

Abstract
Inflammatory bowel disease (IBD), mainly Crohn’s disease (CD) and ulcerative colitis (UC), are characterized by epithelial barrier disruption and altered immune regulation. Colonic Ste20-like proline/alanine-rich kinase (SPAK) plays a role in intestinal inflammation, but its underlying mechanisms need to be defined. Both SPAK-transfected Caco2-BBE cells and villin-SPAK transgenic (TG) FVB/6 mice exhibited loss of intestinal barrier function. Further studies demonstrated that SPAK significantly increased paracellular intestinal permeability to fluorescein isothiocyanate (FITC)-dextran. In vivo studies using the mouse models of colitis induced by dextran sulfate sodium (DSS) and trinitrobenzene sulfonic acid (TNBS) showed that TG FVB/6 mice were more susceptible to DSS and TNBS treatment than wild-type FVB/6 mice, as demonstrated by clinical and histological characteristics and enzymatic activities. Consistent with this notion, we found that SPAK increased intestinal epithelial permeability, which likely facilitated the production of inflammatory cytokines in vitro and in vivo and aggravated bacterial translocation in TG mice under DSS treatment, consequently established a context favorable for the triggering of intestinal inflammation cascades. In conclusion, over-expression of SPAK inhibits maintenance of intestinal mucosal innate immune homeostasis, which makes regulation of SPAK important to attenuate pathological responses in IBD.
Introduction
Inflammatory bowel disease (IBD) is a group of chronic intestinal diseases characterized by inflammation of the bowel, or the large or small intestine (1, 2), which refers Crohn’s disease (CD) and ulcerative colitis (UC). A healthy intestinal barrier is composed of several specific features, including the luminal commensal microflora, the mucus layer, the epithelial cells and the tight junctions, and the intestinal immune system. Any stresses that interfere with these features could eventually cause intestinal barrier loss, leading to inflammation. A strong linkage has been established between increased permeability and intestinal inflammation in both CD and UC patients (3–6) with the involvement of a variety of underlying mechanisms. For example, pathogens and bacterial toxins can alter transepithelial permeability by modulating the expression and activity of tight junction proteins (7); pro-inflammatory cytokines, such as interferon-gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) reduce intestinal barrier function by reorganizing several tight junction proteins in cultured IECs and experimental mouse models (8–11). Also, genetic studies in IBD patients and experimental mouse models of IBD, including the IL-10−/− model (12), the SAMP1/Yit model (13), and the TLR4−/− model (14), highlight the importance of genetic background on intestinal barrier function.
SPAK, a MAP4K, contains an N-terminal series of proline/alanine repeats (PAPA box) followed by a catalytic domain, a nuclear localization signal, a potential caspase-cleavage motif, and a C-terminal regulatory region (15). The colonic isoform of SPAK, which lacks the PAPA box and the F-α helix loop in the catalytic sub-domain IX, was cloned and characterized by our group (16, 17). SPAK plays important roles in several physiological processes including cell differentiation (15), transformation and proliferation (18), and regulation of chloride transport (19). Recently, SPAK was found to be involved in intestinal inflammation(16, 17, 20), but the underlying mechanisms have yet to be defined. In addition, our understanding of the involvement of SPAK in intestinal barrier function is limited.
In the present study, we investigated the effects of SPAK on intestinal barrier function, and the mechanisms thereof. We found that SPAK caused an increase in intestinal permeability and SPAK transgenic mice were more susceptible to experimental colitis. Additionally, increased cytokine production and bacterial translocation were associated with the increased colitis susceptibility. Further studies will be needed to move from this association to causation.
Materials and Methods
Plasmid construction
SPAK/pcDNA6 was cloned in our lab previously (16). SPAK siRNA and scramble siRNA were commercially obtained from Applied Biosystems (Foster City, CA), IL-1β-Luc promoter was a gift from Jesse Roman (Emory University), TNF-α-Luc, IL-17-Luc and IFN-γ-Luc plasmids were purchased from Addgene Inc. (Cambridge, MA).
Cell culture
The human intestinal cell line Caco2-BBE between passages 20 and 30 was cultured according to the standard protocol and transfected using lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to manufactured instructions.
Real-time PCR
Total RNA from Caco2-BBE cells and mouse colon mucosa were reverse transcribed using the Thermoscript RT-PCR System (Invitrogen, Carlsbad, CA) and purified with the RNeasy Mini Kit (Qiagen, Germantown, MD). Real-time PCR was performed using the iQ SYBR Green Supermix kit (Bio-Rad, Hercules, CA) with the iCycler sequence detection system (Bio-Rad, Hercules, CA) with specific primers (Table 1 and Supplemental table s1). Real-time PCR data were presented using delta–delta Ct (ΔΔCt) method (21) with GAPDH or 36B4 gene levels serving as the internal standard.
Table 1.
Primers used for inflammatory mediators’ quantification
| Name | Nucleotide sequence |
|---|---|
| Human SPAK For | 5′ AGAGTTCCTGGGTCAAGTGGTCA 3′ |
| Human SPAK Rev | 5′ TTCGCTCTTCTCATCCATCTCG 3′ |
| Mouse SPAK For | 5′ CGTTGACATTTAAGTTGGCYYCTG 3′ |
| Mouse SPAK Rev | 5′ TCACTTCATCAGGAATCTCCG 3′ |
| GAPDH For | 5′GTCGGAGTCAACGGATTTGG 3′ |
| GAPDH Rev | 5′ AAGCTTCCCGTTCTCAGCCT 3′ |
| 34B4 For | 5′ TCCAGGCTTTGGGCATCA 3′ |
| 36B4 Rev | 5′ CTTTATCAGCTGCACATCACTCAGA-3′ |
| TNF-α For | 5′ AGGCTGCCCCGACTACGT 3′ |
| TNF-α Rev | 5′ GACTTTCTCCTGGTATGAGATAGCAAA 3′ |
| IFN-γ For | 5′ CAGCAACAGCAAGGCGAAA 3′ |
| IFN-γ Rev | 5′ CTGGACCTGTGGGTTGTTGAC 3′ |
| IL-17 For | 5′ CAGGAACCCTCATCCTTCAA 3′ |
| IL-17 Rev | 5′ ATTCCCAAGCCCAGAATCTT 3′ |
| IL-1β For | 5′ GGGCCTCAAGGAAAAGAATC 3′ |
| IL-1β Rev | 5′ AGCTGACTGTCCTGGCTGAT 3′ |
| TNF-α chipFor | 5′ AGCCCATGTTGTAGCAAACC 3′ |
| TNF-α chip Rev | 5′ GGTTGAGGGTGTCTGAAGGA 3′ |
| IL-17 chip For | 5′ CAGGAACCCTCATCCTTCAA 3′ |
| IL-17 chip Rev | 5′ ATTCCCAAGCCCAGAATCTT 3′ |
| IL-1β chip For | 5′ GGACAAGCTGAGGAAGATGC 3′ |
| IL-1β chip Rev | 5′ TCTTTCAACACGCAGGACAG 3′ |
| EUB | 5′ GCTGCCTCCCGTAGGAGT 3′ |
| EUB control | 5′ CGACGGAGGGCATCCTCA 3′ |
Note: TNF-α: Tumor necrosis factor-alpha, IFN-γ: Interferon-gamma, IL-1β: Interleukin-1 beta, IL-17: Interleukin-17, chip: Chromatin immunoprecipitate: EUB: Eubacteria, For: Forward, Rev: Reverse
Western blotting
All Western blots were performed with appropriate antibodies based on standard protocols.
Immunocytochemistry and Immunohistochemistry
Immunostaining assays were performed according to the standard protocol with relevant primary antibodies (SPAK and mucin 2 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), ZO-1, ZO-2, occludin, claudin-1, -2 and -4 antibodies (Invitrogen, Carlsbad, CA)) and Alexa Fluor® 488 secondary antibody (Molecular Probes, Carlsbad, CA), and rhodamine/phalloidin (Molecular Probes, Carlsbad, CA) as described previously (22) to visualize actin. Samples were mounted in Prolong Gold Antifade Reagent and analyzed by ZEISS AXIOSKOP 2 PLUS Microscope (Carl Zeiss MicroImaging, Inc. Thornwood, NY).
In vitro and ex vivo transepithelial resistance (TER) assay
As transepithelial barrier dysfunction is necessary for the development of intestinal inflammation, we studied the effects of SPAK expression on intestinal barrier function by in vitro and ex vivo experiments. For in vitro assay, Caco2-BBE cells grew confluent on snap well filters (Corning Costar, Corning, NY), relative TER was measured with ussing chambers (Physiologic Instruments, San Diego, CA). Ex vivo TER assay with distal colonic mucosa obtained by blunt stripping from muscularis and serosa was the same as in vitro TER assay.
In vitro and in vivo permeability assays
In vitro and in vivo permeability assays were performed using a fluorescein isothiocyanate (FITC)-labeled dextran method to assess barrier function. For in vitro permeability assay, confluent and polarized Caco2-BBE cells grown on filters were treated with FITC-labled dextrans (4-kDa, Sigma-Aldrich, St. Louis, MO) on the upper chamber for 2hrs at 37°C. The medium in the lower chamber was collected. In vivo permeability assays were performed as described previously (20). Briefly, food and water were withdrawn from the mice for 4 h and then gavaged with permeability tracer FITC-labled dextrans (60 mg/100g body weight, Sigma-Aldrich, St. Louis, MO). Serum was collected retro-orbitally 4 hours after gavage; fluorescence intensity of each sample was measured (485Ex/520Em, Cytofluor 2300; Millipore, Waters Chromatography) and FITC-dextran concentrations were determined from standard curves generated by serial dilution of FITC-dextran.
In vitro and ex vivo ion selectivity assays
The ion selectivity of tight junctions was determined by measurement of dilution potentials (Caco2-BBE monolayer) or short-circuit current (mouse colonic mucosa) by replacing either the apical or basolateral solution, while keeping the other side (basolateral or apical) bathed in Kreb’s solution. For 2:1 NaCl dilution potentials/short-circuit current, the 128 mM NaCl solution was replaced with 52 mM NaCl in Kreb’s solution, and osmolarity was maintained with mannitol.
SPAK translocation, immunoprecipitation, and kinase assays
Nuclear proteins were extracted from Caco2-BBE cells as described previously (23). Immunoprecipitation was performed using the Catch and Release Reversible Immunoprecipitation Kit (Millipore, Billerica, MA), according to the manufacturer’s instructions. Exogenous substrate phosphorylation assays with myelin basic protein (MBP; Upstate, Charlottesville, VA) as substrate were performed as described previously (16). To confirm these results, the N-terminus of SPAK was cloned into the vector PCR11 (Invitrogen, Carlsbad, CA) and subjected to TNT T7 Quick Coupled Transcription/Translation System (Promega, Madison, WI) to express the N-terminus protein in vitro for the kinase assay. All kinase assays were visualized by western blot with anti-phospho MBP antibody (Upstate, Charlottesville, VA) and anti-phospho threonine antibody (Sigma-Aldrich, ST. Louis, MO).
Mouse model
FVB/6 mice (8–10 wk, 18–22 g) were obtained from Jackson Laboratories (Bar Harbor, ME). In collaboration with the Transgenic Mouse and Gene Targeting Core Facility (Emory University), we established the SPAK transgenic mouse model in FVB/6 mice. All animal experiments were approved by The Institutional Animal Care and Use Committee of Emory University, Atlanta and were in accordance with the guide for the Care and Use of Laboratory Animal, published by the U.S. Public Health Service.
Induction and assessment of colitis
Colitis was induced by the addition of 3.5 % (wt/vol) dextran sodium sulfate (DSS) (molecular weight 50,000; ICN Biochemicals, Aurora, OH) to the drinking water or by colonic injection of 150 mg/kg body weight of trinitrobenzene sulfonic acid (TNBS; Sigma, St. Louis, MO) dissolved in 50% ethanol. Colitis was assessed 8 days after DSS treatment or 48 hours after TNBS administration as described previously (24). N = 6 mice/group. Direct visualization of colon was performed using the “Coloview system” (Karl Storz Veterinary Endoscopy, Goleta, CA). Neutrophil infiltration into the colon was quantified by measurement of MPO activity, as described previously (17). To study the effects of SPAK on the healing phase of intestinal inflammation, we monitored the survival status of the mice for another week after DSS/TNBS withdrawal.
Array analysis
Microarray hybridizations were performed in collaboration with Dr. Andrew Neish (Emory University) using the Vanderbilt Microarray Shared Resource (VMSR) Human 30k Oligonucleotide Microarray Chip. To confirm and narrow the expression gene targets regulated by SPAK, we used the human inflammatory cytokines and receptor RT2 Profiler PCR array (SABioscience, Frederick, MD) with the iCycler sequence detection system (Bio-Rad, Hercules, CA). To minimize variability, the mean of three independent experiments for each gene was calculated and used for final data clustering. Onlygenes that showed significant change (over 2-fold difference) were selected for further characterization. DNASTAR ArrayStar 2 analytic software packages were used for Scatter plotting.
Chromatin immunoprecipitation (ChIP) assay
ChIP assays identifying binding sites of the pro-inflammatory cytokines TNF-α, IL-1β, IL-17, and IFN-γ were performed using a ChIPassay kit (Upstate, Charlottesville, VA), according to the manufacturer’s instructions using primers listed in Table 1.
Transactivation assay
The transactivation assays were performed as described previously with minor modifications (24). Renilla (phRL-CMV, 5ng), cytokine Luc-promoter constructs (4μg), and SPAK/pcDNA6 (4μg) were co-transfected into Caco2-BBE cells with lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 48 hours, cells were collected with the Dual Luciferase Reporter Assay System (Promega, Madison, WI), and the luminescence was measured in a luminometer (Luminoskan, Thermal Labsystems, MA). Luciferase activity was normalized based on the control renilla luciferase activity. Extracts were analyzed in triplicate, and each experiment was performed at least three times.
Bacterial translocation assay
Colony forming units (CFU) in freshly isolated small intestine, colon, spleen, and liver tissues were determined via homogenization of material in PBS/0.01% Triton X-100 followed by serial dilution plating on nonselective Luria-Bertani agar as described previously(25). In parallel, DNA was prepared with Wizard SV Genomic DNA Purificaton System (Promega, Madison, WI) from 3 mice of each group. DNA was analyzed by PCR for 20 cycles with universal primers directed against a region of the 16S rRNA gene common to most bacteria: forward 5′CCATGAAGTCGGAATCGCTAG 3′ and reverse, 5′ ACTCCCATGGTGTGACGG-3 (bp 1302–1394 in bacteria EU622773). The PCR products were analyzed by electrophoresis.
Fluorescence in situ hybridization (FISH)
The FISH assay was performed using an Alexa Fluor 555-conjugated EUB (bp 337–354); the NON-EUB-Alexa Fluor 555 probe was used as a negative control.
Statistical Analysis
Values are expressed as mean ± SEM with unpaired two-tailed Student’s t test by InStat v3.06 (GraphPad, San Diego, CA) software. P < 0.05 was considered statistically significant.
Results
Colonic SPAK reduces transepithelial resistance (TER) in vitro
Using real-time PCR and Western-blot analyses, we found significant increases in SPAK expression in SPAK/pcDNA6-transfected cells and significant decreases of SPAK expression in siRNA-transfected cells at both mRNA and protein levels, in comparison with controls (Fig. 1A and 1B). Furthermore, immunostaining showed increased SPAK expression in both cytosolic and nuclear pools (Fig. 1C). We have previously shown that SPAK expression was increased in the colonic mucosa of CD (20) and UC (17) patients, and DSS colitic mice (16). Also, SPAK synthesis was increased in IECs treated with pro-inflammatory signals such as TNF-α (17) or hyperosmolarity (20). Therefore it is of interest to study the involvement of SPAK in epithelial barrier function. As shown in Fig. 1D, over-expression of SPAK significantly reduced transepithelial resistance (TER) to 69.6±14.3 ohms.cm2 (SPAK-transfected Caco2-BBE cells) from 132.1±20.1 ohms.cm2 in vector-transfected cells. Knockdown of SPAK expression significantly increased TER, from 119.9±23.6 ohms.cm2 to 234.9±31.0 ohms.cm2. In addition, we found that Caco2-BBE cells over-expressing SPAK did not demonstrate increased levels of apoptosis (data not shown), indicating the increase of intestinal permeability by SPAK expression is not due to apoptosis.
Figure 1. SPAK is involved in the regulation of barrier function in IECs in vitro.

SPAK expression is modulated in Caco2-BBE cells by SPAK/pcDNA6 and SPAK siRNA transient transfection compared with vector pcDNA6 and scramble siRNA (ssiRNA) transient transfection by (A) real time PCR, (B) western blot and (C) immunoflurescrence (SPAK-green, beta-actin-red). (D) Transepithelial resistance (TER) assay with Ussing chamber in Caco2-BBE monolayer: over-expression of SPAK decreases TER, while knock down of SPAK expression by siRNA increases TER. (E) FITC-dextran (4 kDa) was added to the apical side of polarized monolayers of Caco2-BBE cells at 10 mg/ml, and the basolateral reservoir was sampled at 2 h after the addition of FITC-dextran to the apical side. Histograms show mean ± SEM of ng/ml/min FITC-dextran translocation to the basolateral reservoir. (F) The dilution potential was determined by the change of transepithelial voltage upon switching from symmetrical bathing solutions (apical and basolateral side) to a 2:1 NaCl concentration gradient in Ussing chamber. Data are representative of three independent experiments. Error bars represent the means ± SEM. P-values were determined by Student’s t test. *: P<0.05, **: P<0.01.
The total TER is represented by two resistances in parallel: the transcellular resistance (Rtc) and the paracellular resistance (Rp). The paracellular pathway is formed mainly by the resistance of the tight junction (Rt). In the circuit model, TER is represented by: 1/TER= (1/Rtc) + (1/Rt). This value of TER, across a leaky epithelium, such as that of the intestine, reflects principally the resistance afforded by Rt. Since TER is determined by tight junction size and ion selectivity (2, 26, 27), the reduction in TER caused by SPAK may be attributable to alteration of tight junction size or ion selectivity. To further investigate this hypothesis, we examined transepithelial permeability using the 4-kDa (Fig. 1E) FITC-dextran method. As shown in Fig. 1E, vector-transfected cells showed a FITC-dextran flux (ng/ml/min) of 12.1±2.6. In comparison, a ~2-fold increase in FITC-dextran flux was observed in SPAK-transfected cells (23.8±4.03), with no significant change in scrambled-siRNA-transfected cells (13.9±3.2). In addition, a ~3.2-fold decrease in SPAK-siRNA-transfected cells (4.3±1.3) was observed. To further evaluate the effects of SPAK on IEC barrier function, ion selectivity was first examined in vitro. As shown in Fig. 1F, the dilution potential (mV) did not differ on both apical side (Vector:−6.2±1.03; SPAK:−5.7±0.85; siRNA:−6.4±1.93; scrambled siRNA:−5.64±1.33) and basolateral side (Vector: 4.2±1.29; SPAK: 3.6±1.58; siRNA: 3.9±0.48; scrambled siRNA: 3.96±0.97).
Colonic SPAK reduces transepithelial resistance (TER) ex vivo
To investigate whether SPAK operates as a barrier function regulator in vivo, we generated villin-SPAK transgenic (SPAK TG) mice, which targets SPAK over-expression to IECs. SPAK TG mice had a 7-fold increase of mRNA transcripts (Fig.2A), and a marked increase of SPAK protein expression (Fig.2B), in colonic mucosa compared with wild-type (WT) mice. Immunostaining (Fig. 2C) of colon sections detected SPAK mainly in the muscularis, muscularis mucosa, and epithelial layers of WT mice. However, in comparison with WT mice, TG mice showed increased SPAK expression only in the epithelium, including different cell lineages, such as absorptive enterocytes, enteroendorine cells, and goblet cells. Goblet cells are the principal cell lineage to express SPAK in TG mice. A similar degree of expression in the muscularis and muscularis mucosa layers was observed. In ex vivo assays, the mucosa of SPAK TG mice exhibited a TER (214.5±43.2 ohms.cm2) that was significantly lower than that of WT mice (361.9±18.3 ohms.cm2) (Fig. 2D). These data indicate that SPAK affects colonic TER ex vivo. In vivo size selectivity assays using 4 kDa (Fig. 2E) of FITC-dextran yielded similar results as in vitro experiments. WT mice had a flux level of 86.5±21.0 ng/μg protein of FITC-dextran. In comparison, an increased flux (≥2-fold) was observed in TG mice (180.3±40.4 ng/μg protein). These results indicated that SPAK increased transepithelial permeability in vivo. But in ex vivo experiments, the transepithelial current (mA) did not differ significantly between SPAK TG and WT colon mucosa (Fig. 2F) on either the apical (−10.63±4.54 vs −10.01±2.05) or basolateral side (5.47±2.36 vs 5.12±1.22).
Figure 2. SPAK transgenic mice display loss of intestinal barrier function.

SPAK expression is enhanced in colonic mucosa in TG mice by (A) real time PCR, (B) Western blot and (C) (SPAK-green, beta-actin-red). (D) Transepithelial resistance (TER) assay with Ussing chamber in mouse colonic mucosa, TG mice have lower TER compared to WT mice in ex vivo experiments. (E) WT and TG mice were starved for 4 h and then gavaged with FITC-dextran (4 kDa). Serum was collected retro-orbitally 4 hours after gavage; fluorescence intensity of each sample was measured; and FITC-dextran concentrations were determined from standard curves generated by serial dilution of FITC-dextran. (F) In 2:1 NaCl short-circuit current assay in mouse colonic mucosa, the 128 mM NaCl solution was replaced with 52 mM NaCl in Kreb’s solution, and osmolarity was maintained with mannitol. Transepithelial current was monitored and recorded at 10 sec intervals, with the voltage continuously clamped at zero. Data are expressed as means ± SEM (n = 9 mice per group). Statistical differences of TG versus WT mice are reported. Data are pooled from three independent experiments. *: P<.05, **: P<.01.
SPAK over-expression deteriorates experimental colitis
We then examined clinical and histological changes from phenotypic and pathologic perspectives (28). TG mice showed a marked increase in diarrhea and more serious rectal bleeding (Fig. 3A) than WT mice during DSS treatment, body weight decreased significantly in both TG and WT mice, but weight loss was more severe in TG mice (20% vs 6 %) (Fig. 3B). In addition, DSS caused a significant reduction in colon length, as shown in Fig. 3C and 3D, which was more severe in TG mice compared with WT mice. The body weight loss and colon length reduction in TNBS colitis model are consistent with the DSS colitis model (Fig. 3).
Figure 3. SPAK TG mice were more susceptible to DSS- and TNBS-induced mouse colitis.

(A) Phenotypic assay of WT and TG mice with 3.5 % DSS treatment for 10 days or with 150 mg/kg body weight of TNBS treatment for 48 hours. (B) Body weight measurement in WT and TG mice with 3.5 % DSS treatment for 10 days or with 150 mg/kg body weight of TNBS treatment for 48 hours. (C) Colonic phenotypic assay in WT and TG mice with 3.5 % DSS treatment for 10 days or with 150 mg/kg body weight of TNBS treatment for 48 hours. (D) Measurement of mouse colon length in WT and TG mice with 3.5 % DSS treatment for 10 days or with 150 mg/kg body weight of TNBS treatment for 48 hours. Data are expressed as means ± SEM (n = 4 mice per group). Statistical differences of TG versus WT mice are reported. Data are pooled from three independent experiments. *: P<0.05, **: P<0.01.
Histological staining (Fig. 4A) showed an intact epithelium, a well-defined crypt length, no edema, and no neutrophil infiltration into the mucosa or submucosa, and no ulcers or erosions in untreated WT mice. Untreated TG mouse colon sections (Fig. 4A) exhibited an intact epithelium with a shorter crypt length, and a thicker submucosa or lamina propia, noticeable edema, slight neutrophil infiltration into the mucosa, and no ulcers or erosions. Colon tissue from DSS-treated WT mice had extensive inflammatory lesions throughout the mucosa (Fig. 4A), both ulcers and shortening/loss of crypts were focally apparent. In TG mice treated with DSS, ulcers and shortening and loss of crypts progressed to more extensive areas of mucosa; submucosal edema became more severe with DSS treatment, and a large infiltration of immune cells was seen in DSS-treated TG mice. Similar histological characteristics were noticed in the TNBS colitis mouse model (Fig. 4A). Colonoscopic analysis demonstrated that both WT and TG animals showed no evidence of macroscopic inflammation, displaying a semi-translucent mucosa characteristic of a healthy colon (Fig. 4B, left two panels). However, both DSS and TNBS induced a rapid and progressive severe, ulcerating, colonic inflammation with bloody diarrhea in both WT and TG mice (Fig. 4B, right two panels). Furthermore, TG mice exhibited more extensive intestinal inflammation, including increased colonic inflammation with prominent mucosal edema and spontaneous bleeding compared to WT mice.
Figure 4. SPAK TG mice exhibit aggravated inflammation.

(A) Representative photomicrographs of paraffin-embedded, hematoxylin-stained sections of the distal colon. Original magnification 10 × (upper panels) and 20 × (lower panels). (B) Intestinal inflammation was evaluated macroscopically in vivo using a murine miniature endoscope. Representative images of 6 different mice are shown. (C) WT and TG mice were given water or 3.5 % DSS for 10 days or 150 mg/kg body weight of TNBS for 48 hours as described in Figure 3, distal colon tissue was collected and subjected for MPO activity measurement. Data are expressed as means ± SEM (n = 9 mice per group). Statistical analysis was performed using an unpaired two-tailed Student’s t test. *: P<0.05, **: P<0.01. (D) After 10 days of 3.5 % DSS or 48 hours of TNBS, mice were given tap water and followed for mortality during recovery phase.
Myeloperoxidase (MPO) activity was measured as an indicator of tissue damage and the extent of infiltration by inflammatory cells. No significant MPO changes were noticed between untreated WT (0.06±0.03 mUnits/μg protein) and untreated TG mice (0.095±0.05 mUnits/μg protein) (Fig. 4C). MPO values increased in DSS-treated mice compared with controls, but DSS-treated TG mice showed significantly higher MPO values (0.67±0.167 mUnits/μg protein) than DSS-treated WT mice (0.27±0.095 mUnits/μg protein), indicating more neutrophil infiltration into the mucosa and submucosa of TG mice than WT mice. Fig. 4D shows the survival curves of WT and TG mice after DSS and TNBS treatment; 38% of TG mice died during this period, whereasno WT mice succumbed, indicating a higher mortality of TG mice during recovery. The pattern of MPO activities and the pattern of recovery were also seen in TNBS-induced colitis (Fig. 4C and 4D).
SPAK is involved in production of inflammatory cytokines
Microarray analysis showed that SPAK changed the expression of 631 genes greater than 2-fold (Fig. 5A), and 35 of these 631 genes have increased expression by more than 4-fold in comparison with controls, including some pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-17. These data have been deposited in GEO with the accession number GSE25641 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE25641). Given that pro-inflammatory cytokines play central roles in the pathogenesis of IBD, we confirmed these results using a PCR array analysis (Fig. 5B). The white solid squares (A, B, and C) represent cytokines TNFα, IL-1β, and IL-17, respectively. We then performed real-time PCR to monitor the levels of pro-inflammatory cytokines in mouse colon tissue. As shown in Fig. 5C, the levels of IL-1β (a 6.4-fold increase), TNF-α (a 4.8-fold increase), and IL-17 (a 5.3-fold increase) in TG animals were significantly higher than seen in WT mice. But we did not find any of these four inflammatory cytokines detectable by ELISA, which suggests any effect of SPAK on the intestinal barrier in vitro is not mediated by cytokine secretion. No significant difference in IFN-γ level was observed between TG and WT mice. After DSS treatment, production of pro-inflammatory cytokines was significantly increased in both WT and TG mice. The IL-1β transcripts showed a 17.5-fold increase in WT mice and a 20.6-fold increase in TG mice; the TNF-α transcripts increased 7.6-fold in WT mice and 18.8-fold in TG mice; the IL-17 transcripts showed a 2.6-fold increase in WT mice and a 13.7-fold increase in TG mice; and the IFN-γ transcripts increased 11.2-fold in WT mice and 11.8-fold in TG mice.
Figure 5. SPAK facilitates the production of proinflammatory cytokines.

(A) Microarray hybridizations using the Affymetrix GeneChip exhibited alteration of expression of hundreds of different genes in human IECs. The mean of three independent experiments for each gene was calculated and used for data clustering. Only genes that showed significant change (over 2-fold difference) were selected for further characterization. DNASTAR ArrayStar 2 analytic software packages were used for Scatter plotting. (B) PCR array of cytokines demonstrated increase of proinflammatory cytokines, including TNF-α, IL-1β and IL-17 in human IECs. (C) Real time PCR was performed in WT and TG mice for proinflammatory cytokines, including TNF-α, IL-1β and IL-17, with or without DSS treatment. IFN-γ acted as a negative control. Data are expressed as means ± SEM (n = 9 mice per group). Statistical differences of TG versus WT mice are reported *: P<0.05, **: P<0.01.
SPAK N-terminus can undergo translocation into the nucleus and retain kinase activity
Since SPAK increases the production of cytokines TNF-α, IL-1β, and IL-17, we were prompted to study the underlying mechanism. First, we found that N-terminus of SPAK expressed by TNT T7 Quick Coupled Transcription/Translation System can phosphorylate the substrate myelin basic protein (MBP) and autophosphorylate itself (Fig. 6A), which indicates that the N-terminus of SPAK maintains its kinase activity in vitro. Next, we studied the kinase activity of SPAK in the cell nucleus. Immmuno-precipitation by Xpress antibody, kinase assays and Western-blot analyses showed (Fig. 6B) that the N-terminal catalytic domain underwent translocation to the nucleus, and retained kinase activity by phosphorylating MBP and autophosphorylation.
Figure 6. Mechanism assays of SPAK involvement in the production of proinflammatory cytokines.
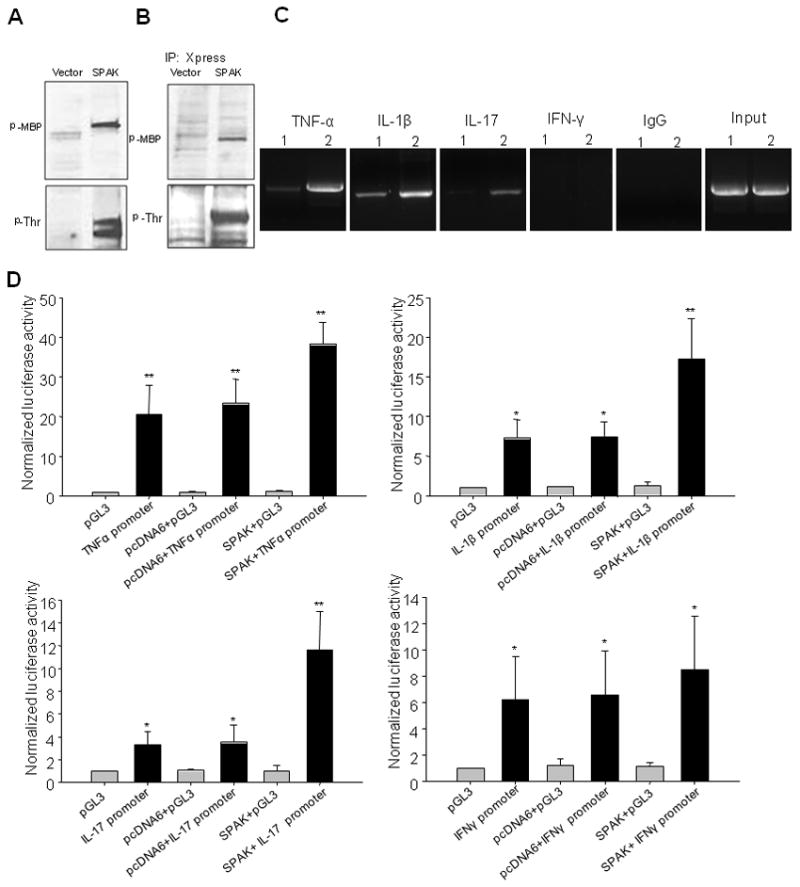
(A) In vitro kinase assay of SPAK N-terminus expressed by TNT Quick Coupled Transcription/Translation System. MBP acts as substrate for kinase assay. The kinase assay complex was subjected to Western blot with anti-phospho MBP and threonine antibody. (B) In vivo kinase assay of N-terminus of SPAK, Immunoprecipitate of Caco2-BBE nuclear protein with Xpress antibody. MBP acted as a substrate for the kinase assay. The kinase assay complex was subjected to Western blot with anti-phospho MPB antibody and threonine antibody. (C) ChIP assay exhibited association of SPAK and cytokine genes, (1) Vector-transfected-Caco2-BBE cells; (2) SPAK-transfected-Caco2-BBE cells. (D) Transient transfection of different constructs into Caco2-BBE cells and transactivation assays of SPAK and the genes related to cytokines TNF-α, IL-1β and IL-17. The results are representative of three independent experiments performed in triplicate, and error bars represent standard deviation analyzed by Student’s t test by InStat v3.06 (GraphPad) software. *: P<0.05, **: P<0.01.
SPAK may associate with and transactivate cytokine genes
To confirm the importance of SPAK in the increased production of TNF-α, IL-1β, and IL-17 in vivo, we performed ChIP analyses. As shown in Fig. 6C, under resting conditions SPAK was associated with genes encoding TNF-α, IL-1β, and IL-17, but not with the IFN-γ gene. Overexpression of SPAK increased the association of the SPAK protein with these genes. To further study the linkage between enhanced pro-inflammatory cytokine production and SPAK expression, we investigated whether SPAK could stimulate transactivation of target cytokines. The results (Fig. 6D) indicate that transfection of cytokine reporters leads to increased basal level reporter activity levels of TNF-α (20-fold), IL-1β (7-fold), and IL-17 (3-fold), compared with empty vector. However, co-transfection of SPAK and cytokine reporter genes results in a marked increased luciferase activity for TNF-α (38-fold), IL-1β (17-fold), and IL-17 (11-fold) compared with co-transfection of control vectors. We used the IFN-γ reporter as a control. These results indicate that SPAK is able to trigger transactivation of certain pro-inflammatory cytokines including TNF-α, IL-1β, and IL-17.
SPAK aggravates commensal bacterial translocation in DSS-induced colitis
The imbalance between innate and adaptive immunity caused by luminal bacteria is thought to be a main contributor to the onset of IBD (29). We examined the translocation of luminal bacteria in TG and WT mice. First, no significant difference in CFU levels was seen in any organs tested in TG and WT mice. However, CFU levels increased markedly in all DSS-treated mice, and TG animals showed significantly higher CFU levels than WT in all organs tested (Fig. 7A). PCR analysis using primers directed toward highly conserved bacterial 16S rRNA sequences confirmed these results. Twenty cycles of amplification did not generate visible products in either untreated WT or TG mice; however, DSS-treated samples yielded strong DNA bands in both WT and TG mice. Furthermore, TG mice exhibited significantly more intense product bands than WT mice (Fig. 7B). We then performed FISH and immunostaining on the same slides to visualize translocated bacteria (Fig. 7C). We obtained results similar to those shown by CFU analysis, with no noticeable bacterial translocation in either untreated TG or WT mice. Bacterial translocation was apparent upon DSS treatment in both TG and WT mice, and was significantly higher in TG mice.
Figure 7. SPAK aggravates the luminal bacteria burden and translocation.
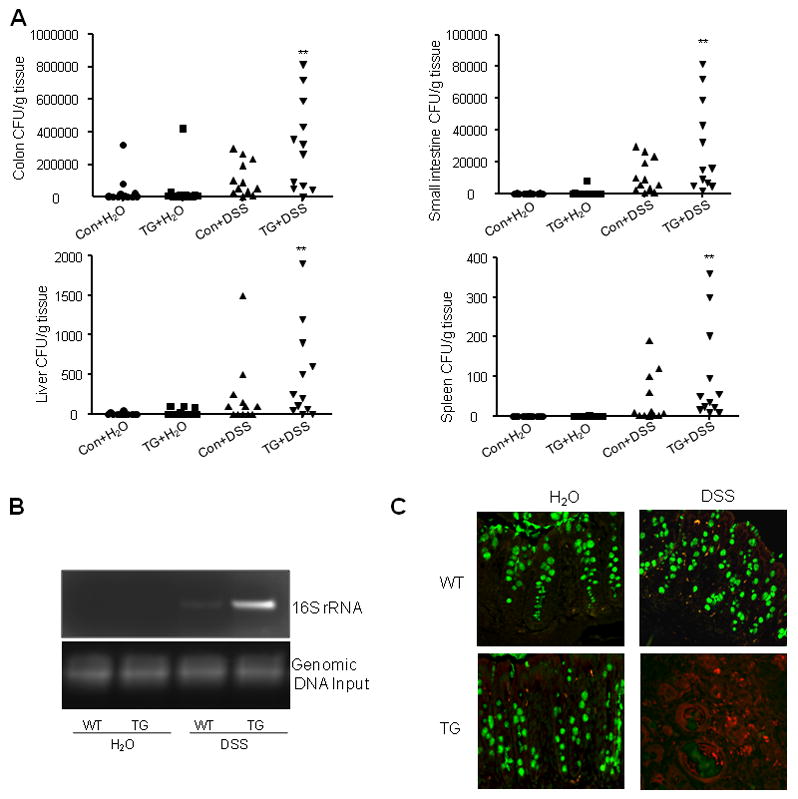
(A) Colon, small intestine, spleen and liver tissue from SPAK TG mice or WT littermates were homogenized and cultured on nonselective media. CFUs were counted as described in Material & Methods. Data are expressed as means ± SEM (n = 9 mice per group). Statistical differences of TG versus WT mice with or without DSS treatment are reported. Data are pooled from three independent experiments. *: P<0.05, **: P<0.01. (B) PCR of bacterial 16S gene using DNA isolated from equal surface areas of colonic mucosa. Amplification was performed for 20 cycles. (C) SPAK facilitates the translocation of luminal bacteria inside the crypt of colon. FISH with the probe of Alexa Fluor 555-conjugated EUB and mucus staining in each slide were performed. There is very sparse presence of bacteria (in red) in untreated WT and TG mice. After DSS treatment, there is a dramatic increase of translocation of bacteria in both WT and TG mice; further, significantly more bacterial colonies were observed in TG mice.
Discussion
In the present study, we have established for the first time, a colonic SPAK TG mouse model to highlight the importance of SPAK in the regulation of intestinal barrier function, which describes substantial new mechanistic insights into the pathogenesis of IBD.
Unlike other experimental colitis models with transgenic IL-17 (30), STAT-4 (31), HLA B27 (32), or Nod2 (33), which are mainly involved in the imbalance of innate and adaptive immune systems, SPAK TG mice primarily cause collapse of barrier function by increasing epithelial permeability, then enhance production of inflammatory cytokines and translocation of luminal bacteria. Similar observations have been reported in junctional adhesion molecule (JAM) knockout mice, which demonstrated increased mucosal permeability with enhanced expression of claudin-10, claudin-15 and inflammatory cytokines in colonic mucosa (34, 35). Additionally, myosin light chain kinase (MLCK) transgenic mice demonstrated significant barrier loss and accelerated onset and severity of immune-mediated colitis (36). The tight junction, unlike the epithelial cells themselves, forms a selectively dynamic permeable barrier with the ability to alter its permeability in response to extracellular stimuli. The sustained enhancement of paracellular permeability under certain circumstances may result in an uncontrolled “leaky” tight junction barrier, facilitating the constant passage of luminal pathogens and/or antigens through the mucosa and leading to inflammation in susceptible individuals (37). In fact, increased intestinal permeability is implicated in the pathogenesis of CD and UC as an initiating factor, which leads to a secondary mucosal inflammatory response (3–6). The SPAK TG mouse model mimics the natural pathogenesis of IBD, primarily affecting the intestinal barrier function, rendering IECs more susceptible to stress factors.
Mechanisms underlying the intestinal barrier dysfunction caused by SPAK are complicated. We found that epithelial barrier dysfunction is the primary effect of SPAK, which then leads to the increased production of inflammatory cytokines (supplemental data s1, s2 and s3). The increased expression of inflammatory cytokines could in turn cause intestinal barrier function collapse and are of great importance not only in the pathogenesis of colitis, but as intervention targets against colitis (9, 38). Another possible mechanism is that SPAK facilitates the translocation of luminal bacteria into mucosa after DSS treatment. It has been accepted that luminal bacteria are involved in innate immune homeostasis and the intestinal barrier function (39). We also observed that ZO-1, ZO-2, Claudin-1, -2, and Claudin-4 were expressed at the same levels in SPAK TG and wild-type animals (supplemental data s4, s5, s7, s8, s9 and supplemental table s1). In contrast, the level of occludin expression (supplemental data s6 and supplemental table s1) was significantly lower in SPAK TG mice compared to WT littermates. These results suggest that the lower level of occludin observed in SPAK TG animals could be responsible for the increase in paracellular flux of small-molecular weight tracers, thus modulating the overall intestinal barrier defect observed in SPAK TG mice. These results are in agreement with studies showing that occludin affected both tight junction size selectivity and electrical resistance (40–43). The studies underlying the involvement of occludin in SPAK overexpression-induced barrier defects are ongoing. In addition, it is known the cytoskeleton factors actin and myosin (44) play important roles in the regulation of intestinal barrier function. Some stress factors, such as hyperosmolarity, which regulate the expression and activity of SPAK, can modulate intestinal barrier function by modulating redistribution of actin and phosphorylation of the myosin light chain (MLC)(44, 45). Since we did not investigate in vivo ion selectivity, we thus cannot draw any definitive conclusions on the in vivo ion selectivity of tight junctions. It is known that ion selectivity of tight junctions plays an important role in the integrity of epithelial cells, thus in TER (27, 40). The claudin tight junction protein family plays an important role in the regulation of ion selectivity to mediate TER (46–48). IFN-γ and IL-1 regulate TER not only through size selectivity but also ion selectivity by regulating claudins (49, 50). TNF-alpha increases size selectivity without altering ion selectivity, whereas IL-13 alters ion selectivity without affecting size selectivity (51). Here, we did not see significant differences in the expression of claudin-1, 2 or 4 (supplemental data s7, s8, s9 and supplemental table s1); however, more studies are needed to determine the claudin expression profile during the alteration of SPAK expression. Also, our data indicate that barrier function loss caused by SPAK does not result from reduced cell viability and increased cytotoxicity, or from decreased proliferation, or increased apoptosis (data not shown), which is consistent with reports that TNF-α causes epithelial dysfunction in an apoptosis-independent manner (52).
Tremendous evidence shows that kinases are involved in the production of pro-inflammatory cytokines. For example, p38 regulates the biosynthesis of pro-inflammatory cytokine IL-1β(53); MAPK 6 (MKK6) can stimulate the production of cytokine IL-6 (54); and MAPK-activated protein kinase 2 (MK2) regulates TNF-α expression (55). Protein kinase C θ (PKCθ) is involved in the expression of IL-2, IFN-γ, IL-6, and IL-17 (56–59). Interestingly, SPAK is the first Ste20-like kinase, to our knowledge, to stimulate the production of the pro-inflammatory cytokines IL-1β, TNF-α, and IL-17. The underlying mechanisms involved in regulating the expression of pro-inflammatory cytokines by kinases could be at the transcriptional level. For example, calmodulin-dependent kinase II (CaMK II) regulates expression of granulocyte macrophage colony-stimulating factor (GM-CSF) by phosphorylating transcription factor Ets1 (60). The data from our ChIP and transactivation assays demonstrated that SPAK could be associated with the IL-1β, TNF-α and IL-17 genes, either directly or indirectly. However, we cannot exclude the possibility that SPAK phosphorylates another protein, perhaps an enzyme, kinase, or transcription factor, which in turn stimulates production of IL-1β, TNF-α, and IL-17.
In summary, SPAK regulates IEC barrier function in vitro and in vivo by modulating the size selectivity of tight junctions. Furthermore, SPAK dramatically facilitates the secretion of proinflammatory cytokines and luminal bacterial translocation in SPAK-transgenic mice in the presence of DSS or TNBS. We believe that SPAK establishes a context favorable for the triggering of intestinal inflammation cascades induced by DSS and TNBS. In conclusion, SPAK TG mice are more susceptible to induced colitis due to deregulated intestinal mucosal innate immune homeostasis.
Supplementary Material
Acknowledgments
This paper is for the memorial of Dr. Shanthi Sitarman-A brilliant scientist, a dedicated physician, a passionate humanitarian, and a dearest friend. We thank Arianne L. Theiss and Andrew T. Gewirtz for critically reading the manuscript.
This work was supported by grants from the Department of Veterans Affairs and the National Institutes of Health of Diabetes and Digestive and Kidney by the grants R24-DK-064399 (center grant), R56DK084987 (to D.M), RO1-DK55850 (to S.S).
Abbreviations used
- SPAK
Ste20-like proline/alanine-rich kinase
- DSS
dextran sodium sulfate
- TNBS
trinitrobenzene sulfonic acid
- FITC
fluorescein isothiocyanate
- MPO
myeloperoxidase
- PCR
polymerase chain reaction
- ChIP
Chromatin immunoprecipitation
- MAP4K
Mitogen-activated protein (MAP) kinase kinase kinase kinase
- FISH
Fluorescence in situ hybridization
- SPAK TG mice
transgenic mice specifically overexpressing SPAK in intestinal epithelial cells
- WT
wild-type
References
- 1.Clayburgh DR, Shen L, Turner JR. A porous defense: the leaky epithelial barrier in intestinal disease. Lab Invest. 2004;84:282–291. doi: 10.1038/labinvest.3700050. [DOI] [PubMed] [Google Scholar]
- 2.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 3.Schmitz H, Barmeyer C, Fromm M, Runkel N, Foss HD, Bentzel CJ, Riecken EO, Schulzke JD. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology. 1999;116:301–309. doi: 10.1016/s0016-5085(99)70126-5. [DOI] [PubMed] [Google Scholar]
- 4.Swidsinski A, Loening-Baucke V, Theissig F, Engelhardt H, Bengmark S, Koch S, Lochs H, Dorffel Y. Comparative study of the intestinal mucus barrier in normal and inflamed colon. Gut. 2007;56:343–350. doi: 10.1136/gut.2006.098160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut. 2007;56:61–72. doi: 10.1136/gut.2006.094375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katz KD, Hollander D, Vadheim CM, McElree C, Delahunty T, Dadufalza VD, Krugliak P, Rotter JI. Intestinal permeability in patients with Crohn’s disease and their healthy relatives. Gastroenterology. 1989;97:927–931. doi: 10.1016/0016-5085(89)91499-6. [DOI] [PubMed] [Google Scholar]
- 7.Berkes J, V, Viswanathan K, Savkovic SD, Hecht G. Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut. 2003;52:439–451. doi: 10.1136/gut.52.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, 2nd, Raleigh DR, Guan Y, Watson AJ, Montrose MH, Turner JR. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189:111–126. doi: 10.1083/jcb.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sartor RB. Cytokines in intestinal inflammation: pathophysiological and clinical considerations. Gastroenterology. 1994;106:533–539. doi: 10.1016/0016-5085(94)90614-9. [DOI] [PubMed] [Google Scholar]
- 10.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–419. doi: 10.1016/s0002-9440(10)62264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–5744. [PubMed] [Google Scholar]
- 12.Kennedy RJ, Hoper M, Deodhar K, Erwin PJ, Kirk SJ, Gardiner KR. Interleukin 10-deficient colitis: new similarities to human inflammatory bowel disease. Br J Surg. 2000;87:1346–1351. doi: 10.1046/j.1365-2168.2000.01615.x. [DOI] [PubMed] [Google Scholar]
- 13.Olson TS, Reuter BK, Scott KG, Morris MA, Wang XM, Hancock LN, Burcin TL, Cohn SM, Ernst PB, Cominelli F, Meddings JB, Ley K, Pizarro TT. The primary defect in experimental ileitis originates from a nonhematopoietic source. J Exp Med. 2006;203:541–552. doi: 10.1084/jem.20050407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, Nast CC, Lechago J, Xu R, Naiki Y, Soliman A, Arditi M, Abreu MT. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1055–1065. doi: 10.1152/ajpgi.00328.2004. [DOI] [PubMed] [Google Scholar]
- 15.Johnston AM, Naselli G, Gonez LJ, Martin RM, Harrison LC, DeAizpurua HJ. SPAK, a STE20/SPS1-related kinase that activates the p38 pathway. Oncogene. 2000;19:4290–4297. doi: 10.1038/sj.onc.1203784. [DOI] [PubMed] [Google Scholar]
- 16.Yan Y, Nguyen H, Dalmasso G, Sitaraman SV, Merlin D. Cloning and characterization of a new intestinal inflammation-associated colonic epithelial Ste20-related protein kinase isoform. Biochim Biophys Acta. 2007;1769:106–116. doi: 10.1016/j.bbaexp.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan Y, Dalmasso G, Nguyen HT, Obertone TS, Charrier-Hisamuddin L, Sitaraman SV, Merlin D. Nuclear factor-kappaB is a critical mediator of Ste20-like proline-/alanine-rich kinase regulation in intestinal inflammation. Am J Pathol. 2008;173:1013–1028. doi: 10.2353/ajpath.2008.080339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Hu J, Vita R, Sun B, Tabata H, Altman A. SPAK kinase is a substrate and target of PKCtheta in T-cell receptor-induced AP-1 activation pathway. Embo J. 2004;23:1112–1122. doi: 10.1038/sj.emboj.7600125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gagnon KB, England R, Delpire E. Characterization of SPAK and OSR1, regulatory kinases of the Na-K-2Cl cotransporter. Mol Cell Biol. 2006;26:689–698. doi: 10.1128/MCB.26.2.689-698.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan Y, Dalmasso G, Nguyen HT, Obertone TS, Sitaraman SV, Merlin D. Ste20-related proline/alanine-rich kinase (SPAK) regulated transcriptionally by hyperosmolarity is involved in intestinal barrier function. PLoS One. 2009;4:e5049. doi: 10.1371/journal.pone.0005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyle JP, Wu XJ, Shoemaker CB, Yoshino TP. Using RNA interference to manipulate endogenous gene expression in Schistosoma mansoni sporocysts. Mol Biochem Parasitol. 2003;128:205–215. doi: 10.1016/s0166-6851(03)00078-1. [DOI] [PubMed] [Google Scholar]
- 22.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105:15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan Y, Dalmasso G, Sitaraman S, Merlin D. Characterization of the human intestinal CD98 promoter and its regulation by interferon-gamma. Am J Physiol Gastrointest Liver Physiol. 2007;292:G535–545. doi: 10.1152/ajpgi.00385.2006. [DOI] [PubMed] [Google Scholar]
- 24.Tulasne D, Paumelle R, Weidner KM, Vandenbunder B, Fafeur V. The multisubstrate docking site of the MET receptor is dispensable for MET-mediated RAS signaling and cell scattering. Mol Biol Cell. 1999;10:551–565. doi: 10.1091/mbc.10.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vijay-Kumar M, Sanders CJ, Taylor RT, Kumar A, Aitken JD, Sitaraman SV, Neish AS, Uematsu S, Akira S, Williams IR, Gewirtz AT. Deletion of TLR5 results in spontaneous colitis in mice. J Clin Invest. 2007;117:3909–3921. doi: 10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madara JL. Maintenance of the macromolecular barrier at cell extrusion sites in intestinal epithelium: physiological rearrangement of tight junctions. J Membr Biol. 1990;116:177–184. doi: 10.1007/BF01868675. [DOI] [PubMed] [Google Scholar]
- 27.Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 29.Macdonald TT, Monteleone G. Immunity, inflammation, and allergy in the gut. Science. 2005;307:1920–1925. doi: 10.1126/science.1106442. [DOI] [PubMed] [Google Scholar]
- 30.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wirtz S, Finotto S, Kanzler S, Lohse AW, Blessing M, Lehr HA, Galle PR, Neurath MF. Cutting edge: chronic intestinal inflammation in STAT-4 transgenic mice: characterization of disease and adoptive transfer by TNF- plus IFN-gamma-producing CD4+ T cells that respond to bacterial antigens. J Immunol. 1999;162:1884–1888. [PubMed] [Google Scholar]
- 32.Hammer RE, Maika SD, Richardson JA, Tang JP, Taurog JD. Spontaneous inflammatory disease in transgenic rats expressing HLA-B27 and human beta 2m: an animal model of HLA-B27-associated human disorders. Cell. 1990;63:1099–1112. doi: 10.1016/0092-8674(90)90512-d. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z, I, Fuss J, Watanabe T, Asano N, Davey MP, Rosenbaum JT, Strober W, Kitani A. NOD2 transgenic mice exhibit enhanced MDP-mediated down-regulation of TLR2 responses and resistance to colitis induction. Gastroenterology. 2007;133:1510–1521. doi: 10.1053/j.gastro.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, Dermody TS, Nusrat A, Parkos CA. JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med. 2007;204:3067–3076. doi: 10.1084/jem.20071416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vetrano S, Rescigno M, Cera MR, Correale C, Rumio C, Doni A, Fantini M, Sturm A, Borroni E, Repici A, Locati M, Malesci A, Dejana E, Danese S. Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology. 2008;135:173–184. doi: 10.1053/j.gastro.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 36.Su L, Shen L, Clayburgh DR, Nalle SC, Sullivan EA, Meddings JB, Abraham C, Turner JR. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology. 2009;136:551–563. doi: 10.1053/j.gastro.2008.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 38.Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology. 1994;107:1643–1652. doi: 10.1016/0016-5085(94)90803-6. [DOI] [PubMed] [Google Scholar]
- 39.Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;8:411–420. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- 40.Balda MS, Whitney JA, Flores C, Gonzalez S, Cereijido M, Matter K. Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol. 1996;134:1031–1049. doi: 10.1083/jcb.134.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, 2nd, Raleigh DR, Guan Y, Watson AJ, Montrose MH, Turner JR. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189:111–126. doi: 10.1083/jcb.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Utech M, Bruwer M, Nusrat A. Tight junctions and cell-cell interactions. Methods Mol Biol. 2006;341:185–195. doi: 10.1385/1-59745-113-4:185. [DOI] [PubMed] [Google Scholar]
- 43.Severson EA, Kwon M, Hilgarth RS, Parkos CA, Nusrat A. Glycogen Synthase Kinase 3 (GSK-3) influences epithelial barrier function by regulating Occludin, Claudin-1 and E-cadherin expression. Biochem Biophys Res Commun. 2010 doi: 10.1016/j.bbrc.2010.05.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clayburgh DR, Barrett TA, Tang Y, Meddings JB, Van Eldik LJ, Watterson DM, Clarke LL, Mrsny RJ, Turner JR. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J Clin Invest. 2005;115:2702–2715. doi: 10.1172/JCI24970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. 1998;273:29745–29753. doi: 10.1074/jbc.273.45.29745. [DOI] [PubMed] [Google Scholar]
- 46.Van Itallie C, Rahner C, Anderson JM. Regulated expression of claudin-4 decreases paracellular conductance through a selective decrease in sodium permeability. J Clin Invest. 2001;107:1319–1327. doi: 10.1172/JCI12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colegio OR, Van Itallie CM, McCrea HJ, Rahner C, Anderson JM. Claudins create charge-selective channels in the paracellular pathway between epithelial cells. Am J Physiol Cell Physiol. 2002;283:C142–147. doi: 10.1152/ajpcell.00038.2002. [DOI] [PubMed] [Google Scholar]
- 48.Yu AS, Enck AH, Lencer WI, Schneeberger EE. Claudin-8 expression in Madin-Darby canine kidney cells augments the paracellular barrier to cation permeation. J Biol Chem. 2003;278:17350–17359. doi: 10.1074/jbc.M213286200. [DOI] [PubMed] [Google Scholar]
- 49.Madsen KL, Lewis SA, Tavernini MM, Hibbard J, Fedorak RN. Interleukin 10 prevents cytokine-induced disruption of T84 monolayer barrier integrity and limits chloride secretion. Gastroenterology. 1997;113:151–159. doi: 10.1016/s0016-5085(97)70090-8. [DOI] [PubMed] [Google Scholar]
- 50.Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, Johnson LG. Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell. 2002;13:3218–3234. doi: 10.1091/mbc.E02-03-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weber CR, Raleigh DR, Su L, Shen L, Sullivan EA, Wang Y, Turner JR. Epithelial myosin light chain kinase activation induces mucosal interleukin-13 expression to alter tight junction ion selectivity. J Biol Chem. 2010;285:12037–12046. doi: 10.1074/jbc.M109.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brown SJ, Abreu MT. Antibodies to tumor necrosis factor-alpha in the treatment of Crohn’s disease. Curr Opin Drug Discov Devel. 2005;8:160–168. [PubMed] [Google Scholar]
- 53.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 54.Yoshizawa T, Hammaker D, Boyle DL, Corr M, Flavell R, Davis R, Schett G, Firestein GS. Role of MAPK kinase 6 in arthritis: distinct mechanism of action in inflammation and cytokine expression. J Immunol. 2009;183:1360–1367. doi: 10.4049/jimmunol.0900483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thuraisingam T, Xu YZ, Moisan J, Lachance C, Garnon J, Di Marco S, Gaestel M, Radzioch D. Distinct role of MAPKAPK-2 in the regulation of TNF gene expression by Toll-like receptor 7 and 9 ligands. Mol Immunol. 2007;44:3482–3491. doi: 10.1016/j.molimm.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 56.Pfeifhofer C, Kofler K, Gruber T, Tabrizi NG, Lutz C, Maly K, Leitges M, Baier G. Protein kinase C theta affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J Exp Med. 2003;197:1525–1535. doi: 10.1084/jem.20020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Salek-Ardakani S, So T, Halteman BS, Altman A, Croft M. Protein kinase Ctheta controls Th1 cells in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:7635–7641. doi: 10.4049/jimmunol.175.11.7635. [DOI] [PubMed] [Google Scholar]
- 58.Healy AM, Izmailova E, Fitzgerald M, Walker R, Hattersley M, Silva M, Siebert E, Terkelsen J, Picarella D, Pickard MD, LeClair B, Chandra S, Jaffee B. PKC-theta-deficient mice are protected from Th1-dependent antigen-induced arthritis. J Immunol. 2006;177:1886–1893. doi: 10.4049/jimmunol.177.3.1886. [DOI] [PubMed] [Google Scholar]
- 59.Tan SL, Zhao J, Bi C, Chen XC, Hepburn DL, Wang J, Sedgwick JD, Chintalacharuvu SR, Na S. Resistance to experimental autoimmune encephalomyelitis and impaired IL-17 production in protein kinase C theta-deficient mice. J Immunol. 2006;176:2872–2879. doi: 10.4049/jimmunol.176.5.2872. [DOI] [PubMed] [Google Scholar]
- 60.Liu H, Holm M, Xie XQ, Wolf-Watz M, Grundstrom T. AML1/Runx1 recruits calcineurin to regulate granulocyte macrophage colony-stimulating factor by Ets1 activation. J Biol Chem. 2004;279:29398–29408. doi: 10.1074/jbc.M403173200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.