

Abstract
Background
The association between secondhand smoke (SHS) exposure and bladder cancer is inconclusive. Epigenetic alterations in bladder tumors have been linked to primary cigarette smoking and could add to the biological plausibility of an association between SHS exposure and bladder cancer.
Hypothesis
SHS exposure is associated with DNA methylation in bladder tumors.
Methods
Using an array-based approach, we profiled DNA methylation from never smoking cases of incident bladder cancer. Analyses examined associations between individual loci’s methylation with SHS variables (exposure in adulthood, childhood, occupationally and total exposure). A canonical pathway analysis was used to find pathways significantly associated with each SHS exposure type.
Results
There is a trend towards increased methylation of numerous CpG loci with increasing exposure to adulthood, occupational and total SHS. Discrete associations between methylation extent of several CpG loci and SHS exposures demonstrated significantly increased methylation of these loci across all types of SHS exposure. CpGs with SHS-related methylation alterations were associated with genes in pathways involved in carcinogenesis, immune modulation and immune signaling.
Interpretation
Exposure to SHS in adulthood, childhood, occupationally and in total are each significantly associated with changes in DNA methylation of several CpG loci in bladder tumors, adding biological plausibility to SHS as a risk factor for bladder cancer.
Keywords: Bladder Cancer, Second Hand Smoking, DNA Methylation, Methylation Profiling, Array-based Methodologies
Introduction
Bladder cancer is the ninth most incident form of cancer in the United States. Genetics, epigenetics, dietary factors, occupational exposures, environmental and lifestyle factors are all known to play important roles in bladder cancer initiation (1–5). The majority of bladder cancer cases are non-invasive and highly treatable, although these patients have great morbidity related to the need for intensive cystoscopic follow-up. Approximately 25% of cases present as muscle-invasive with high mortality rates (6–7). The biggest risk factor for bladder cancer is cigarette smoking, accounting for approximately 65% of disease risk in men and a smaller proportion in women (8). Tobacco smoke contains 2-naphthylamine, 4-aminobiphenyl and other arylamines amongst hundreds of other chemicals which experimental evidence has implicated as being bladder carcinogens (9–14). Environmental tobacco smoke or secondhand smoke (SHS) also contains these chemicals. Although the level of exposure may be less than in active smoking, the carcinogens present in SHS may make a contribution to bladder carcinogenesis, especially among people with higher susceptibility to tobacco toxicants (13). In fact, these toxicants have been detected in the urine of SHS-exposed individuals (15–16). However, only one case-control study among many epidemiological investigations found a positive association between exposure to SHS and risk of bladder cancer; and that risk was found to be present only in women (17–20).
Epigenetic mechanisms provide a heritable and stable form of gene expression control beyond the DNA sequence (21–22). A number of genes have been found to undergo epigenetic silencing through DNA methylation in bladder cancer (5, 23–24). Changes in the DNA methylation pattern of the promoter region of several cancer-related genes have also been associated with specific risk factors for bladder cancer, including smoking (5, 23–24).
Despite reported associations between DNA methylation and active smoking, the potential effect of SHS exposure on DNA methylation in bladder cancer remains unclear. Identifying associations between DNA methylation in tumors and SHS exposure would provide biological plausibility to the observed associations between SHS exposure and bladder cancer risk. Hence, we used an array-based DNA methylation profiling approach to examine the potential association between SHS exposure and DNA methylation in 1413 CpG loci, associated with 773 genes, in primary human bladder tumors from non-smoking individuals. Our aim was to conduct a pilot study examining these potential associations using a discovery-based platform in a small sample of bladder cancer cases. Identification of specific genes whose epigenetic alteration is associated specifically with SHS exposure may provide insight into important biological differences of these tumors, and suggest potential pathways for targeted chemotherapeutic or chemopreventative measures.
Materials and Methods
Subjects
A description of the study design appears in earlier reports (25–26). Briefly, bladder cancer cases were drawn from subjects enrolled in two stages of a non-consecutive, population-based case-control study of bladder cancer in New Hampshire, conducted from 1994–1998 and from 2001–2004. Cases of incident bladder cancer were identified from the state cancer registry and a standardized histopathologic review was conducted by a single study pathologist (A.R.S.) to verify the diagnosis and histopathology of the cases. Formalin-fixed, paraffin-embedded (FFPE) tumor tissue was obtained from the cases in the overall study. For the analyses presented here, the case group was restricted to non-smoking cases, with second hand smoke exposure (SHS) data, and excluded cases that were diagnosed as carcinoma in situ due to small numbers. We obtained promoter methylation data from all non-smoking cases in our sample giving us a final sample of 43 cases whose characteristics are presented in Table 1. We noted no significant differences in the demographics or clinical characteristics of these non-smoking cases with SHS exposure data as compared to the remaining non-smoking cases without SHS exposure data. We also conducted additional sensitivity analyses on former and current smoking cases. For those analyses, the case group was restricted to cases having smoking status data and promoter methylation data and excluded cases that were diagnosed as carcinoma in situ due to small numbers. All procedures and study materials were approved by the appropriate Institutional Review Boards.
Table 1.
Selected characteristics of bladder cancer cases with second hand smoking status data
| Characteristics | Bladder Cancer Cases (N=43) |
|---|---|
| Gender, n (%) | |
| Female | 14 (32.6%) |
| Male | 29 (67.4%) |
| Age, mean (SD) | 63 (11.5) |
| Tumor Stage* | |
| Non-invasive | 30 (75.0%) |
| Invasive | 10 (25.0%) |
| Second hand smoking variables, mean(SD)† | |
| Adulthood Passive †† | 19.5 (24.5) |
| Childhood Passive ± | 18.6 (19.5) |
| Occupational Passive ‖ | 81.9 (151.1) |
| Total Passive: Childhood, Adulthood, Occupational | 119.9 (153.8) |
not available on 3 subjects
Weighted by years of exposure
Sum of the years spent at each adult residence multiplied by the number of smokers in each residence
Sum of the years spent at each childhood residence (up to age 18 years) multiplied by the number of smokers in each residence
Sum of the years spent at the longest job multiplied by the number of Smokers
Personal Interview and Second Hand Smoking measurement
Consenting subjects underwent a detailed in-person interview, usually at their home, which assessed sociodemographic information, occupational history, detailed information about the use of tobacco products (such as history of cigarette smoking) and medical history prior to the reference or diagnosis date. Never smokers were defined as cases who had smoked less than 100 cigarettes over their lifetime. Among never smokers, SHS exposure was assessed through a questionnaire asking participants about the number of people who smoked around them everywhere they lived for at least 2 years duration since the age of 10 years and at the longest job they held for at least 6 months since the age of 16 years (25). Four types of second hand smoking exposure were assessed namely; childhood, adulthood, occupational, and total SHS exposure (cumulative exposure among childhood, adulthood and occupational). SHS exposures were calculated using the following measures: years spent living with ≥ 1 smoker in childhood (age ≤ 18), cumulative residential exposure in adulthood (the sum of the total number of smokers in each residence multiplied by the time spent in each residence over the person’s lifetime), and the cumulative occupational SHS exposure (measured by the duration of time in years spent working with ≥ 1 smoker for the longest job that the subject held (as used in 25). Figure 1 shows the distribution of second hand smoke (SHS) exposure by each type of SHS exposure using boxplots.
Figure 1.
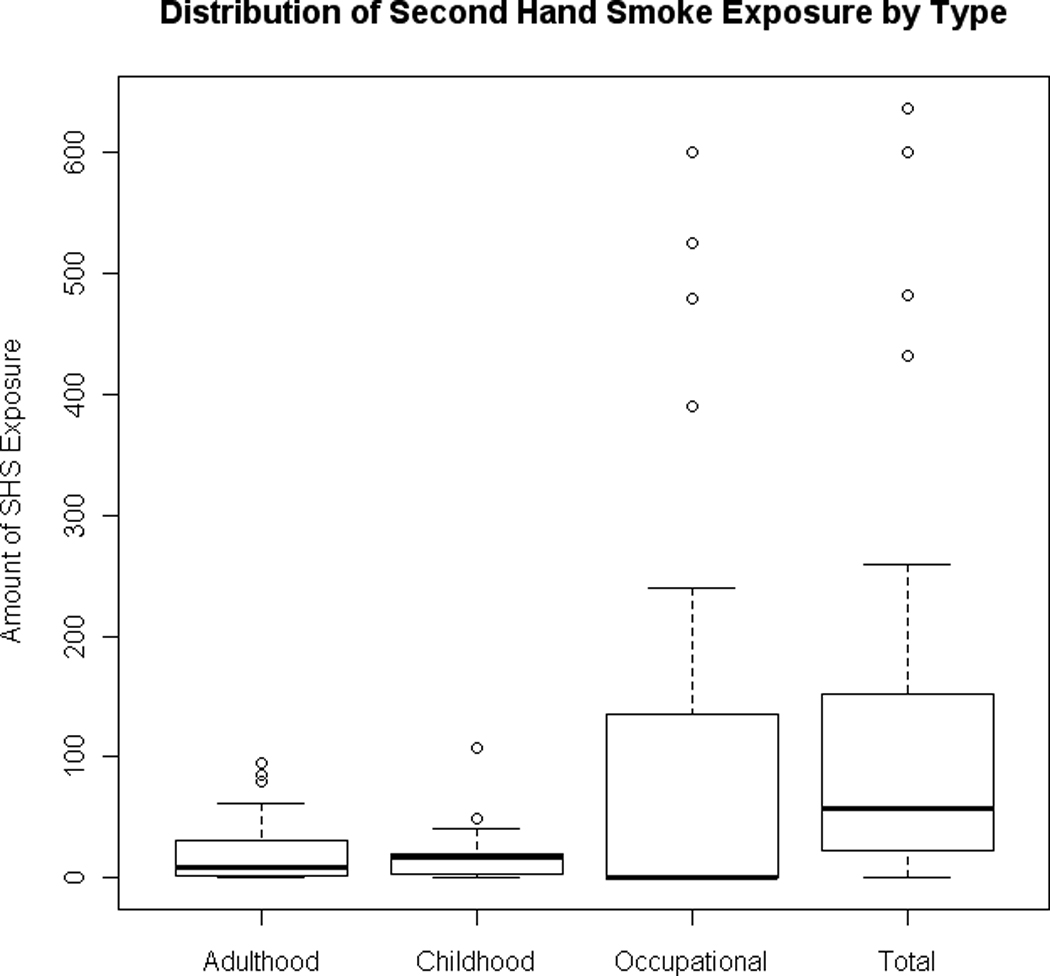
The distribution of second hand smoke (SHS) exposure by each type of SHS exposure is shown using boxplots. A boxplot is a graphical technique used to depict the range of a variable, with the 5th and the 95th percentiles of the variable being shown as the whiskers of the box and with the lower quartile (25th percentile) and the upper quartile (75th percentile) being shown as the edges of the box. The median or 50th percentile is shown by the horizontal line through the box. A boxplot can not only show the range of the data but also gives us a sense of potential outlying observations of a certain variable.
DNA Methylation Analysis
DNA was extracted from the FFPE tumor samples as previously described (27). One µg of DNA was then subjected to sodium bisulfite modification using the EZ DNA Methylation Kit (Zymo Research, Orange, CA) following the manufacturer’s protocol. Methylation profiling was performed using the Illumina GoldenGate Methylation Bead Array at the UCSF Institute for Human Genetics Genomic Core Facility as previously described (28–29). All array data points were represented by fluorescent signals from both methylated (Cy5) and unmethylated (Cy3) alleles to create the average methylation (β) value derived from about 30 replicate methylation measurements, and which ranged from 0 (fully unmethylated) to 1 (fully methylated). The data was then assembled with BeadStudio methylation software from Illumina. Quality assurance and quality control (QA/QC) was performed and 8 poor performing loci (with >25% of samples having a detection p-value >1.0×10−5 at that locus) were removed. In addition, only autosomal CpG loci were considered, leaving 1413 CpG loci associated with 773 genes for analysis. In our previous work, we validated the results of the array using a bisulfite pyrosequencing, and observed strong correlation between the results of the array and this gold-standard method (30).
Statistical Methods
We calculated spearman correlations between each type of SHS exposure; spearman ranks were used to account for the potential nonparametric and nonlinear correlations between each type of SHS exposure. To identify specific loci whose differential methylation was associated with SHS exposure, we fit individual generalized linear models between the methylation beta value for each locus and the exposure. The beta distribution of average beta values was accounted for with a quasi-binomial logit link with an estimated scale parameter constraining the mean between 0 and 1, as previously described (31). We accounted for multiple comparisons using false discovery rate estimation to calculate Q-values through the qvalue package in R (32). We also conducted individualized generalized linear models between the methylation beta value for each locus and former and current smoking status. To identify the cellular pathways represented by the genes associated with the differentially methylated loci, we conducted a canonical pathways analysis using the Ingenuity Pathway Analysis (Ingenuity Systems, 33). CpG loci that were found to be significantly associated with either adulthood, childhood, occupational or total second hand smoking exposure in the locus-by-locus analysis at the Q<0.05 level were considered for pathways analysis where all autosomal genes on the GoldenGate array were used as the referent gene set. The significance of gene-locus enrichment within canonical pathways was measured using a Fisher’s exact test at the p<0.05 level. Data were analyzed by use of R, version 2.10.
Results
The characteristics of the cases used in these analyses, including SHS exposures are shown in Table 1, with the distribution of second hand smoke (SHS) exposure by each type of SHS exposure shown in Figure 1, using boxplots. In examining the correlation between the individual types of SHS exposure, we identified relatively modest correlations between adulthood, childhood, and occupational exposures ranging from −0.2 to 0.2 (Supplementary Table 1). As expected the correlation between each of the measures and the total exposure was greatest, as this is a cumulative measure of all exposure. Occupational exposures appeared to be the most correlated with total exposure, having a correlation coefficient of 0.8.
We examined whether each SHS exposure type was associated with specific methylation changes in the 1413 CpG loci in a locus-by-locus analysis. Figure 2 shows the −log(p-values) versus regression coefficients from locus-by-locus analysis of CpG methylation by type of exposure (Adulthood, Childhood, Occupational or Total) to SHS. A greater number of CpG loci show a significant, positive association between their methylation beta value and weighted measures of SHS exposure for all four types of exposure.
Figure 2.

Volcano plots volcano plot of the −log (p-values) plotted against the quasi binomial regression coefficients from the locus-by-locus analysis of 1413 CpG methylation values by (A) adulthood second hand smoking, (B) childhood second hand smoking, (C) occupational second hand smoking, and (D) total second hand smoking. The horizontal blue dotted line intercepts the y-axis to illustrate significance above the line (before correction for multiple comparisons).
Those specific loci identified with the strongest associations (FDR Q<0.05) between methylation and SHS exposure are shown in Table 2. As expected from Figure 2, all of the loci reaching a Q<0.05 had increased methylation with increasing exposure to SHS. A total of 23 CpG loci were found to be significantly associated with adulthood exposure to SHS, while only 7 loci were found to be significantly associated with both childhood and total exposure to SHS. Only 3 CpG loci were found to be significantly associated with occupational exposure to SHS. Among CpGs with significant SHS associations, across exposure groups there were no CpGs in common.
Table 2.
Significant loci (at the Q-value<0.05 level) by second hand smoking exposure type:
| Genes | Specific CpG loci | Q-value | Type of second hand smoking exposure |
|---|---|---|---|
| FLT1 | FLT1_E444_F | 0.036 | Total |
| PTPN6 | PTPN6_E171_R | 0.036 | Total |
| CTSH | CTSH_P238_F | 0.001 | Total |
| EPHB2 | EPHB2_P165_R | 0.001 | Total |
| CD9 | CD9_E14_R | 0.001 | Total |
| JAK2 | JAK2_P772_R | 0.000 | Total |
| CTGF | CTGF_E156_F | 0.000 | Total |
| FLT1 | FLT1_E444_F | 0.004 | Occupational |
| JAK2 | JAK2_P772_R | 0.001 | Occupational |
| CTGF | CTGF_E156_F | 0.000 | Occupational |
| SHB | SHB_P473_R | 0.037 | Childhood |
| SMARCA3 | SMARCA3_P109_R | 0.037 | Childhood |
| EPHA1 | EPHA1_E46_R | 0.028 | Childhood |
| VAMP8 | VAMP8_P114_F | 0.028 | Childhood |
| LIF | LIF_E208_F | 0.027 | Childhood |
| IGF2R | IGF2R_P396_R | 0.000 | Childhood |
| RRAS | RRAS_P100_R | 0.000 | Childhood |
| BCL3 | BCL3_P1038_R | 0.029 | Adulthood |
| TGFA | TGFA_P642_R | 0.022 | Adulthood |
| TYK2 | TYK2_P494_F | 0.022 | Adulthood |
| ITPR3 | ITPR3_P1112_F | 0.021 | Adulthood |
| YES1 | YES1_P216_F | 0.021 | Adulthood |
| LIG4 | LIG4_P194_F | 0.015 | Adulthood |
| FHIT | FHIT_P93_R | 0.012 | Adulthood |
| IL12A | IL12A_E287_R | 0.012 | Adulthood |
| PPARG | PPARG_E178_R | 0.011 | Adulthood |
| PKD2 | PKD2_P336_R | 0.007 | Adulthood |
| RET | RET_seq_54_S260_F | 0.007 | Adulthood |
| MGMT | MGMT_P272_R | 0.005 | Adulthood |
| NR2F6 | NR2F6_E375_R | 0.002 | Adulthood |
| TYRO3 | TYRO3_P501_F | 0.002 | Adulthood |
| IGF1R | IGF1R_E186_R | 0.002 | Adulthood |
| INHA | INHA_P1144_R | 0.002 | Adulthood |
| EPHB2 | EPHB2_P165_R | 0.001 | Adulthood |
| CDH3 | CDH3_E100_R | 0.001 | Adulthood |
| DDR1 | DDR1_E23_R | 0.000 | Adulthood |
| PTPN6 | PTPN6_E171_R | 0.000 | Adulthood |
| CTSH | CTSH_P238_F | 0.000 | Adulthood |
| CD9 | CD9_E14_R | 0.000 | Adulthood |
| EFNA1 | EFNA1_P591_R | 0.000 | Adulthood |
To compare our results of epigenetic alterations associated with SHS exposure, we also looked at those specific loci identified with the strongest associations (p<0.05) between methylation and former and current smoking status in Supplemental Tables 2 and 3. After adjusting for multiple comparisons using FDR, only two loci were found to be marginally associated (at the Q=0.06 level) with current smoking status. Neither of those two overlapped with any of the specific loci that were identified with the strongest associations (Q<0.05) between methylation and any type of SHS exposure. However, two specific CpG loci (EPHA1_E46_R and INHA_P1144_R) that were found to be associated at the p<0.05 level with former smoking status were also associated with Childhood and Adulthood exposure to SHS respectively. Additionally, one specific CpG loci (TGFA_P642_R) that was found to be associated at the p<0.05 level with current smoking status was also associated with Adulthood exposure to SHS. In addition, members of the SFRP family were found to be significantly associated (at the p<0.05 level) with former and current smoking status consistent with our previous work in another subset of bladder cancer cases from New Hampshire (27).
We performed a pathways analysis to determine which cellular pathways were over-represented amongst CpG loci identified in the locus-by-locus analysis as significantly (Q<0.05) associated with adulthood, childhood, occupational or total SHS exposure. For CpG loci significantly associated with adulthood SHS exposure we found that cancer-related, JAK/Stat, IL-4, and renin-angiotensin signaling pathways were significantly over-represented (Table 3). For CpG loci significantly associated with exposure to SHS in childhood we found that the ERK5, glioma, and T-Cell receptor signaling pathways were significantly over-represented (Table 3). Among CpGs significantly associated with occupational SHS exposure, the hepatic fibrosis/hepatic stellate cell activation pathway was significantly over-represented (Table 3). Lastly, for CpG loci significantly associated with total SHS exposure, the growth hormone, erythropoietin, JAK/Stat, IL-3, IL-4, and the CTLA4 signaling in cytotoxic T lymphocytes pathways were over-represented (Table 3).
Table 3.
Pathways over-represented amongst loci whose methylation was significantly associated with second hand smoking exposure
| Pathway | Type of second hand smoke exposure |
p-value | Genes |
|---|---|---|---|
| Non-Small Cell Lung Cancer Signaling | Adulthood | 0.004 | FHIT, NRAS, ITPR3, TGFA |
| Thyroid Cancer Signaling | Adulthood | 0.014 | PPARG, NRAS, RET |
| JAK/Stat Signaling | Adulthood | 0.020 | PTPN6, NRAS, TYK2 |
| IL-4 Signaling | Adulthood | 0.023 | PTPN6, NRAS, TYK2 |
| Renin-Angiotensin Signaling | Adulthood | 0.040 | PTPN6, NRAS, ITPR3 |
| ERK5 Signaling | Childhood | 0.036 | LIF, RRAS |
| Glioma Signaling | Childhood | 0.048 | RRAS, IGF2R |
| T Cell Receptor Signaling | Childhood | 0.048 | SHB, RRAS |
| Hepatic Fibrosis / Hepatic Stellate Cell Activation | Occupational | 0.027 | CTGF, FLT1 |
| JAK/Stat Signaling | Total | 0.033 | PTPN6, JAK2 |
| Growth Hormone Signaling | Total | 0.034 | PTPN6, JAK2 |
| Erythropoietin Signaling | Total | 0.035 | PTPN6, JAK2 |
| IL-4 Signaling | Total | 0.036 | PTPN6, JAK2 |
| IL-3 Signaling | Total | 0.037 | PTPN6, JAK2 |
| CTLA4 Signaling in Cytotoxic T Lymphocytes | Total | 0.046 | PTPN6, JAK2 |
Discussion
This study aimed to investigate the association between SHS exposure and DNA methylation of a large number of CpG loci bladder cancer cases who had never smoked. We tested the hypothesis that epigenetic changes in bladder tumors are associated with SHS exposure. Our finding of specific CpG loci methylation being associated with either adulthood, childhood, occupational or total SHS exposure, provides considerable additional evidence for the biological plausibility for the association between SHS exposure and bladder cancer risk, and may aid in explaining the incidence of bladder cancer amongst non-primary smoking individuals. CpG loci with statistically significant associations between their methylation status and SHS exposures demonstrated exclusively greater extents of methylation suggesting that SHS exposure may induce and/or select for specific hypermethylation events in bladder cancer.
Previous work has shown associations between methylation of the promoter region of several cancer-related genes in bladder tumors and primary tobacco smoking, particularly identifying associations between current and recent former smoking and hypermethylation of the CDKN2A and RASSF1A genes (23). A propensity for hypermethylation of a panel of tumor suppressor genes was also significantly more pronounced in current smokers (23–24). Our results suggest that SHS exposure may also lead to hypermethylation of key tumor suppressor genes. Interestingly, we did not identify those loci whose hypermethylation was previously found to be associated with primary smoking exposures, including CDKN2A, RASSF1A, MDM2, or the SFRP family (23, 27, 34). This was confirmed when we ran individualized generalized linear models between the methylation beta value for each locus and former and current smoking status, where we found no overlap with any of the specific loci that were identified with the strongest associations (FDR Q<0.05) between methylation and any type of SHS exposure. We did find that two specific CpG loci that were found to be associated at the p<0.05 level with former smoking status were also associated with Childhood and Adulthood exposure to SHS respectively, and that one specific CpG loci that was found to be associated at the p<0.05 level with current smoking status was also associated with Adulthood exposure to SHS. This may suggest that the types of exposure encountered by the bladders of SHS-exposed individuals are different than those of primary smoking individuals, and thus contribute different selective pressures resulting in differential methylation. In addition, the fact that the genes whose CpGs were significantly associated with either adulthood, childhood or occupational SHS exposure were all distinct from one another, suggest that the specific type or timing of SHS exposure is important epigenetically. Indeed, there may be variability in the way the different SHS exposures were experienced; this could lead to stochastic epigenetic changes due to the less consistent way individuals may experience these individual types of SHS exposures. These SHS exposures are themselves likely to be less intense and less consistent than those experienced by primary smokers. We also checked the correlation between our SHS exposures and key bladder cancer risk factors (namely age, gender, tumor grade and stage) and found that these covariates largely are not associated with the change in methylation observed between exposed and unexposed individuals (reflected by low correlation coefficients).
A number of CpG loci associated with genes in various cellular receptor kinase families were found to have significantly greater methylation associated with individual types of SHS exposure. The insulin-like growth factor-I receptor (IGF1R) gene and the insulin-like growth factor-II receptor (IGF2R) gene have opposite roles in oncogenesis with IGF1R acting as a mediator in tumor cell growth (as shown in pancreatic cancer) by inhibiting tumor apoptosis, while IGF2R acts as a tumor suppressor decreasing cellular proliferation and increasing apoptosis (35–36). IGF2R has been found to be hypermethylated in ovarian and breast tumors showing that both IGF1R and IGF2R could potentially be good targets for alterations by the components of SHS exposure (36). Other receptor kinases found to be hypermethylated with exposure to SHS were the fms-related tyrosine kinase 1 (FLT1), the EPH receptor B2 (EPHB2) and the ephrin-A1 (EFNA1) gene (37–39). All three have been shown to have aberrant methylation in tumors, with FLT1 showing CpG island methylator phenotype (CIMP)-associated DNA hypermethylation in colorectal tumors, EPHB2 showing altered methylation of its promoter regulatory sequences in colorectal tumors, and EFNA1 being hypermethylated in leukemia cells (37–39). Also of particular interest was the hypermethylation of O6-methylguanine-DNA methyltransferase (MGMT) associated with adulthood SHS exposure. MGMT is involved in DNA repair and has been found to be aberrantly hypermethylated in tumors such as breast cancer (40). Its methylation state in glioma serves as a clinically useful marker of susceptibility to the chemotherapy temozolamide (41). Finally, the connective tissue growth factor (CTGF) was found to be hypermethylated after exposure to both occupational and total SHS. CTGF is a secreted protein belonging to the CCN family shown to be hypermethylated in ovarian cancer; it may therefore be a factor in the carcinogenesis of ovarian cancer (42).
Of the pathways found to be significantly enriched amongst genes associated with any type of exposure to second hand smoking in our canonical pathways analysis, the JAK/Stat, IL-3 IL-4, CTLA4, T-cell receptor, and erythropoietin signaling pathways were all found to be significantly associated with adulthood and total exposure to SHS. The pathways are of interest, both in that they have been linked to tumorigenesis, and are critical regulators of the immune response (43–48). The role of the immune system in bladder cancer is of great interest as patients with localized disease are often treated with Bacillus Calmette-Guérin therapy, which is thought to illicit a local inflammatory response against the bladder tumor. Epigenetic alteration of key regulators of these immune responses may help to explain differential response to this treatment.
The extracellular-signal-regulated kinase 5 (ERK5) and the renin-angiotensin signaling pathways were also enriched amongst genes whose methylation status was associated with SHS exposure (49–50). This suggests that these exposures may be influencing vascular remodeling and angiogenic pathways with may be critical in sustaining or enhancing tumor growth (49–50).
Strengths of this study included the population-based nature of the study, as well as the use of the Illumina GoldenGate Methylation Bead Array for methylation profiling. Limitations of this study include the small sample size and the use of a questionnaire to ascertain second hand smoking exposure of study participants, as their responses may have been subject to recall bias. Future studies with larger sample sizes and adequately detailed SHS data are warranted.
In summary, this study demonstrates that passive smoking, like active smoking is associated with DNA methylation across numerous CpG loci in bladder cancer, but appears to be targeting different genes than those that have been associated with active smoking. The novelty of these results lies in the use of array-based methodologies to examine methylation of a large number of CpG loci in relation to SHS to help further clarify the potential association and biological plausibility of the risk of bladder cancer and SHS exposure. These findings indicate that SHS exposure has effects on methylation levels of genes and in turn affect important biological pathways which may have impacts on overall bladder cancer risk. However, due to our small sample size, we recognize the necessity to replicate and extend these results in a larger sample in the future.
Supplementary Material
Acknowledgments
We would like to thank Dr. Dominique Michaud for helpful comments and suggestions for this work.
Funding: This work was supported by the Flight Attendant Medical Research Institute (YCSA 052341 to C.J.M.); and the National Institutes of Health (R01CA121147, and P42ES007373 and R01CA057494).
Footnotes
Conflicts of interest and financial disclosures: The authors declared no conflicts of interest and no financial disclosures.
References
- 1.American Cancer Society, Cancer facts and Figures 2009. Atlanta, Ga: American Cancer Society; 2009. [Google Scholar]
- 2.Parkin MP. The global burden of urinary bladder cancer. Scand J Urol Nephrol Suppl. 2008;218:12–20. doi: 10.1080/03008880802285032. [DOI] [PubMed] [Google Scholar]
- 3.Karagas MR, Tosteson TD, Morris JS, Demidenko E, Mott LA, Heaney J, et al. Incidence of transitional cell carcinoma of the bladder and arsenic exposure in New Hampshire. Cancer Causes Control. 2004;15(5):465–472. doi: 10.1023/B:CACO.0000036452.55199.a3. [DOI] [PubMed] [Google Scholar]
- 4.Boffetta P. Human cancer from environmental pollutants: The epidemiological evidence. Mutat Res. 2006;608(2):157–162. doi: 10.1016/j.mrgentox.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 5.Wolff EM, Liang G, Jones PA. Mechanisms of Disease: genetic and epigenetic alterations that drive bladder cancer. Nat Clin Pract Urol. 2005;2(10):502–510. doi: 10.1038/ncpuro0318. [DOI] [PubMed] [Google Scholar]
- 6.Kaufman DS, Shipley WU, Feldman AS. Bladder cancer. Lancet. 2009;374(9685):239–249. doi: 10.1016/S0140-6736(09)60491-8. [DOI] [PubMed] [Google Scholar]
- 7.Dinney CP, McConkey DJ, Millikan RE, Wu X, Bar-Eli M, Adam L, et al. Focus on bladder cancer. Cancer Cell. 2004;6(2):111–116. doi: 10.1016/j.ccr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Silverman DT, Devesa SS, Moore LL. Bladder cancer. In: Schottenfeld D, Fraumeni JF Kr, editors. Cancer Epidemiology and Prevention. New York, NY: Oxford University Press; 2006. pp. 1101–1127. [Google Scholar]
- 9.Jones PA, Ross RK. Prevention of bladder cancer. N Engl J Med. 1999;340(18):1424–1426. doi: 10.1056/NEJM199905063401810. [DOI] [PubMed] [Google Scholar]
- 10.Vineis P, Pirastu R. Aromatic amines and cancer. Cancer Causes Control. 1997;8(3):346–355. doi: 10.1023/a:1018453104303. [DOI] [PubMed] [Google Scholar]
- 11.Skipper PL, Tannenbaum SR, Ross RK, Yu MC. Nonsmoking-related arylamine exposure and bladder cancer risk. Cancer Epidemiol Biomarkers Prev. 2003;12(6):503–507. [PubMed] [Google Scholar]
- 12.Anderson KE, Carmella SG, Ye M, Bliss RL, Le C, Murphy L, et al. Metabolites of a tobacco-specific lung carcinogen in nonsmoking women exposed to environmental tobacco smoke. J Natl Cancer Inst. 2001;93:378–381. doi: 10.1093/jnci/93.5.378. [DOI] [PubMed] [Google Scholar]
- 13.Vineis P, Alavanja M, Garte S. Dose-response relationship in tobacco-related cancers of bladder and lung: a biochemical interpretation. Int J Cancer. 2004;108:2–7. doi: 10.1002/ijc.11467. [DOI] [PubMed] [Google Scholar]
- 14.Talaska G. Aromatic amines and human urinary bladder cancer: exposure sources and epidemiology. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2003;21(1):29–43. doi: 10.1081/GNC-120021372. [DOI] [PubMed] [Google Scholar]
- 15.Mohtashamipur E, Norpoth K, Lieder F. Urinary excretion of mutagens in smokers of cigarettes with various tar and nicotine yields, black tobacco, and cigars. Cancer Lett. 1987;34:103–112. doi: 10.1016/0304-3835(87)90079-6. [DOI] [PubMed] [Google Scholar]
- 16.Riedel K, Scherer G, Engl J, Hagedorn HW, Tricker AR. Determination of three carcinogenic aromatic amines in urine of smokers and nonsmokers. J Anal Toxicol. 2006;30(3):187–195. doi: 10.1093/jat/30.3.187. [DOI] [PubMed] [Google Scholar]
- 17.Burch JD, Rohan TE, Howe GR, Risch HA, Hill GB, Steele R, et al. Risk of bladder cancer by source and type of tobacco exposure: a case-control study. Int J Cancer. 1989;44:622–628. doi: 10.1002/ijc.2910440411. [DOI] [PubMed] [Google Scholar]
- 18.Zeegers MP, Goldbohm RA, van den Brandt PA. A prospective study on active and environmental tobacco smoking and bladder cancer risk (the Netherlands) Cancer Causes Control. 2002;13:83–90. doi: 10.1023/a:1013954932343. [DOI] [PubMed] [Google Scholar]
- 19.Sandler DP, Everson RB, Wilcox AJ. Passive smoking in adulthood and cancer risk. Am J Epidemiol. 1985;121:37–48. doi: 10.1093/oxfordjournals.aje.a113980. [DOI] [PubMed] [Google Scholar]
- 20.Jiang X, Yuan JM, Skipper PL. A case-control study of passive smoking and bladder cancer risk among lifelong nonsmokers in Los Angeles. Proc Amer Assoc Cancer Res. 2005;46:2210. [Google Scholar]
- 21.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6(2):107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 22.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 23.Marsit CJ, Karagas MR, Danaee H, Liu M, Andrew A, Schned A, et al. Carcinogen exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis. 2006;27(1):112–116. doi: 10.1093/carcin/bgi172. [DOI] [PubMed] [Google Scholar]
- 24.Marsit CJ, Houseman EA, Schned AR, Karagas MR, Kelsey KT. Promoter hypermethylation is associated with current smoking, age, gender and survival in bladder cancer. Carcinogenesis. 2007;28(8):1745–1751. doi: 10.1093/carcin/bgm116. [DOI] [PubMed] [Google Scholar]
- 25.Baris D, Karagas MR, Verrill C, Johnson A, Andrew AS, Marsit CJ, et al. A case-control study of smoking and bladder cancer risk: emergent patterns over time. J Natl Cancer Inst. 2009;101(22):1553–1561. doi: 10.1093/jnci/djp361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karagas MR, Tosteson TD, Blum J, Morris JS, Baron JA, Klaue B. Design of an epidemiologic study of drinking water arsenic exposure and skin and bladder cancer risk in a U.S. population. Environ Health Perspect. 1998;106 Suppl 4:1047–1050. doi: 10.1289/ehp.98106s41047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marsit CJ, Karagas MR, Andrew A, Liu M, Danaee H, Schned AR, et al. Epigenetic inactivation of SFRP genes and TP53 alteration act jointly as markers of invasive bladder cancer. Cancer Res. 2005;65(16):7081–7085. doi: 10.1158/0008-5472.CAN-05-0267. [DOI] [PubMed] [Google Scholar]
- 28.Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006;16(3):383–393. doi: 10.1101/gr.4410706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christensen BC, Kelsey KT, Zheng S, Houseman EA, Marsit CJ, Wrensch MR, et al. Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 2010;6(7) doi: 10.1371/journal.pgen.1001043. e1001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marsit CJ, Houseman EA, Christensen BC, Gagne L, Wrensch MR, Nelson HH, et al. Identification of methylated genes associated with aggressive bladder cancer. PLoS One. 2010;5(8) doi: 10.1371/journal.pone.0012334. pii: e12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsiung DT, Marsit CJ, Houseman EA, Eddy K, Furniss CS, McClean MD, et al. Global DNA methylation level in whole blood as a biomarker in head and neck squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2007;16(1):108–114. doi: 10.1158/1055-9965.EPI-06-0636. [DOI] [PubMed] [Google Scholar]
- 32.Storey J, Taylor J, Siegmund D. Strong control, conservative point estimation and simulteanous conservative consistency of false discovery rates: a unified approach. J Royal Stat Soc Series B. 2004:187–205. [Google Scholar]
- 33.Ingenuity Pathways Analysis application. 2008 6.3; build 54960 ed: Ingenuity Systems. [Google Scholar]
- 34.Schlott T, Quentin T, Korabiowska M, Budd B, Kunze E. Alteration of the MDM2-p73-P14ARF pathway related to tumour progression during urinary bladder carcinogenesis. Int J Mol Med. 2004;14(5):825–836. [PubMed] [Google Scholar]
- 35.Schayek H, Bentov I, Sun S, Plymate SR, Werner H. Progression to metastatic stage in a cellular model of prostate cancer is associated with methylation of the androgen receptor gene and transcriptional suppression of the insulin-like growth factor-I receptor gene. Exp Cell Res. 2010;316(9):1479–1488. doi: 10.1016/j.yexcr.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang Z, Wen Y, Shandilya R, Marks JR, Berchuck A, Murphy SK. High throughput detection of M6P/IGF2R intronic hypermethylation and LOH in ovarian cancer. Nucleic Acids Res. 2006;34(2):555–563. doi: 10.1093/nar/gkj468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hinoue T, Weisenberger DJ, Pan F, Campan M, Kim M, Young J. Analysis of the association between CIMP and BRAF in colorectal cancer by DNA methylation profiling. PLoS One. 2009;4(12):e8357. doi: 10.1371/journal.pone.0008357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alazzouzi H, Davalos V, Kokko A, Domingo E, Woerner SM, Wilson AJ, et al. Mechanisms of inactivation of the receptor tyrosine kinase EPHB2 in colorectal tumors. Cancer Res. 2005;65(22):10170–10173. doi: 10.1158/0008-5472.CAN-05-2580. [DOI] [PubMed] [Google Scholar]
- 39.Kuang SQ, Bai H, Fang ZH, Lopez G, Yang H, Tong W, et al. Aberrant DNA methylation and epigenetic inactivation of Eph receptor tyrosine kinases and ephrin ligands in acute lymphoblastic leukemia. Blood. 2010;115(12):2412–2419. doi: 10.1182/blood-2009-05-222208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharma G, Mirza S, Parshad R, Srivastava A, Gupta SD, Pandya P, et al. Clinical significance of promoter hypermethylation of DNA repair genes in tumor and serum DNA in invasive ductal breast carcinoma patients. Life Sci. 2010;87(3–4):83–91. doi: 10.1016/j.lfs.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 41.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 42.Kikuchi R, Tsuda H, Kanai Y, Kasamatsu T, Sengoku K, Hirohashi S, et al. Promoter hypermethylation contributes to frequent inactivation of a putative conditional tumor suppressor gene connective tissue growth factor in ovarian cancer. Cancer Res. 2007;67(15):7095–7105. doi: 10.1158/0008-5472.CAN-06-4567. [DOI] [PubMed] [Google Scholar]
- 43.Aaronson DS, Horvath CM. A road map for those who don't know JAK-STAT. Science. 2002;296(5573):1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 44.Sokol CL, Barton GM, Farr AG, Medzhitov R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat Immunol. 2008;9(3):310–318. doi: 10.1038/ni1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang YC, Ciarletta AB, Temple PA, Chung MP, Kovacic S, Witek-Giannotti JS, et al. Human IL-3 (multi-CSF): identification by expression cloning of a novel hematopoietic growth factor related to murine IL-3. Cell. 1986;47(1):3–10. doi: 10.1016/0092-8674(86)90360-0. [DOI] [PubMed] [Google Scholar]
- 46.Maeda A, Maeda T, Liang Y, Yenerel M, Saperstein DA. Effects of cytotoxic T lymphocyte antigen 4 (CTLA4) signaling and locally applied steroid on retinal dysfunction by recoverin, cancer-associated retinopathy antigen. Mol Vis. 2006;12:885–891. [PubMed] [Google Scholar]
- 47.Govers C, Sebestyén Z, Coccoris M, Willemsen RA, Debets R. T cell receptor gene therapy: strategies for optimizing transgenic TCR pairing. Trends Mol Med. 2010;16(2):77–87. doi: 10.1016/j.molmed.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 48.Tóvári J, Pirker R, Tímár J, Ostoros G, Kovács G, Döme B. Erythropoietin in cancer: an update. Curr Mol Med. 2008;8(6):481–491. doi: 10.2174/156652408785747979. [DOI] [PubMed] [Google Scholar]
- 49.Roberts OL, Holmes K, Müller J, Cross DA, Cross MJ. ERK5 and the regulation of endothelial cell function. Biochem Soc Trans. 2009;37(Pt 6):1254–1259. doi: 10.1042/BST0371254. [DOI] [PubMed] [Google Scholar]
- 50.Zhao Y, Chen X, Cai L, Yang Y, Sui G, Fu S. Angiotensin II / Angiotensin II type I receptor (AT1R) signaling promotes MCF-7 breast cancer cells survival via PI3-kinase/Akt pathway. J Cell Physiol. 2010;225(1):168–173. doi: 10.1002/jcp.22209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.