

Abstract
Background
While many of the contributing cell types and mediators of allergic asthma are known, less well understood are the factors that induce allergy in the first place. Amongst the mediators speculated to affect initial allergen sensitization and the development of pathogenic allergic responses to innocuous inhaled antigens and allergens are exogenously- or endogenously-generated reactive oxygen species (ROS) and reactive nitrogen species (RNS).
Scope of Review
The interactions between ROS/RNS, dendritic cells (DCs), and CD4+ T cells, as well as their modulation by lung epithelium, are of critical importance for the genesis of allergies that later manifest in allergic asthma. Therefore, this review will primarily focus on the initiation of pulmonary allergies and the role that ROS/RNS may play in the steps therein, using examples from our own work on the roles of NO2 exposure and airway epithelial NF-κB activation.
Major Conclusions
Endogenously-generated ROS/RNS and those encountered from environmental sources interact with epithelium, DCs, and CD4+ T cells to orchestrate allergic sensitization through modulation of the activities of each of these cell types, which quatitiatively and qualitatively dictate the degree and type of the allergic asthma phenotype.
General Significance
Knowledge of the effects of ROS/RNS at the molecular and cellular levels has the potential to provide powerful insight into the balance between inhalational tolerance (the typical immunologic response to an innocuous inhaled antigen) and allergy, as well as to potentially provide mechanistic targets for the prevention and treatment of asthma.
Keywords: asthma, epithelium, dendritic cells, CD4+ T cells, reactive oxygen species, reactive nitrogen species, NF-kappaB
Allergic asthma
Allergic asthma afflicts more than 23 million Americans (1) and costs the nation over 18 billion dollars annually (2), making it a primary public health concern. The incidence of asthma has been steadily rising in the United States over the past 20 years (3) and there are many potential reasons for this increase. Allergic asthma in humans is typically characterized by airway inflammation with eosinophils and lymphocytes, increased levels of Th2 cytokines, circulating IgE, airway goblet (mucus-producing) cell metaplasia, and airway hyperreponsiveness, which is defined as bronchoconstriction in response to inhalation of a specific (allergen) or non-specific (methacholine, cold air) agonist (4). Allergic asthma is the result of an inappropriate adaptive immune response to an inhaled antigen. Antigens include protein allergens with enzymatic activity or with structural features capable of inducing innate immune responses, innocuous protein antigens encountered in the presence of other agents capable of inducing innate immune responses (such as environmental oxidants and ambient particulate matter), and low molecular weight chemicals acting as haptens via their electrophilic properties that allow them to form a complex with proteins that are able to initiate an immune response. The immune response in allergic asthma is, driven primarily by CD4+ T helper type 2 (Th2) lymphocytes (4). Activation of Th2 cells has been shown to be both necessary (5) and sufficient (6) to induce all of the features of allergic asthma in mice. These Th2 cells produce IL-4, IL-5, and IL-13, resulting in IgE production, eosinophilia, and mucus production within the lung, respectively (4). A more recently described subset of CD4+ T helper cells, named Th17 cells, produce IL-17A, IL-17F, and IL-22, and seem to be involved in severe asthma involving neutrophilia (7). Despite knowing much about effector mechanisms in allergic asthma that promote airway hypersensitivity, eosinophilia, IgE, and mucus production, the mechanisms that allow for initial allergen sensitization are poorly understood. Sensitization, the act or process of inducing an acquired allergy, requires activation of innate immune cells, such as dendritic cells, that are then capable of activating naïve CD4+ T cells (8) and influencing their differentiation into Th2 or Th17 cells.
Mouse models of allergic asthma
Mouse models of allergic asthma enable investigation of the genesis of allergic sensitization because of the ability to manipulate both the mouse genome and environment. The most extensively employed protocol for eliciting symptoms of allergic asthma in mice involves the use of the Th2-skewing adjuvant, aluminum hydroxide (Alum), and the antigen ovalbumin (Ova), which are combined and administered to the mice by an intraperitoneal (i.p.) injection. The animals are then ‘challenged’ with aerosolized or intranasal Ova and subsequently analyzed. Pulmonary and immunologic alterations induced by antigen challenge include the production of the Ova-specific Th2 immunoglobulins IgE and IgG1, Th2 cytokines, including IL-4, IL-5, and IL-13, mucus production and airway hyperresponsiveness (9). While this model produces robust allergic sensitization and manifestation of features of allergic asthma similar to those observed in humans, it is limited in that it does not recapitulate the genesis of allergic asthma in humans, as allergen sensitization most often occurs via inhalation. Therefore, over the past several years, additional models of allergic asthma have been developed that involve inhalational sensitization to authentic allergens such as house dust mite (10) or Apergillus fumigatus (11), low molecular weight electrophilic chemicals (reviewed in (12)), or the antigen Ova encountered in the lung accompanied by environmental molecules with adjuvant-like activities (9, 13–26), since innocuous inhaled antigens alone, such as Ova, normally induce inhalational tolerance (27).
The recent revelation that, in addition to the well-described effects of Th2 cells in allergic asthma, Th17 cells contribute to a severe form of the syndrome (7) associated with a steroid-unresponsive asthma phenotype in mouse models (28) has altered the view of how CD4+ T cell populations dictate the pathology of allergic asthma and how IL-17-producing cells are generated. There is considerable plasticity in CD4+ T cells, and no longer are they and their progeny considered to be as committed to a specific phenotype as was once thought (29). IL-17-producing CD4+ T cells can be generated in a number of ways but are strongly influenced by inflammatory cytokines, including IL-1β (30–35).
Exogenous sources of oxidants that contribute to the pathogenesis of allergic asthma
By definition, an oxidant is a chemical compound that readily transfers oxygen atoms, or gains electrons in a redox chemical reaction. In biological systems, some of these oxidants typically have an oxygen- or nitrogen-based unpaired electron. Classical examples of these are O2•−.(radical anion superoxide), •OH (hydroxyl radical), and.• NO (nitric oxide). Their reaction with metals, other oxidants, and reductants found both in the atmosphere and in the intracellular milieu generates many other reactive species. Because of the complex chemistry in which these species are involved, the terms reactive oxygen species (ROS) and reactive nitrogen species (RNS) will be used in this review to refer to the species that are derived from oxygen or nitrogen, respectively.
ROS and RNS most certainly contribute to the pathological features of asthma, from inflammation, to bronchoconstriction, to remodeling. A recent review by Comhair and Erzurum (36) elegantly details the pro- and antioxidant systems in the lung and the mechanisms by which oxidants modulate the pathophysiology of asthma. Less well understood is how and why in an otherwise healthy lung, a cascade of events is initiated to allow allergens or innocuous inhaled antigens to initiate an allergic reaction that can later, upon subsequent reexposure to antigen, manifest in the pathophysiological features of allergic asthma. ROS and RNS may contribute substantially to this process as well, either through enhanced exposure or generation, or through deficiencies in major lung antioxidant systems, such as glutathione or superoxide dismutase (36). A typical example of enhanced exposure to ROS/RNS is in the case of nitrogen dioxide (NO2). NO2 is a pollutant generated during combustion processes, such as motor vehicle exhaust and biomass burning, and can be visualized as a reddish-brown layer over urban areas (37). Since there is constant growth in industrial the centers around the world, it is not surprising that the levels of tropospheric NO2 levels are also on the rise globally (38). Concentrations of NO2 above 5ppm cause lung damage (39, 40), whereas lower concentrations (100–400ppb) contribute to poor respiratory health (41) and exacerbate existing asthma (42, 43). In mouse models of allergic asthma, exposure to NO2 increases both the degree and duration of the allergic inflammatory response (44). Additionally, human asthmatics experience an enhanced reaction to inhaled allergen in the presence of NO2 (45) and living in areas with high ambient NO2 concentrations is correlated with an increased likelihood for developing asthma (46). With the rise in allergic asthma (3) and ambient levels of NO2 reaching 2ppm in certain settings, such as those with substantial industrialization and heavy motor vehicle usage (47), understanding the mechanisms underlying the correlation between NO2 exposure and respiratory wellbeing is of direct interest to public health. Other environmental oxidants, especially ozone and ambient particles that possess the capacity to redox cycle and themselves generate ROS and RNS, also contribute to the pathogenesis of allergic asthma (13–17, 21–25).
Endogenous sources of ROS and RNS
In addition to exposure originating from exogenous sources, ROS and RNS are also encountered as a consequence of endogenous production. ROS generated in the lung include O2•−, •OH, and hydrogen peroxide (H2O2), whereas RNS include •NO, peroxynitrite (ONOO−), and NO2. ROS and RNS generation in the lung is typically induced as part of a defensive reaction intended to clear infectious and environmental threats to homeostasis, including inhaled microbial agents, particles, and gasses. The generation of ROS and RNS is initiated by both cells of non-hematopoietic and hematopoietic origin. The airway and alveolar epithelium, along with alveolar macrophages, constitute a first line of defense against inhaled material and are themselves capable of ROS and RNS production. The resident and inflammatory hematopoietic-derived cells in the lung possess oxidant-generating enzyme systems, including NADPH oxidase, activity of which is mediated through the catalytic subunit gp91phox (NOX2) (48). This NADPH oxidase catalytic subunit functions in a coordinated manner with other oxidase subunits and the GTPase Rac1 to assemble a functional holoenzyme capable of generating the ROS, O2•−, which spontaneously or enzymatically dismutates to H2O2 to further induce oxidation. More recently, the important role of non-hematopoietic ROS generation has become appreciated, which is mediated by a plethora of enzymes, including the non-phagocytic oxidase subunits, NOX1, NOX3, and NOX4, which function distinct from gp91phox to generate O2•− (49). In addition, epithelial cells have recently been described to produce active Duox enzymes capable of basally and inducibly generating hydrogen peroxide (H2O2) (49). •NO is produced by the respiratory epithelium, neutrophils, and macrophages in response to viral infection via the inducible nitric oxide synthase (iNOS) (50). Induction of iNOS and an increased concentration of •NO in exhaled breath have been observed in patients with asthma, correlating •NO with inflammation (50). In response to infection, neutrophils also produce O2•− via NADPH oxidase (51). •NO and O2•− combine in the lung (52) to form ONOO−, which ultimately can produce molecules with a reactivity profile similar to that of NO2 (53). •NO is highly diffusible, allowing it to potentially form ONOO− in areas spatially separated from the site of •NO synthesis, limited only by its potent capacity to react with macromolecules (54). In addition, the reaction of •NO with molecular oxygen (O2) yields nitrite (NO2−), which can be oxidized by hemeperoxidases to form NO2, thereby perpetuating the capacity for NO2 reactivity. NO2 is also formed when eosinophil peroxidase and myeloperoxidase, from eosinophils and neutrophils, respectively, consume •NO and H2O2 (53, 55). Epidemiologic data suggest that Respiratory Syncytial Virus (RSV) infection early in life increases the risk of developing asthma (56), providing a potential correlation between •NO/NO2 generation and increased allergic sensitization. Furthermore, animal data suggest that respiratory viral infections (•NO/NO2 production) and increased asthma are causally related, with the viral infection acting on pulmonary leukocytes and structural cells to enhance antigen presentation and inflammatory cell recruitment (57). Inhaled NO2 primarily interacts with airway surface macromolecules, forming stable footprints of reactivity including the protein tyrosine modifications nitrotyrosine and dityrosine (53) that can alter protein function. In addition, ONOO− or NO2 can decompose to form •HO and H2O2, which can facilitate further oxidation and participate in intracellular signaling events. Therefore, exogenous or endogenously-generated ROS and RNS may directly and indirectly affect pulmonary cells to participate in the processes of allergic sensitization leading to allergic asthma.
Nitrogen dioxide-promoted allergic sensitization
Our laboratory developed a mouse model of NO2 exposure followed by inhalation of Ova to study the effects of ROS and RNS on allergic sensitization. We have reported that NO2 acts as an adjuvant, promoting the development of allergic asthma features including airway eosinophilia, airway hyperreponsiveness, antigen-specific IgE and IgG1, and antigen-specific CD4+ T cells that exhibit a biased Th2 (9) and Th17 cytokine profile (19). We have also shown that NO2 inhalation substantially impacts pulmonary CD11c+ dendritic cells, as shown by increased cytokine production, upregulation of maturation markers, increased antigen uptake, migration to the lung-draining lymph node, and improved ability to stimulate naïve CD4+ T cells (19). Additionally, CD11c+ cells are critical for NO2-promoted allergic sensitization, as depletion of these cells during sensitization diminishes multiple features of allergic asthma in mice (19). It is now appreciated that certain environmental agents, including those such as NO2 and ozone (20, 21) that are themselves ROS and RNS, as well as respiratory viral infections (57) and ambient particles that can induce the endogenous generation of ROS and RNS (13–17, 22, 23, 25), are capable of functioning as adjuvants, promoting pulmonary allergic responses. Furthermore, gaseous pollutants have also been shown to augment the allergenicity of certain allergens (45) through oxidative modification of proteins, suggesting that ROS and RNS also have the capacity to alter antigens themselves in addition to enhancing cell responses to them. However, to initiate an allergic response requires a coordinated interplay between several cell types in the lung and draining lymph node, each of which are affected by and capable of generating ROS and RNS.
Pulmonary dendritic cells
Dendritic cells (DCs) form a complex network along the airway and in the alveolus that interdigitate between epithelial cells to allow sampling of the airspace lumen to ingest and process inhaled and endogenously-generated material (58). In this manner, DCs can be directly activated by respirable materials, including microbes, oxidant gasses, and particulate matter. DCs are strongly implicated as having a causal role in allergic asthma due to their potent ability to activate naïve CD4+ T cells, thereby serving an essential role in allergen sensitization (59). A unique attribute of airway mucosal dendritic cells is their rapid turnover under homeostatic conditions (58) and their capacity for rapid increase following a local inflammatory response (60). This pulmonary DC pool is comprised of both myeloid DCs (mDC) and plasmacytoid DCs (pDC), but the myeloid subset generally dominates in airway mucosa (61). Immature airway mucosal dendritic cells (AMDCs) are highly phagocytic and are strategically positioned for antigen uptake both within and directly beneath the epithelium, surveying the lung through prosthetic extensions into the airway lumen (8, 62, 63). Upon stimulation of specific receptors by microbe-associated molecular patterns or endogenous molecules, the DCs mature, lowering their phagocytic capacity and increasing expression of MHCII and the co-stimulatory molecules required for naïve CD4+ T cell activation (64). Activated DCs home to the draining lymph node where antigen presentation to naïve T cells occurs in the context of complex lymphoid architecture that provides microenvironmental support for the expansion of the antigen-specific CD4+ T cell populations (59).
Multiple subsets of DCs have been identified, having both distinct and overlapping functions. In the lung, it can be challenging to distinguish these different DC subsets from one another, as well as to distinguish DCs from pulmonary macrophages, as they share many cell surface markers routinely used for flow cytometry. A recent review by Jakubzick and Randolph elegantly describes methods for pulmonary DC extraction, tracking, and identification using flow cytometry (65). Mouse myeloid DCs are classified by the expression of CD11c (the integrin-αx) and moderate to high levels of MHCII, which can be further elevated after activation (64). These features also correlate with their potent ability to induce T cell proliferation (64) and polarization. In support of a role for myeloid DCs in allergen sensitization, Ova-pulsed myeloid DCs administered intratracheally induce allergic sensitization in mice (66). Plasmacytoid DCs (pDCs), another subset of DCs found within the lung (67), have been shown to have an anti-inflammatory role, decreasing both the ability of mDCs to generate effector T cells as well as inducing the proliferation of T-regulatory cells (Tregs) (68). This pDC population expresses B220 (CD45RB) as well as low levels of Gr-1, CD11c, and MHC II (64). In pDC-depleted mice, accomplished by the administration of an anti-Gr-1 antibody, inhalational tolerance to Ova is abolished, but can be re-established with the adoptive transfer of FMS-related tyrosine kinase 3 ligand (FLT3L) cultured bone marrow derived pDCs (68). Also, the Th2 immune response elicited to RSV infection is exacerbated when pDCs are depleted (69). It is thought that pDCs may convert to mDCs during early viral infection, reducing the number of pDCs in the lung (56). These data suggest that pDCs are important in maintaining tolerance in the lung, implying that depletion of or damage to these cells could lead to aberrant immune responses, such as that seen in allergy.
Redox regulation of DC and T lymphocyte activities
DCs are capable of skewing the T helper cell response through expression of distinct patterns of co-stimulatory molecules as well as the production of cytokines that create an environment for T cell polarization (70). Expression of the co-stimulatory molecules CD86 and OX40L have been shown to promote naïve CD4+ T cells to develop a Th2 phenotype (70–72). DCs further regulate Th2 cell differentiation and expansion through the production of IL-6 (73, 74). Th17 cells may also be induced by the production of IL-6 in combination with TGFβ or by IL-23 (73, 75, 76), IL-1 (30–35), or prostaglandin E2 (PGE2) (77–80), whereas IL-12 alone results in a Th1 response (81). Thus, DCs are critical regulators of CD4+ mediated T cell responses through their capacity to present antigen in the draining lymph node, provide co-stimulation, and secrete polarizing cytokines. ROS can participate in the induction of Th2 responses through several cellular and molecular mechanisms. In response to injection of cysteine proteases, often an enzymatic component of allergens that enable activation of innate immune responses, ROS are generated by dermal epithelium and DCs, as well as by DCs in draining lymph nodes. These ROS inhibit IL-12 production and induce the recruitment to lymph nodes of basophils that provide IL-4 to drive Th2 responses (82). Furthermore, depletion of the major antioxidant, glutathione, causes antigen-presenting cells to preferentially induce Th2 responses and decrease Th1 responses (83). In contrast, it has been demonstrated that augmenting intracellular thiols via treatment with N-acetylcysteine (NAC) inhibited Th1 and enhanced Th2 responses via the modulation of antigen-presenting cell activities. Apparently this effect is not due increases in intracellular glutathione levels (84). Whereas ROS are generally considered capable of contributing to the induction of allergic responses (85), this is not strictly the case. For example, the ROS, O2•−, but not H2O2, can act on DCs to induce their maturation (86). This study underscores the selectivity of specific ROS in the activities that they modulate. An additional signal proposed to be critical for naïve CD4+ T cell stimulation is the generation of ROS and RNS from activated antigen-presenting cells (APCs), which results in enhanced adaptive immune immune responses. On the other hand, inhibition of ROS and RNS production by APCs using catalytic antioxidants (87) or selenium-containing antioxidant ebselen (88), results in substantially diminished T cell stimulation and cytokine production, consequently attenuating the immune response.
Efficient antigen uptake and processing are critical for the initiation of adaptive immune responses. In this regard, DCs are particularly well-equipped to sample the external cellular environment, taking up antigenic material (including proteins) via phagocytosis/endocytosis and proteolytic processing into peptide fragments appropriate for presentation to CD4+ T cells via MHCII or for cross-presentation to CD8+ T cells via MHCI. In phagocytic neutrophils and macrophages, antigen proteolysis is a very efficient process accomplished by lysosomal proteases in the fused phagolysosome that have an optimal activity at acidic pH. In these cells, proteins are rapidly processed to complete degradation, thereby providing proficient microbicidal activity. Despite the fact that protons required for endosomal acidification can be consumed through the activity of NADPH oxidase during the oxidative burst in neutrophils and macrophages, this process is limited in duration and efficient proteolysis continues nearly unabated. In contrast, phagocytic dendritic cells induce NADPH oxidase (NOX2) assembly on the phagolysosome membrane to generate O2•− and the decomposition product H2O2 over a protracted timecourse that effectively prevents acidification of the phagolysosome (89). As a consequence, the DCs more effectively present antigenic peptides to T cells when NADPH oxidase activity is unhindered (89). Therefore, ROS substantially affect the capacity of DCs to function optimally and enhanced NADPH-dependent ROS generation in these cells may augment antigen presentation activity and may allow for allergic sensitization to occur.
ROS, especially O2•− and H2O2, are also generated by T lymphocytes themselves upon T cell-receptor (TCR) engagement (88, 90–92). T cells have been demonstrated to express functional NADPH oxidase, which is required for optimal T cell activation (93). A recent report described that T cells from mice with a p47phox mutation that prevents NOX assembly and activity exhibit a Th17-skewed phenotype upon stimulation (94). Furthermore, these NOX-deficient mice were more prone to developing a Th17-mediated autoimmune disease and were protected from developing a Th1-mediated autoimmune disease (94), although their susceptibility to developing allergic airway disease was not examined. Nonetheless, this study clearly provides compelling support for the important role of intrinsic NOX regulation of T cell differentiation. Indicative of their central roles in the induction of T cell responses, specific ROS and RNS participate in APC maturation, modulate the capacity of APCs to stimulate CD4+ T cells, and are generated within T lymphocytes during their activation and modulate polarization. Thus, ROS and RNS may allow for antigen sensitization via directly or indirectly regulating the activities of DCs and T lymphocytes. In light of what is known about ROS and RNS in these cells and activation/differentiation pathways, there is clearly much need for additional studies that address ROS and RNS effects during allergic sensitization.
Recognition of pulmonary danger and damage to induce allergy
DC maturation occurs mainly in response to recognition of Microbe Associated Molecular Patterns (MAMPs) by Pattern Recognition Receptors (PRRs), such as the Toll-like Receptors (TLRs) (95). Stimulation of TLRs promotes the activation of NOX enzymes leading to the generation of ROS (96), especially H2O2, which can directly modify proteins and modulate intracellular signaling (36). Mature DCs can also generate RNS, such as ONOO−, that can cause protein tyrosine nitration and modification of tryptophans, leading to presentation of chemically-modified antigenic peptides that are recognized by a distinct population of CD4+ T cells that does not recognize unmodified antigen (97). While many TLR ligands are derived from infectious microorganisms (98), it has more recently been recognized that some PRRs are stimulated by endogenous ‘danger’ signals, such as products released from necrotic cells, including heat shock proteins, uric acid, and ATP (95, 99–101). These Danger Associated Molecular Patterns (DAMPs) are also capable of inducing DC maturation (102). Under conditions of cellular stress, ATP can be released by lytic and non-lytic mechanisms from many cell types, including structural and inflammatory cells (95, 103). While little is known about the immunologic contribution of ATP in the asthmatic airway, it has been shown to modify the recruitment and activation state of myeloid dendritic cells (95). Furthermore, intracellular proteins such as IL-1α and high mobility group box (HMGB)1 (104, 105) have potent functions as cytokines when released from damaged and dying cells, acting as “alarmins” to promote DC maturation and CD4+ T cell polarization (106). Similarly, IL-33 functions as a “necrokine” to activate the T1/ST2 receptor on CD4+ T cells and facilitate the secretion of Th2 cytokines (104, 107). These data show that endogenous molecules, released by damaged or ‘alarmed’ cells, may lead to the activation of DCs and Th2 cells. Since ROS and RNS are known to induce cell death when encountered at high concentrations, release of alarmins and necrokines may provide a potential mechanism through which oxidative stress may participate in allergic sensitization.
More recently, another class of PRRs, the NOD-Like Receptors (NLRs), has become appreciated for its role in recognition of exogenous and endogenous molecules, as well as for the generation of inflammatory and adaptive immune responses. NLRs respond to microbial molecules and endogenous danger signals within the cytosol (108). In particular, NOD-Like Receptor containing a Pyrin domain (NLRP)3 (NALP3, cryopyrin) is one of several NLRs involved in the assembly of a multi-protein scaffold, termed the inflammasome, which ultimately activates caspase-1 and allows for the cleavage and secretion of IL-β and IL-18 in itstheir active forms (109). The best-studied inflammasome-assembling platform is that comprised of the NOD-Like Receptor containing a Pyrin domain (NLRP)3, which recruits caspase-1 via an adaptor protein termed Apoptosis-related Speck-like complex (ASC), also known as Pycard because it contains both a pyrin and a Caspase Activation and Recruitment Domain (CARD), which can interact with the pyrin and CARD domains of NLRP3 and caspase-1, respectively, through intermolecular homotypic protein-protein interactions (110). NLRP3 is considered to be a cytoplasmic sensor of infectious, environmental, and endogenous molecules that have gained inappropriate access to this intercellular compartment. These molecules include products of intracellular microbes or those that have escaped the endocytic machinery, the consequences of endosome rupture and release of active cathepsins by crystalline or amyloidogenic particles, or those induced subsequent to ATP-dependent activation of the purinergic P2×7 membrane receptor and the opening of larger pannexin channels (108, 110, 111). Additionally, the commonly used adjuvant, Alum, is now recognized to principally function to induce Th2 responses through activation of the NLRP3 inflammasome, leading to the production of IL-1β, the generation of an inflammatory response, CD11c+ DC maturation, and CD4+ T cell proliferation (112, 113). Intraperitoneal injection of Alum/Ova induces a rapid accumulation of inflammatory monocytes in the peritoneal cavity (113), which are immediate precursors for DCs (114, 115), followed by a significant increase in the number of myeloid DCs (113). In addition to recruitment of these cells, a strong neutrophilic response was seen, associated with an increase in CXCL1 (KC), CCL2 (MCP-1), IL-1β, and IL-18, similar to the response seen when the endogenous danger signal uric acid is injected into the peritoneal cavity (116, 117). Alum, similar to or through the generation of uric acid, activates caspase-1 and leads to the release of IL-1β, providing a potential mechanism for inflammatory DC activation (113). Furthermore, injection of Alum promoted antigen uptake by the recruited monocytes and induced their conversion to CD11c+ cells in lymph nodes, which were required for allergic sensitization (113).
The activation of NLRP3 by such a wide array of seemingly dissimilar molecules has led to the proposal that there may be a common mechanism involved. Many of the agents that induce NLRP3 inflammasome activation also cause ROS and RNS generation, which are beginning to be considered important for NLRP3 inflammasome activation. It has recently been reported that P2×7 is required for allergic sensitization leading to contact dermatitis (118), which involves IL-1β secretion subsequent to TLR2, TLR4, and NLRP3 activation. It is well documented that activation of P2×7 is accompanied by production of ROS, produced at least in part by NADPH oxidases (119). Several studies using antioxidants support a model in which ROS production by NLRP3 agonists drive inflammasome assembly (120). However, the mechanisms of production and the nature of ROS and RNS involvement in inflammasome activation remain the subject of intense scrutiny. It was recently demonstrated (121) that NLRP3 can be directly regulated by thioredoxin inhibitory protein (TXNIP), which itself modulates activity of the intracellular antioxidant protein thioredoxin (TRX). In resting cells, TXNIP interacts with TRX and is therefore unable to activate NLRP3. Upon oxidative stress, TXNIP is released from oxidized TRX and in turn directly binds the leucine-rich repeat region of NLRP3 leading to inflammasome assembly and activation. Mitochondria represent an additional source of intracellular ROS that contribute to NLRP3 activation (122). Intriguingly, mitochondrial dysfunction has been reported in models of allergic asthma (123, 124) and can be induced by exposure to environmental oxidants, including ozone (125) and ultrafine particles (126). Whereas oxidative stress may facilitate NLRP3 activation (120, 127), there are also conflicting data that suggest the early oxidative stress must be balanced with a later antioxidant effect to facilitate IL-1β secretion (128). These reports implicate that NLRP3 is redox modulated in a biphasic manner. Understanding the biochemical events involved in NLRP3 activation is applicable to–allergic asthma because Alum, the best-studied adjuvant used to promote Th2 responses, functions–in part through its capacity to activate the NLRP3 inflammasome (113, 129). In addition, since NLRP3 activation regulates the secretion of IL-1β, an important cytokine that enables naïve (34), Foxp3+ regulatory (30, 33), effector/memory Th2 (32) CD4+ T cells, and γδT cells (35) to generate IL-17 and other Th17-associated cytokines, the potential for control of NLRP3 by ROS and RNS is an emerging research area that may be important for understanding the pathogenesis of severe allergic asthma.
Airway epithelium in allergic sensitization
In addition to DCs and macrophages, epithelial cells are also known to express PRRs, and may also play a significant role in the detection of microbial infection, danger (such as by inhaled oxidant gasses and respirable particulate matter), and cellular damage through release of pro-inflammatory cytokines and chemokines that activate other innate and adaptive immune cells (130). The airway epithelium is capable of influencing DC recruitment, maturation, and CD4+ T cell polarization by producing various cytokines and chemokines that promote allergic sensitization and subsequent inflammation. For instance, airway epithelium constitutively and inducibly expresses IL-6 (131), a cytokine that can promote Th2 polarization of CD4+ T cells (73, 74). IL-6-deficient mice exhibit diminished IL-5, IL-13, and IL-17 expression, as well as decreased mucus cell metaplasia in an Aspergillus fumigatus model of allergic asthma (132). In response to inflammatory stimuli, airway epithelial cells (AECs) secrete chemokines to recruit DCs (133, 134), including CCL20 (MIP-3α) and β-defensins, known ligands of CCR2/CCR6 present on immature DCs (67). It has recently been demonstrated that, in mice, CCR2 is a predominant receptor involved in recruiting Th2 inducing DCs to the lung (135).
AECs can also produce DC-activating cytokines including thymic stromal lymphopoietin (TSLP), GM-CSF, IL-1, IL-33, osteopontin, and IL-25 (67). TSLP is produced by epithelial cells in the lungs, gut, and skin (136). Recent work has shown that TSLP levels are increased at sights of inflammation, such as in the airway epithelium of asthmatics (137, 138). TSLP is transcriptionally regulated by NF-κB, and its production by human AECs is inducible by IL-1β and TNF-α (139). TSLP also activates human CD11c+ myeloid DCs (136, 140). These TSLP-activated DCs induce an inflammatory Th2 response through increasing expression of the co-stimulatory molecule OX40L (141). CD4+ T cells primed by TSLP-treated DCs produce Th2 cytokines, including IL-4, 5, and 13, indicating a role for TSLP in promoting Th2 allergic responses (140). Lastly, when TSLP is administered to mice intranasally, lymphocytes accumulate in the lung with an associated spike in Th2 cytokines and circulating IgE (142). This TSLP-driven inflammation is dramatically reduced by the administration of anti-OX40L–blocking mAb, indicating that OX40L is important in TSLP-driven atopic inflammation (142). Following allergen challenge, AECs also produce IL-25, a member of the IL-17 cytokine family that drives Th2 cell differentiation (143). Blocking IL-25 reduces airway inflammation and Th2 cytokine production in an allergen-induced asthma model using Aspergillus oryzae (143). Furthermore, IL-25 augments Th2 responses by enhancing the function of Th2 memory cells, thereby increasing the magnitude and duration of allergic inflammation (143).
It was recently demonstrated using irradiated chimeric mice that house dust mite (HDM) extracts induce allergic asthma via TLR4 expressed on airway structural cells, not DCs (144). The absence of TLR4 on structural cells, but not on hematopoietic cells, abolished HDM-driven allergic airway inflammation. Signaling through TLR4 on airway structural cells resulted in production of the innate pro-allergic cytokines TSLP, GM-CSF, IL-25 and IL-33 (144). Thus, it is possible that allergic sensitization in the context of ROS and RNS occurs due to the inappropriate activation of AECs, leading to production of cytokines and chemokines capable of recruiting and activating mDCs. Interestingly, it was also recently reported that the Derp2 allergen in HDM extract has structural homology to the TLR4-associating molecule, MD-2, and can interact with TLR4 directly and not as a consequence of contaminating endotoxin (145). As a consequence, Derp2 is able to initiate TLR4-dependent signals necessary for allergic sensitization and eventual manifestation of allergic airway disease. These studies further underscore the interrelationships between the epithelium, DCs, and CD4+ T cells in the initiation of allergic sensitization.
NF-κB: a redox-regulated transcription factor
While there is a growing appreciation for the interplay between the lung epithelium and DCs in modulating allergic airway disease (67, 146–149), the role of ROS and RNS in these processes that facilitate allergic sensitization remains relatively understudied. ROS and RNS generation in the lung can be triggered by a multitude of agents and events that manifest in a) frank injury and easily-detectable macromolecule oxidation and nitration when at high levels or b) nuanced alterations in molecular and cellular function when at low-levels. In the later case, transient macromolecular alterations induced by ROS are reflected by induction of intracellular signaling cascades. Among one of the most early-described transcription factors modified in its activity as a consequence of ROS or RNS exposure is nuclear factor-kappaB (NF-κB). Dependent upon cell type and the concentration and type of ROS or RNS, NF-κB can become activated or repressed (150). Activation of both the canonical and non-canonical NF-κB cascade is typically manifest as a consequence of activation of an upstream kinase, termed an IkappaB kinase (IKK), which itself is inducibly phosphorylated by upstream kinases. It was described over two decades ago that exposure to H2O2 promotes NF-κB activity, as indicated by expression of NF-κB-regulated genes, NF-κB-dependent reporter gene expression assays, NF-κB nuclear translocation, inhibitor of NF-κB (IκB) phosphorylation and degradation, and IKK phosphorylation (150, 151). We have reported that NF-κB is active in the airway epithelium of mice exposed to NO2 (9, 152) and have found that NO2 exposure induces NF-κB activity in airway epithelial cells in vitro, as reflected by IKK activity and NF-κB-dependent reporter gene expression (our unpublished results).
Most interestingly, the effects of NO2 inhalation on initiation of an antigen-specific immune response (9, 19) are also manifest simply by activating NF-κB in the airway epithelium and exposing to inhaled Ova (153). For these studies, we have used a mouse in which IKK activity can be induced exclusively in non-ciliated airway epithelium, which in turn promotes transient NF-κB activation in those cells (154). While airway epithelial IKK and NF-κB activation are the initial signaling events in this model, there are most certainly additional cells and signaling cascades activated as a consequence of IKK transgene expression. As an example of the complex interplay between and within cells, similar to exposing mice to inhaled NO2, transiently activating NF-κB in airway epithelial cells induced a mixed Th2/Th17 immune response to the inhaled antigen, as well as eosinophilic, neutrophilic, and lymphocytic airway inflammation accompanied by methacholine hyperresponsiveness following aerosolized antigen challenge (153). These alterations took place at a time at which the transgene was no longer expressed in the airway epithelium, implicating the induced adaptive immune response as the causal pathogenic mediator at this time point. Furthermore, transient activation of airway epithelial NF-κB did not alter epithelial permeability or induce pulmonary expression of the Th2-skewing cytokines IL-6, TSLP, IL-25, or IL-33, but did induce expression of CCL20, GM-CSF, SAA3, and the Th17-supporting cytokine IL-23, as well as promote maturation and migration of pulmonary DCs (153).
It has been previously demonstrated that H2O2 modulates activity of the NF-κB pathway in airway epithelial cells, but target molecules directly modified by H2O2 were not identified (155). Later studies revealed that the NF-κB subunit, p65, as well as the regulatory kinase, IKKβ, are post-translationally modified by S-glutathionylation (156) or S-nitrosylation (157, 158), which are downstream effects of ROS and RNS. Therefore, exogenous and endogenously-generated oxidants participate in the initiation of allergic responses via both direct and indirect mechanisms. Whether ROS and RNS are directly or indirectly facilitating NF-κB activation as a consequence of NO2 inhalation or whether ROS and RNS are generated as a consequence of airway epithelial NF-κB activation remain to be explored in our models.
Modulation of DC responses to ROS and RNS
Immature dendritic cells can be recruited to the airway as a consequence of epithelial-derived signals that are released subsequent to exposure to environmental agents including allergens, particles, and gasses. Along with the release of chemotactic signals, epithelial cells can release ROS and RNS generated from a number of sources, including mitochondria and peroxisomes (159), as well as non-phagocytic oxidases (49), and nitric oxide synthases (160–162), which may modulate epithelial NF-κB activities and participate in the maintenance of epithelial barrier integrity and regulation of genes controlling the expression of DC chemokines. Recruited DCs are exposed to epithelial-derived ROS and RNS and themselves generate ROS and RNS to regulate a myriad of transcription factors, including NF-κB, that modulate DC maturation and CD4+ T cell activation. As an additional consequence of ROS and RNS generation and exposure, cells generally initiate a program to counter the detrimental effects of ROS and RNS by inducing expression of antioxidant genes. Several of these antioxidant genes are regulated at the transcriptional level by DNA sequences termed Antioxidant Response Elements (AREs), which are recognized by the basic leucine zipper transcription factor, Nuclear erythroid 2 p45-related factor 2 (Nrf2) (163). Nrf2 normally resides in the cytosol and responds during oxidative stress by detaching from its inhibitor, Kelch-like ECH-associated protein 1 (Keap1), translocating to the nucleus, and binding AREs to induce expression of antioxidant genes (163). Mice deficient in Nrf2 display remarkable sensitivity to the deleterious effects of ROS and exhibit an enhanced allergic response to pulmonary antigen challenge following allergic sensitization (164), including augmented activation of NF-κB. Whereas this study demonstrates the effects of oxidants in driving the pathology of allergic airway disease during allergen challenge, it has been further elucidated that the activities of Nrf2 modulate sensitivity to the genesis of allergic responses upon coexposure to antigen and ambient particles, which themselves induce the generation of ROS and RNS and activate Nrf2 in macrophages and epithelial cells (165), and are epidemiologically and mechanistically linked to the processes of allergic sensitization (163). Since ROS and RNS can modulate the maturation of DCs and shape the adaptive immune responses initiated as a consequence of DC activities, the role of Nrf2 in the response of DCs to ambient particulate matter was investigated. In these studies (166), the absence of Nrf2 led to increased oxidative stress, inflammatory cytokine production, and expression of maturation markers by DCs. Interestingly, Nrf2-deficient DCs displayed an enhanced capacity to promote Th2 polarization of antigen-specific naïve CD4+ T cells. Furthermore, the capacity of ragweed extracts, which can themselves induce ROS production through their own NADPH oxidase-like activity (167), to induce DC maturation is regulated by Nrf2 (168). These studies clearly implicate an important role for ROS and RNS, their intracellular sensing, and their detoxification in DCs for controlling inappropriate responses to inducers of allergic disease.
Concluding remarks
ROS and RNS are active participants in complex biological processes, including the allergic sensitization that predisposes to the development of allergic asthma. Whether ROS and RNS are encountered environmental factors or are generated endogenously, they can affect several steps involved in allergic sensitization (Figure 1). It is critical to be mindful that individual cells and multicellular organisms have developed intricate mechanisms through which to respond to and utilize ROS and RNS to modulate homeostasis and respond to threats. Therefore, generalized therapeutic and prophylactic approaches to modulate ROS and RNS generation and reactivity may not represent a realistic target to combat allergic asthma. Therefore, a better understanding of the sequence of events leading to allergic sensitization, the involvement of ROS and RNS, and the potential molecular targets of oxidative modification, may provide crucial knowledge for the future development of therapeutic interventions. Indeed, while antioxidant trials in asthmatic subjects have largely fallen short of the hypothesized benefit, there is some potential for dietary antioxidant supplementation, aimed at achieving the levels present in normal subjects, to have prophylactic effects in certain populations (169–171). In addition, the cells and intracellular signaling events affected by ROS and RNS may represent even more efficacious therapeutic targets. While several laboratories are working hard to understand these ROS- and RNS-mediated, as well as the ROS- and RNS-modulating mechanisms, additional efforts are required to successfully integrate the many processes involved in ROS and RNS regulation of allergic sensitization and asthma.
Figure 1. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) in allergic sensitization.
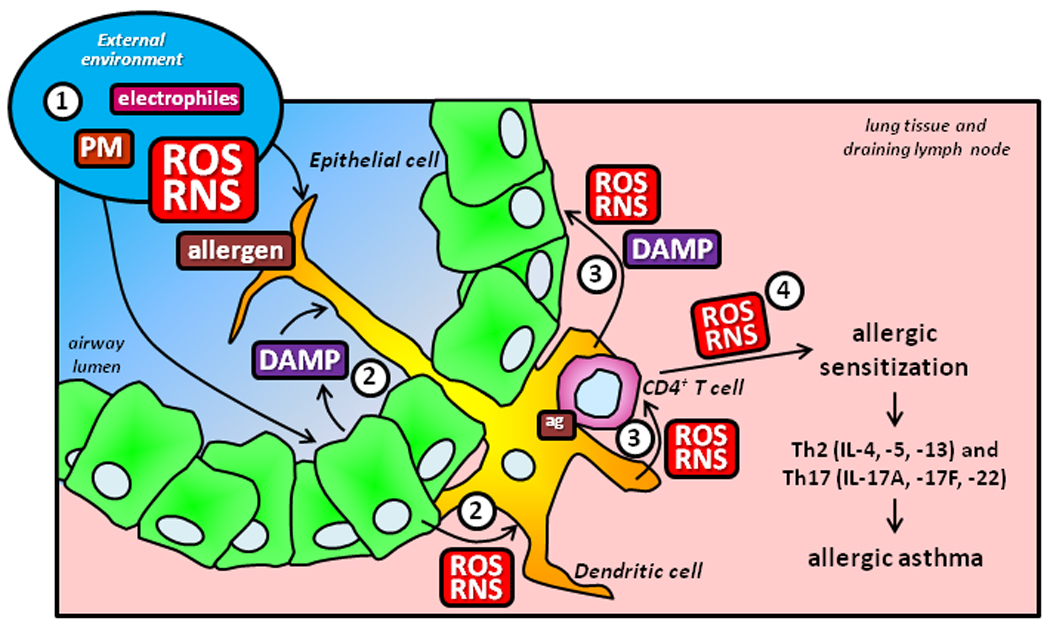
1) Environmental (gaseous, particle-associated, and allergen-generated) and endogenous (from resident and inflammatory cells or as a consequence of infection) ROS, RNS, and Danger-Associated Molecular Patterns (DAMPs, including respirable particulate matter and the products from damaged cells) in the lung may directly and indirectly affect resident pulmonary epithelial cells and dendritic cells, inducing their activation and the potential to generate additional ROS and RNS. 2) Mediators derived from activated epithelial cells, including cytokines and ROS/RNS enzymatically generated from NOXs and DUOXs, can also promote dendritic cell activation. 3) Activated dendritic cells undergo maturation and, in the presence of an environmental antigen, gain the capacity to present antigenic peptides and stimulate CD4+ T cells. Dendritic cell-derived ROS impact upon CD4+ T cell activation and can also further activate pulmonary epithelium, perpetuating the stimulatory state in the lung. 4) Activated CD4+ T cells use NOX-derived ROS as a component of the intracellular signaling cascade induced subsequent to TCR stimulation, signals that participate in their capacity to differentiate into Th2 and Th17 effector cells that secrete cytokine mediators contributing to the pathogenesis and pathophysiology of allergic asthma.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.United States Center for Disease Control. Summary Health Statistics for U.S. Adults: National Health Interview Survey. 2007 [Google Scholar]
- 2.Asthma and Allergy Foundation of America. The Costs of Asthma. 2000 [Google Scholar]
- 3.Mannino DM, Homa DM, Pertowski CA, Ashizawa A, Nixon LL, Johnson CA, Ball LB, Jack E, Kang DS. Surveillance for asthma--United States, 1960–1995. MMWR CDC Surveill Summ. 1998;47:1–27. [PubMed] [Google Scholar]
- 4.Herrick CA, Bottomly K. To respond or not to respond: T cells in allergic asthma. Nat Rev Immunol. 2003;3:405–412. doi: 10.1038/nri1084. [DOI] [PubMed] [Google Scholar]
- 5.Corry DB, Grunig G, Hadeiba H, Kurup VP, Warnock ML, Sheppard D, Rennick DM, Locksley RM. Requirements for allergen-induced airway hyperreactivity in T and B cell-deficient mice. Mol Med. 1998;4:344–355. [PMC free article] [PubMed] [Google Scholar]
- 6.Lambrecht BN, Hammad H. Taking our breath away: dendritic cells in the pathogenesis of asthma. Nat Rev Immunol. 2003;3:994–1003. doi: 10.1038/nri1249. [DOI] [PubMed] [Google Scholar]
- 7.Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol. 2010;72:495–516. doi: 10.1146/annurev-physiol-021909-135926. [DOI] [PubMed] [Google Scholar]
- 8.Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nat Rev Immunol. 2008;8:142–152. doi: 10.1038/nri2236. [DOI] [PubMed] [Google Scholar]
- 9.Bevelander M, Mayette J, Whittaker LA, Paveglio SA, Jones CC, Robbins J, Hemenway D, Akira S, Uematsu S, Poynter ME. Nitrogen dioxide promotes allergic sensitization to inhaled antigen. J Immunol. 2007;179:3680–3688. doi: 10.4049/jimmunol.179.6.3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke AH, Thomas WR, Rolland JM, Dow C, O'Brien RM. Murine allergic respiratory responses to the major house dust mite allergen Der p 1. Int Arch Allergy Immunol. 1999;120:126–134. doi: 10.1159/000024230. [DOI] [PubMed] [Google Scholar]
- 11.Paveglio SA, Allard J, Foster Hodgkins SR, Ather J, Bevelander M, Mayette Campbell J, Whittaker Leclair LA, McCarthy SM, van der Vliet A, Suratt BT, Boyson JE, Uematsu S, Akira S, Poynter ME. Airway Epithelial Indoleamine 2,3-Dioxygenase Inhibits CD4+ T Cells During Aspergillus fumigatus Antigen Exposure. Am J Respir Cell Mol Biol. 2010 doi: 10.1165/rcmb.2009-0167OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boverhof DR, Billington R, Gollapudi BB, Hotchkiss JA, Krieger SM, Poole A, Wiescinski CM, Woolhiser MR. Respiratory sensitization and allergy: current research approaches and needs. Toxicol Appl Pharmacol. 2008;226:1–13. doi: 10.1016/j.taap.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Arras M, Huaux F, Vink A, Delos M, Coutelier JP, Many MC, Barbarin V, Renauld JC, Lison D. Interleukin-9 reduces lung fibrosis and type 2 immune polarization induced by silica particles in a murine model. Am J Respir Cell Mol Biol. 2001;24:368–375. doi: 10.1165/ajrcmb.24.4.4249. [DOI] [PubMed] [Google Scholar]
- 14.Bleck B, Tse DB, Curotto de Lafaille MA, Zhang F, Reibman J. Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturation and polarization via thymic stromal lymphopoietin. J Clin Immunol. 2008;28:147–156. doi: 10.1007/s10875-007-9149-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan RC, Wang M, Li N, Yanagawa Y, Onoe K, Lee JJ, Nel AE. Prooxidative diesel exhaust particle chemicals inhibit LPS-induced dendritic cell responses involved in T-helper differentiation. J Allergy Clin Immunol. 2006;118:455–465. doi: 10.1016/j.jaci.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 16.de Haar C, Kool M, Hassing I, Bol M, Lambrecht BN, Pieters R. Lung dendritic cells are stimulated by ultrafine particles and play a key role in particle adjuvant activity. J Allergy Clin Immunol. 2008;121:1246–1254. doi: 10.1016/j.jaci.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Diaz-Sanchez D, Garcia MP, Wang M, Jyrala M, Saxon A. Nasal challenge with diesel exhaust particles can induce sensitization to a neoallergen in the human mucosa. J Allergy Clin Immunol. 1999;104:1183–1188. doi: 10.1016/s0091-6749(99)70011-4. [DOI] [PubMed] [Google Scholar]
- 18.Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J Exp Med. 2002;196:1645–1651. doi: 10.1084/jem.20021340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodgkins SR, Ather JL, Paveglio SA, Allard JL, LeClair LA, Suratt BT, Boyson JE, Poynter ME. NO2 inhalation induces maturation of pulmonary CD11c+ cells that promote antigen-specific CD4+ T cell polarization. Respir Res. 2010;11:102. doi: 10.1186/1465-9921-11-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koike E, Kobayashi T. Ozone exposure enhances antigen-presenting activity of interstitial lung cells in rats. Toxicology. 2004;196:217–227. doi: 10.1016/j.tox.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Koike E, Watanabe H, Kobayashi T. Exposure to ozone enhances antigen-presenting activity concentration dependently in rats. Toxicology. 2004;197:37–46. doi: 10.1016/j.tox.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 22.Lambert AL, Dong W, Selgrade MK, Gilmour MI. Enhanced allergic sensitization by residual oil fly ash particles is mediated by soluble metal constituents. Toxicol Appl Pharmacol. 2000;165:84–93. doi: 10.1006/taap.2000.8932. [DOI] [PubMed] [Google Scholar]
- 23.Lambert AL, Dong W, Winsett DW, Selgrade MK, Gilmour MI. Residual oil fly ash exposure enhances allergic sensitization to house dust mite. Toxicol Appl Pharmacol. 1999;158:269–277. doi: 10.1006/taap.1999.8709. [DOI] [PubMed] [Google Scholar]
- 24.Li N, Harkema JR, Lewandowski RP, Wang M, Bramble LA, Gookin GR, Ning Z, Kleinman MT, Sioutas C, Nel AE. Ambient ultrafine particles provide a strong adjuvant effect in the secondary immune response: implication for traffic-related asthma flares. Am J Physiol Lung Cell Mol Physiol. 2010;299:L374–L383. doi: 10.1152/ajplung.00115.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stevens T, Cho SH, Linak WP, Gilmour MI. Differential potentiation of allergic lung disease in mice exposed to chemically distinct diesel samples. Toxicol Sci. 2009;107:522–534. doi: 10.1093/toxsci/kfn248. [DOI] [PubMed] [Google Scholar]
- 26.Wilson RH, Whitehead GS, Nakano H, Free ME, Kolls JK, Cook DN. Allergic sensitization through the airway primes Th17-dependent neutrophilia and airway hyperresponsiveness. Am J Respir Crit Care Med. 2009;180:720–730. doi: 10.1164/rccm.200904-0573OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ostroukhova M, Seguin-Devaux C, Oriss TB, Dixon-McCarthy B, Yang L, Ameredes BT, Corcoran TE, Ray A. Tolerance induced by inhaled antigen involves CD4(+) T cells expressing membrane-bound TGF-beta and FOXP3. J Clin Invest. 2004;114:28–38. doi: 10.1172/JCI20509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKinley L, Alcorn JF, Peterson A, Dupont RB, Kapadia S, Logar A, Henry A, Irvin CG, Piganelli JD, Ray A, Kolls JK. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181:4089–4097. doi: 10.4049/jimmunol.181.6.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 30.Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee WW, Kang SW, Choi J, Lee SH, Shah K, Eynon EE, Flavell RA, Kang I. Regulating human Th17 cells via differential expression of IL-1 receptor. Blood. 2010;115:530–540. doi: 10.1182/blood-2009-08-236521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y-H, Voo KS, Liu B, Chen C-Y, Uygungil B, Spoede W, Bernstein JA, Huston DP, Liu Y-J. A novel subset of CD4+ TH2 memory/effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J Exp Med. 2010 doi: 10.1084/jem.20101376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L, Kim J, Boussiotis VA. IL-1{beta}-Mediated Signals Preferentially Drive Conversion of Regulatory T Cells but Not Conventional T Cells into IL-17-Producing Cells. J Immunol. 2010;185:4148–4153. doi: 10.4049/jimmunol.1001536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 36.Comhair SA, Erzurum SC. Redox control of asthma: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2010;12:93–124. doi: 10.1089/ars.2008.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.United States Environmental Protection Agency. NOx: How nitrogen oxides affect the way we live and breathe. Office of Air Quality Living and Standards. 1998 [Google Scholar]
- 38.Richter A, Burrows JP, Nuss H, Granier C, Niemeier U. Increase in tropospheric nitrogen dioxide over China observed from space. Nature. 2005;437:129–132. doi: 10.1038/nature04092. [DOI] [PubMed] [Google Scholar]
- 39.Barth PJ, Muller B, Wagner U, Bittinger A. Quantitative analysis of parenchymal and vascular alterations in NO2-induced lung injury in rats. Eur Respir J. 1995;8:1115–1121. doi: 10.1183/09031936.95.08071115. [DOI] [PubMed] [Google Scholar]
- 40.United States Department of Labor, Occupational Health and Safety Administration. OSHA Permissible Exposure Limit (PEL) for General Industry. 2004 29 CFR 1910.1000 Z-1. [Google Scholar]
- 41.Beelen R, Hoek G, van den Brandt PA, Goldbohm RA, Fischer P, Schouten LJ, Jerrett M, Hughes E, Armstrong B, Brunekreef B. Long-term effects of traffic-related air pollution on mortality in a Dutch cohort (NLCS-AIR study) Environ Health Perspect. 2008;116:196–202. doi: 10.1289/ehp.10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kattan M, Gergen PJ, Eggleston P, Visness CM, Mitchell HE. Health effects of indoor nitrogen dioxide and passive smoking on urban asthmatic children. J Allergy Clin Immunol. 2007;120:618–624. doi: 10.1016/j.jaci.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 43.Tunnicliffe WS, Burge PS, Ayres JG. Effect of domestic concentrations of nitrogen dioxide on airway responses to inhaled allergen in asthmatic patients. Lancet. 1994;344:1733–1736. doi: 10.1016/s0140-6736(94)92886-x. [DOI] [PubMed] [Google Scholar]
- 44.Poynter ME, Persinger RL, Irvin CG, Butnor KJ, van Hirtum H, Blay W, Heintz NH, Robbins J, Hemenway D, Taatjes DJ, Janssen-Heininger Y. Nitrogen dioxide enhances allergic airway inflammation and hyperresponsiveness in the mouse. Am J Physiol Lung Cell Mol Physiol. 2006;290:L144–L152. doi: 10.1152/ajplung.00131.2005. [DOI] [PubMed] [Google Scholar]
- 45.Strand V, Rak S, Svartengren M, Bylin G. Nitrogen dioxide exposure enhances asthmatic reaction to inhaled allergen in subjects with asthma. Am J Respir Crit Care Med. 1997;155:881–887. doi: 10.1164/ajrccm.155.3.9117021. [DOI] [PubMed] [Google Scholar]
- 46.Gauderman WJ, Avol E, Lurmann F, Kuenzli N, Gilliland F, Peters J, McConnell R. Childhood asthma and exposure to traffic and nitrogen dioxide. Epidemiology. 2005;16(6):737–743. doi: 10.1097/01.ede.0000181308.51440.75. [DOI] [PubMed] [Google Scholar]
- 47.Blomberg A, Krishna MT, Bocchino V, Biscione GL, Shute JK, Kelly FJ, Frew AJ, Holgate ST, Sandstrom T. The inflammatory effects of 2 ppm NO2 on the airways of healthy subjects. Am J Respir Crit Care Med. 1997;156:418–424. doi: 10.1164/ajrccm.156.2.9612042. [DOI] [PubMed] [Google Scholar]
- 48.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 49.van der Vliet A. NADPH oxidases in lung biology and pathology: host defense enzymes, and more. Free Radic Biol Med. 2008;44:938–955. doi: 10.1016/j.freeradbiomed.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Janssen-Heininger YM, Persinger RL, Korn SH, Pantano C, McElhinney B, Reynaert NL, Langen RC, Ckless K, Shrivastava P, Poynter ME. Reactive nitrogen species and cell signaling: implications for death or survival of lung epithelium. Am J Respir Crit Care Med. 2002;166:S9–S16. doi: 10.1164/rccm.2206008. [DOI] [PubMed] [Google Scholar]
- 51.Gaston B, Drazen JM, Loscalzo J, Stamler JS. The biology of nitrogen oxides in the airways. Am J Respir Crit Care Med. 1994;149:538–551. doi: 10.1164/ajrccm.149.2.7508323. [DOI] [PubMed] [Google Scholar]
- 52.Kato M, Hayashi Y, Kimura H. Oxygen radicals in inflammation and allergy related to viral infections. Curr Drug Targets Inflamm Allergy. 2005;4:497–501. doi: 10.2174/1568010054526377. [DOI] [PubMed] [Google Scholar]
- 53.Brennan ML, Wu W, Fu X, Shen Z, Song W, Frost H, Vadseth C, Narine L, Lenkiewicz E, Borchers MT, Lusis AJ, Lee JJ, Lee NA, Abu-Soud HM, Ischiropoulos H, Hazen SL. A tale of two controversies: defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidase-deficient mice, and the nature of peroxidase-generated reactive nitrogen species. J Biol Chem. 2002;277:17415–17427. doi: 10.1074/jbc.M112400200. [DOI] [PubMed] [Google Scholar]
- 54.Lancaster JR, Jr, Gaston B. NO and nitrosothiols: spatial confinement and free diffusion. Am J Physiol Lung Cell Mol Physiol. 2004;287:L465–L466. doi: 10.1152/ajplung.00151.2004. [DOI] [PubMed] [Google Scholar]
- 55.van der Vliet A, Eiserich JP, Shigenaga MK, Cross CE. Reactive nitrogen species and tyrosine nitration in the respiratory tract: epiphenomena or a pathobiologic mechanism of disease? Am J Respir Crit Care Med. 1999;160:1–9. doi: 10.1164/ajrccm.160.1.9807044. [DOI] [PubMed] [Google Scholar]
- 56.Hansbro NG, Horvat JC, Wark PA, Hansbro PM. Understanding the mechanisms of viral induced asthma: New therapeutic directions. Pharmacol Ther. 2008;117:313–353. doi: 10.1016/j.pharmthera.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Rijt LS, van Kessel CH, Boogaard I, Lambrecht BN. Respiratory viral infections and asthma pathogenesis: a critical role for dendritic cells? J Clin Virol. 2005;34:161–169. doi: 10.1016/j.jcv.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 58.Holt PG, Haining S, Nelson DJ, Sedgwick JD. Origin and steady-state turnover of class II MHC-bearing dendritic cells in the epithelium of the conducting airways. J Immunol. 1994;153:256–261. [PubMed] [Google Scholar]
- 59.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 60.Stumbles PA, Thomas JA, Pimm CL, Lee PT, Venaille TJ, Proksch S, Holt PG. Resting respiratory tract dendritic cells preferentially stimulate T helper cell type 2 (Th2) responses and require obligatory cytokine signals for induction of Th1 immunity. J Exp Med. 1998;188:2019–2031. doi: 10.1084/jem.188.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jahnsen FL, Moloney ED, Hogan T, Upham JW, Burke CM, Holt PG. Rapid dendritic cell recruitment to the bronchial mucosa of patients with atopic asthma in response to local allergen challenge. Thorax. 2001;56:823–826. doi: 10.1136/thorax.56.11.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 63.Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, Littman DR, Reinecker HC. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 64.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 65.Jakubzick C, Randolph GJ. Methods to study pulmonary dendritic cell migration. Methods Mol Biol. 2010;595:371–382. doi: 10.1007/978-1-60761-421-0_24. [DOI] [PubMed] [Google Scholar]
- 66.Lambrecht BN, De Veerman M, Coyle AJ, Gutierrez-Ramos JC, Thielemans K, Pauwels RA. Myeloid dendritic cells induce Th2 responses to inhaled antigen, leading to eosinophilic airway inflammation. J Clin Invest. 2000;106:551–559. doi: 10.1172/JCI8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol. 2008;8:193–204. doi: 10.1038/nri2275. [DOI] [PubMed] [Google Scholar]
- 68.de Heer HJ, Hammad H, Soullie T, Hijdra D, Vos N, Willart MA, Hoogsteden HC, Lambrecht BN. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J Exp Med. 2004;200:89–98. doi: 10.1084/jem.20040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, Kastner DL. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A. 2006;103:9982–9987. doi: 10.1073/pnas.0602081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lewkowich IP, Lajoie S, Clark JR, Herman NS, Sproles AA, Wills-Karp M. Allergen uptake, activation, and IL-23 production by pulmonary myeloid DCs drives airway hyperresponsiveness in asthma-susceptible mice. PLoS ONE. 2008;3:e3879. doi: 10.1371/journal.pone.0003879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deurloo DT, van Berkel MA, van Esch BC, Hofhuis F, Nijkamp FP, Oosterwegel MA, van Oosterhout AJ. CD28/CTLA4 double deficient mice demonstrate crucial role for B7 co-stimulation in the induction of allergic lower airways disease. Clin Exp Allergy. 2003;33:1297–1304. doi: 10.1046/j.1365-2222.2003.01757.x. [DOI] [PubMed] [Google Scholar]
- 72.Hoshino A, Tanaka Y, Akiba H, Asakura Y, Mita Y, Sakurai T, Takaoka A, Nakaike S, Ishii N, Sugamura K, Yagita H, Okumura K. Critical role for OX40 ligand in the development of pathogenic Th2 cells in a murine model of asthma. Eur J Immunol. 2003;33:861–869. doi: 10.1002/eji.200323455. [DOI] [PubMed] [Google Scholar]
- 73.Krishnamoorthy N, Oriss TB, Paglia M, Fei M, Yarlagadda M, Vanhaesebroeck B, Ray A, Ray P. Activation of c-Kit in dendritic cells regulates T helper cell differentiation and allergic asthma. Nat Med. 2008;14:565–573. doi: 10.1038/nm1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rincon M, Anguita J, Nakamura T, Fikrig E, Flavell RA. Interleukin (IL)-6 directs the differentiation of IL-4-producing CD4+ T cells. J Exp Med. 1997;185:461–469. doi: 10.1084/jem.185.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 76.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 77.Boniface K, Bak-Jensen KS, Li Y, Blumenschein WM, McGeachy MJ, McClanahan TK, McKenzie BS, Kastelein RA, Cua DJ, de Waal Malefyt R. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med. 2009;206:535–548. doi: 10.1084/jem.20082293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen H, Qin J, Wei P, Zhang J, Li Q, Fu L, Li S, Ma C, Cong B. Effects of leukotriene B4 and prostaglandin E2 on the differentiation of murine Foxp3+ T regulatory cells and Th17 cells. Prostaglandins Leukot Essent Fatty Acids. 2009;80:195–200. doi: 10.1016/j.plefa.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 79.Chizzolini C, Chicheportiche R, Alvarez M, de Rham C, Roux-Lombard P, Ferrari-Lacraz S, Dayer JM. Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood. 2008;112:3696–3703. doi: 10.1182/blood-2008-05-155408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Khayrullina T, Yen JH, Jing H, Ganea D. In vitro differentiation of dendritic cells in the presence of prostaglandin E2 alters the IL-12/IL-23 balance and promotes differentiation of Th17 cells. J Immunol. 2008;181:721–735. doi: 10.4049/jimmunol.181.1.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Macatonia SE, Hosken NA, Litton M, Vieira P, Hsieh CS, Culpepper JA, Wysocka M, Trinchieri G, Murphy KM, O'Garra A. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J Immunol. 1995;154:5071–5079. [PubMed] [Google Scholar]
- 82.Tang H, Cao W, Kasturi SP, Ravindran R, Nakaya HI, Kundu K, Murthy N, Kepler TB, Malissen B, Pulendran B. The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling. Nat Immunol. 2010;11:608–617. doi: 10.1038/ni.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peterson JD, Herzenberg LA, Vasquez K, Waltenbaugh C. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc Natl Acad Sci U S A. 1998;95:3071–3076. doi: 10.1073/pnas.95.6.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Monick MM, Samavati L, Butler NS, Mohning M, Powers LS, Yarovinsky T, Spitz DR, Hunninghake GW. Intracellular thiols contribute to Th2 function via a positive role in IL-4 production. J Immunol. 2003;171:5107–5115. doi: 10.4049/jimmunol.171.10.5107. [DOI] [PubMed] [Google Scholar]
- 85.Riedl MA, Nel AE. Importance of oxidative stress in the pathogenesis and treatment of asthma. Curr Opin Allergy Clin Immunol. 2008;8:49–56. doi: 10.1097/ACI.0b013e3282f3d913. [DOI] [PubMed] [Google Scholar]
- 86.Kantengwa S, Jornot L, Devenoges C, Nicod LP. Superoxide anions induce the maturation of human dendritic cells. Am J Respir Crit Care Med. 2003;167:431–437. doi: 10.1164/rccm.200205-425OC. [DOI] [PubMed] [Google Scholar]
- 87.Tse HM, Milton MJ, Schreiner S, Profozich JL, Trucco M, Piganelli JD. Disruption of innate-mediated proinflammatory cytokine and reactive oxygen species third signal leads to antigen-specific hyporesponsiveness. J Immunol. 2007;178:908–917. doi: 10.4049/jimmunol.178.2.908. [DOI] [PubMed] [Google Scholar]
- 88.Matsue H, Edelbaum D, Shalhevet D, Mizumoto N, Yang C, Mummert ME, Oeda J, Masayasu H, Takashima A. Generation and function of reactive oxygen species in dendritic cells during antigen presentation. J Immunol. 2003;171:3010–3018. doi: 10.4049/jimmunol.171.6.3010. [DOI] [PubMed] [Google Scholar]
- 89.Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, Seabra MC, Raposo G, Amigorena S. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. 2006;126:205–218. doi: 10.1016/j.cell.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 90.Devadas S, Zaritskaya L, Rhee SG, Oberley L, Williams MS. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J Exp Med. 2002;195:59–70. doi: 10.1084/jem.20010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol. 2002;3:1129–1134. doi: 10.1038/ni1202-1129. [DOI] [PubMed] [Google Scholar]
- 92.Williams MS, Kwon J. T cell receptor stimulation, reactive oxygen species, and cell signaling. Free Radic Biol Med. 2004;37:1144–1151. doi: 10.1016/j.freeradbiomed.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 93.Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol. 2004;5:818–827. doi: 10.1038/ni1096. [DOI] [PubMed] [Google Scholar]
- 94.Tse HM, Thayer TC, Steele C, Cuda CM, Morel L, Piganelli JD, Mathews CE. NADPH oxidase deficiency regulates Th lineage commitment and modulates autoimmunity. J Immunol. 2010;185:5247–5258. doi: 10.4049/jimmunol.1001472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, Muskens F, Hoogsteden HC, Luttmann W, Ferrari D, Di Virgilio F, Virchow JC, Jr, Lambrecht BN. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med. 2007;13:913–919. doi: 10.1038/nm1617. [DOI] [PubMed] [Google Scholar]
- 96.Basu S, Fenton MJ. Toll-like receptors: function and roles in lung disease. Am J Physiol Lung Cell Mol Physiol. 2004;286:L887–L892. doi: 10.1152/ajplung.00323.2003. [DOI] [PubMed] [Google Scholar]
- 97.Herzog J, Maekawa Y, Cirrito TP, Illian BS, Unanue ER. Activated antigen-presenting cells select and present chemically modified peptides recognized by unique CD4 T cells. Proc Natl Acad Sci U S A. 2005;102:7928–7933. doi: 10.1073/pnas.0502255102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, Liu YJ. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–869. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gu Y, Hershfield MS, Cohen A. The danger within. N Engl J Med. 2004;350:2721–2722. doi: 10.1056/NEJM200406243502622. author reply 2721–2722. [DOI] [PubMed] [Google Scholar]
- 100.Skoberne M, Beignon AS, Bhardwaj N. Danger signals: a time and space continuum. Trends Mol Med. 2004;10:251–257. doi: 10.1016/j.molmed.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 101.Adriaensen D, Timmermans JP. Purinergic signalling in the lung: important in asthma and COPD? Curr Opin Pharmacol. 2004;4:207–214. doi: 10.1016/j.coph.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 102.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114–119. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 103.Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5'-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. doi: 10.1016/j.pharmthera.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 104.Haraldsen G, Balogh J, Pollheimer J, Sponheim J, Kuchler AM. Interleukin-33 - cytokine of dual function or novel alarmin? Trends Immunol. 2009;30:227–233. doi: 10.1016/j.it.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 105.Yang D, Tewary P, de la Rosa G, Wei F, Oppenheim JJ. The alarmin functions of high-mobility group proteins. Biochim Biophys Acta. 2010;1799:157–163. doi: 10.1016/j.bbagrm.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Willart MA, Hammad H. Alarming dendritic cells for allergic sensitization. Allergol Int. 2010;59:95–103. doi: 10.2332/allergolint.09-RAI-0162. [DOI] [PubMed] [Google Scholar]
- 107.Zhao W, Hu Z. The enigmatic processing and secretion of interleukin-33. Cell Mol Immunol. 2010;7:260–262. doi: 10.1038/cmi.2010.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14:10–22. doi: 10.1038/sj.cdd.4402038. [DOI] [PubMed] [Google Scholar]
- 110.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 111.Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A. 2008;105:8067–8072. doi: 10.1073/pnas.0709684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kool M, Petrilli V, De Smedt T, Rolaz A, Hammad H, van Nimwegen M, Bergen IM, Castillo R, Lambrecht BN, Tschopp J. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol. 2008;181:3755–3759. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 113.Kool M, Soullie T, van Nimwegen M, Willart MA, Muskens F, Jung S, Hoogsteden HC, Hammad H, Lambrecht BN. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med. 2008;205:869–882. doi: 10.1084/jem.20071087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Randolph GJ, Inaba K, Robbiani DF, Steinman RM, Muller WA. Differentiation of phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 1999;11:753–761. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- 115.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 116.Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116:2262–2271. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 118.Weber FC, Esser PR, Muller T, Ganesan J, Pellegatti P, Simon MM, Zeiser R, Idzko M, Jakob T, Martin SF. Lack of the purinergic receptor P2×7 results in resistance to contact hypersensitivity. J Exp Med. 2010 doi: 10.1084/jem.20092489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010;40:616–619. doi: 10.1002/eji.200940168. [DOI] [PubMed] [Google Scholar]
- 121.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 122.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 123.Aguilera-Aguirre L, Bacsi A, Saavedra-Molina A, Kurosky A, Sur S, Boldogh I. Mitochondrial dysfunction increases allergic airway inflammation. J Immunol. 2009;183:5379–5387. doi: 10.4049/jimmunol.0900228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mabalirajan U, Dinda AK, Kumar S, Roshan R, Gupta P, Sharma SK, Ghosh B. Mitochondrial structural changes and dysfunction are associated with experimental allergic asthma. J Immunol. 2008;181:3540–3548. doi: 10.4049/jimmunol.181.5.3540. [DOI] [PubMed] [Google Scholar]
- 125.Servais S, Boussouar A, Molnar A, Douki T, Pequignot JM, Favier R. Age-related sensitivity to lung oxidative stress during ozone exposure. Free Radic Res. 2005;39:305–316. doi: 10.1080/10715760400011098. [DOI] [PubMed] [Google Scholar]
- 126.Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, Wang M, Oberley T, Froines J, Nel A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect. 2003;111:455–460. doi: 10.1289/ehp.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 128.Tassi S, Carta S, Delfino L, Caorsi R, Martini A, Gattorno M, Rubartelli A. Altered redox state of monocytes from cryopyrin-associated periodic syndromes causes accelerated IL-1beta secretion. Proc Natl Acad Sci U S A. 2010;107:9789–9794. doi: 10.1073/pnas.1000779107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Iliev ID, Matteoli G, Rescigno M. The yin and yang of intestinal epithelial cells in controlling dendritic cell function. J Exp Med. 2007;204:2253–2257. doi: 10.1084/jem.20062535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Takizawa H, Ohtoshi T, Ohta K, Hirohata S, Yamaguchi M, Suzuki N, Ueda T, Ishii A, Shindoh G, Oka T, et al. Interleukin 6/B cell stimulatory factor-II is expressed and released by normal and transformed human bronchial epithelial cells. Biochem Biophys Res Commun. 1992;187:596–602. doi: 10.1016/0006-291x(92)91236-j. [DOI] [PubMed] [Google Scholar]
- 132.Neveu WA, Allard JB, Dienz O, Wargo MJ, Ciliberto G, Whittaker LA, Rincon M. IL-6 is required for airway mucus production induced by inhaled fungal allergens. J Immunol. 2009;183:1732–1738. doi: 10.4049/jimmunol.0802923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stumbles PA, Strickland DH, Pimm CL, Proksch SF, Marsh AM, McWilliam AS, Bosco A, Tobagus I, Thomas JA, Napoli S, Proudfoot AE, Wells TN, Holt PG. Regulation of dendritic cell recruitment into resting and inflamed airway epithelium: use of alternative chemokine receptors as a function of inducing stimulus. J Immunol. 2001;167:228–234. doi: 10.4049/jimmunol.167.1.228. [DOI] [PubMed] [Google Scholar]
- 134.Reibman J, Hsu Y, Chen LC, Bleck B, Gordon T. Airway epithelial cells release MIP-3alpha/CCL20 in response to cytokines and ambient particulate matter. Am J Respir Cell Mol Biol. 2003;28:648–654. doi: 10.1165/rcmb.2002-0095OC. [DOI] [PubMed] [Google Scholar]
- 135.Robays LJ, Maes T, Lebecque S, Lira SA, Kuziel WA, Brusselle GG, Joos GF, Vermaelen KV. Chemokine receptor CCR2 but not CCR5 or CCR6 mediates the increase in pulmonary dendritic cells during allergic airway inflammation. J Immunol. 2007;178:5305–5311. doi: 10.4049/jimmunol.178.8.5305. [DOI] [PubMed] [Google Scholar]
- 136.Reche PA, Soumelis V, Gorman DM, Clifford T, Liu M, Travis M, Zurawski SM, Johnston J, Liu YJ, Spits H, de Waal Malefyt R, Kastelein RA, Bazan JF. Human thymic stromal lymphopoietin preferentially stimulates myeloid cells. J Immunol. 2001;167:336–343. doi: 10.4049/jimmunol.167.1.336. [DOI] [PubMed] [Google Scholar]
- 137.Ying S, O'Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, Robinson D, Zhang G, Zhao J, Lee TH, Corrigan C. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol. 2005;174:8183–8190. doi: 10.4049/jimmunol.174.12.8183. [DOI] [PubMed] [Google Scholar]
- 138.Kato A, Favoreto S, Jr, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol. 2007;179:1080–1087. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lee HC, Ziegler SF. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proc Natl Acad Sci U S A. 2007;104:914–919. doi: 10.1073/pnas.0607305104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, Gilliet M, Ho S, Antonenko S, Lauerma A, Smith K, Gorman D, Zurawski S, Abrams J, Menon S, McClanahan T, de Waal-Malefyt Rd R, Bazan F, Kastelein RA, Liu YJ. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- 141.Ito T, Wang YH, Duramad O, Hori T, Delespesse GJ, Watanabe N, Qin FX, Yao Z, Cao W, Liu YJ. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005;202:1213–1223. doi: 10.1084/jem.20051135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Seshasayee D, Lee WP, Zhou M, Shu J, Suto E, Zhang J, Diehl L, Austin CD, Meng YG, Tan M, Bullens SL, Seeber S, Fuentes ME, Labrijn AF, Graus YM, Miller LA, Schelegle ES, Hyde DM, Wu LC, Hymowitz SG, Martin F. In vivo blockade of OX40 ligand inhibits thymic stromal lymphopoietin driven atopic inflammation. J Clin Invest. 2007;117:3868–3878. doi: 10.1172/JCI33559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Angkasekwinai P, Park H, Wang YH, Chang SH, Corry DB, Liu YJ, Zhu Z, Dong C. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15:410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R, Thorne PS, Wills-Karp M, Gioannini TL, Weiss JP, Karp CL. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature. 2009;457:585–588. doi: 10.1038/nature07548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hamilton LM, Davies DE, Wilson SJ, Kimber I, Dearman RJ, Holgate ST. The bronchial epithelium in asthma--much more than a passive barrier. Monaldi Arch Chest Dis. 2001;56:48–54. [PubMed] [Google Scholar]
- 147.Holgate ST, Lackie P, Wilson S, Roche W, Davies D. Bronchial epithelium as a key regulator of airway allergen sensitization and remodeling in asthma. Am J Respir Crit Care Med. 2000;162:S113–S117. doi: 10.1164/ajrccm.162.supplement_2.ras-12. [DOI] [PubMed] [Google Scholar]
- 148.Holgate ST, Lackie PM, Davies DE, Roche WR, Walls AF. The bronchial epithelium as a key regulator of airway inflammation and remodelling in asthma. Clin Exp Allergy. 1999;29 Suppl 2:90–95. doi: 10.1046/j.1365-2222.1999.00016.x. [DOI] [PubMed] [Google Scholar]
- 149.Lambrecht BN, Hammad H. The role of dendritic and epithelial cells as master regulators of allergic airway inflammation. Lancet. 2010;376:835–843. doi: 10.1016/S0140-6736(10)61226-3. [DOI] [PubMed] [Google Scholar]
- 150.Janssen-Heininger YM, Poynter ME, Aesif SW, Pantano C, Ather JL, Reynaert NL, Ckless K, Anathy V, van der Velden J, Irvin CG, van der Vliet A. Nuclear factor kappaB, airway epithelium, and asthma: avenues for redox control. Proc Am Thorac Soc. 2009;6:249–255. doi: 10.1513/pats.200806-054RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Pantano C, Reynaert NL, van der Vliet A, Janssen-Heininger YM. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxid Redox Signal. 2006;8:1791–1806. doi: 10.1089/ars.2006.8.1791. [DOI] [PubMed] [Google Scholar]
- 152.Ather JL, Alcorn JF, Brown AL, Guala AS, Suratt BT, Janssen-Heininger YM, Poynter ME. Distinct functions of airway epithelial nuclear factor-kappaB activity regulate nitrogen dioxide-induced acute lung injury. Am J Respir Cell Mol Biol. 2010;43:443–451. doi: 10.1165/rcmb.2008-0416OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ather JL, Hodgkins SR, Janssen-Heininger YM, Poynter ME. Airway Epithelial NF-{kappa}B Activation Promotes Allergic Sensitization to an Innocuous Inhaled Antigen. Am J Respir Cell Mol Biol. 2010 doi: 10.1165/rcmb.2010-0106OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pantano C, Ather JL, Alcorn JF, Poynter ME, Brown AL, Guala AS, Beuschel SL, Allen GB, Whittaker LA, Bevelander M, Irvin CG, Janssen-Heininger YM. Nuclear factor-kappaB activation in airway epithelium induces inflammation and hyperresponsiveness. Am J Respir Crit Care Med. 2008;177:959–969. doi: 10.1164/rccm.200707-1096OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Korn SH, Wouters EF, Vos N, Janssen-Heininger YM. Cytokine-induced activation of nuclear factor-kappa B is inhibited by hydrogen peroxide through oxidative inactivation of IkappaB kinase. J Biol Chem. 2001;276:35693–35700. doi: 10.1074/jbc.M104321200. [DOI] [PubMed] [Google Scholar]
- 156.Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, Heintz NH, Heim J, Ho YS, Matthews DE, Wouters EF, Janssen-Heininger YM. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kelleher ZT, Matsumoto A, Stamler JS, Marshall HE. NOS2 regulation of NF-kappaB by S-nitrosylation of p65. J Biol Chem. 2007;282:30667–30672. doi: 10.1074/jbc.M705929200. [DOI] [PubMed] [Google Scholar]
- 158.Reynaert NL, Ckless K, Korn SH, Vos N, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc Natl Acad Sci U S A. 2004;101:8945–8950. doi: 10.1073/pnas.0400588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Moldovan L, Moldovan NI. Oxygen free radicals and redox biology of organelles. Histochem Cell Biol. 2004;122:395–412. doi: 10.1007/s00418-004-0676-y. [DOI] [PubMed] [Google Scholar]
- 160.Bove PF, van der Vliet A. Nitric oxide and reactive nitrogen species in airway epithelial signaling and inflammation. Free Radic Biol Med. 2006;41:515–527. doi: 10.1016/j.freeradbiomed.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 161.Bove PF, Wesley UV, Greul AK, Hristova M, Dostmann WR, van der Vliet A. Nitric oxide promotes airway epithelial wound repair through enhanced activation of MMP-9. Am J Respir Cell Mol Biol. 2007;36:138–146. doi: 10.1165/rcmb.2006-0253SM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Olson N, Greul AK, Hristova M, Bove PF, Kasahara DI, van der Vliet A. Nitric oxide and airway epithelial barrier function: regulation of tight junction proteins and epithelial permeability. Arch Biochem Biophys. 2009;484:205–213. doi: 10.1016/j.abb.2008.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Li N, Nel AE. Role of the Nrf2-mediated signaling pathway as a negative regulator of inflammation: implications for the impact of particulate pollutants on asthma. Antioxid Redox Signal. 2006;8:88–98. doi: 10.1089/ars.2006.8.88. [DOI] [PubMed] [Google Scholar]
- 164.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, Slaughter N, Killeen E, Wang X, Huang A, Wang M, Miguel AH, Cho A, Sioutas C, Nel AE. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol. 2004;173:3467–3481. doi: 10.4049/jimmunol.173.5.3467. [DOI] [PubMed] [Google Scholar]
- 166.Williams MA, Rangasamy T, Bauer SM, Killedar S, Karp M, Kensler TW, Yamamoto M, Breysse P, Biswal S, Georas SN. Disruption of the transcription factor Nrf2 promotes pro-oxidative dendritic cells that stimulate Th2-like immunoresponsiveness upon activation by ambient particulate matter. J Immunol. 2008;181:4545–4559. doi: 10.4049/jimmunol.181.7.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Boldogh I, Bacsi A, Choudhury BK, Dharajiya N, Alam R, Hazra TK, Mitra S, Goldblum RM, Sur S. ROS generated by pollen NADPH oxidase provide a signal that augments antigen-induced allergic airway inflammation. J Clin Invest. 2005;115:2169–2179. doi: 10.1172/JCI24422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rangasamy T, Williams MA, Bauer S, Trush MA, Emo J, Georas SN, Biswal S. Nuclear erythroid 2 p45-related factor 2 inhibits the maturation of murine dendritic cells by ragweed extract. Am J Respir Cell Mol Biol. 2010;43:276–285. doi: 10.1165/rcmb.2008-0438OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Allan K, Kelly FJ, Devereux G. Antioxidants and allergic disease: a case of too little or too much? Clin Exp Allergy. 2010;40:370–380. doi: 10.1111/j.1365-2222.2009.03413.x. [DOI] [PubMed] [Google Scholar]
- 170.Hartert TV, Peebles RS. Dietary antioxidants and adult asthma. Curr Opin Allergy Clin Immunol. 2001;1:421–429. doi: 10.1097/01.all.0000011055.59442.37. [DOI] [PubMed] [Google Scholar]
- 171.Riccioni G, Barbara M, Bucciarelli T, di Ilio C, D’Orazio N. Antioxidant vitamin supplementation in asthma. Ann Clin Lab Sci. 2007;37:96–101. [PubMed] [Google Scholar]