stained by immunoperoxidase using mAbs to CD4, CD8 or isotype controls (1, 2).
Quantitative Real-Time PCR (qPCR)
RNA was extracted using TRIzol reagent (Life Technologies), and RT of RNA samples (2 μg) using random hexamers was performed with an ABI PRISM 5700 unit (Applied Biosystems, Foster City, CA). Specific primer and probe sequences for target genes were used for qRT-PCR amplification of total cDNA (50 ng) with an ABI PRISM 5700 machine (TaqMan PDAR; Applied Biosystems); samples were normalized with primers to rRNA (18).
Flow Cytometry of Adoptively Transferred CFSE-Labeled Cells
Alloreactive T cells were generated by i.v. injection of 20 million CFSE-labeled C57BL/6 (H-2b) spleen and lymph node cells into C57BL/6xDBA F1 (H-2b/d) recipients, a parent->F1 MHC mismatch in which only donor cells respond (19). Splenocytes from F1 mice were harvested at day 3 and assayed for CFSE-dilution in CD4+ and CD8+ T cells that did not express H-2kd and anti-H-2dd (adoptively transferred alloreactive cells).
RESULTS
Production of hamster anti-mouse CXCR3 mAbs
We immunized Armenian hamsters with a peptide encompassing the first 37 amino acids of mouse CXCR3 (mCXCR3) given that Qin et al. had successfully generated neutralizing mAbs to the human receptor using a similar peptide from human CXCR3 (20). By screening hybridoma supernatants for reactivity against BaF3 cells engineered for mCXCR3 expression (BaF3/mCXCR3), we identified three hybridomas producing mCXCR3-reactive mAbs, of which CXCR3-173 mAb had the most favorable properties. Purified CXCR3-173 mAb stained BaF3/mCXCR3 cells (Fig. 1A), but not untransfected control BaF3 cells (Fig 1B). More importantly, this mAb stained ConA-stimulated splenocytes from wild-type mice (Fig. 1C), but not ConA-stimulated splenocytes from CXCR3-/- mice (Fig 1D). As with many mAbs that detect the native protein but not denatured forms, we were unable to use CXCR3-173 to detect CXCR3 by Western blotting (data not shown). Thus, the CXCR3-173 mAb is specific for mouse CXCR3 naturally expressed on mouse cells.
Figure 1. CXCR3-173 mAb binding to mouse CXCR3 blocks CXCL10 and CXCL11 binding and chemotaxis.
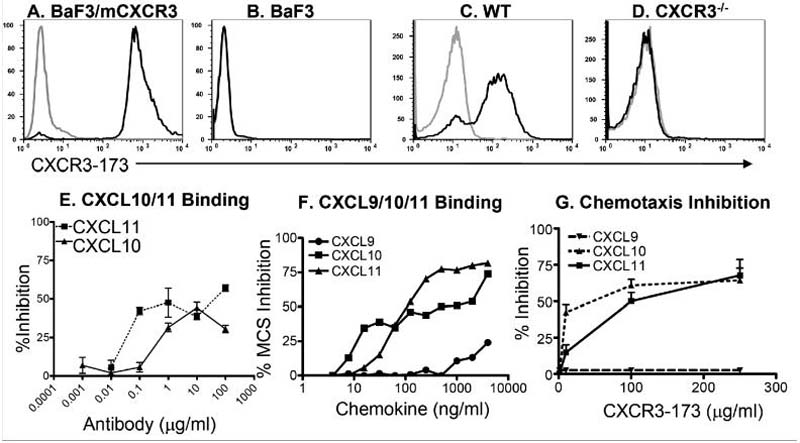
(A) BaF3/mCXCR3 cells, or (B) untransfected BaF3 cells, (C) ConA stimulated C57BL/6 splenocytes or (D) ConA stimulated CXCR3-/- splenocytes were stained with CXCR3-173 mAb. Grey lines represent isotype control staining for each antibody. (E) The BaF3/ mCXCR3 cells were pre-incubated with CXCR3-173 mAb or isotype-matched control mAb and used as targets in ligand binding assays with radiolabeled hCXCL10 (filled squares) or hCXCL11 (filled triangles). The percent inhibition of binding was calculated using specific binding seen with isotype control treatment as 100% binding; data are representative of at least two experiments. (F) CXCR3-173 mAb binding to BaF3/mCXCR3 cells is blocked by mCXCL10 and mCXCL11 but not by mCXCL9. BaF3/mCXCR3 cells were pre-incubated with increasing concentrations of either mCXCL9, mCXCL10 or mCXCL11 and subsequently stained with CXCR3-173 mAb (all steps at 4 °C). The percent inhibition of mean channel shift (MCS) was calculated by the decrease in CXCR3-173 mAb binding relative to staining of chemokine untreated cells; data are representative of three independent experiments. (G) Dose dependent inhibition of chemotaxis by CXCR3-173 mAb was analyzed on ConA stimulated splenocytes; data are representative of 2-5 experiments with each point performed in triplicate.
Inhibition of Ligand Binding by CXCR3-173
We next examined whether CXCR3-173 blocked receptor-mediated ligand binding and chemotaxis. Radiolabeled forms of mouse CXCR3 ligands are not available. Therefore, we used commercially available forms of human CXCL10 and CXCL11 which have been shown by others to represent suitable substitute ligands to study mouse CXCR3 (15). Consistent with previous reports (15), we found that 125I-hCXCL10 and 125I-hCXCL11 bound to mouse cells engineered for mCXCR3-expression in a specific and saturable manner with KDs of 1.4 nM and 0.4 nM, respectively (data not shown). Pretreatment of BaF3/mCXCR3 cells with varying concentrations of CXCR3-173 mAb inhibited hCXCL10 or hCXCL11 binding in a dose-dependent manner (Fig. 1E). CXCR3-173 mAb inhibited a maximum of 60% of hCXCL10 binding and 41% of hCXCL11 binding. Higher doses of CXCR3-173 mAb did not cause more inhibition (data not shown). Thus, CXCR3-173 mAb displayed significant but partial blockade of hCXCL10 and hCXCL11 binding to CXCR3.
To assess whether the mAb could also compete for binding of mouse CXCR3 ligands to mCXCR3, we developed a FACS based assay that monitored the capacity of unlabeled mouse chemokines to inhibit binding of APC-conjugated CXCR3-173 to BaF3/mCXCR3 cells (Fig. 1F). Pre-incubation of BaF3/mCXCR3 cells with either mCXCL10 or mCXCL11 inhibited CXCR3-173 binding in a dose-dependent manner. mCXCL10 inhibited a maximum of 74% of mAb binding and mCXCL11 inhibited up to of 81% of mAb binding. In contrast, mCXCL9 did not significantly affect mAb binding except at the highest 2 and 4 μg/ml doses, where 13 and 24% mAb binding inhibition was achieved, respectively. The same preparation of mCXCL9 caused significant transmigration of cells at 200-500 ng/ml. Thus, binding of CXCR3-173 to mCXCR3 inhibits receptor binding of mCXCL10 and mCXCL11 but not mCXCL9.
Inhibition of Splenocyte Chemotaxis by CXCR3-173
CXCR3-173 also inhibited the CXCR3-dependent chemotaxis of activated mouse splenocytes to mCXCL10 and mCXCL11 but not mCXCL9 (Fig. 1G) up to a maximum of 61% and 67%, respectively. This result is consistent with both the current view that CXCR3-dependent chemotaxis to CXCL9 involves regions of the receptor that are different from those required to manifest CXCL10 or CXCL11 function (5, 21), as well as our own finding that CXCL9 did not inhibit CXCR3-173 binding to CXCR3. Hence, CXCR3-173 mAb recognizes an epitope of CXCR3 expressed on the surface of activated mouse splenocytes and significantly inhibits binding and chemotaxis to CXCL10 and CXCL11 but not CXCL9.
CXCR3 expression on mouse lymphocyte subsets
We next asked whether CXCR3-173 could be used to detect CXCR3 expression by normal mouse lymphocyte subsets (Fig. 2). In naive mice, CXCR3 was detected on 11.8% of total CD3+ cells that were also CD4+ (i.e. 23% of splenic CD4+ CD3+ T cells, Fig. 2A), 18% of total CD3+ cells that were also CD8+ (i.e. 44% of CD8+ CD3+ T cells, Fig. 2B) and 0.78% of total CD3- cells that were also NK1.1+ (i.e. 24% of NK1.1+ CD3- NK cells, Fig. 2C). In contrast, all splenic NK1.1+CD3+ NKT cells were CXCR3+ (data not shown). Since the spleen has limited numbers of NKT cells, we repeated the analysis on NK1.1+ CD3+ cells from the liver and confirmed that essentially all NKT cells expressed CXCR3 (Fig. 2D).
Figure 2. CXCR3-173 mAb staining of mouse splenocytes.
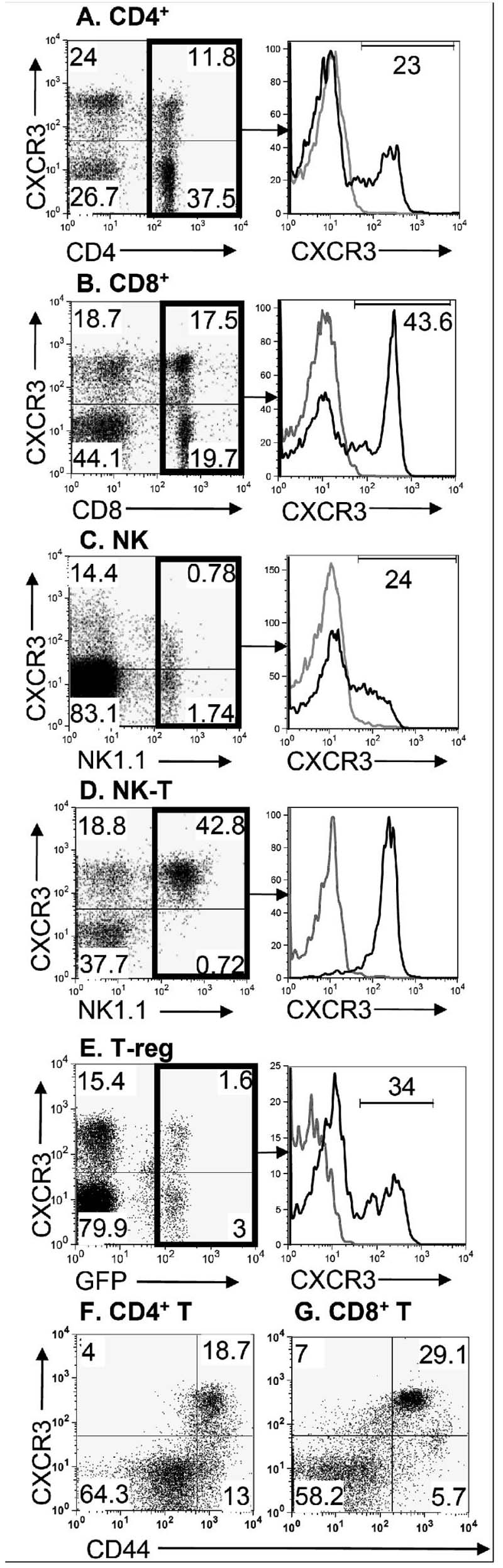
Splenocytes from C57BL/6 mice were analyzed for CXCR3 expression (y axis on all dot plots) on (A, left panel) CD4+ T cells by gating on CD3+ cells; (B, left panel) CD8+ T cells by gating on CD3+ cells; (C, left panel) NK cells by gating on CD3- cells and using the NK1.1 marker; (D, left panel) liver NKT cells by gating on CD3+ cells and using the NK1.1 marker; and (E, left panel) T-regs in Foxp3gfp splenocytes by first gating on the CD4 surface marker and subsequently analyzing GFP positive cells for CXCR3. (A-E, right panel histograms) The expression of CXCR3 (black line) on specific subsets of splenocytes is shown versus isotype control (gray line). (F) Gated CD4+ cells were analyzed for CD44 and CXCR3 staining. (G) Gated CD8+ cells were analyzed for CD44 and CXCR3 staining. Data are representative of 2 (Foxp3gfp) or 3 (all other) experiments.
Analyses of unstimulated CD4+ T cells showed that CXCR3 expression was limited to two distinct subpopulations. One of these were regulatory T cells (i.e., CD4+CD25+ Foxp3+ Tregs) which were distinguishable in this study because they were derived from a mouse in which the coding region of one allele of the Treg specific FoxP3 transcription factor had been replaced with green fluorescent protein (Foxp3gfp mice) (13). FoxP3 expressing Tregs were 34% positive for CXCR3 expression (Fig. 2E). Similar results were obtained using Tregs from wild type mice identified by their co-expression of cell surface CD25 and intracellular Foxp3 as detected by flow cytometry (data not shown). The second was a population of memory phenotype CD4+ T cells, that constitute 30% of the total CD4+ T cell population and which were identified by their expression of CD44. These memory phenotype CD4+ T cells were 59% positive for CXCR3 (i.e., this population accounted for 19% of the total CD4+ T cells) (Fig. 2F). Thus, in CD4+ T cells, CXCR3 was expressed on cells which may have opposing functions: Tregs and memory phenotype T-effector cells. Similar analyses of CD8+ T cells from naïve mice showed that the vast majority of those expressing CXCR3 also expressed the CD44 effector/memory T cell marker (Fig. 2G). Significantly less CXCR3 was seen on the other CD8+ T cells. Taken together, these results suggest that most of the CXCR3 expression by CD8+ T cells is on the memory phenotype subpopulation.
CXCR3-173 prolongs allograft survival
Before testing the effects of CXCR3-173 mAb in vivo, we established that the mAb displayed a half-life of 4.5 days and, like other hamster mAbs, had negligible immunogenicity in mice (data not shown). We then tested the capacity of CXCR3-173 mAb to block rejection of fully MHC-mismatched mouse cardiac allografts (BALB/c->C57BL/6), a disease process known to be dependent on CD4+ T cells and the CXCL10/CXCR3 pathway but not CD8+ T cells (i.e. rejection is blocked by treatment with anti-CD4 but is unaffected in either wild-type mice treated with anti-CD8 or in CD8-/- mice)(1, 2, 22-24). Consistent with published data, PIP control mAb-treated mice rejected cardiac allografts by 6-7 days post-transplant (Fig. 3A). In contrast, allograft rejection was significantly delayed until day 16-18 (p<0.0067, log-rank analysis of Kaplan-Meier curves (2)) in recipients treated with CXCR3-173 mAb using a dosing schedule of either 500 μg mAb every day or 200 μg mAb administered every other day (qod). At a lower 100 μg dose qod, CXCR3-173 mAb had no significant effect on allograft rejection. The CXCR3-173 mAb-induced prolongation of cardiac allograft survival observed in the current experiments was similar to that achieved in an earlier study using a rat IgM anti-CXCR3 that depleted CXCR3 expressing cells (2). No prolongation of survival was seen using the mAb in CXCR3-/- recipients (data not shown).
Figure 3. CXCR3-173 mAb therapy prolongs cardiac and islet allograft survival.
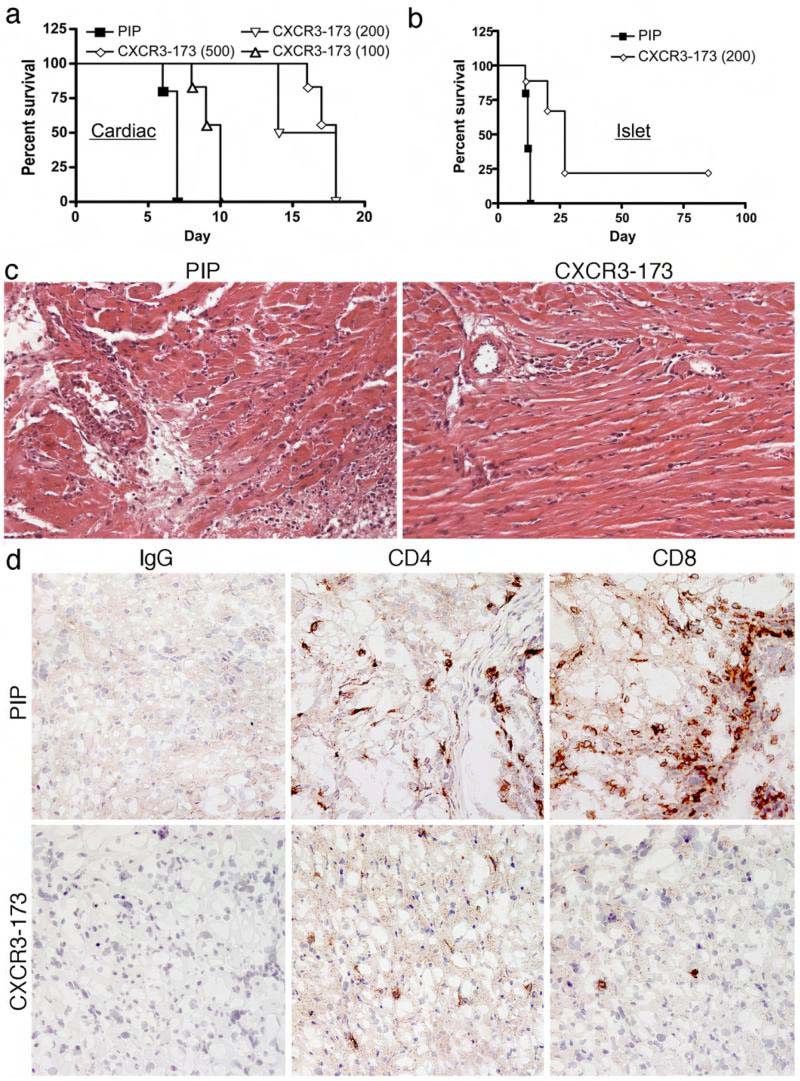
Full MHC mismatched (A) cardiac or (B) islet allografts (BALB/c->C57BL/6) were performed and recipient mice (6 allografts/group) were treated with the doses of CXCR3-173 or control PIP mAb every other day at the doses (μg) indicated. Percent survival was determined by daily palpation of cardiac allografts and by serum glucose measurements for islet allografts. In cardiac allograft recipients, use of CXCR3-173 mAb at 200 or 500 μg every other day was induced statistically significant prolongation of allograft survival compared to PIP control (p<0.0067, log-rank analysis of Kaplan-Meier curves), whereas the effect of 100 μg every other day of CXCR3-173 was not statistically significant. In islet allograft recipients, CXCR3-173 (200 μg, qod) also led to a statistically significant prolongation of allograft survival compared to use of PIP control (p<0.05). (C) Histologic examination of cardiac allografts harvested at day 7 post-transplant (n=4/group) showed PIP control mAb therapy did not affect development of acute cellular rejection, with interstitial mixed leukocytic infiltration, arteritis and focal myocyte necrosis. By contrast, examination of allografts from CXCR3-173 mAb-treated recipients at 7 days post-transplant showed only minor mononuclear cell infiltrates, well-preserved vessels and normal myocardium. (H&E-stained paraffin sections, original magnifications ×300). (D) Immunoperoxidase staining of corresponding snap-frozen samples showed that CXCR3-173 mAb markedly reduced intragraft accumulation of CD4 and CD8 T cells. (Cryostat sections, hematoxylin counterstain, ×300 original magnifications).
We extended these findings to a model of islet allograft transplantation where, similar to cardiac allografts, islet allograft survival was shown to be prolonged in mice lacking CXCR3 (25). CXCR3-173 mAb administered at 200 μg mAb qod in the peri-transplant period was able to significantly delay islet allograft rejection from 12.5 days to 21 days median survival (p<0.05, Fig. 3B). This delay is similar to that seen when transplants were performed with CXCR3-/- mice. These data show that, in two models of CXCR3-dependent allograft rejection, CXCR3-173 mAb recapitulates findings described using CXCR3-/- mice.
Immunopathology
Histologic examination of cardiac allografts (n=4/group) harvested at the time of rejection in control mAb-treated mice (7 days) showed acute cellular rejection, with interstitial cell infiltrates and arteritis and widespread myocyte necrosis (Fig. 3C). In contrast, analysis of allografts harvested at day 7 post-transplant from CXCR3-173 mAb-treated mice showed only minor mononuclear cell infiltrates and intact myocardium and vasculature (Fig. 3D). Immunoperoxidase staining of corresponding snap-frozen tissues showed marked interstitial and vascular wall infiltration by CD4+ and CD8+ T cells in control mAb-treated recipients, whereas CXCR3-173 mAb therapy was associated with a marked reduction in both CD4+ and CD8+ T cell infiltration. While allografts in CXCR3-173 mAb-treated mice were progressively lost due to acute cellular rejection by about day 16 post-transplant onwards, these studies suggest an important benefit of anti-CXCR3 mAb therapy. The reduced infiltration led us to consider whether the CXCR3-173 mAb depleted the critical CD4+ T cells or dampened their recruitment in the initial post-transplant period.
CXCR3-173 does not deplete the critical CD4+ T cells required for allograft rejection
We tested whether in vivo treatment with CXCR3-173 mAb resulted in depletion of CD4+ T cells required for cardiac allograft rejection, by treating WT mice with increasing amounts of CXCR3-173 mAb and 4 days later assessing lymphocyte depletion. CXCR3 expressing CD4+ T cells (Fig. 4A) were not depleted from spleen, blood or lymph nodes (data not shown for blood and lymph nodes) in mice following administration of a single 40 μg, 200 μg or 1 mg dose of CXCR3-173. In addition, no differences in the numbers of total splenocytes between control and CXCR3-173 treated mice were observed (data not shown). To test the possibility that CXCR3-173 depleted only activated effector CD4+ T cell populations, we performed similar analyses in a model of parent→F1 adoptive transfer where CFSE-labeled CD4+ T cells were monitored for activation and potential depletion by CXCR3-173 (19). The donor T cell proliferation was unaffected by control mAb treatment (Fig. 4B). When recipient mice were treated with CXCR3-173, the upregulation of cell surface CXCR3 on dividing cells was observed using an anti-hamster secondary antibody for detection. However, no depletion of CD4+ T cells was observed, thus confirming that CXCR3-173 does not deplete the cells that are critical for allograft rejection. Taken together these results clearly distinguish the current study from earlier studies that employed the depleting IgM anti-CXCR3 (2) and thus directly point to the utility of CXCR3-173 in probing CXCR3 dependent mechanisms in allograft rejection.
Figure 4. CXCR3-173 mAb does not deplete CD4+T cells.
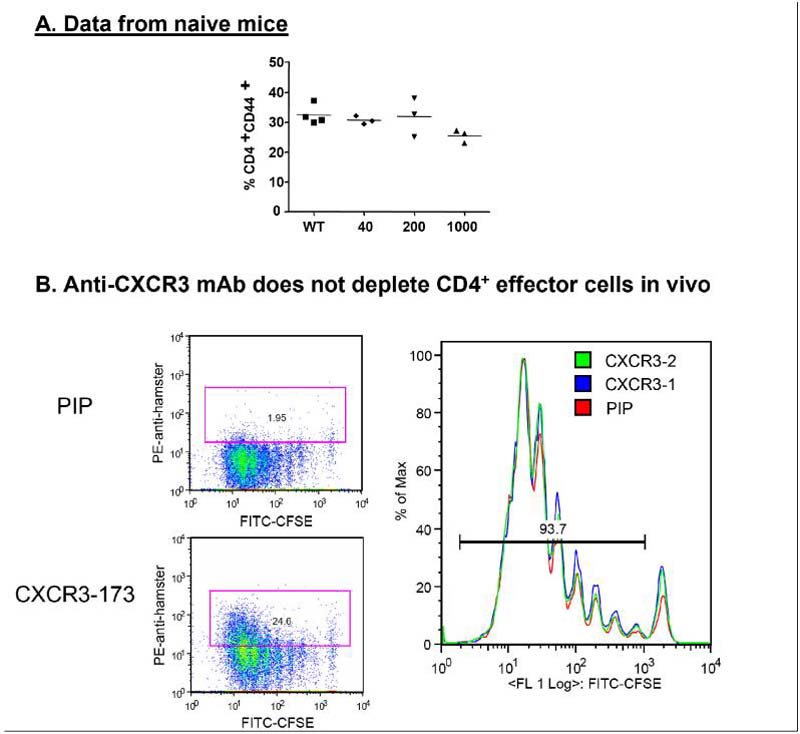
Naïve C57BL/6 mice were treated with 40 μg, 200 μg, or 1 mg of CXCR3-173 mAb. Five days later splenocytes were analyzed for (A) percentage of CD4+CD44+ T cells of total CD4+ cells. No significant differences in splenocyte numbers were noted (data not shown). Each symbol represents a single mouse, and data are representative of two experiments. (B) A parent->F1 transfer of CFSE-labeled cells was performed with mice receiving PIP control or CXCR3-173 mAb. 72 hours later cells were harvested and CD4+ T cells were analyzed for CFSE dilution and secondarily stained with an anti-hamster antibody to examine CXCR3-173 mAb binding to cell surfaces.
Intragraft mRNA expression following CXCR3-173 treatment
Previous analysis of intragraft events showed that early induction of CXCL10 post-transplant was required for the rapid recruitment of CXCR3+ cells that mediated acute cardiac allograft rejection (1). In the current study, we found by qPCR analysis that rejecting grafts showed induction of IL-2, IFN-γ, CXCL9 and CXCL10 (Fig. 5). In addition, CXCR3 and CCR5 mRNAs were present in rejecting grafts. In contrast, cardiac allografts in mice treated with CXCR3-173 (but not in mice treated with control mAb) displayed reduced expression of the IL-2 and IFNγ cytokines, as well as reduced expression of CXCL9, CCR5 and CXCR3. However, CXCL10 mRNA was not significantly reduced in the allografts, consistent with data that the very early induction of CXCL10 is related to cardiac ischemia/reperfusion (1) and is therefore not altered by reduced IFN-γ production. The current data obtained with CXCR3-173 treated wild type mice are highly consistent with findings using this cardiac allograft model using either CXCR3-/- or CXCL10-/- mice (1, 2).
Figure 5. Intragraft cytokine and chemokine mRNA expression.
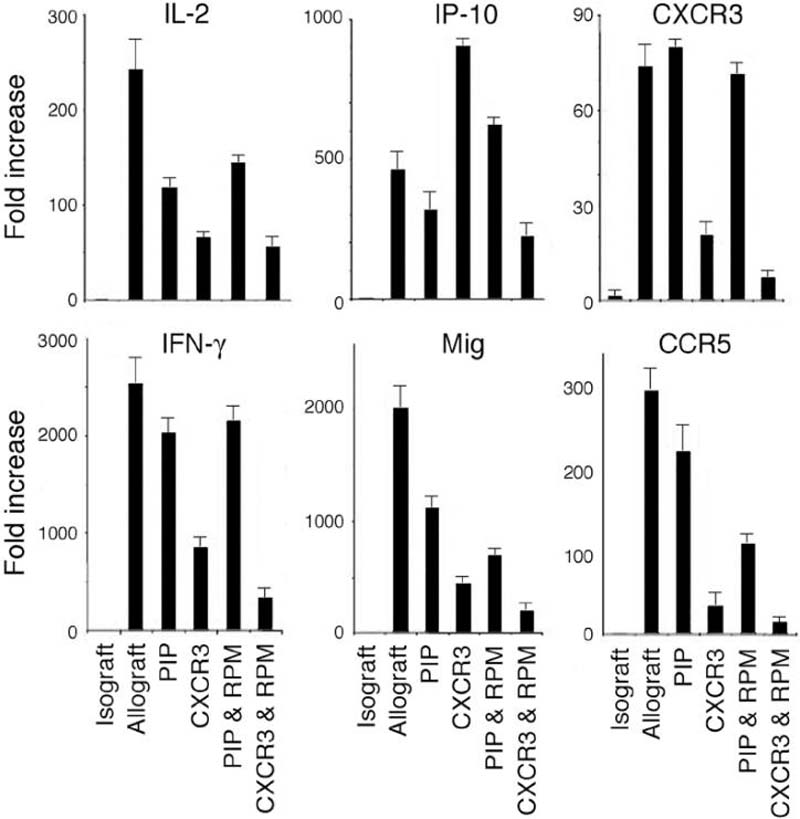
Comparison using qPCR of cytokine (IL-2, IFN-γ), chemokine (IP-10 (CXCL10), Mig (CXCL9)) and chemokine receptor (CXCR3, CCR5) mRNA expression in cardiac isografts or allografts (4/group) harvested at day 7 post-transplant; data are presented as fold increase (mean ± SD) over levels in corresponding isografts. The groups indicated were treated with PIP control mAb, CXCR3-173 mAb, PIP plus sirolimus (RPM), or CXCR3-173 mAb plus RPM. Data from CXCR3-173 mAb plus RPM were statistically significant (Student's t test) from that of the group receiving PIP plus RPM: IL-2 (p<0.01), IFN-γ (p<0.001), IP-10 (p<0.01), Mig (p<0.05), CXCR3 (p<0.001) and CCR5 (p<0.01).
CXCR3-173 plus rapamycin therapy induces long-term allograft survival
Combined use of a low dose of cyclosporine and CXCR3+ cell depletion by an IgM rat anti-mCXCR3 mAb was previously shown to result in long-term engraftment of fully MHC-mismatched cardiac allografts (2). In the current study, we tested whether low-dose rapamycin in conjunction with non-depleting CXCR3-173 blockade would produce a similar effect. Mice treated with low dose rapamycin (RPM) alone, or with control PIP mAb plus RPM, rejected cardiac allografts 2-3 days after control mice that were treated only with vehicle (Fig. 6A). Treatment of mice with 500 μg doses of CXCR3-173 mAb alone prolonged allograft survival to 16-18 days (Fig. 3A). In contrast, mice that received a combination of 14 days of therapy with CXCR3 mAb (500 μg) and low dose rapamycin retained their cardiac allografts up to 100 days after transplant (Fig. 6A). When CXCR3-173 and rapamycin were combined, expression of CXCR3 and CCR5 transcripts in the graft were lower than those detected in mice treated with either agent alone, consistent with a synergistic therapeutic effect of these two approaches (Fig. 5).
Figure 6. Combined CXCR3-173 and rapamycin therapy prolongs cardiac and islet engraftment.
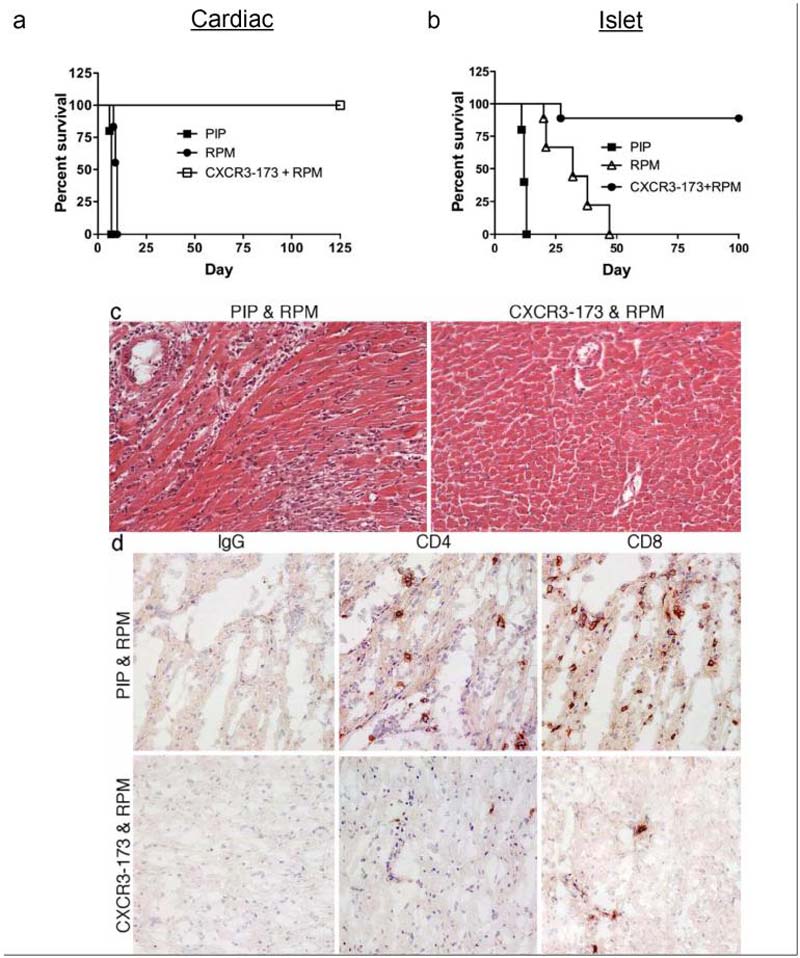
Full MHC mismatched cardiac allografts (A) or islet allografts (B) were performed as in Fig. 3. In cardiac allograft recipients, combined administration of CXCR3-173 mAb at 500 μg every other day, plus daily rapamcyin (RPM, 0.1 mg/kg/d, ip), for 14 days from the time of transplantation, induced statistically significant prolongation of allograft survival compared to PIP control or sirolimus (p<0.001, log-rank analysis of Kaplan-Meier curves). In islet allograft recipients, CXCR3-173 mAb (200 μg, qod) plus rapamcyin (0.1 mg/kg/d, ip) for 14 days also led to a statistically significant prolongation of allograft survival compared to use of RPM alone (p<0.01) or PIP control (p<0.001). (C) Histologic examination of cardiac allografts harvested at 10-12 days post-transplant (n=4/group) showed PIP control mAb plus RPM therapy did not affect development of acute cellular rejection, with interstitial mixed leukocytic infiltration, arteritis and focal myocyte necrosis. By contrast, examination of allografts in recipients treated with CXCR3-173 mAb and RPM showed essentially normal graft histology. (H&E-stained paraffin sections, original magnifications ×300). (D) Immunoperoxidase staining of corresponding snap-frozen samples showed that CXCR3-173 mAb plus RPM markedly reduced intragraft accumulation of CD4 and CD8 T cells. (Cryostat sections, hematoxylin counterstain, ×300 original magnifications).
Using a similar protocol, mice bearing islet allografts were treated with rapamycin alone, or rapamycin plus 200 μg qod of CXCR3-173 mAb. As shown in Fig. 3B, mAb therapy alone significantly delayed islet allograft rejection and similarly, rapamycin alone caused a delay of rejection up to 47 days (Fig. 6B). However, when mice bearing islet allografts were treated with a combination of rapamycin and CXCR3 blockade, long-term engraftment of 80% of the allografts was observed (Fig. 6B). Thus, the combination of low dose rapamycin and CXCR3 blockade results in highly successful allograft survival in two different models.
Histologic analysis of cardiac allografts (n= 4/group) harvested at the time of rejection in mice treated with PIP control mAb plus rapamycin (~10-12 days post-transplant) showed acute cellular rejection, with mixed leukocytic infiltrates, arteritis and focal myocyte necrosis, whereas grafts harvested at 10-14 days post-transplant in mice treated with CXCR3-173 mAb plus rapamycin showed essentially normal histology (Fig. 6C). Immunoperoxidase studies confirmed a marked reduction in CD4 and CD8 T cell infiltration by CXCR3-173 mAb plus rapamycin therapy compared to the control group (Fig. 6D).
DISCUSSION
We describe the generation and characterization of a blocking mAb to mouse CXCR3. Using this mAb (CXCR3-173), we identified a hitherto unrecognized pattern of expression of CXCR3 on NK cells, Tregs and CD8+CD44+ memory phenotype T cells, in addition to expression by subsets of CD4+ and CD8+ T cells. In addition, we show that CXCR3-173 was capable of delaying cardiac allograft rejection in wild-type mice, which was previously shown using either CXCR3-/- mice or a depleting rat anti-mCXCR3 IgM mAb. However, in contrast to previous studies that employed a depleting anti-mCXCR3 IgM mAb, we show that the inhibitory action of our novel hamster anti-mCXCR3 IgG mAb reflected its ability to block chemokine binding rather then its ability to deplete the critical CD4+ T cells key to the pathogenesis of cardiac allograft rejection (1, 2). We also demonstrated that CXCR3-173 in combination with low-dose rapamycin resulted in prolonged cardiac allograft survival. We extended these findings in an islet allograft model and, similar to the cardiac model, showed enhanced allograft survival with CXCR3-173 treatment, and long-term islet allograft survival as a result of combined mAb and low-dose rapamycin treatment. Thus, in two different allograft models, long-term survival was observed with combined chemokine receptor and anti-proliferative blockade, a combination not previously reported in these models.
Analysis of the inhibitory function of CXCR3-173 mAb showed that mAb binding to transfected cells was blocked by mouse CXCL10 and CXCL11 but not by CXCL9. In addition, CXCR3-173 mAb blocked CXCL10- or CXCL11-induced chemotaxis of primary activated T cells but did not inhibit CXCL9-induced chemotaxis. These data are consistent with the reported functional domain structure of human CXCR3 where the receptor amino terminus was required for CXCL10 and CXCL11 function but not CXCL9 function (5). In practice, since CXCR3-173 mAb reacts with the amino terminal portion of the receptor, it may permit undiminished interaction of CXCL9 with the receptor's extracellular loops, thereby permitting CXCL9 binding and chemotaxis.
Although many studies of CXCR3 expression on human cells have been conducted, there is less information available about receptor expression on mouse cells, due to the scarcity of appropriate and well-characterized mAbs. Our analysis of CXCR3 expression confirmed that, in mice, a proportion of memory phenotype CD4+ and the majority of memory phenotype CD8+ T cells, as defined by CD44 staining, expressed CXCR3. We also found that, in contrast to human CD8+ T cells (26) naïve CD8+ T cells in C57BL/6 mice do not express CXCR3 constitutively. Although the current study is restricted to the examination of CXCR3 expression on primary mouse splenocytes, future work will examine CXCR3 expression during antigen-specific memory T cell development.
We extended the analysis of CXCR3 expression on CD4+ T cells by utilizing the Foxp3gfp mouse to identify Tregs and found that one third of regulatory T cells express this receptor. Multiple chemokine receptors including CXCR4, CCR4, CCR5 and CCR8 have been identified as having important roles in Treg function (27-32). However, in a model of Treg induced colitis, lack of CXCR3 expression by Tregs did not affect their ability to regulate development of inflammatory bowel disease induced by co-transferred T cells (33), and we have found that Tregs from CXCR3-/- mice displayed normal suppressive ability using standard in vitro assays (data not shown). Thus, CXCR3 expression on Tregs may be redundant, irrelevant or nonfunctional. Our studies suggest that despite the expression of CXCR3 on Tregs, blockade of CXCR3 function remains a viable therapeutic option for establishing long-term allograft survival.
We assessed whether CXCR3-173 mAb was able to inhibit a model of cardiac allograft rejection known to be dependent on expression of CXCR3 and CXCL10 (1, 2). In this model, it was shown that allografts produce CXCL10 and thus recruit lymphocytes in a CXCR3-dependent manner, leading to rejection. However, use of an anti-CXCR3 IgM mAb that depleted CXCR3-expressing lymphocytes complicated interpretation of the results and required the use of the CXCR3-/- mice for confirmation. Consistent with the earlier study, we found that the CXCR3-173 IgG mAb also delayed allograft rejection, and as shown only in the current study, when combined with low-dose rapamycin, promoted permanent engraftment. Additional confirmation that CXCR3-173 faithfully replicated findings from CXCR3-/- mice came from analysis of intragraft cytokines and chemokines. Since the CXCR3-dependence in the cardiac allograft model has been attributed to CD4+ T cells, we assessed and found that mAb treatment did not eliminate these cells in CXCR3-173 treated wild-type mice or in a parent-to-F1 transfer model. Thus, the effect of our antibody in the transplant model points to a likely blockade of the directed migration of these cells to the allograft (1, 24). Finally, a previous study demonstrated the enhanced survival of islet allografts in CXCR3-/- mice (from 12.7 days in wild type mice to 20 days in CXCR3-/- mice) (25). Using CXCR3-173 mAb, a similar prolongation of allograft survival was observed in our study. However, our study is the first to show that combination therapy with chemokine blockade and low-dose rapamycin results in permanent engraftment.
In summary, we have generated and characterized a hamster IgG mAb that unequivocally identifies and functionally interferes with naturally expressed mouse CXCR3. The use of this mAb in established models of cardiac and islet allograft rejection provides strong evidence of its functional utility. Perhaps most importantly, the availability of this mAb will facilitate efforts by us and others to better define the physiologic roles of CXCR3 in mouse models of host defense to infectious agents and tumors, as well as in immunopathologic states.
ACKNOWLEDGEMENTS
We thank A. Y. Rudensky for Foxp3gfp mice, J. Michael White, Tiffany Stephans and Jessica Archambault for mouse care and advice, and Gavin P. Dunn for critical reading and discussion of the manuscript.
RU is supported by NCI K08 CA090403. JDB was supported by a CRI post-doctoral fellowship. CG is supported by NIH grant HL51366. WWH is supported by NIH grants AI54720, AI51724 and AI68061, and by the Biesecker Center for Pediatric Liver Disease. RDS is supported by NIH grants CA43059 and CA107527. The work cited in this publication was performed in a facility supported by NCRR grant C06 RR012466. The authors all declare no conflict of interest in this research.
References
- 1.Hancock WW, Gao W, Csizmadia V, Faia KL, Shemmeri N, Luster AD. Donor-derived IP-10 initiates development of acute allograft rejection. J Exp Med. 2001;193(8):975. doi: 10.1084/jem.193.8.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hancock WW, Lu B, Gao W, et al. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med. 2000;192(10):1515. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan IA, MacLean JA, Lee FS, et al. IP-10 is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity. 2000;12(5):483. doi: 10.1016/s1074-7613(00)80200-9. [DOI] [PubMed] [Google Scholar]
- 4.Martin-Fontecha A, Thomsen LL, Brett S, et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5(12):1260. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- 5.Xanthou G, Williams TJ, Pease JE. Molecular characterization of the chemokine receptor CXCR3: evidence for the involvement of distinct extracellular domains in a multi-step model of ligand binding and receptor activation. Eur J Immunol. 2003;33(10):2927. doi: 10.1002/eji.200324235. [DOI] [PubMed] [Google Scholar]
- 6.Salazar-Mather TP, Hamilton TA, Biron CA. A chemokine-to-cytokine-to-chemokine cascade critical in antiviral defense. J Clin Invest. 2000;105(7):985. doi: 10.1172/JCI9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lasagni L, Francalanci M, Annunziato F, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med. 2003;197(11):1537. doi: 10.1084/jem.20021897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strieter RM, Belperio JA, Phillips RJ, Keane MP. CXC chemokines in angiogenesis of cancer. Semin Cancer Biol. 2004;14(3):195. doi: 10.1016/j.semcancer.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Burns JM, Summers BC, Wang Y, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203(9):2201. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sierro F, Biben C, Martinez-Munoz L, et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A. 2007;104(37):14759. doi: 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haskova Z, Izawa A, Contreras AG, Flynn E, Boulday G, Briscoe DM. Organ-Specific Differences in the Function of MCP-1 and CXCR3 During Cardiac and Skin Allograft Rejection. Transplantation. 2007;83(12):1595. doi: 10.1097/01.tp.0000266892.69117.9a. [DOI] [PubMed] [Google Scholar]
- 12.Hancock WW, Wang L, Ye Q, Han R, Lee I. Chemokines and their receptors as markers of allograft rejection and targets for immunosuppression. Curr Opin Immunol. 2003;15(5):479. doi: 10.1016/s0952-7915(03)00103-1. [DOI] [PubMed] [Google Scholar]
- 13.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22(3):329. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 14.Sheehan KC, Calderon J, Schreiber RD. Generation and characterization of monoclonal antibodies specific for the human IFN-gamma receptor. J Immunol. 1988;140(12):4231. [PubMed] [Google Scholar]
- 15.Lu B, Humbles A, Bota D, et al. Structure and function of the murine chemokine receptor CXCR3. Eur J Immunol. 1999;29(11):3804. doi: 10.1002/(SICI)1521-4141(199911)29:11<3804::AID-IMMU3804>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 16.Sheehan KC, Ruddle NH, Schreiber RD. Generation and characterization of hamster monoclonal antibodies that neutralize murine tumor necrosis factors. J Immunol. 1989;142(11):3884. [PubMed] [Google Scholar]
- 17.Wang L, Han R, Lee I, et al. Permanent survival of fully MHC-mismatched islet allografts by targeting a single chemokine receptor pathway. J Immunol. 2005;175(10):6311. doi: 10.4049/jimmunol.175.10.6311. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Han R, Hancock WW. Programmed cell death 1 (PD-1) and its ligand PD-L1 are required for allograft tolerance. Eur J Immunol. 2007;37(10):2983. doi: 10.1002/eji.200737583. [DOI] [PubMed] [Google Scholar]
- 19.Lee I, Wang L, Wells AD, et al. Blocking the monocyte chemoattractant protein-1/CCR2 chemokine pathway induces permanent survival of islet allografts through a programmed death-1 ligand-1-dependent mechanism. J Immunol. 2003;171(12):6929. doi: 10.4049/jimmunol.171.12.6929. [DOI] [PubMed] [Google Scholar]
- 20.Qin S, Rottman JB, Myers P, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101(4):746. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colvin RA, Campanella GS, Manice LA, Luster AD. CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol Cell Biol. 2006;26(15):5838. doi: 10.1128/MCB.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campos L, Naji A, Deli BC, et al. Survival of MHC-deficient mouse heterotopic cardiac allografts. Transplantation. 1995;59(2):187. [PubMed] [Google Scholar]
- 23.Han WR, Murray-Segal LJ, Mottram PL. Assessment of peripheral tolerance in anti-CD4 treated C57BL/6 mouse heart transplants recipients. Transpl Immunol. 1999;7(1):37. doi: 10.1016/s0966-3274(99)80017-3. [DOI] [PubMed] [Google Scholar]
- 24.Krieger NR, Yin DP, Fathman CG. CD4+ but not CD8+ cells are essential for allorejection. J Exp Med. 1996;184(5):2013. doi: 10.1084/jem.184.5.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baker MS, Chen X, Rotramel AR, et al. Genetic deletion of chemokine receptor CXCR3 or antibody blockade of its ligand IP-10 modulates posttransplantation graft-site lymphocytic infiltrates and prolongs functional graft survival in pancreatic islet allograft recipients. Surgery. 2003;134(2):126. doi: 10.1067/msy.2003.213. [DOI] [PubMed] [Google Scholar]
- 26.Rabin RL, Park MK, Liao F, Swofford R, Stephany D, Farber JM. Chemokine receptor responses on T cells are achieved through regulation of both receptor expression and signaling. J Immunol. 1999;162(7):3840. [PubMed] [Google Scholar]
- 27.Bystry RS, Aluvihare V, Welch KA, Kallikourdis M, Betz AG. B cells and professional APCs recruit regulatory T cells via CCL4. Nat Immunol. 2001;2(12):1126. doi: 10.1038/ni735. [DOI] [PubMed] [Google Scholar]
- 28.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 29.Iellem A, Mariani M, Lang R, et al. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4(+)CD25(+) regulatory T cells. J Exp Med. 2001;194(6):847. doi: 10.1084/jem.194.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee I, Wang L, Wells AD, Dorf ME, Ozkaynak E, Hancock WW. Recruitment of Foxp3+ T regulatory cells mediating allograft tolerance depends on the CCR4 chemokine receptor. J Exp Med. 2005;201(7):1037. doi: 10.1084/jem.20041709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, et al. Critical role for CCR5 in the function of donor CD4+CD25+ regulatory T cells during acute graft-versus-host disease. Blood. 2005;106(9):3300. doi: 10.1182/blood-2005-04-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou L, Barnett B, Safah H, et al. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 2004;64(22):8451. doi: 10.1158/0008-5472.CAN-04-1987. [DOI] [PubMed] [Google Scholar]
- 33.Kristensen NN, Gad M, Thomsen AR, Lu B, Gerard C, Claesson MH. CXC chemokine receptor 3 expression increases the disease-inducing potential of CD4+ CD25- T cells in adoptive transfer colitis. Inflamm Bowel Dis. 2006;12(5):374. doi: 10.1097/01.MIB.0000217337.15442.e1. [DOI] [PubMed] [Google Scholar]