

Abstract
The CXCR4 chemokine receptor is a G protein-coupled receptor (GPCR) that signals in T lymphocytes by forming a heterodimer with the T cell antigen receptor (TCR). CXCR4 and TCR functions are consequently highly cross-regulated, affecting T cell immune activation, cytokine secretion, and T cell migration. The CXCR4-TCR heterodimer stimulates T cell migration and activation of the ERK MAP kinase and downstream AP-1-dependent cytokine transcription in response to SDF-1, the sole chemokine ligand of CXCR4. These responses require Gi-type G proteins as well as TCR ITAM domains and the ZAP-70 tyrosine kinase, thus indicating that the CXCR4-TCR heterodimer signals to integrate GPCR-associated and TCR-associated signaling molecules in response to SDF-1. Yet, the phospholipase C (PLC) isozymes responsible for coupling the CXCR4-TCR heterodimer to distinct downstream cellular responses are incompletely characterized. Here, we demonstrate that PLC activity is required for SDF-1 to induce ERK activation, migration, and CXCR4 endocytosis in human T cells. SDF-1 signaling via the CXCR4-TCR heterodimer uses PLC-β3 to activate the Ras-ERK pathway and increase intracellular Ca2+ concentrations, while PLC-γ1 is dispensable for these outcomes. In contrast, PLC-γ1, but not PLC-β3, is required for SDF-1-mediated migration, via a mechanism independent of LAT. These results increase understanding of the signaling mechanisms employed by the CXCR4-TCR heterodimer, characterize new roles for PLC-β3 and PLC-γ1 in T cells, and suggest that multiple PLCs may also be activated downstream of other chemokine receptors in order to distinctly regulate migration versus other signaling functions.
INTRODUCTION
CXCR4, a G-protein coupled receptor (GPCR), initiates migration, calcium mobilization, ERK activation, survival signals, gene transcription, and cytokine production upon binding it’s ligand, SDF-1 (also called CXCL12). CXCR4 signaling in multiple cell types is important for embryonic development, homeostasis, vascularization, immune regulation, HIV-1 infection, and cancer progression (1, 2). CXCR4 utilizes numerous signal transduction strategies to regulate these downstream events, including activation of several G proteins and heterodimerization with other types of cell-surface receptors (3–5). The molecular mechanisms involved in mediating these diverse signaling outcomes have begun to be characterized. Of particular interest is the identification of signaling molecules that could be therapeutically targeted to disrupt SDF-1-mediated migration without altering SDF-1-induced gene transcription, because this would be helpful for treating the numerous diseases involving CXCR4. Here, we show that migration and ERK activation in response to SDF-1/CXCR4 signaling in T cells is mediated by two distinct phosphatidyl inositol-specific phospholipase C (PLC) isozymes.
We previously showed that upon SDF-1 binding to CXCR4 in T cells, CXCR4 heterodimerizes with the TCR in order to stimulate increased calcium ion concentrations ([Ca2+]i), prolonged ERK activation, gene transcription, and cytokine production. We further showed that these outcomes of CXCR4-TCR signal transduction occur via pathways that utilize several traditional TCR signaling molecules including ZAP-70 and SLP-76 as well as G proteins (6–8). Others have similarly demonstrated that CXCR4 signaling in T cells utilizes the TCR to mediate CXCR4 endocytosis and SDF-1-mediated migration via p52Shc (9). Thus, the SDF-1 induced heterodimerization of CXCR4 with the TCR is critical for many T cell functions and allows CXCR4 to access additional signaling molecules to achieve CXCR4’s diverse signaling outcomes. Although both the TCR and GPCRs including CXCR4 have been shown to signal via various PLC isoforms, the particular PLC isoforms involved in mediating the distinctive signaling pathways arising from SDF-1 signaling via the CXCR4-TCR heterodimer in T cells have not been fully characterized.
PLC enzymes, upon their activation by signals derived from upstream receptors, catalyze the cleavage of phosphatidylinositol 4,5-bisphophate into 1,4,5-inositol triphospate (IP3) and diacylglycerol (DAG), and these second messengers subsequently lead to calcium mobilization and activation of protein kinase C (PKC). Mammals express eleven PLC isoforms all of which contain the X and Y domains that comprise the catalytic core of the enzyme. Different PLC isoforms vary in the types of regulatory domains they contain which allow the upstream receptors to signal to activate only particular PLC isoforms (10). For example, lysophosphatidic acid (LPA) receptors signal to activate PLC-β3 while bradykinin signals to activate PLC-β1, with these PLC isoforms being activated by distinct upstream signaling mediators that bind their distinct PDZ-binding motifs in order to mediate their respective signaling functions within the same cell type (11).
The predominant PLC isoforms expressed in T lymphocytes include PLC-γ1, PLC-β2, and PLC-β3. PLC-γ isoforms were originally thought to be only activated downstream of receptor tyrosine kinases, while PLC-β isoforms are predominantly activated by GPCRs (10). Direct stimulation of the TCR activates PLC-γ1, which is responsible for the subsequent calcium mobilization and DAG production leading to the ERK activation, gene transcription, cytokine production and proliferation necessary for T cell development and T cell activation (12). PLC-β3 has been shown to be phosphorylated in response to SDF-1 in leukemia cells (13), and mice lacking both PLC-β2 and PLC-β3 display a partial defect in migration in response to SDF-1 (14). PLC-γ1 was recently shown to be required for T cell migration in response to signaling by the CCR7 chemokine receptor (15) and PLC-γ2 was shown to be required for B cell migration in response to SDF-1 (16), suggesting that chemokine receptors can also utilize PLC-γ isoforms for signaling leading to cellular migration. Yet, the particular PLC isoforms required for CXCR4 signaling apart from migration are poorly understood, and the role(s) for PLC-γ1 in SDF-1 mediated signaling in T cells has not been defined.
Here we demonstrate that the CXCR4-TCR heterodimer utilizes multiple PLCs, each with a distinct role, to mediate the diverse signaling outcomes of SDF-1 treatment of T cells. In addition, we describe new roles for both PLC-γ1 and PLC-β3 in CXCR4-TCR signaling. These findings identify molecules that can regulate one pathway from a receptor without altering another, and thus may allow for the selective therapeutic targeting of different CXCR4-derived responses.
MATERIAL AND METHODS
Cells, stimulation conditions, reagents and statistical analysis
Jurkat T cells were maintained as described (6, 8). Jurkat sublines deficient in PLC-γ1 (J.gamma1, 100 % deficient in PLC-γ1, P98, >90 % deficient in PLC-γ1) (17), or 100% deficient in LAT (ANJ-3) (18) were gifts of R. Abraham (Pfizer, Pearl River, NY). Stimulations were performed at 37 °C with 5 × 10−8 M SDF-1α (R&D Systems, Minneapolis, MN). Where indicated, cells were pretreated with 5 μM U73122 (Calbiochem, San Diego, CA) or vehicle (ethanol) for 15–30 min. CXCR4 (R&D Systems, Minneapolis, MN) and TCR (CD3ε, BD Biosciences, San Diego, CA) mAbs were used for flow cytometry. Statistical analysis was via two-tailed t-test (Microsoft Excel). The means of two distributions were considered significantly different if p ≤ 0.05.
Assays of active, phosphorylated ERK1 and ERK2, and shRNA-mediated protein depletion
For analysis of active ERK, cells were stimulated and assayed via flow cytometry or western blot as described (8). Raw data from representative flow cytometric determinations of ERK activation is shown in Supplementary Fig. 1. Where indicated, 72 hr prior to assay, cells were transiently-transfected as described (8) with either a control plasmid or a plasmid encoding an H1-promoter-driven shRNA specific for human PLC-β3. The shRNA encoding plasmid also encoded either m.Cherry or GFP via a separate SV40 promoter. Flow cytometric data was gated to compare ERK activation in cells expressing similar levels of m.Cherry. The PLC-β3 shRNA-encoding plasmid was made by annealing and ligating DNA oligonucleotides into either pCMS4.H1p.mCherry (5) or the p1012 plasmid vector, a modified version of pCMS4.eGFP.H1p (19) (gifts of D. Billadeau, Mayo Clinic, Rochester, MN) lacking the FLAG tag. PLC-β3 shRNA oligos were GATCCCCGGTCTGAGGAGCTATTCAATTCAAGAGATTGAATAGCTCCTCAGACCTTTTTGGAAA and AGCTTTTCCAAAAAGGTCTGAGGAGCTATTCAATCTCTTGAATTGAATAGCTCCTCAGACCGGG. Protein depletion was confirmed by immunoblotting whole cell lysates with anti-PLC-β3 (Santa Cruz Biotechnology, Santa Cruz, CA). Controls were the same blots re-immunoblotted for total ERK2 (Santa Cruz Biotechnology).
Migration Assay
Where indicated, Jurkat T cells were transfected with either p1012 or p1012 encoding shRNA specific for PLC-β3. 72 hr later, migration assays were performed utilizing the GFP fluorescence expressed by these plasmids to detect migrating cells. For migration assays utilizing mutant Jurkat sublines, the cells were stained with 2 μM calcein-AM (Invitrogen, Carlsbad, CA) for 30 min, washed, and assayed for migration. All migration assays used 96-well Chemotx Chemotaxis plates (Neuroprobe, Gaithersburg, MD) with 5 μm-pore filters coated with fibronectin (Invitrogen). Cells (500,000) were diluted in Migration Buffer (RPMI without phenol red supplemented with 0.5 % BSA and 1 % DMSO) and placed in the upper chamber of each well. After migrating at 37 °C for 1 hr towards lower chambers containing the indicated concentrations of SDF-1, cells remaining in the upper chambers were removed, and cells that migrated into the lower chambers were quantified by measuring GFP or calcein-AM (485 nm ex/530 nm em) fluorescence using a Cytofluor 4000 spectrometer (PerSeptive Biosystems, Framingham, MA).
Assays of active, GTP-bound N-Ras and K-Ras, CXCR4-TCR interaction by FRET, and [Ca2+]i
Active, GTP-bound Ras was measured using an activated Ras assay kit (Upstate, Waltham, MA) and immunoblotting with specific antibodies to N-Ras or K-Ras (Santa Cruz Biotechnology). As controls, cell lysates were immunoblotted for N-Ras or actin. The interaction between CXCR4 and TCR in response to 20 min of SDF-1 treatment was assayed by fluorescence resonance energy transfer (FRET) as described (8, 20), with gating on GFP+ cells following transfection with P1012. [Ca2+]i assays were performed as described (6).
Immunoprecipitation of PLC-γ1 and LAT
Cells were stimulated with SDF-1 for the indicated time, lysed in Buffer A (25 mM Tris HCl, 150 mM NaCl, 5 mM EDTA, 50 mM β-glycerophosphate, and 1% each of NP40, leupeptin, microcystin-LR, aprotinin, and sodium orthovanadate), and endogenous signaling proteins were immunoprecipitated with antibodies to PLC-γ1 or LAT (Santa Cruz Biotechnology). Immunoprecipitated proteins were separated by SDS-PAGE and immunoblotted with P-Tyr mAb (4G10; Upstate), LAT-phospho-171 (Cell Signaling, Danvers, MA), PLC-γ1, or LAT.
RESULTS
SDF-1/CXCR4 signaling requires a PLC to stimulate the ERK MAP kinase pathway, T cell migration, and internalization of CXCR4
We previously demonstrated that SDF-1 induces CXCR4 to complex with the TCR in order to prolong ERK activation, and we showed that these events lead to gene expression and cytokine production in activated T cells (7, 8). In addition, the SDF-1-mediated migration of T cells has been shown to require a mechanism involving CXCR4 utilization of the TCR (9). Yet, the PLC isoforms utilized by the CXCR4-TCR heterodimer to mediate these distinct outcomes have not been fully characterized. First, we used the PLC inhibitor, U73122, to identify signaling outcomes affected by the loss of PLC activity. Jurkat T cells treated with U73122 activated significantly less ERK in response to SDF-1 as compared to vehicle-treated cells (Fig. 1A). Similarly, inhibiting PLC activity with U73122 substantially decreased the percent of cells migrating in response to SDF-1 (Fig. 1B). Furthermore, the internalization of CXCR4 in response to SDF-1 was also inhibited by pretreatment with U73122 (Fig. 1C&D). Together, these results indicate that PLC activity is required for SDF-1-induced signaling following formation of the CXCR4-TCR complex that mediates ERK activation and T cell migration as well as CXCR4 internalization.
Fig. 1. SDF-1/CXCR4 signaling requires a PLC to stimulate the ERK MAP kinase pathway, T cell migration, and internalization of CXCR4.

Jurkat T cells were treated with either the vehicle, ethanol, or 5 μM U73122 for 15–30 min. A) The treated cells were then stimulated with SDF-1 and assayed for ERK activation via flow cytometry. Bars denote the fold-increase in ERK activation of SDF-1-stimulated as compared to unstimulated cells ± S.E.M., n = 3; *, significantly different from control, p<0.05. B) Following pre-treatment with either vehicle or U73122, Jurkat cells were assayed for migration in response to the indicated concentrations of SDF-1. Each point in B) denotes the mean % of cells migrated ± S.D., n = 4. Results shown are representative of 2 independent experiments. C) Jurkat T cells were pretreated as indicated and the endocytosis of endogenous CXCR4 was stimulated by treating cells with SDF-1 for 20 min. Cells were stained with fluorescently conjugated CXCR4 mAb and analyzed by flow cytometry. D) Summary of results obtained as in C. Bars denote the mean results for each condition ± S.E.M., n = 3; *, significantly different from unstimulated cells; **, significantly different from SDF-1 treated vehicle control, p < 0.05.
SDF-1/CXCR4 signaling does not require PLC-γ1 to mobilize calcium, to activate N-Ras, K-Ras, or ERK, or to internalize CXCR4
We previously showed that calcium mobilization in response to SDF-1 requires the TCR, ZAP-70, and SLP-76 (6, 8). PLC-γ1, which is known to utilize these same signaling molecules to mobilize calcium in response to direct TCR stimulation (21), seemed likely to participate in mediating calcium mobilization in response to SDF-1. Yet the PLC-γ1-deficient cell line, J.gamma1, mobilized calcium in response to SDF-1 in a manner similar to the parental wild-type Jurkat T cell line (Fig. 2A). Moreover, ERK activation in response to SDF-1, which also requires the TCR, ZAP-70, and SLP-76 (6, 8), was not defective in J.gamma1 cells as assayed by a flow cytometric ERK activation assay. In fact, J.gamma1 cells showed more than a 50 % increase in ERK activation in response to SDF-1 as compared to the parental Jurkat T cell line (Fig. 2B). The enhanced SDF-1-induced ERK activation in the J.gamma1 cells was nevertheless sensitive to inhibition with U73122, suggesting a role for a different PLC in this pathway (Fig. 2B). ERK activation assayed by western blot similarly showed no defect in SDF-1-dependent ERK activation in J.gamma1 cells as compared to normal Jurkat cells (Fig. 2C). To determine which Ras isoforms are potentially mediating the increase in ERK activation in PLC-γ1 deficient cells, we examined the activation of the Ras isoforms, N-Ras and K-Ras. We recently showed that these Ras isoforms are responsible for mediating signaling from the CXCR4-TCR heterodimer in response to SDF-1 (submitted for publication). N-Ras was activated in response to SDF-1 treatment of the J.gamma1 cells to levels similar to that seen in the parental Jurkat cell line (Fig. 2D), and K-Ras activation was actually increased in the J.gamma1 cell line (Fig. 2D). Thus, PLC-γ1 is not required for either N-Ras or K-Ras activation in response to SDF-1. Moreover, compared to wild-type cells, CXCR4 on J.gamma1 cells internalized normally in response to SDF-1 (Fig. 2E&F). In addition, PLC-γ1 is not required for the SDF-1-dependent signaling that leads to formation of the CXCR4-TCR heterodimer. Endogenous, cell-surface CXCR4 and TCR molecules were labeled via mAbs, stimulated with SDF-1, and assayed for fluorescence resonance energy transfer (FRET) signals via flow cytometry as in (8, 20). The results show that PLC-γ1-deficient Jurkat cells increased CXCR4-TCR FRET (FL3) fluorescence in response to SDF-1 in a manner similar to that observed in the parental Jurkat cell line (Fig. 2G&H). Thus, the results in Figs. 1&2 show that PLC-γ1 is not required for CXCR4-TCR complex formation in response to SDF-1, and that PLC-γ1 is not the PLC required for calcium mobilization, activation of N-Ras, K-Ras, or ERK, or for internalization of CXCR4 in response to SDF-1 treatment of T cells.
Fig. 2. SDF-1/CXCR4 signaling does not require PLC-γ1 to mobilize calcium, to activate N-Ras, K-Ras, or ERK, or to internalize CXCR4.

A) Flow cytometric [Ca2+]i assays of the indicated cell lines in response to SDF-1 (arrow). Fluor. Ratio, the 405 nm:495 nm fluorescence ratio that correlates with [Ca2+]i. Results shown are typical of three independent experiments. Inset: Whole cell lysates immunoblotted for PLC-γ1. The same membrane was immunoblotted for total ZAP-70 as a control. B) Jurkat cells or the PLC-γ1-deficient Jurkat cell line, J.gamma1, were pretreated with either vehicle or U73122 for 15 min, stimulated with SDF-1 for 8 min, and assayed for ERK activation as in Fig. 1a. Bars denote the fold-increase in ERK activation of SDF-1-stimulated as compared to unstimulated cells ± S.E.M., n = 3; *, significantly different from unstimulated control, p < 0.05. **, significantly different from wild-type Jurkat, p < 0.05. C) Jurkat or J.gamma1 cells were assayed for active, phosphorylated ERK by immunoblotting (upper gel). The same blots were stripped and immunoblotted with actin as a control (lower gel). D) Jurkat or J.gamma1 cells were stimulated with SDF-1 for 8 min and assayed for active, GTP-bound Ras isoforms by affinity purification. For controls, whole cell lysates were immunoblotted for either total N-Ras or actin. E&F) Jurkat or J.gamma1 cells were stimulated with SDF-1 and assayed for endogenous cell-surface CXCR4 levels as in Fig. 1C&D. Bars denote the mean results of each condition ± S.E.M., n = 3; *, significantly different from unstimulated cells, p < 0.05. G) Example of a flow cytometric FRET assay of endogenous CXCR4 and TCR forming CXCR4-TCR complexes in response to SDF-1. Both CXCR4-PE and TCR-CD3-ε-APC mAbs were bound to cell-surface receptors. Cells were stimulated with SDF-1 and assayed for increases in FRET (F3) fluorescence. H) Summary of the results of multiple experiments as in G). Each bar denotes the mean SDF-1-dependent FRET response ± S.E.M. as a percent of control cell responses, n = 3. The J.gamma1 cells did not respond significantly different from the Jurkat parental cell line, p > 0.05.
PLC-β3 mediates SDF-1-induced calcium mobilization and activation of N-Ras, K-Ras and ERK but not internalization of CXCR4
We next looked at the role of PLC-β3 in mediating the downstream signaling events of SDF-1. Jurkat T cells were transfected with either a control plasmid or a plasmid encoding shRNA targeted against PLC-β3 which yielded an 80–95 % depletion of the PLC-β3 protein compared to vector-transfected cells (Fig. 3A). Both plasmids also express the fluorescent protein, m.Cherry, allowing detection and assay of shRNA-containing cells. Compared to cells transfected with the control plasmid, cells transfected with the plasmid encoding PLC-β3 shRNA displayed significantly lower SDF-1-dependent calcium mobilization (Fig. 3A), as well as lower SDF-1-mediated ERK activation as assayed by flow cytometry and western blot (Fig. 3B&C). Consistent with the lower ERK activation in these cells, PLC-β3 deficient Jurkat cells also displayed defective activation of N-Ras and K-Ras in response to SDF-1 (Fig. 3D). In contrast to its effects on Ras and ERK activation, PLC-β3 depletion did not alter the internalization of CXCR4 in response to SDF-1 (Fig. 3E&F). Furthermore, PLC-β3 depletion did not inhibit the SDF-1-mediated CXCR4-TCR complex formation, as assayed by FRET (Fig. 3G&H). Thus, PLC-β3 is required for ERK activation downstream of the formation of the CXCR4-TCR heterodimeric complex in response to SDF-1. Together, these results indicate that PLC-β3 is required for SDF-1-mediated calcium mobilization and for the activation of N-Ras, K-Ras, and ERK in response to SDF-1, while indicating that other PLCs participate in mediating the internalization of CXCR4 in response to SDF-1.
Fig. 3. PLC-β3 mediates SDF-1-induced calcium mobilization and activation of N-Ras, K-Ras and ERK, but not internalization of CXCR4.

Jurkat cells were transiently-transfected with either a control plasmid (Vector) or a plasmid encoding shRNA targeted against PLC-β3. A) [Ca2+]i was assayed as in Fig. 2A. Inset: Whole cell lysates immunoblotted for PLC-β3. The same membranes were immunoblotted for total ERK2 as a control. B) ERK activation was assayed as in Fig. 1A. Bars denote the fold-increase in ERK activation of SDF-1-stimulated as compared to unstimulated cells ± S.E.M., n = 3; *, significantly different from control, p<0.05. C) ERK activation assayed by western blot as in Fig. 2C. D) N-Ras and K-Ras activation were assayed as in Fig. 2C, n = 3. E&F) Down-regulation of endogenous CXCR4 in response to SDF-1 was assayed and quantitated as in Fig. 1C&D, n = 3. Bars denote the mean results of each condition ± S.E.M., n = 3; *, significantly different from unstimulated cells, p < 0.05. G) Example of a flow cytometric FRET assay of endogenous CXCR4 and TCR forming CXCR4-TCR complexes in response to SDF-1 as in Fig. 2G. H) Summary of the results of multiple experiments as in G). Each bar denotes the mean SDF-1-dependent FRET response ± S.E.M. as a percent of control cell responses, n = 3. The cells depleted of PLC-β3 did not respond significantly different from the vector control transfected cells, p > 0.05.
PLC-γ1, but not PLC-β3, is required for the migration of T cells in response to SDF-1
We next determined the effect of depleting PLC-β3 on the migration of Jurkat T cells in response to SDF-1. Despite the requirement for PLC-β3 in the signaling pathways that lead to calcium mobilization and ERK activation in response to SDF-1 (Fig. 3), cells deficient in PLC-β3 did not show a defect in migration in response to SDF-1. In fact, the Jurkat cells transfected with the PLC-β3 shRNA showed a slight increase in migration in response to SDF-1 compared to cells transfected with the vector control (Fig. 4A). We next assayed the role of PLC-γ1 in migration, by utilizing the PLC-γ1-deficient cell line, J.gamma1. In contrast to PLC-β3 deficiency, PLC-γ1 deficiency in the J.gamma1 cell line resulted in a substantial decrease in migration in response to SDF-1 (Fig. 4B). To confirm this finding, we also used the P98 cell line, an independently-obtained somatic mutant of Jurkat that is only approximately 90 % deficient in PLC-γ1, in contrast to the J.gamma1 cell line which is 100 % deficient in PLC-γ1 (17). As seen for J.gamma1 cells, P98 cells displayed defective migration in response to SDF-1 (Fig. 4C). The defective migration of PLC-γ1-deficient cells did not arise from deficient cell-surface levels of CXCR4 (Fig. 4B, inset histograms). Cell-surface TCR levels were lower on J.gamma1 but not P98 cells (Fig. 4B&C, inset histograms), however, these PLC-γ1-deficient cells were similarly defective in migration towards SDF-1 (Fig. 4A–C). Together these results indicate that, PLC-γ1, but not PLC-β3, is essential for the SDF-1-mediated migration of T cells.
Fig. 4. PLC-γ1, but not PLC-β3, is required for the migration of T cells in response to SDF-1.

A) Jurkat T cells were transiently-transfected with a plasmid encoding vector control or shRNA targeting PLC-β3. Migration was assayed in response to SDF-1. Each point in A) denotes the mean % of cells migrated ± S.D., n = 3. Results shown are representative of 3 independent experiments. Inset (A–C): The indicated cell lines were assayed for CXCR4 and TCR cell-surface levels by flow cytometry before SDF-1 treatment. B&C) Jurkat or the PLC-γ1-deficient cell lines, J.gamma1 and P98, were stimulated with SDF-1 and assayed for migration as in Fig. 1B, n = 3–10. D&E) Jurkat cells were stimulated with SDF-1, lysed, and either PLC-γ1 or LAT were immunoprecipitated and immunoblotted with either phospho-Tyr mAb or LAT-phosphoTyr-171. The same blots were stripped and immunoblotted for PLC-γ1 or total LAT as controls, n = 3. F) Jurkat or a LAT-deficient Jurkat cell line were assayed for migration as in Fig. 1B, n = 3–10.
PLC-γ1 has not previously been shown to mediate T cell migration in response to SDF-1. Upon direct stimulation of the TCR, PLC-γ1 is recruited to the plasma membrane via the multi-molecular signaling complex nucleated by LAT and SLP-76, and is then rapidly phosphorylated by Itk which allows PLC-γ1 to mediate its downstream signaling outcomes (21). Since migration in response to SDF-1 has been shown to depend on the transactivation of the TCR by CXCR4 (9), we determined if SDF-1 treatment induces the phosphorylation of PLC-γ1. In contrast to the results of directly stimulating the TCR with anti-CD3 mAb, SDF-1 treatment failed to detectably increase the tyrosine phosphorylation of PLC-γ1 (Fig. 4D). The phosphorylation of LAT on Tyr171 is involved in PLC-γ1 activation and downstream signaling in response to direct stimulation of the TCR (22), thus we also assayed LAT phosphorylation on Tyr171 in response to SDF-1. Tyr171 of LAT failed to be inducibly phosphorylated in response to SDF-1 treatment, despite being phosphorylated in response to direct TCR stimulation in the same experiment (Fig. 4E). To confirm that LAT is not involved in PLC-γ1-dependent migration in response to SDF-1, we also assayed migration of a LAT-deficient somatic mutant of the Jurkat T cell line. Indeed, LAT-deficient Jurkat cells migrated in a manner similar to wild-type Jurkat cells (Fig. 4F). Thus, SDF-1 utilizes PLC-γ1, not PLC-β3, to mediate migration of T cells via a mechanism that is distinct from that arising from direct ligation of the TCR and that does not require tyrosine phosphorylation of either PLC-γ1 or LAT-Tyr171 and is independent of LAT protein expression.
SDF-1 signaling via the CXCR4-TCR heterodimer requires different PLCs in order to mediate distinct cellular outcomes
Based on our results presented here, and together with results reported previously by our group and others, we propose that CXCR4-TCR heterodimer signals in T cells in response to SDF-1 via the signaling pathway shown in Fig. 5. We propose that SDF-1 treatment induces the formation of the CXCR4-TCR heterodimer which mediates the activation of Gi proteins and ZAP-70 (6, 8, 23, 24), and that this event either directly or indirectly leads to the activation of several PLCs including both PLC-γ1 and PLC-β3. PLC-γ1 activity is required to mediate the migration of T cells via a mechanism that does not require either LAT (shown here) or SLP-76 (24) and is also not impaired by the lack of PLC-β3. Based on others’ observations, PLC-β2 (not shown) may also mediate migration via signaling crosstalk with PLC-γ1 (14). In contrast to PLC-γ1 activation, PLC-β3 activation leads to ERK activation and gene transcription via a mechanism that previous results show does require SLP-76 but is independent of LAT (6). Thus, PLC-γ1 and PLC-β3 perform distinct, non-redundant functions in response to SDF-1. In addition to PLC-γ1 and PLC-β3, our results also indicate that SDF-1 treatment of T cells activates one or more other PLCs that are responsible for mediating CXCR4 endocytosis and the down-regulation of CXCR4 from the cell-surface in response to SDF-1.
Fig. 5. SDF-1 signaling via the CXCR4-TCR heterodimer requires different PLCs in order to mediate distinct cellular outcomes.
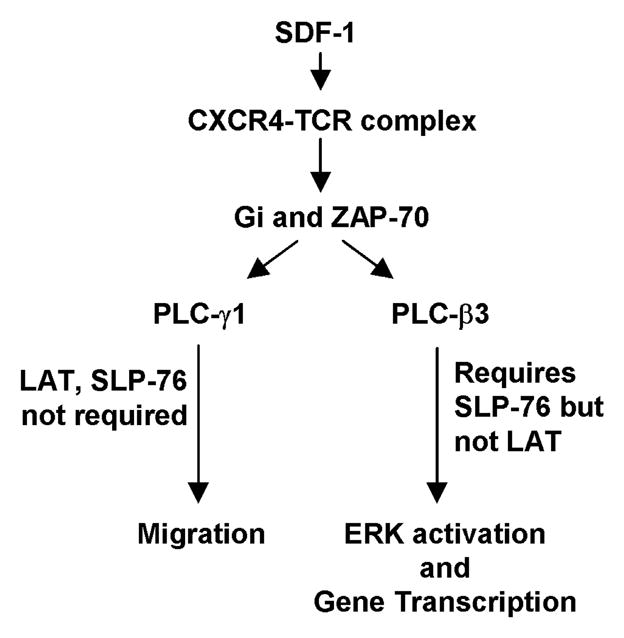
Based on our results presented here, and together with results reported previously by our group and others, we propose that CXCR4-TCR heterodimer appears to signal in T cells in response to SDF-1 via the signal transduction pathway model shown in Fig. 5. SDF-1 treatment induces the formation of the CXCR4-TCR heterodimer which mediates the activation of Gi proteins and ZAP-70 and this either directly or indirectly leads to the activation of several PLCs including both PLC-γ1 and PLC-β3. PLC-γ1 activity is required to mediate the migration of T cells via a mechanism that does not require either LAT (shown here) or SLP-76 and is also independent of PLC-β3. In contrast, PLC-β3 activation leads to ERK activation and gene transcription via a mechanism that previous results show does require SLP-76 but is independent of LAT. In addition to PLC-γ1 and PLC-β3, the CXCR4-TCR heterodimer appears to activate a different PLC(s) to participate in regulating CXCR4 endocytosis in response to SDF-1.
DISCUSSION
Since its discovery in 1994 (25), CXCR4 has been shown to be one of the widest-expressed and most biologically critical of all the chemokine receptors. CXCR4 mediates a diverse array of signaling pathways in a large number of cell types, regulating critical developmental, immunological, and pathological cues by activating a wide range of signaling molecules (1, 26). One strategy that CXCR4 utilizes to diversify its signal transduction mechanisms among these different cell types is heterodimerization with other types of cell-surface receptors; in T cells, CXCR4 signals by heterodimerizing with the T cell antigen receptor (TCR) (8). In addition, we demonstrate here that the CXCR4-TCR heterodimer further diversifies its downstream signaling within T cells by utilizing distinct PLC isoforms in order to mediate different signal transduction outcomes.
We show here that upon ligation by SDF-1, the CXCR4-TCR heterodimer signals to activate one PLC isozyme for calcium mobilization and ERK activation, another PLC for migration, and yet another PLC for internalization of CXCR4. We found that ERK activation, migration, and internalization of CXCR4 in response to SDF-1 are inhibited by a PLC inhibitor drug, indicating that PLC activity is required for all of these events. We further showed that PLC-β3 expression is required for SDF-1-mediated calcium mobilization, activation of N-Ras and K-Ras, as well as for activation of ERK, while also showing that PLC-γ1 expression is not required for these events. In contrast, we showed that PLC-γ1 expression is required for T cell migration in response to SDF-1 while depletion of PLC-β3 actually enhances migration. Moreover, we found that neither PLC-β3 nor PLC-γ1 was required for SDF-1 treatment to induce the internalization and downregulation of CXCR4 from the cell-surface, indicating a role for other PLCs. Finally, we showed that PLC-γ1 mediates migration in response to SDF-1 in a manner that is independent of LAT, thus indicating that the CXCR4-TCR heterodimer couples to PLC-γ1 in a manner distinct from that arising from direct ligation of the TCR. Our results presented here, combined with previous work, provide further insight into the molecular mechanisms that permit the CXCR4-TCR heterodimer to integrate the signaling of traditional GPCR-linked and traditional TCR-linked signaling mediators. Both pertussis toxin-sensitive Gi-type G proteins and ZAP-70 have been shown to be required for calcium mobilization, ERK activation, and migration in response to SDF-1 treatment of T cells (6, 8, 24, 26). Thus, our results indicate that, in response to SDF-1, both PLC-β3 and PLC-γ1 require upstream Gi proteins and ZAP-70 for their regulation in T cells.
SDF-1-induced calcium mobilization and ERK activation require some of the same signaling molecules, the TCR, ZAP-70, and SLP-76 (6, 8), that the direct ligation of the TCR requires to activate these pathways (27). We therefore investigated if, like the TCR, the CXCR4-TCR heterodimer signals via PLC-γ1. Yet we found that PLC-β3, not PLC-γ1, is required for SDF-1 to induce calcium mobilization and ERK activation, a result indicating that PLC-β3 is able to integrate signaling from ZAP-70 and SLP-76 as well as G proteins. The Gβγ subunit of Gi proteins has been shown to bind directly to and mediate the activation of PLC-β3 downstream of GPCR signaling (10, 28). PLC-β3 has also been shown to bind various PDZ-containing scaffold proteins, including Nherf-2 and Shank-2 that mediate the interactions of numerous signaling molecules (11, 29, 30). Nherf family members are implicated in regulating T cell activation (31), suggesting that Nherf or related molecules may mediate interactions between PLC-β3 and TCR signaling molecules.
Similarly, the requirement for PLC-γ1 in mediating migration in response to SDF-1 suggests that PLC-γ1 is able to integrate signals from Gi proteins and possibly other G proteins (32) as well as traditional TCR signaling molecules such as ZAP-70. However, SDF-1 clearly regulates PLC-γ1 in a manner independent of other key mediators that are required for signal transduction arising from direct TCR stimulation. Following direct ligation of the TCR, LAT and SLP-76 are both required for the recruitment and activation of PLC-γ1 (27). In contrast, we show here that SDF-1-mediated migration requires PLC-γ1 but occurs independently of LAT expression, and others have shown that SDF-1-dependent T cell migration also does not involve SLP-76 (24). GPCR kinase interacting protein-1 (GIT1) has been shown to regulate PLC-γ1 signaling downstream of other GPCRs, and is also activated by the TCR in a manner independent of SLP-76 (33, 34). SDF-1 signaling via the CXCR4-TCR heterodimer may therefore utilize GIT1 as an alternative mechanism of PLC-γ1 activation in order to regulate T cell migration. Furthermore and as we show here, PLC-γ1 can mediate cellular functions without the commonly-seen inducible phosphorylation of its tyrosines (35). Bach et al. have shown that depletion of both PLC-β2 and PLC-β3 in mouse T cells partially inhibits T cell migration in response to SDF-1 (14). PLC-γ1 may therefore function to regulate migration via a mechanism that integrates its signals with PLC-β2, possibly via forming a PLC dimer as has been seen for other PLC isoforms (36, 37). Thus, CXCR4-TCR signaling utilizes a distinct mechanism to integrate the functions of GPCR-associated and TCR-associated signaling molecules in order to mediate PLC-γ1-dependent migration.
Internalization of CXCR4 has been extensively studied because the functions of CXCR4 critically depend on its cell-surface expression levels. A key mechanism regulating this pathway includes the phosphorylation of CXCR4 by G-protein-coupled receptor kinases (GRKs) and PKC followed by the recruitment ofβ-arrestins (38). A role for PLC activity in the internalization of CXCR4 has not previously been described. Inhibition of Gi proteins (23) or the depletion of ZAP-70 (unpublished observations) failed to inhibit internalization of CXCR4 in response to SDF-1, consistent with neither PLC-γ1 nor PLC-β3 being required for this process. Further studies will be required to determine which PLC isoforms are critically required for the internalization of CXCR4 in response to SDF-1.
Together, the results in this paper indicate that multiple PLCs are able to integrate signaling from the CXCR4-TCR heterodimer in order to act as key mediators of SDF-1-mediated T cell functions. The PLC-dependent T cell functions mediated by SDF-1 described here are critical for many immunological events. Both PLC-β3 deficient mice and mice specifically deficient in T cell PLC-γ1 display spontaneous autoimmune phenotypes involving mononuclear infiltrates particularly into the skin and ear (12, 39). Furthermore, T cell specific PLC-γ1 deficient mice were characterized by a paucity of peripheral T cells, possibly arising from abnormal T cell migration, as well as by the impaired development and function of regulatory T cells (12). We previously demonstrated that SDF-1 costimulates IL-10 production in regulatory T cells by activating prolonged ERK and AP-1-mediated transcription (7). Thus, it seems possible that the SDF-1 signaling that is impaired by the lack of PLC-γ1 may inhibit the appropriate migration of regulatory T cells, while SDF-1 signaling impaired by the lack of PLC-β3 inhibits the costimulation of IL-10 production by regulatory T cells. In addition, both CXCR4 and PLC-γ1 have been implicated as regulators of the metastasis of various cancer cell types (2, 40–42). Our results therefore raise the possibility that PLC-γ1 helps mediate the SDF-1-induced metastasis of these tumoral cells. The novel integration of multiple PLCs with distinct functions downstream of SDF-1 mediated signaling described here may thereby be responsible for the diverse roles CXCR4 plays in immunology and pathology.
Supplementary Material
Acknowledgments
We are grateful to Dr. R. Abraham for providing mutant Jurkat sublines and Dr. D. Billadeau for the pCMS4.mCherry.H1p and pCMS4.eGFP.H1p constructs.
Footnotes
This work was supported in part by Alma B. Stevenson Endowment Fund for Medical Research, and by N.I.H Grants #R01GM59763 (to K.E.H), R25GM055252 (to A.O.B.), and R25GM075148 (to A.G.M.).
References
- 1.Busillo JM, Benovic JL. Regulation of CXCR4 signaling. Biochimica et biophysica acta. 2007;1768:952–963. doi: 10.1016/j.bbamem.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res. 2010;16:2927–2931. doi: 10.1158/1078-0432.CCR-09-2329. [DOI] [PubMed] [Google Scholar]
- 3.Salanga CL, O’Hayre M, Handel T. Modulation of chemokine receptor activity through dimerization and crosstalk. Cell Mol Life Sci. 2009;66:1370–1386. doi: 10.1007/s00018-008-8666-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soede RD, I, Zeelenberg S, Wijnands YM, Kamp M, Roos E. Stromal cell-derived factor-1-induced LFA-1 activation during in vivo migration of T cell hybridoma cells requires Gq/11, RhoA, and myosin, as well as Gi and Cdc42. J Immunol. 2001;166:4293–4301. doi: 10.4049/jimmunol.166.7.4293. [DOI] [PubMed] [Google Scholar]
- 5.Kumar A, Kremer KN, Dominguez D, Tadi M, Hedin KE. G{alpha}13 and Rho Mediate Endosomal Trafficking of CXCR4 into Rab11+ Vesicles upon Stromal Cell-Derived Factor-1 Stimulation. J Immunol. 2011;186:951–958. doi: 10.4049/jimmunol.1002019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kremer KN, Humphreys TD, Kumar A, Qian NX, Hedin KE. Distinct role of ZAP-70 and Src homology 2 domain-containing leukocyte protein of 76 kDa in the prolonged activation of extracellular signal-regulated protein kinase by the stromal cell-derived factor-1 alpha/CXCL12 chemokine. J Immunol. 2003;171:360–367. doi: 10.4049/jimmunol.171.1.360. [DOI] [PubMed] [Google Scholar]
- 7.Kremer KN, Kumar A, Hedin KE. Haplotype-independent costimulation of IL-10 secretion by SDF-1/CXCL12 proceeds via AP-1 binding to the human IL-10 promoter. J Immunol. 2007;178:1581–1588. doi: 10.4049/jimmunol.178.3.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar A, Humphreys TD, Kremer KN, Bramati PS, Bradfield L, Edgar CE, Hedin KE. CXCR4 physically associates with the T cell receptor to signal in T cells. Immunity. 2006;25:213–224. doi: 10.1016/j.immuni.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 9.Patrussi L, Ulivieri C, Lucherini OM, Paccani SR, Gamberucci A, Lanfrancone L, Pelicci PG, Baldari CT. p52Shc is required for CXCR4-dependent signaling and chemotaxis in T cells. Blood. 2007;110:1730–1738. doi: 10.1182/blood-2007-01-068411. [DOI] [PubMed] [Google Scholar]
- 10.Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi JW, Lim S, Oh YS, Kim EK, Kim SH, Kim YH, Heo K, Kim J, Kim JK, Yang YR, Ryu SH, Suh PG. Subtype-specific role of phospholipase C-beta in bradykinin and LPA signaling through differential binding of different PDZ scaffold proteins. Cell Signal. 2010;22:1153–1161. doi: 10.1016/j.cellsig.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 12.Fu G, Chen Y, Yu M, Podd A, Schuman J, He Y, Di L, Yassai M, Haribhai D, North PE, Gorski J, Williams CB, Wang D, Wen R. Phospholipase C{gamma}1 is essential for T cell development, activation, and tolerance. J Exp Med. 2010;207:309–318. doi: 10.1084/jem.20090880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haribabu B, Richardson RM, Fisher I, Sozzani S, Peiper SC, Horuk R, Ali H, Snyderman R. Regulation of human chemokine receptors CXCR4. Role of phosphorylation in desensitization and internalization. J Biol Chem. 1997;272:28726–28731. doi: 10.1074/jbc.272.45.28726. [DOI] [PubMed] [Google Scholar]
- 14.Bach TL, Chen QM, Kerr WT, Wang Y, Lian L, Choi JK, Wu D, Kazanietz MG, Koretzky GA, Zigmond S, Abrams CS. Phospholipase cbeta is critical for T cell chemotaxis. J Immunol. 2007;179:2223–2227. doi: 10.4049/jimmunol.179.4.2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shannon LA, Calloway PA, Welch TP, Vines CM. CCR7/CCL21 migration on fibronectin is mediated by phospholipase Cgamma1 and ERK1/2 in primary T lymphocytes. J Biol Chem. 2010;285:38781–38787. doi: 10.1074/jbc.M110.152173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Gorter DJ, Beuling EA, Kersseboom R, Middendorp S, van Gils JM, Hendriks RW, Pals ST, Spaargaren M. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity. 2007;26:93–104. doi: 10.1016/j.immuni.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 17.Irvin BJ, Williams BL, Nilson AE, Maynor HO, Abraham RT. Pleiotropic contributions of phospholipase C-gamma1 (PLC-gamma1) to T-cell antigen receptor-mediated signaling: reconstitution studies of a PLC-gamma1-deficient Jurkat T-cell line. Mol Cell Biol. 2000;20:9149–9161. doi: 10.1128/mcb.20.24.9149-9161.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang W, Irvin BJ, Trible RP, Abraham RT, Samelson LE. Functional analysis of LAT in TCR-mediated signaling pathways using a LAT-deficient Jurkat cell line. Int Immunol. 1999;11:943–950. doi: 10.1093/intimm/11.6.943. [DOI] [PubMed] [Google Scholar]
- 19.Gomez TS, McCarney SD, Carrizosa E, Labno CM, Comiskey EO, Nolz JC, Zhu P, Freedman BD, Clark MR, Rawlings DJ, Billadeau DD, Burkhardt JK. HS1 functions as an essential actin-regulatory adaptor protein at the immune synapse. Immunity. 2006;24:741–752. doi: 10.1016/j.immuni.2006.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar A, Kremer KN, Sims OL, Hedin KE. Measuring the proximity of T-lymphocyte CXCR4 and TCR by fluorescence resonance energy transfer (FRET) Methods in enzymology. 2009;460:379–397. doi: 10.1016/S0076-6879(09)05219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin J, Weiss A. Identification of the minimal tyrosine residues required for linker for activation of T cell function. J Biol Chem. 2001;276:29588–29595. doi: 10.1074/jbc.M102221200. [DOI] [PubMed] [Google Scholar]
- 23.Amara A, Gall SL, Schwartz O, Salamero J, Montes M, Loetscher P, Baggiolini M, Virelizier JL, Arenzana-Seisdedos F. HIV coreceptor downregulation as antiviral principle: SDF-1alpha-dependent internalization of the chemokine receptor CXCR4 contributes to inhibition of HIV replication. J Exp Med. 1997;186:139–146. doi: 10.1084/jem.186.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ottoson NC, Pribila JT, Chan AS, Shimizu Y. Cutting edge: T cell migration regulated by CXCR4 chemokine receptor signaling to ZAP-70 tyrosine kinase. J Immunol. 2001;167:1857–1861. doi: 10.4049/jimmunol.167.4.1857. [DOI] [PubMed] [Google Scholar]
- 25.Loetscher M, Geiser T, O’Reilly T, Zwahlen R, Baggiolini M, Moser B. Cloning of a human seven-transmembrane domain receptor, LESTR, that is highly expressed in leukocytes. J Biol Chem. 1994;269:232–237. [PubMed] [Google Scholar]
- 26.Patrussi L, Baldari CT. Intracellular mediators of CXCR4-dependent signaling in T cells. Immunology letters. 2008;115:75–82. doi: 10.1016/j.imlet.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Koretzky GA, Abtahian F, Silverman MA. SLP76 and SLP65: complex regulation of signalling in lymphocytes and beyond. Nat Rev Immunol. 2006;6:67–78. doi: 10.1038/nri1750. [DOI] [PubMed] [Google Scholar]
- 28.Rebres RA, Roach TI, Fraser ID, Philip F, Moon C, Lin KM, Liu J, Santat L, Cheadle L, Ross EM, Simon MI, Seaman WE. Synergistic Ca2+ responses by G{alpha}i- and G{alpha}q-coupled G-protein-coupled receptors require a single PLC{beta} isoform that is sensitive to both G{beta}{gamma} and G{alpha}q. J Biol Chem. 2011;286:942–951. doi: 10.1074/jbc.M110.198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang JI, Kim HS, Lee JR, Kim E, Ryu SH, Suh PG. The interaction of phospholipase C-beta3 with Shank2 regulates mGluR-mediated calcium signal. J Biol Chem. 2005;280:12467–12473. doi: 10.1074/jbc.M410740200. [DOI] [PubMed] [Google Scholar]
- 30.Suh PG, Hwang JI, Ryu SH, Donowitz M, Kim JH. The roles of PDZ-containing proteins in PLC-beta-mediated signaling. Biochem Biophys Res Commun. 2001;288:1–7. doi: 10.1006/bbrc.2001.5710. [DOI] [PubMed] [Google Scholar]
- 31.Itoh K, Sakakibara M, Yamasaki S, Takeuchi A, Arase H, Miyazaki M, Nakajima N, Okada M, Saito T. Cutting edge: negative regulation of immune synapse formation by anchoring lipid raft to cytoskeleton through Cbp-EBP50-ERM assembly. J Immunol. 2002;168:541–544. doi: 10.4049/jimmunol.168.2.541. [DOI] [PubMed] [Google Scholar]
- 32.Tan W, Martin D, Gutkind JS. The Galpha13-Rho signaling axis is required for SDF-1-induced migration through CXCR4. J Biol Chem. 2006;281:39542–39549. doi: 10.1074/jbc.M609062200. [DOI] [PubMed] [Google Scholar]
- 33.Phee H, Abraham RT, Weiss A. Dynamic recruitment of PAK1 to the immunological synapse is mediated by PIX independently of SLP-76 and Vav1. Nature immunology. 2005;6:608–617. doi: 10.1038/ni1199. [DOI] [PubMed] [Google Scholar]
- 34.Haendeler J, Yin G, Hojo Y, Saito Y, Melaragno M, Yan C, Sharma VK, Heller M, Aebersold R, Berk BC. GIT1 mediates Src-dependent activation of phospholipase Cgamma by angiotensin II and epidermal growth factor. J Biol Chem. 2003;278:49936–49944. doi: 10.1074/jbc.M307317200. [DOI] [PubMed] [Google Scholar]
- 35.Everett KL, Bunney TD, Yoon Y, Rodrigues-Lima F, Harris R, Driscoll PC, Abe K, Fuchs H, de Angelis MH, Yu P, Cho W, Katan M. Characterization of phospholipase C gamma enzymes with gain-of-function mutations. J Biol Chem. 2009;284:23083–23093. doi: 10.1074/jbc.M109.019265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singer AU, Waldo GL, Harden TK, Sondek J. A unique fold of phospholipase C-beta mediates dimerization and interaction with G alpha q. Nat Struct Biol. 2002;9:32–36. doi: 10.1038/nsb731. [DOI] [PubMed] [Google Scholar]
- 37.Guo S, Zhang X, Seaton BA, Roberts MF. Role of helix B residues in interfacial activation of a bacterial phosphatidylinositol-specific phospholipase C. Biochemistry. 2008;47:4201–4210. doi: 10.1021/bi702269u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M, Benovic JL. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem. 2010;285:7805–7817. doi: 10.1074/jbc.M109.091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- 40.Kassis J, Lauffenburger DA, Turner T, Wells A. Tumor invasion as dysregulated cell motility. Semin Cancer Biol. 2001;11:105–117. doi: 10.1006/scbi.2000.0362. [DOI] [PubMed] [Google Scholar]
- 41.Sala G, Dituri F, Raimondi C, Previdi S, Maffucci T, Mazzoletti M, Rossi C, Iezzi M, Lattanzio R, Piantelli M, Iacobelli S, Broggini M, Falasca M. Phospholipase Cgamma1 is required for metastasis development and progression. Cancer Res. 2008;68:10187–10196. doi: 10.1158/0008-5472.CAN-08-1181. [DOI] [PubMed] [Google Scholar]
- 42.Davies G, Martin TA, Ye L, Lewis-Russell JM, Mason MD, Jiang WG. Phospholipase-C gamma-1 (PLCgamma-1) is critical in hepatocyte growth factor induced in vitro invasion and migration without affecting the growth of prostate cancer cells. Urol Oncol. 2008;26:386–391. doi: 10.1016/j.urolonc.2007.06.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.