

Abstract
Cancer cell molecular mimicry of stem cells (SC) imbues neoplastic cells with enhanced proliferative and renewal capacities. In support, numerous mediators of SC self-renewal have been evinced to exhibit oncogenic potential. We have recently reported that shRNA-mediated knockdown of the embryonic stem cell (ESC) self-renewal gene NANOG significantly reduced the clonogenic and tumorigenic capabilities of various cancer cells. In this study, we sought to test the potential pro-tumorigenic functions of NANOG, particularly, in prostate cancer (PCa). Using quantitative RT-PCR, we first confirmed that PCa cells expressed NANOG mRNA primarily from the NANOGP8 locus on chromosome 15q14. We then constructed a lentiviral promoter reporter in which the -3.8 kb NANOGP8 genomic fragment was used to drive the expression of green fluorescence protein (GFP). We observed that NANOGP8-GFP+ PCa cells exhibited cancer stem cell (CSC) characteristics such as enhanced clonal growth and tumor regenerative capacity. To further investigate the functions and mechanisms of NANOG in tumorigenesis, we established tetracycline-inducible NANOG overexpressing cancer cell lines, including both prostate (Du145 and LNCaP) and breast (MCF-7) cancer cells. NANOG induction promoted drug-resistance in MCF-7 cells, tumor regeneration in Du145 cells, and, most importantly, castration-resistant tumor development in LNCaP cells. These pro-tumorigenic effects of NANOG were associated with key molecular changes, including an upregulation of molecules such as CXCR4, IGFBP5, CD133 and ALDH1. The present gain-of-function studies, coupled with our recent loss-of-function work, establish the integral role for NANOG in neoplastic processes and shed light on its mechanisms of action.
Keywords: Nanog, prostate cancer, cancer stem cells, castration resistance, self-renewal
Introduction
Immortality is among the fundamental biological properties of malignant cells attributable to the presence of a distinct subset of stem-like cells within tumors. Self-renewing neoplastic cells have been hypothesized to underlie the long-term proliferative potential of cancer cells, often sharing gene expression profiles with embryonic stem cells (ESCs) or primitive somatic SCs (Ben-Porath et al., 2008; Liu et al., 2009; Pece et al., 2010; Wong et al., 2008). Numerous studies over the past several years have provided solid evidence for the existence of such cancer sustaining stem-like cells, generally termed ‘cancer stem cells’ (CSCs) or ‘tumor-initiating cells’(Al-Hajj et al., 2003; Boiko et al., 2010; O'Brien et al., 2007; Patrawala et al., 2006; Singh et al., 2003). However, the molecular framework within which cancer cells fit into the SC paradigm and how this relates to the development of clinical manifestations of late stage disease, including resistance to conventional therapeutics, tumor relapse and metastasis, remains obscure.
Infinitely self-renewing cells (i.e., CSCs) could conceivably account for the immortal nature of tumor cells at the population level, and accumulating evidence supports the notion that ESC self-renewal and pluripotency genes, including the transcription factor triad OCT-3/4 (i.e., OCT-4), SOX-2, and, of particular interest, NANOG, serve as neoplastic engines driving oncogenesis (Gangemi et al., 2009; Hochedlinger et al., 2005; Jeter et al., 2009; Liu et al., 2009; Piestun et al., 2006; Po et al., 2010; Zbinden et al., 2010). Previously, we evaluated the causal relationship between the expression of NANOG in human epithelial cancer cells and tumor development (Jeter et al., 2009). We found that NANOG mRNA in somatic cancer cells appeared to originate predominantly from NANOGP8, a retrogene derivative distinct from the NANOG1 locus expressed in ESCs and that NANOGP8 mRNA was enriched in CD44+ prostate cancer (PCa) stem/progenitor cells and in MCF7 side population (SP) cells. Further, we showed that NANOG protein was heterogeneously expressed as a ‘gradient’ in xenograft as well as primary prostate tumor cells. Of particular importance, we observed that NANOG knockdown decreased the clonogenic growth and tumorigenicity of breast (MCF-7), colon (Colo320) and several prostate (PC3, Du145 and LAPC-9) cancer cell lines and xenografts (Jeter et al., 2009). These results raise an important question of whether NANOG expression actively promotes neoplasia and/or tumor progression. Here, we address this question by further investigating the origins of NANOG in cancer cells, particularly, in PCa, and exploring its oncogenic potential and associated mechanisms.
Results
NANOGP8 mRNA expression in PCa, including primary patient tumors (HPCa)
Our first aim in the present study was to determine whether cancer cells expressing endogenous NANOG are intrinsically unique compared to their non-expressing (or low-expressing) counterparts. To this end, we conceived a promoter reporter strategy to track and isolate cells differentially expressing NANOG. However, one complication is that at least two highly similar NANOG mRNA species have been reported originating from two distinct loci (Figure 1a), including the NANOG1, located on chromosome 12p13, in ESCs, and its retrogene NANOGP8, located on chromosome 15q14 (Booth and Holland, 2004). Given their remarkable similarity (highly conserved over the entire length of the processed transcripts with only 6 nucleotide substitutions in the coding regions and 99% identical at the amino acid level) (Jeter et al., 2009), theoretically, NANOG1 and/or NANOGP8 could account for the presence of NANOG mRNA in cancer cells. We previously provided preliminary evidence that NANOGP8 was the predominant source of NANOG mRNA in epithelial cancer cells, including PCa (Jeter et al., 2009); however, due to methodological limitations we could not eliminate NANOG1 as an important contributor. Thus, we set out to further investigate the origin of NANOG mRNA using quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis.
Figure 1. NANOG genomic loci and NANOG mRNA expression in PCa cells.
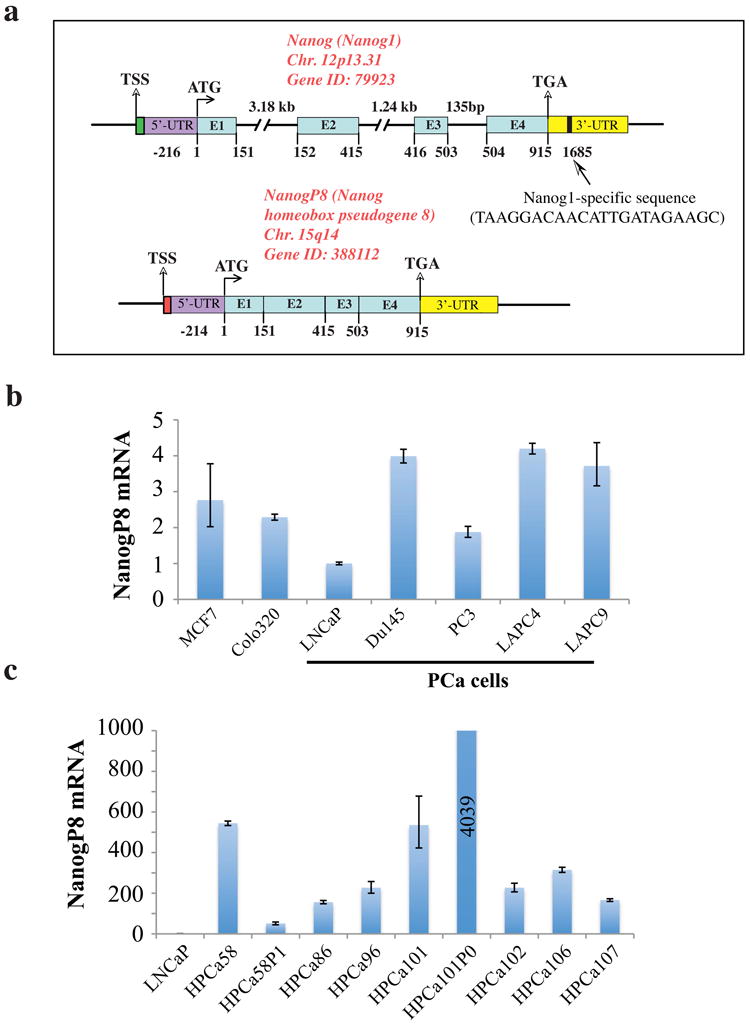
a) Schematic of NANOG1 and NANOGP8 gene structures. Chr, chromosome; E, exon; UTR, untranslated region. The 22-bp region unique to NANOG1 (vertical bar) was used to design primers/probes for NANOG1 and NANOGP8 specific PCR. b) Quantitative PCR (qPCR) detection of NANOGP8 mRNA expression in various cancer cells, normalized to GAPDH. The normalized NANOGP8 mRNA levels in LNCaP cells were set at 1 and NANOGP8 mRNA levels in breast (MCF7), colon (Colo320) and PCa cell lines (LNCaP, Du145 and PC3) and xenografts (LAPC-4 and LAPC-9) were presented relative to LNCaP. c) NANOGP8 mRNA levels in a cohort of primary patient tumors (HPCa) as determined by qPCR. HPCa56P1 and HPCa101P0 are two early-passage (P) xenografts established in our lab.
We designed NANOG1 and NANOGP8 specific qRT-PCR primers and probes (Supplemental Table S1) by targeting a unique 22-base pair (bp) region in the NANOG1 3′-untranslated region (3′-UTR; Figure 1a). NANOGP8 mRNA was readily detectable in a range of cancer cell lines including those derived from breast (MCF7), colon (Colo320) and prostate (LNCaP, Du145 and PC3) cancers, as well as in LAPC4 and LAPC9 PCa xenografts (Figure 1b). As might be expected, LNCaP, the most benign PCa line, expressed the least NANOGP8 mRNA (Figure 1b). In contrast, NANOG1 qRT-PCR in the same panel of cancer cells (using identical template used in NANOGP8 amplifications) was generally below the detection limit (i.e., cycle-threshold >38) although these same primers robustly amplified product from N-TERA teratocarcinoma cells (cycle-threshold of 21) (data not shown). Importantly, a cohort of 7 primary PCa patient samples (HPCa) and two early xenografts (i.e., HPCa58P1 and HPCa101P0) also expressed readily detectable NANOGP8 mRNA (Figure 1c) whereas NANOG1 was undetectable (i.e., cycle threshold >38) in all samples (not shown). Consistent with earlier findings (Jeter et al., 2009), primary PCa specimens expressed higher relative levels of NANOGP8 mRNA than cultured PCa cells (compare Figure 1c and Figure 1b). Interestingly, the two HPCa samples (i.e., HPCa58 and HPCa101) that successfully gave rise to xenografts upon transplantation in NOD/SCID mice expressed higher levels of NANOGP8 mRNA than the other five HPCa samples (Figure 1c). Upon in vivo passaging, the HPCa58P1 showed reduced whereas the HPCa101P0 showed increased NANOGP8 mRNA levels (Figure 1c), suggesting that passaging of HPCa tumors in mice may alter the expression levels of NANOGP8 mRNA.
A NANOGP8-GFP promoter reporter tracks subsets of PCa cells enriched in clonogenic and tumorigenic properties
The above qRT-PCR analyses are consistent with our previous semi-quantitative RT-PCR data (Jeter et al., 2009) and confirm that NANOGP8 mRNA is the predominant NANOG mRNA species in PCa (and some other cancer) cells. Consequently, we replaced the constitutive and ubiquitous phosphoglycerate kinase (PGK) promoter in the RRL-PGK-GFP lentiviral vector (Zufferey et al., 1998) with the ∼3.8 kb region (nucleotides – 3 764 to + 58) upstream of the NANOGP8 Transcription Start Site (TSS) to construct the RRL-NP8-GFP vector (Figure 2a). FACS analysis of PGK-GFP transduced PCa cells revealed that in LNCaP and Du145 cells, 80-90% cells were GFP+ at 2-μl virus and only slightly more cells became GFP+ at 20-μl virus (Figure 2b; Supplemental Figure S1a). PC3 cells infected with the PGK-GFP vector, on the other hand, showed a viral titer-dependent increase in GFP+ cells (Figure 2b). In comparison, only a very small percentage of PCa cells were GFP+ upon transduction with 2-μl of NP8-GFP lentivirus, prepared in parallel (Figure 2b; Supplemental Figure S1a). Even at 20-μl virus, only ∼19% LNCaP cells and ∼34% Du145 cells were NP8-GFP+ (Figure 2b). For PC3 cells, even at 100-μl virus, <10% cells infected with the NP8-GFP were GFP+ (Figure 2b). These results suggest that as we previously reported (Jeter et al., 2009), NANOG-expressing PCa cells are generally rare and NANOGP8 mRNA levels in cultured PCa cells are low, consistent with the preceding qRT-PCR analysis (Figure 1b). Of note, Du145 cells expressed NP8-GFP as a clear gradient with increasing volumes of virus, reaching ∼80% GFP+ cells at 100-μl virus (Figure 2b). These observations parallel our previous reports of NANOG protein expression as a gradient and NANOG protein being more readily detectable in Du145 cells relative to PC3 or LNCaP cells (Jeter et al., 2009). Immunofluorescence (IF) staining of LNCaP cells transduced with NP8-GFP revealed concordant expression of NANOG and GFP in these cells (Figure 2c). Using the denaturation immunostaining protocol (Jeter et al., 2009), we observed ∼10-fold enrichment of NANOG-positive cells in purified NP8-GFP+ LNCaP cells compared to bulk LNCaP cells (not shown). Moreover, qRT-PCR analysis revealed NANOG mRNA levels in NP8-GFP+ versus NP8-GFP- PC3 cells to be 2.02% versus 0.11% relative to the levels in N-TERA human embryonal carcinoma cells, indicating a 18-fold enrichment of NANOG mRNA in NP8-GFP+ PC3 cells. Taken together, these results suggest that PCa cells expressing the promoter constructs are enriched in NANOG mRNA and protein expression.
Figure 2. NANOGP8-expressing PCa cells possess CSC properties.
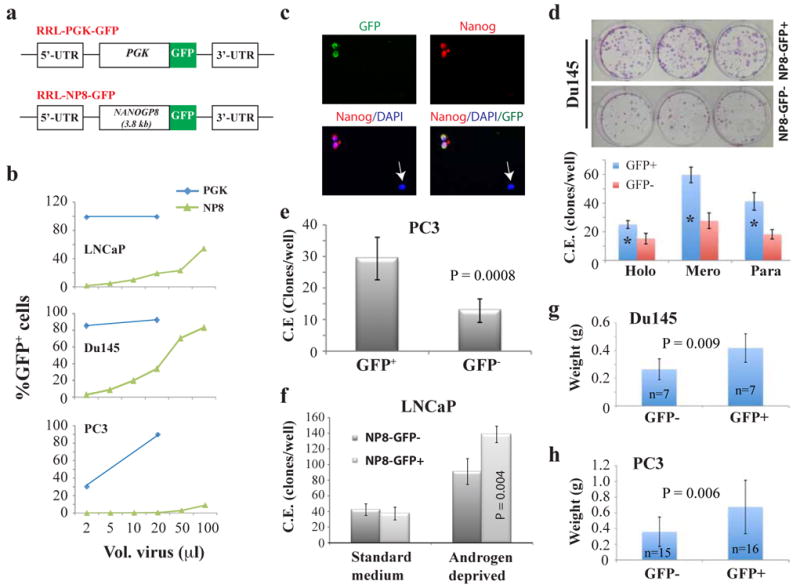
a) Schematic of RRL-GFP lentiviral reporter constructs. GFP, green fluorescence protein; PGK, phosphoglycerate kinase; NP8, NANOGP8 promoter. b) FACS analysis of the percentage of GFP+ cells following transduction with the promoter reporter lentiviruses. 50K cells/well in 12-well dishes plated 1-day earlier were transduced with the indicated volumes of lentivirus. Cultured cells were maintained in fresh media and passaged until FACS analysis ∼ 5 d post-transduction. c) Immunostaining of NP8-GFP transduced LNCaP cells for NANOG (red) demonstrates co-localization of GFP and NANOG expression. Original magnifications, x200. (d-f) FACS purified viable (7AAD-) NP8-GFP+ PCa cells transduced (7-10 d prior) with the NP8-GFP lentivirus exhibit enhanced cloning efficiency (C.E.). d) NP8-Du145 cells (200 cells/well in 6-well culture plate) scored 14 d post-plating: holo, holoclones; mero, meroclones; para, paraclones. *P <0.001. e) NP8-PC3 cells (200 cells/well in 6-well culture plate) scored 14 d post-plating. f) LNCaP cells assayed in parallel at clonal density in androgen-deprived conditions (CDSS + 20 μM bicalutamide) versus standard growth media (2K cells/well and 200 cells/well, respectively) scored 14 d post-plating. NP8-GFP+ LNCaP cells displayed increased C.E. only in androgen-deprivation conditions. g-h) NP8-GFP+ PCa cells are more tumorigenic than NP8-GFP- cells. FACS-purified GFP+/- cells were injected s.c in Matrigel in NOD/SCID-γ recipients. In g), 5K each of NP8-GFP+/- Du145 cells were injected and tumors harvested at ∼2 months. In h), 1K each of purified NP8+/- PC3 cells were injected s.c in the 1° and 2° tumor transplantation experiments (harvested at d 56 and day 49, respectively). Shown are the pooled data.
Next, we set out to test the biological characteristics of RRL transduced and sorted Du145, PC3 and LNCaP cells. Du145 NP8-GFP+ cells, compared to NP8-GFP− cells, generated more holoclones, meroclones and paraclones (Figure 2d), morphologically distinct classes generally thought to reflect the differentiation state of the originating cell with the holoclones containing the long-term self-renewing CSCs (Li et al., 2008). In contrast, purified PGK-GFP+ and PGK-GFP- Du145 cells exhibited similar cloning efficiency (C.E) (Supplemental Figure S1b). PC3 NP8-GFP+ cells also exhibited enhanced C.E. (Figure 2e) and further, gave rise to larger clones compared to NP8-GFP− PC3 cells (3 949 ± 1 462 versus 2 278 ± 912, respectively; P = 0.04). Interestingly, NP8-GFP+ and NP8-GFP− LNCaP cells exhibited similar C.E. in standard serum-containing media. However, in androgen-deprived conditions, i.e., in media containing charcoal dextran-stripped serum (CDSS) and/or antiandrogen bicalutamide, purified NP8-GFP+ LNCaP cells exhibited significantly higher C.E. than the NP8-GFP− cells (Figure 2f; Supplemental Figure S2a). These latter observations in LNCaP cells provided the first evidence that NANOG might be associated with resistance to androgen deprivation.
Finally, we assayed the quintessential biological property of tumor cells, i.e., tumor development. NP8-GFP transduced Du145 cells, initially 30% GFP+, were sorted into 7-aminoactinomycin D (7AAD) negative (i.e., viable) NP8-GFP− and NP8-GFP+ subsets and injected subcutaneously (s.c) into NOD/SCID mice. The primary (1°) Du145-NP8-GFP+ tumors were not only larger than those derived from Du145-NP8-GFP− cells (Figure 2g) but also comprised a mixture of GFP+ (41%) and GFP− cells (not shown), thus recapitulating the heterogeneity of the starting population and suggesting that the NP8-GFP+ cells could divide asymmetrically to give rise to NP8-GFP− cells. NP8-GFP+ Du145 cells purified from the 1° tumors also regenerated tumors ∼60% larger than NP8-GFP− cell derived tumors (not shown). Histological analysis revealed more readily detectable NANOG-expressing and Ki-67+ cells in tumors derived from NP8-GFP+ Du145 cells (Supplemental Figure S2b). Similar to NP8-GFP+ Du145 cells, the NP8-GFP+ PC3 cells also generated larger tumors than their NP8-GFP− counterparts in both 1° and 2° tumor assays (Figure 2h). Collectively, the promoter-tracking studies demonstrate that the NANOGP8-expressing PCa cells possess higher clonal and tumor-regenerating capacities, characteristics associated with and expected of CSCs.
Establishing an inducible NANOG-overexpressing lentiviral system
The second principal aim of this study was to determine the cellular and molecular mechanisms underpinning the biological functions of NANOG in cancer cells. Since NANOG is normally expressed at low levels in cultured epithelial cancer cells relative to ESCs (Jeter et al., 2009), we engineered a tetracycline (doxycycline, dox) inducible lentiviral system to overexpress NANOG (Figure 3a). Cancer cells (MCF7, Du145 and LNCaP) initially infected with the pLVX-TetON lentivirus (Figure 3a) and expressing high levels of the dox-inducible transactivator (TetR/VP16) were clonally derived via analysis of expression of a Tetracycline Responsive Element (TRE) luciferase reporter (data not shown). Stable clones were then subjected to 2° transductions with either pLVX empty vector or the pLVX-TRE-Nanog1/NanogP8 lentiviral vectors in which the NANOG1 or NANOGP8 cDNA was cloned downstream of the TRE (Figure 3a). Western blot analysis revealed dox-induced upregulation of NANOG protein in a dose-dependent fashion (Figure 3b-c; note that most commercial anti-NANOG Abs were generated against NANOG1 and recognize both NANOG1 and NANOGP8 on Western and IF; Jeter et al., 2009). The induced NANOG protein (for the sake of simplicity, both NANOG1 and NANOGP8 proteins will frequently be termed NANOG henceforward due to nearly identical amino acid sequences) was mostly detected in the nucleus (Figure 3d-e, Supplemental Figure S3), a subcellular localization expected for a transcription factor. In the absence of dox, NANOG-expressing Du145 (Figure 3d) and MCF7 (Supplemental Figure S3) cells were rare and NANOG-expressing LNCaP cells were even more rare (Figure 3e), consistent with our earlier results (Figure 1b; Figure 2b; Jeter et al., 2009) and our observations that the endogenous NANOG protein was only detectable by Western upon longer exposure (Figure 3b-c and data not shown).
Figure 3. Establishment and characterization of a dox-inducible NANOG overexpression system.
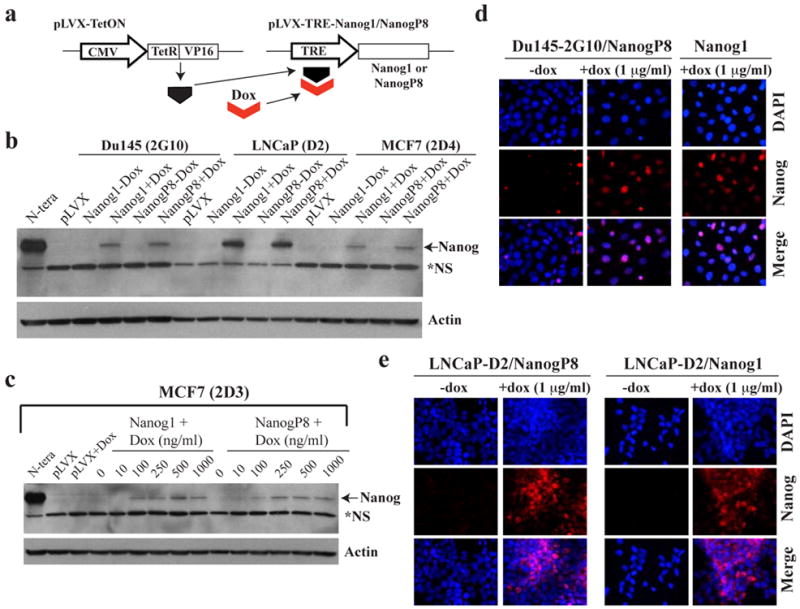
a) Schematic of pLVX-TetON (Clontech) binary system to express inducible NANOG1 or NANOGP8: CMV, cytomegalovirus promoter; Dox, doxycycline; TRE, tetracycline responsive element. b-c) Western blot analysis of protein extracts (80 μg whole cell lysate) demonstrates dox-inducible NANOG overexpression. N.S, non-specific band. b) Western analysis of NANOG in extracts derived from control pLVX MCF-7, Du145 and LNCaP cells or respective pLVX-NANOG1/P8 clones, either untreated or induced with doxycycline (500 ng/ml; 48 h). c) Doxycycline titration in MCF-7 pLVX cells as indicated. Note that in both Western analyses, the endogenous NANOG protein was barely detectable due to NANOG-expressing cells being very rare (see below). d-e) NANOG IF staining using a Rb polyclonal anti-NANOG antibody (H-155; Santa Cruz) revealed nuclear NANOG expression in the majority of clonally-derived cells following dox induction in pLVX-NANOG1/P8 Du145 (d) or LNCaP (e) cells.
To evaluate whether the overexpressed NANOG protein is functional, we performed NANOG chromatin immunoprecipitation (ChIP) assays followed by PCR using primers against several reported gene targets of NANOG1 in hESCs including c-Myc, Oct-4, E-cadherin (E-cad), FGF4, Gli1, and HoxC13 (Medeiros et al., 2009; Supplemental Figure S4). We first performed ChIP assays in N-TERA cells and the results revealed differential NANOG protein binding to the individual gene promoters (Figure 4a). ChIP assays in NANOG1 and NANOGP8 overexpressing (i.e., pLVX-Nanog1/NanogP8) LNCaP cells revealed that dox-induced NANOG also bound to the promoter regions of these genes (Figure 4b) although the levels of binding, after normalizing to RbIgG ChIP, was expectedly lower than in N-TERA cells (compare Figure 4b-c vs. Figure 4a). Similar ChIP assays did not reveal significant binding of either NANOG1 or NANOGP8 at the GAPDH promoter (Figure 4c). Altogether, he IF and ChIP assays indicate that the overexpressed NANOG1/NANOGP8 proteins in our inducible systems localize to the nucleus and are functional in binding the expected targets.
Figure 4. Overexpressed NANOG1 and NANOGP8 proteins in cancer cells bind the expected molecular targets.
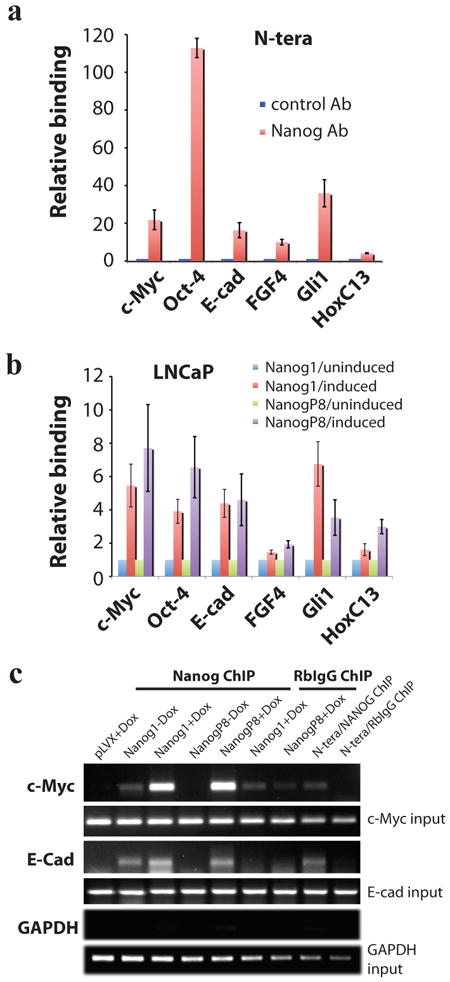
a) ChIP assays in N-TERA human embryonal carcinoma cells. Chromatin prepared from N-TERA cells was used in immunoprecipitations with a Rb polyclonal anti-NANOG Ab or, as control, Rb IgG. DNA co-precipitated in ChIP was then used in PCR analysis of the promoter region of the indicated molecules. The results were expressed as NANOG binding to the targets relative to RbIgG (plotted as 1). b) pLVX-NANOG1/NANOGP8 LNCaP cells (clone D2) were either untreated or induced with dox (500 ng/ml) for 72 h. Chromatin was prepared and ChIP assays were performed similarly to those in N-TERA cells. c) Examples of ChIP assays with GAPDH ChIP as negative control.
Short-term NANOG induction does not promote cancer cell proliferation
Having successfully established an inducible system to overexpress functional NANOG, we next sought to determine the biological responses to elevated NANOG protein in cancer cells. As NANOG overexpression had been previously reported to enhance proliferation in several cell types, including NIH3T3 cells (Zhang et al., 2005), ESCs (Ma et al., 2009) and colorectal cancer cells (Meng et al., 2010), we first performed short-term assays in which our inducible-NANOG cell lines were stimulated with doxycycline for 3-4 d prior to counting the total cell number. To our surprise, under standard culture conditions, no significant difference was apparent in pLVX-control versus pLVX-Nanog1/P8 LNCaP, Du145 or MCF-7 cells (Supplemental Figure S5a; data not shown). BrdU (bromodeoxyuridine) incorporation assays also did not reveal any differences (Supplemental Figure S5b), suggesting that increased NANOG expression short-term did not promote cell proliferation in these cancer cells. Consistent with this observed lack of phenotype, Western blot analysis of short-term (∼3-4 d) doxycycline-treated Du145, LNCaP and MCF-7 cells revealed that despite a clear induction of NANOG protein, there was no detectable change in the levels of several positive regulators of the cell cycle, including CDC25A and CDK6, previously reportedly to be transcriptionally upregulated in response to NANOG overexpression in human ESCs cells (Zhang et al., 2009), as well as cyclin D1 and the c-Myc oncogene (Supplemental Figure S5c-d). Similar to these cell-cycle regulators, several SC molecules including Sox2, Oct4, Gli1, Nkx3.1, and CD44, as well as two molecules associated with EMT (epithelial-mesenchymal transition), i.e., Snail and E-cad, also did not show significant changes upon short-term NANOG1/NANOGP8 induction (Supplemental Figure S5c-d).
NANOG overexpression promotes castration-resistant (androgen-independent) phenotypes and tumor regeneration in LNCaP cells
Although short-term induction of NANOG did not promote cancer cell proliferation, it promoted anchorage-independent survival and sphere formation in LNCaP cells in androgen-deficient conditions, generally assayed longer term (2-4 weeks). Two independent experiments revealed that pLVX-NanogP8 but not pLVX-Nanog1 LNCaP cells generated ∼2× more spheres in methylcellulose than pLVX-control cells (Figure 5a). On the other hand, both pLVX-Nanog1 and pLVX-NanogP8 LNCaP cells cultured in CDSS, compared to pLVX control cells, formed more androgen-independent (AI) foci and floating spheroids (Figure 5b-c), and resulted in higher cell numbers (Figure 5d).
Figure 5. NANOG overexpression promotes AI phenotypes in LNCaP cells.
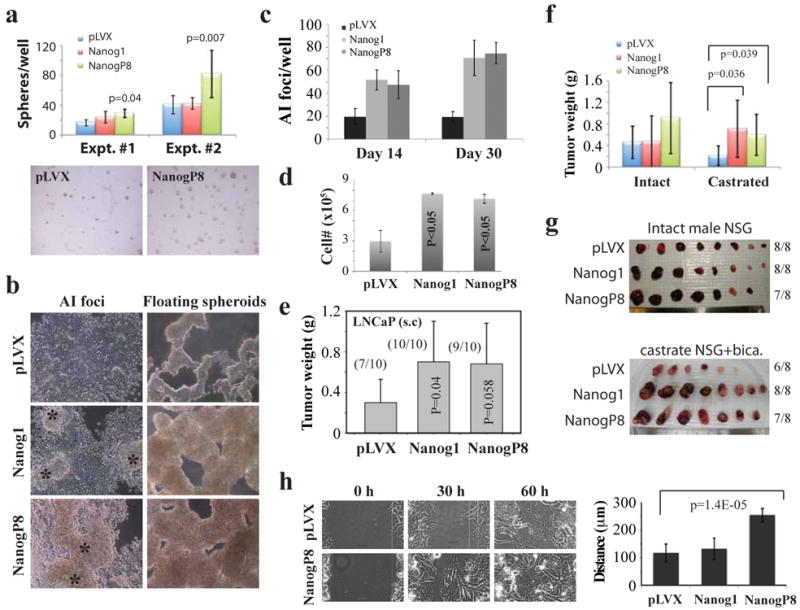
a-d) Enhanced androgen-independent (AI) growth of NANOG-overexpressing LNCaP cells relative to the empty vector (pLVX) control. a) pLVX- NANOGP8 expression promotes LNCaP sphere formation. 2.5K cells (Expt. #1) or 1K (Expt. #2) cells were plated in a 24-well dish in Methocult supplemented with B27 and N2 and 500 ng/ml doxycycline. Spheres (> 100 μM) were scored ∼2 weeks later. Shown below are two representative microphotographs of spheres formed in methycellulose. b-d) NANOG1 and NANOGP8 overexpression promotes the outgrowth of AI LNCaP cells. The three types of LNCaP cells (2K cells/well, plated in a 6-well TC dish) were cultured in CDSS-containing media for up to 30 d, feeding (+ 500 ng/ml doxycycline) every 3-5 d. Bright-field microscopy was used to capture AI foci and floating spheres (b). Shown in c) is quantification of AI-foci at two different time points and in d) is the total number of live cells/well at d30. e) NANOG-overexpressing LNCaP cells (1 × 106/injection) implanted s.c in NOD/SCID-γ males generated larger tumors than the pLVX-control cells (all harvested at d35). f-g) NANOG1/P8 overexpression promotes castration-resistant LNCaP tumor regeneration. The three types of pLVX LNCaP cells were implanted (10K/injection) s.c in either intact or fully castrated (i.e., surgical castration plus bicalutamide treatment) NOD/SCID-γ mice and tumors were harvested 41 (intact) and 61 (castrated) days after implantation. Shown are average tumor weight (f) and tumor images (g). h) NANOGP8-expressing LNCaP cells derived from AI tumors exhibit enhanced migratory properties as assessed by time-lapse video microscopy-assisted ‘scratch’ assays. Shown are representative time-lapse images for pLVX and pLVX-NANOGP8 LNCaP cells (left panels) and quantification of cell migration in all three groups (right).
Next, we determined the impact of NANOG overexpression on LNCaP tumor development in either intact (i.e., androgen-proficient) or castrated (i.e., surgical castration plus bicalutamide treatment; see Methods) NOD/SCID-γ mice. We observed that NANOG overexpression promoted LNCaP tumor regeneration in intact hosts, but in a somewhat inconsistent manner. For example, pLVX-Nanog1 cells developed larger tumors than pLVX cells in one (Figure 5e) but not the other (Figure 5f) experiment. pLVX-NanogP8 cells, on the other hand, regenerated larger tumors than pLVX cells but in both experiments the difference were just below statistical significance (Figure 5e-f). In contrast, both pLVX-NanogP8 and pLVX-Nanog1 cells developed significantly larger tumors than the control cells in castrated NOD/SCID-γ mice (Figure 5f-g). Of note, qRT-PCR and IHC analysis revealed overall similar levels of NANOG mRNA and protein expression in the pLVX-NANOG1 and pLVX-NANOGP8 LNCaP tumors developed in intact (Supplemental Figure S6a, left; Supplemental Figure S6b) or castrated (not shown) hosts. Interestingly, when we derived tumor cells from the LNCaP AI tumors and analyzed their migratory capacities by time-lapse video microscopy, we observed that NANOGP8 expressing AI cells possessed a higher migration rate such that they closed the induced ‘wounds’ faster than the pLVX control or pLVX-Nanog1 cells (Figure 5h).
Molecular changes that accompany NANOG overexpression in LNCaP cells
Given that NANOG-overexpressing LNCaP cells both in vitro and in vivo exhibited enhanced hormone refractory growth, we wondered what molecular mechanisms could account for this resistance to androgen-deprivation? IHC analysis of AI tumors developed in castrated hosts (Figure 5g) revealed significantly more Ki-67+ cells in both pLVX-Nanog8 and pLVX-Nanog1 tumors (Figure 6a), suggesting that NANOG overexpression promotes cell proliferation in vivo. Consistent with this conclusion, the pLVX-Nanog1 AI tumors, and, in particular, the pLVX-NanogP8 AI tumors, showed higher levels of c-Myc compared to the control tumors (Figure 6b). In contrast, we did not observe obvious differences in Ki-67+ cells in the LNCaP tumors developed in intact male mice (Supplemental Figure S6c). Previously, we provided preliminary evidence that NANOG and androgen receptor (AR) seemed to be reciprocally expressed in HPCa cells in patient tumors (Jeter et al., 2009). IHC (Figure 6a) and Western blotting analysis (Figure 6c) detected reduced AR levels in pLVX-Nanog1/NanogP8 LNCaP AI tumors. In fact, even in short-term in vitro assays, both clone B2 (Figure 6d) and clone D2 (Supplemental Figure S5c) LNCaP cells, upon NANOG1/NANOGP8 induction, demonstrated subtle but reproducible decreases in AR. Consistent with reduced AR levels, the pLVX-Nanog1/NanogP8 LNCaP AI tumors also showed reduced levels of PSA (Figure 6a). Pilot experiments with a PSA promoter-driven GFP reporter also revealed that the pLVX-Nanog1/NanogP8 LNCaP cell cultures had ∼20% and 18%, respectively, less GFP+ cells than pLVX control LNCaP cells (data not shown). In principle, increased cell proliferation (i.e., increased Ki-67+ cells associated with c-Myc upregulation) and inhibition of differentiation (i.e., reduced AR and PSA) could help explain NANOG1/NANOGP8-promoted LNCaP AI tumor regeneration.
Figure 6. Molecular changes associated with NANOG-promoted AI phenotypes in LNCaP cells.
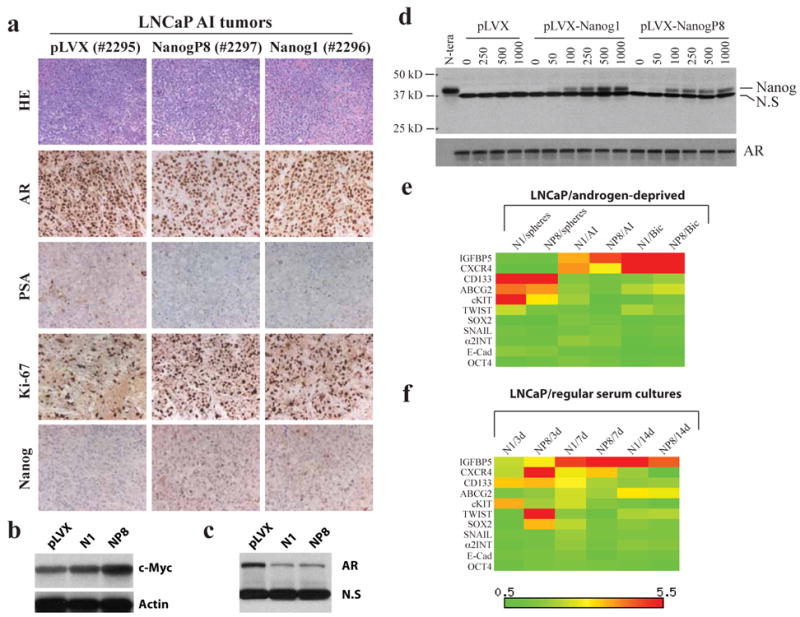
a) H&E staining and IHC analysis of the molecules indicated in three LNCaP AI tumors (tumor tag numbers indicated in parentheses). b-c) Western blotting analysis of c-Myc (b) and AR (c) in AI LNCaP tumors. N1, pLVX-NANOG1 LNCaP AI tumor; NP8, pLVX-NANOGP8 LNCaP AI tumor. N.S, non-specific band (served as loading control). d) Western blotting analysis of Nanog (top; N.S, non-specific band) and AR in clone B2 LNCaP cells treated by the indicated concentrations of dox. e-f) Heatmap presentation of gene expression changes of 11 molecules in LNCaP cells cultured in either androgen-deprived conditions (e) or regular serum cultures (f). The mRNA levels for these and other molecules (see Supplemental Table S1 and S3 for details) were determined by qRT-PCR and the heatmaps generated using the Matrix2.png software. The color-coded bars (scale below) indicate the mRNA level in the NANOG overexpressing cells cultured under the indicated conditions relative to pLVX control cells cultured under the same conditions.
To further explore the potential molecular mechanisms of NANOG-promoted castration-resistant properties in LNCaP cells, we analyzed, by qRT-PCR, the mRNA levels of a panel of molecules (Supplemental Table S1 and S3), which encompasses reported NANOG targets in ESCs (OCT-4 and SOX-2), CSC markers (CD133, CD44, c-KIT/CD117, ALDH1 and ABCG-2/BCRP), and molecules previously implicated in PCa castration resistance (IGFBP5, CXCR4 and Bcl-2). As shown in Figure 6e, insulin growth factor binding-protein 5 (IGFBP-5) and chemokine receptor CXCR4 mRNAs were significantly upregulated in response to NANOG overexpression in androgen-deprived conditions. The relative CXCR4 mRNA levels were elevated ∼12× and ∼10× and IGFBP5 was elevated ∼5× and ∼7×, respectively, in bicalutamide-treated pLVX-Nanog1 and pLVX-NanogP8 LNCaP cells relative to pLVX control cells (Figure 6e; Supplemental Table S3). Three CSC surface markers, i.e., CD133, ABCG2, and c-KIT, generally increased in response to NANOG overexpression, particularly, in spheres (Figure 6e). Several other molecules examined also showed some alterations in androgen-deficient conditions (Figure 6e), although these differences were generally less than two-fold.
Somewhat surprisingly, when we examined the same panel of genes in LNCaP cells cultured in regular serum-containing media and treated with doxycycline for 3-14 days, many of the same genes were also dynamically upregulated in response to NANOG1/NANOGP8 induction, which included both IGFBP5 and CXCR4 as well as CD133, ABCG2, and c-KIT (Figure 6f). These results suggest that the alterations of these genes are also likely involved in mediating the general pro-tumorigenic effects in addition to the AI-specific effects of NANOG.
Further evidence that Nanog overexpression promotes CSC phenotypes and CSC properties
To further evaluate the cellular and molecular consequences of NANOG overexpression in other contexts, we continued the in vitro and in vivo studies using our inducible Du145 and MCF7 cells. NANOGP8 induction in Du145 cells, which are AI PCa cells and lack AR expression, promoted tumor regeneration in both ectopic (i.e., s.c; Figure 7a) or orthotopic (i.e., dorsal prostate or DP; Figure 7b) sites. Curiously, pLVX-Nanog1 Du145 cells did not regenerate significantly larger tumors than pLVX control cells (Figure 7a-b) despite overall similar expression levels of NANOG mRNA (Supplemental Figure S6a, right) and protein (not shown) in pLVX-Nanog1 and pLVX-NanogP8 Du145 tumors. pLVX-NanogP8 Du145 cell-derived orthotopic tumors had higher levels of c-Myc protein in comparison to either pLVX-Nanog1 or pLVX-control cells (Figure 7c), perhaps partially accounting for the protumorigenic activity of NANOGP8.
Figure 7. NANOG overexpression promotes CSC characteristics.
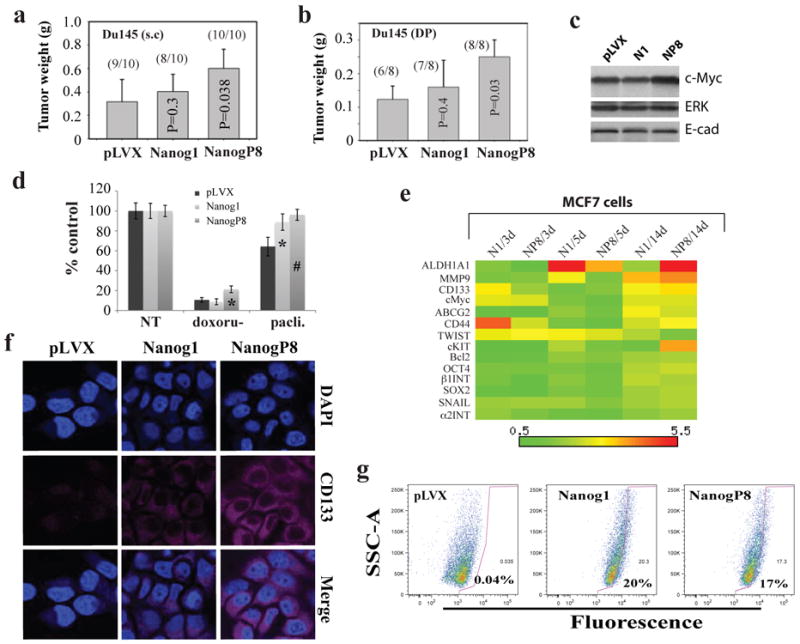
a-b) NANOGP8-overexpressing Du145 cells generate larger tumors than the pLVX-control cells. a) Subcutaneous (s.c) injection of the three types of pLVX Du145 cells (500K) in Matrigel into NOD/SCID hosts, harvested at d 79. b) Orthotopic transplantation of pLVX Du145 cells (1 million) in Matrigel into the dorsal prostate (D.P) of NOD/SCID-γ mice, harvested at d 40. c) Western blot analysis of c-Myc protein levels in the orthotopic Du145 tumors (above). The ERK and E-cadherin levels were unchanged and shown as loading controls. d) NANOG-overexpressing pLVX-MCF7 cells were resistant to the chemotherapeutic drugs doxorubicine (200 nM) and paclitaxel (25 nM). 35K cells plated per well (n = 3) in 6-well dish were induced with 500 ng/ml dox for 48 h followed by drug treatment (96 h). Cells were trypsinized and counted and the number of cells was presented as a percentage relative to the control. *P<0.01; #P<0.001 (Student's t-test). e) Heatmap presentation of gene expression changes of 14 molecules in the pLVX-Nanog1 (N1) or pLVX-NanogP8 (NP8) MCF7 cells induced with dox (500 ng/ml) for 3, 5, and 14 d. The mRNA levels for these and other molecules (see Supplemental Table S1 and S3 for details) were determined by qRT-PCR and the heatmaps generated using the Matrix2.png software. f) IF staining of CD133 protein in pLVX MCF7 cells as indicated. Images were captured on a confocal microscope. g) NANOG1/P8 induction increases the ALDEFLUOR-positive cells. The three types of pLVX-MCF7 cells were treated with dox (500 ng/ml, 10 d) and then used in Aldefluor assays. The mean percentages of ALDEFLUOR-positive cells were indicated.
MCF-7 cells overexpressing NANOG, particularly NANOGP8, exhibited increased resistance to the chemotherapeutic drugs doxorubicine and paclitaxel, consistent with previous findings by others (Bourguignon et al., 2008). To explore the potential mechanisms underlying the NANOG-conferred drug tolerance in MCF7 cells, we also employed qRT-PCR quantification of more than a dozen genes (Figure 7e). The results revealed dynamic upregulation by NANOG1/NANOGP8 of several prosurvival/detoxification genes including Bcl-2, ABCG2, CD133, and ALDH1A1 (Figure 7e; Supplemental Table S3). IF staining confirmed increased CD133 protein in NANOG-overexpressing MCF7 cells (Figure 7f). ALDH1A1 (aldehyde dehydrogenase 1, isoform A1) is the primary ALDH isoform mediating the ALDEFLUOR phenotype (Ginestier et al., 2007). Consistent with the upregulation of the ALDH1A1 mRNA, NANOG1/NANOGP8 overexpression greatly increased the percentage of ALDEFLUOR-positive MCF7 cells (Figure 7g). Significantly, both NANOG1 and NANOGP8 also increased the abundance of ALDEFLUOR+ Du145 cells (Supplemental Figure S6d).
In addition to the above 4 molecules, NANOG1/NANOGP8 overexpression in MCF7 cells also upregulated, to lower levels, many of the other molecules examined including c-Myc, OCT-4, SOX2, CD44, c-KIT, MMP9, TWIST, and SNAIL (Figure 7e).
Discussion
NANOGP8-expressing PCa cells possess CSC properties
In the present study, we first utilize differential qRT-PCR analysis to show that cultured PCa (and some other cancer) cells, and xenograft as well as patient tumors all predominantly express NANOGP8 mRNA with NANOG1 mRNA generally below the detection threshold. These results corroborate our earlier preliminary report (Jeter et al., 2009) and are consistent with NANOG1 being restricted to pluripotent cells and downregulated during differentiation (Chambers et al., 2003; Chambers and Tomlinson, 2009). NANOG1 gene in PCa cells such as PC3 seems to be epigenetically silenced as treatment with TSA, a histone deacetylase inhibitor, but not 5′-aza-deoxycytidine, a DNA methytransferase inhibitor, partially ‘restores’ NANOG1 expression (Jeter et al, 2009). It is intriguing that somatic cancer cells, rather than reactivating the endogenous NANOG1 locus, express NANOG from the NANOGP8 locus. We are currently investigating how the NANOGP8 retrogene is transcriptionally activated in cancer cells.
Knocking down endogenous NANOG in cancer cells significantly diminishes their tumorigenic capacity (Jeter et al., 2009). Since tumor initiation is one of the defining characteristics of CSCs, our loss-of-function results (Jeter et al., 2009) suggest that NANOGP8-expressing cancer cells might possess CSC properties, This premise is born out in the present promoter-tracking studies: NANOGP8-expressing PCa cells are intrinsically unique in comparison to isogenic non-expressing cells, exhibiting enhanced clonogenicity in vitro and tumorigenicity in vivo. Notably, NANOGP8-GFP PCa cells can regenerate tumors that contain GFP- cells as the majority, suggesting that NANOGP8+ PCa cells can self-renew in vivo.
NANOG overexpression promotes CSC phenotypes and properties in vitro and in vivo
Our current NANOG gain-of-function studies further complement the earlier knockdown findings (Jeter et al., 2009) and indicate that NANOG overexpression is sufficient to confer cancer cells certain CSC properties and phenotypes. Thus, NANOG induction promotes clonogenic survival in LNCaP cells, drug resistance in MCF7 cells, and, most importantly, tumor development in LNCaP and Du145 cells. Accompanying the manifestation of such biological properties of CSCs, NANOG overexpression enhances the expression of many CSC–associated molecules, including, among others, CD133, ABCG2, ALDH1A1, and CD44.
NANOG overexpression increases CD133 expression in both LNCaP and MCF7 cells. NANOG expression has been reported to be enriched in CD133+ cells in a variety of tumors, including those in the brain (Heddleston et al., 2009; Po et al., 2010; Zbinden et al., 2010), oral mucosa (Chiou et al., 2008) and liver (Machida et al., 2009). Of particular relevancy, CD133 has been proposed to enrich for human prostatic CSCs (Collins et al., 2005) and we have detected elevated levels of NANOG mRNA in CD133+ human PCa cells (Jeter et al., 2009). We have also observed that CD133+ LAPC4 PCa cells possess higher sphere-forming and tumor-initiating capacities than CD133- LAPC4 cells (Patrawala and Tang, unpublished observations). Curiously, CD133 also appears to be preferentially expressed in transient, reversible drug tolerant subsets of PC9 non-small cell lung carcinoma cells that represent only ∼2% of the parental population (Sharma et al., 2010).
NANOG induction also increases ABCG2 mRNA expression in both LNCaP and MCF7 cells. ABCG2 is a membrane transporter that effluxes toxicants and mediates the side-population (SP) phenotype. Both MCF7 (Engelmann et al., 2008) and LAPC9 PCa (Patrawala et al., 2005) SP cells are enriched in tumor-initiating cells and NANOG mRNA is enriched in the MCF7 SP (Jeter et al., 2009). Reciprocally, ABCG2 has been proposed to mark normal prostate SCs and prostate CSCs (Huss et al., 2005) and prospectively purified ABCG2+ PCa cells consistently demonstrate ∼10× higher tumorigenic potential than the corresponding ABCG2- cells (Patrawala et al., 2005). NANOG overexpression further induces the expression of ALDH1A1, another detoxification protein, in MCF7 cells. ALDH1A1 is the primary ALDH isoform mediating the ALDEFLUOR phenotype, which has been shown to enrich for normal SCs as well as CSCs (Ginestier et al., 2007) including prostate CSCs (Li et al., 2010; van den Hoogen et al., 2010). Accompanying the increase in ALDH1A1 mRNA levels, NANOG dramatically increases the abundance of ALDEFLUOR-hi MCF7 and Du145 cells. Prospectively purified ALDEFLUOR-hi Du145, PC3, and LAPC9 cells all possess much higher tumor-initiating capacities in limiting dilution tumor assays than the corresponding ALDEFLUOR-lo cells (Liu et al., unpublished observations).
Finally, NANOG induces CD44 mRNA in some cancer cells. CD44, individually or in combination with other molecules, is one of the most widely used CSC markers. For example, the first report on solid tumor CSCs in human breast cancer used the CD44+CD24-/lo profile (Al-Hajj et al., 2005). We have shown that the CD44+ PCa cell population is highly enriched in tumor-initiating and metastatic cells (Patrawala et al., 2006, 2007). Knocking down of CD44 significantly inhibits tumor regeneration and/or metastasis in multiple PCa models (Liu et al., 2011). Notably, NANOG seems to be enriched in CD44+ breast (Bourguignon et al., 2008), ovarian (Zhang et al., 2008), and prostate (Jeter et al., 2009; Klarmann et al., 2009) cancer cells.
Simultaneous induction by NANOG of such CSC markers/molecules as CD133, CD44, ABCG2, and ALDH1A1 has likely contributed to the NANOG-conferred CSC phenotypes/properties and further supports NANOG as an important regulator of CSCs.
NANOG overexpression promotes AI phenotypes and castration-resistant PCa regeneration
One of the most significant findings from our study is that NANOG expression is associated with AI and castration resistance in PCa cells. The first hint for such an association comes from the promoter-tracking studies, in which we find that NANOGP8-expressing LNCaP cells possess extended and much enhanced survivability in androgen-deprived conditions. Subsequent gain-of-function studies provide convincing evidence that NANOG overexpression promotes clonogenicity and tumor regeneration of LNCaP cells in androgen-deprived conditions/hosts. These results suggest that NANOG might contribute to PCa progression to a hormone-refractory state. In support, whole-genome cDNA microarray analysis shows that NANOG is preferentially expressed in PCa cells that express low levels of PSA, the population naturally resistant to androgen blockade (Qin et al., unpublished observations). In further support, NANOG overexpression also enhances tumor regeneration in AR- and AI Du145 cells.
What mechanisms might be responsible for NANOG-conferred AI phenotypes and castration resistance? One mechanism is likely related to NANOG-promoted CSC phenotypes and properties discussed above. Prostate CSCs seem to express low levels of AR and thus are intrinsically less dependent on AR signaling (Collins et al., 2005; Li et al., 2009; Patrawala et al., 2006). In support of this connection, we have previously reported reciprocal nuclear expression between AR and NANOG (Jeter et al., 2009) and we have shown here that NANOG-overexpressing LNCaP cells express lower levels of AR, especially in tumors developed in castrated hosts.
NANOG promotes the expression of other molecules such as Bcl-2, IGFBP-5 and CXCR4, which might also be involved in NANOG-mediated castration resistance in LNCaP cells. Bcl-2 is an anti-apoptotic protein and its overexpression has been strongly implicated in the development of castration-resistant PCa (McDonell et al., 1992). IGFBP-5 mRNA and protein are upregulated in response to castration and exogenous IGFBP-5 stimulates the proliferation of immortalized prostate epithelial cells (Xu et al., 2007). Also, overexpression of IGFBP-5 accelerates the proliferation of LNCaP cells in androgen-deprived conditions in a PI3K-dependent manner (Miyake et al., 2000). IGFBP-5 is known to regulate IGF-1 receptor signaling, which has been implicated in LNCaP (and LAPC-9) PCa progression to androgen independence (Nickerson et al., 2001) and is required for the emergence of the CD133+ drug-resistant PC9 cells (Sharma et al., 2010). Finally, CXCR4 and its cognate chemokine CXCL12 have been implicated in PCa cell migration and invasion (Frigo et al., 2009; Singh et al., 2004). A functional consequence of NANOG-promoted CXCR4 expression may be the enhanced migratory capacity of NANOGP8-expressing LNCaP AI tumor cells.
The mechanisms underlying the NANOG-mediated castration resistance are likely multifaceted and our ongoing research seeks to further address whether some of these CSC and AI-associated molecules represent direct NANOG targets and exactly how NANOG promotes PCa AI progression.
NANOGP8 vs. NANOG1: Similarities and distinctions
Throughout the preceding discussions, we mostly used the term NANOG to ‘generically’ refer to the proteins encoded by both NANOG1 and NANOGP8, mainly because the predicted NANOG1 and NANOGP8 proteins are ∼99% identical (Jeter et al., 2009). Multiple lines of evidence support the overt similarities between NANOG1 and NANOGP8 proteins. FIRST, most currently used commercial anti-NANOG Abs are raised against NANOG1 but generally recognize the recombinant NANOGP8 proteins equally well (Jeter et al., 2009). SECOND, both NANOG1 and NANOGP8 are induced, remarkably, to very similar levels in cancer cells (Figure 3b-c). It is worth mentioning that the induced NANOG1 and NANOGP8 are both at much lower levels than endogenous NANOG1 levels in N-TERA cells (Figure 7b-c), suggesting that somatic cancer cells might not be able to tolerate very high levels of NANOG as a result of unique biochemical mechanisms regulating NANOG turnover. THIRD, importantly, NANOG1 and NANOGP8 demonstrate similar biological effects in many experimental settings. For example, both NANOG1 and NANOGP8 promote castration-resistant LNCaP tumor development (Figure 5f-g), increase c-Myc and decrease AR in AI tumors (Figure 6a-c), and enhance the expression of many same genes such as CD133, IGFBP-5, and ABCG2 (Figure 6e-f; Figure 7e).
On the other hand, NANOGP8 is the predominant ‘isoform’ expressed in cancer cells and may, therefore, have evolved new functions distinct from those of NANOG1 in ESCs. In support, we have observed, in most assays, much more consistent biological effects with NANOGP8 overexpression compared to NANOG1 overexpression. For example, only NANOGP8 promotes, in LNCaP cells, AI sphere and AI foci formation, castration-resistant tumor development, and cell migration (Figure 5). Likewise, NANOGP8, but not NANOG1, promotes Du145 tumor development and c-Myc upregulation. At the molecular levels, NANOGP8 and NANOG1 seem to bind to chromatin-associated targets with different affinities (Figure 4). Moreover, NANOGP8 and NANOG1 frequently induce different gene alterations even in the same cell types maintained under the identical conditions (Figure 6e-f; Figure 7e). Finally, whole-genome ChIP-Seq analysis has revealed that, although NANOG1 and NANOGP8 share ∼75% genomic binding sites, the two proteins demonstrate ∼25% distinct binding sites in the genome (Jeter et al., unpublished observations). Further analysis of the ChIP-Seq data, together with our efforts to elucidate the unique biochemical properties and signaling partners of NANOG in cancer cells and the interrelationship between AR/androgen signaling and NANOG, should greatly advance our understanding of the molecular mechanisms underlying the immortality of cancer cells and development of castration-resistant PCa.
Materials and methods
Primary prostate tumor (HPCa) specimens and tumor processing
HPCa samples were obtained at radical prostatectomy with patients' consent. Tumor pieces, designated as adenocarcinoma by pathological examination and classified as Gleason 6-9, were frozen in liquid nitrogen. For first-round xenotransplantation, small (∼1-2 mm2) tumor pieces were implanted subcutaneously into NOD-SCID-γ mice. Early passage HPCa tumor tissues were subject to enzymatic digestion (type I collagenase, 50 U/ml DNase, 12 h), followed by trypsin digestion and discontinuous Percoll gradient purification. Lineage positive cells including hematopoietic, endothelial, and stromal cells were removed using an antibody cocktail and MACS (Miltenyi Biotec, Auburn, CA, USA) as previously described (Jeter et al., 2009).
Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from cultured cancer cells or tumor pieces (frozen in liquid nitrogen) using an RNeasy RNA-purification kit (Qiagen, Valencia, CA, USA). The ABI High-Capacity cDNA Archive Kit (Applied Biosystems, Carlsbad, CA, USA) and random hexamers were used for cDNA synthesis. Quantitative PCR was performed by the M.D. Anderson Science-Park Molecular Biology Core Facility using an ABI Prism 7900HT (Applied Biosystems). File Builder 3.1 software (Applied Biosystems) was used to design NANOG1 and NANOGP8 specific primers and probes (Supplemental Table S1) required to discriminate between RNA species arising from either locus. A panel of gene-specific primer pairs was also used for further expression profiling by the SYBR® Green method (Supplemental Table S1). The experimental Ct (cycle threshold) was calibrated against that of GAPDH control product. All amplifications were performed in duplicate. The ΔΔCt method was used to determine the amount of NANOG product relative to that expressed by the most benign PCa line, LNCaP (1-fold, 100%). For comparative gene expression profiling of pLVX cell lines, the RNA levels of a particular gene in the pLVX-NANOG1/P8 cells were presented relative to the pLVX control cells (1-fold, 100%).
Lentiviral constructs, packaging and transduction
The ∼3.8-kb human NANOGP8 promoter fragment was amplified from LNCaP cell genomic DNA using the 5′ primer CTC GAG CAT AGC TGC ATT GGC AAA GA, incorporating a XhoI restriction site, and the 3′ primer GGA TCC ATG AGG CAA CCA GCT CAG TC, incorporating a BamHI restriction site,, subcloned into pCR2.1 (Invitrogen, Carlsbad, CA, USA), and inserted into RRL-PGK-GFP1 (Zufferey et al., 1998) as a XhoI – BamH1 fragment before the GFP transgene to generate RRL-NANOGP8-GFP. The RRL-based lentivirus was prepared as previously described (Jeter et al., 2009). In brief, G418 selected 293FT cells (12×106/15 cm dish) were transfected with the RRE (6 μg), REV (4 μg) and VSVg (4 μg) packaging plasmids, along with a lentiviral vector (6 μg) using Fugene 6 at a 1:2.7 ratio of DNA (μg) to transfection reagent (μl). Viral supernatants were ultracentrifuged to produce concentrated viral stocks. Cells were plated 24 h earlier and infected with the indicated volumes of virus, prepared in parallel, at approximately 50% cell density.
To create the pLVX-TetON-NANOG constructs, the NANOG1 cDNA from N-TERA cells or NANOGP8 coding region derived from HPCa5 (Jeter et al., 2009) in pCR2.1 (Invitrogen) was subcloned into the pLVX-TetON expression vector (Clontech, Mountain View, CA, USA) via 5′ NotI and 3′ MluI restriction enzyme excision and ligation. All newly generated lentiviral vectors were characterized by restriction digestion and sequencing. The pLVX-based lentivirus was prepared according to the Lenti-X Tet-ON Advanced Inducible Expression System (Clontech) manufacturer's protocol and by using the Lenti-X packaging system.
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed by following the manufacturer's instructions (Upstate, Charlottesville, VA, USA). Cultured pLVX LNCaP cells (i.e., cells expressing pLVX, pLVX-Nanog1, or pLVX-NanogP8) were treated with doxycycline (500 ng/ml) for 3-4 d prior to harvest. After cells were fixed, washed, and collected, their DNA was sheared by sonication in 200 μl of lysis buffer. Then, 20 μl of sonicated DNA from each sample was purified and used as an input DNA control and the remainder of the DNA was pre-cleared. The chromatin-bound DNA was immunoprecipitated with an anti-NANOG antibody (Santa Cruz, Santa Cruz, CA, USA; H-155) and protein-A beads, and the DNA was eluted from the beads. The precipitated DNA was analyzed by PCR for 1 cycle at 95°C for 2 min; 35 cycles at 95°C for 20 s, 55°C for 20 s, and 72°C for 30 s; and 1 cycle at 72°C for 10 min. The PCR primers (see Supplemental Figure S4) included distal E-cadherin promoter (-2273 to -1889 bp, 5′-atcagagaggttggttccagtcc-3′ and 5′-gcaatggtgtgttcttggctcac-3′), FGF4 promoter (-1567 to -1250 bp, 5′-caacacagcaagtgagggatgg-3′ and 5′-ttgagggctcttctctgaggtg-3′), Gli1 promoter (-2991 to -2645 bp, 5′-ctttccattctgccgtcttcag-3′ and 5′-cgctttacccatctcccaacac-3′), Hoxc13 promoter (-1116 to -781 bp, 5′-ctgaatctcccccattttccac-3′ and 5′-acactctgctttacctcgctgg-3′), Oct4 promoter (-1928 to -1519 bp, 5′-cagacagacaaacatcatccctcac-3′ and 5′-tcacacgagatccccttcagagac-3′), c-Myc promoter (-1303 to -968 bp, 5′-gaagcctgagcaggcggggcagg-3′ and 5′-gctttgatcaagagtcccag-3′), and GAPDH promoter (5′-actctgctctgggtggtcattg-3′ and 5′-gggtgctgaacacttgtaaggaag-3′).
Supplementary Material
Acknowledgments
We thank P. Whitney for FACS, N. Otto and the Histology Core for help in IHC, J. Moore for assistance in animal breeding and experiments, and other members of the Tang lab for support and helpful discussions. This work was supported, in part, by grants from NIH (R01-ES015888, R21-ES015893, R21-CA150009), Department of Defense (W81XWH-07-1-0616, W81XWH-08-1-0472), and Elsa Pardee Foundation (D.G.T) and by two Center Grants (CCSG-5 P30 CA016672-34 and ES007784). C. Jeter and C. Liu were supported in part by fellowships from AUA Foundation and DOD, respectively.
Abbreviations
- AD
androgen dependent
- AI
androgen independent
- CE
cloning efficiency
- ChIP
chromatin immunoprecipitation
- CSC
cancer stem cells
- dox
doxycycline
- DP
dorsal prostate
- EMT
epithelial mesenchymal transition
- ESCs
embryonic stem cells
- HPCa
human prostate cancer patient samples
- NOD/SCID
non-obese severe combined immunodeficient mice
- PCa
prostate cancer
- qRT-PCR
quantitative reverse-transcription polymerase chain reaction
- s.c
subcutaneous
- SP
side population
- TSS
transcription start site
- UTR
untranslated region
Footnotes
Conflict of Interest: Authors declare no conflict of interest.
Supplementary Information, including Supplemental Methods, Supplemental References, Supplemental Figure Legends, six Supplemental Figures, and three Supplemental Tables accompanies the paper on the ONCOGENE website (http;//www.nature.com/onc).
References
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466:133–7. doi: 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth HA, Holland PW. Eleven daughters of NANOG. Genomics. 2004;84:229–38. doi: 10.1016/j.ygeno.2004.02.014. [DOI] [PubMed] [Google Scholar]
- Bourguignon LY, Peyrollier K, Xia W, Gilad E. Hyaluronan-CD44 iN-TERAction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J Biol Chem. 2008;283:17635–51. doi: 10.1074/jbc.M800109200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–55. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- Chambers I, Tomlinson SR. The transcriptional foundation of pluripotency. Development. 2009;1362:311–22. doi: 10.1242/dev.024398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou SH, Yu CC, Huang CY, Lin SC, Liu CJ, Tsai TH, et al. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res. 2008;14:4085–95. doi: 10.1158/1078-0432.CCR-07-4404. [DOI] [PubMed] [Google Scholar]
- Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–51. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- Engelmann K, Shen H, Finn OJ. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. 2008;68:2419–26. doi: 10.1158/0008-5472.CAN-07-2249. [DOI] [PubMed] [Google Scholar]
- Frigo DE, Sherk AB, Wittmann BM, Norris JD, Wang Q, Joseph JD, et al. Induction of Kruppel-like factor 5 expression by androgens results in increased CXCR4-dependent migration of prostate cancer cells in vitro. Mol Endocrinol. 2009;23:1385–96. doi: 10.1210/me.2009-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009;27:40–8. doi: 10.1634/stemcells.2008-0493. [DOI] [PubMed] [Google Scholar]
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–67. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle. 2009;8:3274–84. doi: 10.4161/cc.8.20.9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–77. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- Huss WJ, Gray DR, Greenberg NM, Mohler JL, Smith GJ. Breast cancer resistance protein-mediated efflux of androgen in putative benign and malignant prostate stem cells. Cancer Res. 2005;65:6640–50. doi: 10.1158/0008-5472.CAN-04-2548. [DOI] [PubMed] [Google Scholar]
- Jeter CR, Badeaux M, Choy G, Chandra D, Patrawala L, Liu C, et al. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells. 2009;27:993–1005. doi: 10.1002/stem.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarmann GJ, Hurt EM, Mathews LA, Zhang X, Duhagon MA, Mistree T, et al. Invasive prostate cancer cells are tumor initiating cells that have a stem cell-like genomic signature. Clin Exp Metastasis. 2009;26:433–46. doi: 10.1007/s10585-009-9242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chen X, Calhoun-Davis T, Claypool K, Tang DG. PC3 human prostate carcinoma cell holoclones contain self-renewing tumor-initiating cells. Cancer Res. 2008;68:1820–5. doi: 10.1158/0008-5472.CAN-07-5878. [DOI] [PubMed] [Google Scholar]
- Li H, Jiang M, Honorio S, Patrawala L, Jeter CR, Calhoun-Davis T, et al. Methodologies in assaying prostate cancer stem cells. Methods Mol Biol. 2009;568:85–138. doi: 10.1007/978-1-59745-280-9_7. [DOI] [PubMed] [Google Scholar]
- Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011 Jan 16; doi: 10.1038/nm.2284. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Su Y, Mei Y, Leng Q, Leng B, et al. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients' outcome. Lab Invest. 2010;90:234–44. doi: 10.1038/labinvest.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Clem B, Zuba-Surma EK, El-Naggar S, Telang S, Jenson AB, et al. Mouse fibroblasts lacking RB1 function form spheres and undergo reprogramming to a cancer stem cell phenotype. Cell Stem Cell. 2009;4:336–47. doi: 10.1016/j.stem.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Wang Z, Guo Y, Pei D. The C-terminal pentapeptide of Nanog tryptophan repeat domain iN-TERActs with Nac1 and regulates stem cell proliferation but not pluripotency. J Biol Chem. 2009;284:16071–81. doi: 10.1074/jbc.M109.005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida K, Tsukamoto H, Mkrtchyan H, Duan L, Dynnyk A, Liu HM, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci USA. 2009;106:1548–53. doi: 10.1073/pnas.0807390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, et al. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 1992;52:6940–4. [PubMed] [Google Scholar]
- Medeiros RB, Papenfuss KJ, Hoium B, Coley K, Jadrich J, Goh SK, et al. Novel sequential ChIP and simplified basic ChIP protocols for promoter co-occupancy and target gene identification in human embryonic stem cells. BMC Biotechnol. 2009;9:59. doi: 10.1186/1472-6750-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng HM, Zheng P, Wang XY, Liu C, Sui HM, Wu SJ, et al. Overexpression of nanog predicts tumor progression and poor prognosis in colorectal cancer. Cancer Biol Ther. 2010;9 doi: 10.4161/cbt.9.4.10666. [DOI] [PubMed] [Google Scholar]
- Miyake H, Nelson C, Rennie PS, Gleave ME. Overexpression of insulin-like growth factor binding protein-5 helps accelerate progression to androgen-independence in the human prostate LNCaP tumor model through activation of phosphatidylinositol 3′-kinase pathway. Endocrinology. 2000;141:2257–65. doi: 10.1210/endo.141.6.7520. [DOI] [PubMed] [Google Scholar]
- Nickerson T, Chang F, Lorimer D, Smeekens SP, Sawyers CL, Pollak M. In vivo progression of LAPC-9 and LNCaP prostate cancer models to androgen independence is associated with increased expression of insulin-like growth factor I (IGF-I) and IGF-I receptor (IGF-IR) Cancer Res. 2001;61:6276–80. [PubMed] [Google Scholar]
- O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Patrawala L, Calhoun T, Schneider-Broussard R, Li H, Bhatia B, Tang S, et al. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene. 2006;25:1696–708. doi: 10.1038/sj.onc.1209327. [DOI] [PubMed] [Google Scholar]
- Patrawala L, Calhoun T, Schneider-Broussard R, Zhou J, Claypool K, Tang DG. Side population is enriched in tumorigenic, stem-like cancer cells, whereas ABCG2+ and ABCG2- cancer cells are similarly tumorigenic. Cancer Res. 2005;65:6207–19. doi: 10.1158/0008-5472.CAN-05-0592. [DOI] [PubMed] [Google Scholar]
- Patrawala L, Calhoun-Davis T, Schneider-Broussard R, Tang DG. Hierarchical organization of prostate cancer cells in xenograft tumors: the CD44+alpha2beta1+ cell population is enriched in tumor-initiating cells. Cancer Res. 2007;67:6796–805. doi: 10.1158/0008-5472.CAN-07-0490. [DOI] [PubMed] [Google Scholar]
- Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Piestun D, Kochupurakkal BS, Jacob-Hirsch J, Zeligson S, Koudritsky M, Domany E, et al. Nanog transforms NIH3T3 cells and targets cell-type restricted genes. Biochem Biophys Res Commun. 2006;343:279–85. doi: 10.1016/j.bbrc.2006.02.152. [DOI] [PubMed] [Google Scholar]
- Po A, Ferretti E, Miele E, De Smaele E, Paganelli A, Canettieri G, et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. 2010;29:2646–58. doi: 10.1038/emboj.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Singh UP, Grizzle WE, Lillard JW., Jr CXCL12-CXCR4 iN-TERActions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab Invest. 2004;84:1666–76. doi: 10.1038/labinvest.3700181. [DOI] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- van den Hoogen C, van der Horst G, Cheung H, Buijs JT, Lippitt JM, et al. High aldehyde dehydrogenase activity identifies tumor-initiating and metastasis-initiating cells in human prostate cancer. Cancer Res. 2010;70:5163–73. doi: 10.1158/0008-5472.CAN-09-3806. [DOI] [PubMed] [Google Scholar]
- Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–44. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Graf LF, Fazli L, Coleman IM, Mauldin DE, Li D, et al. Regulation of global gene expression in the bone marrow microenvironment by androgen: androgen ablation increases insulin-like growth factor binding protein-5 expression. Prostate. 2007;67:1621–9. doi: 10.1002/pros.20655. [DOI] [PubMed] [Google Scholar]
- Zbinden M, Duquet A, Lorente-Trigos A, Ngwabyt SN, Borges I, Ruiz i Altaba A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010;29:2659–74. doi: 10.1038/emboj.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wang X, Chen B, Suo G, Zhao Y, Duan Z, et al. Expression of Nanog gene promotes NIH3T3 cell proliferation. Biochem Biophys Res Commun. 2005;338:1098–102. doi: 10.1016/j.bbrc.2005.10.071. [DOI] [PubMed] [Google Scholar]
- Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311–20. doi: 10.1158/0008-5472.CAN-08-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Neganova I, Przyborski S, Yang C, Cooke M, Atkinson SP, et al. A role for NANOG in G1 to S transition in human embryonic stem cells through direct binding of CDK6 and CDC25A. J Cell Biol. 2009;184:67–82. doi: 10.1083/jcb.200801009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873–80. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.