

Abstract
The evolutionarily conserved stress-inducible HSP70 molecular chaperone plays a central role in maintaining protein quality control in response to various forms of stress. Constitutively elevated HSP70 expression is a characteristic of many tumor cells and contributes to their survival. We recently identified the small-molecule 2-phenylethyenesulfonamide (PES) as a novel HSP70 inhibitor. Here we present evidence that PES-mediated inhibition of HSP70-family proteins in tumor cells results in an impairment of the two major protein degradation systems, namely the autophagy-lysosome system as well as the proteasome pathway. HSP70-family proteins work closely with the HSP90 molecular chaperone to maintain the stability and activities of their many client proteins and PES causes a disruption in the HSP70/HSP90 chaperone system. As a consequence, many cellular proteins, including known HSP70/HSP90 substrates, accumulate in detergent-insoluble cell fractions, indicative of aggregation and functional inactivation. Overall, PES simultaneously disrupts several cancer-critical survival pathways, supporting the idea of targeting HSP70 as a potential approach for cancer therapeutics.
Keywords: HSP70, HSP90, HSC70, PES, autophagy, proteasome, lysosome
INTRODUCTION
Cell survival relies on protein quality control systems to assure the proper folding, localization and turnover of proteins. While some level of protein misfolding occurs in all cells, the frequency of such events increases in response to various environmental and physiologic stresses. Misfolded proteins can impair function, promote inappropriate interactions among macromolecules, and can lead to the formation of potentially toxic aggregates (1–3). Relative to their normal counterparts, tumor cells generally exhibit a greater "stress phenotype" associated with altered nutrient availability and energy metabolism, modified signaling pathways, dysregulated growth, replicative stress, an accumulation of mutant proteins and/or oncogene activation. As such, transformed cells are particularly dependent on quality control networks to cope with these adverse conditions. Important factors in this system are molecular chaperones, including the evolutionarily conserved families of heat shock, or stress, proteins (HSPs). Classified according to molecular weight, families of molecular chaperones generally comprise multiple members that may exhibit different expression patterns or reside in different cellular compartments (4–6). In mammalian cells the HSP70-family has more than eight members, and includes the major cytoplasmic forms called HSC70 (heat shock cognate 70, HSP73, HSPA8) and HSP70 (HSP72, HSPA1A/A1B), as well as the mitochondrial protein GRP75 and the endoplasmic-reticulum-localized GRP78 (5, 7, 8).
HSC70 is a constitutively and abundantly expressed protein, while the very closely related HSP70 generally is found at very low or undetectable levels in unstressed normal cells. However, in response to a variety of exogenous and physiologic stresses, the HSP70 gene is rapidly upregulated transcriptionally to generate more protein. The broad quality-control activities of HSP70 and HSC70 include aiding with the proper folding, cellular localization, and assembly of its more than one-hundred client proteins. They also mediate the proper folding of nascent proteins, the refolding of misfolded proteins and the formation and disassembly of multi-protein complexes (5–8). The activities of these molecular chaperones are closely coordinated with the two major protein degradation systems in mammalian cells, the autophagy-lysosome and proteasome pathways, to aid in the elimination of conformationally-altered, or otherwise unwanted, proteins and other macromolecules (3, 9).
Of note, most cancer cells exhibit elevated basal expression of HSP70, consistent with the idea that this molecular chaperone represents a "cancer critical" survival factor that helps to maintain protein homeostasis in the presence of enhanced stress. The elevated expression of the stress-inducible HSP70 protein in cancer cells has been implicated in disease progression, chemotherapy resistance and generally poor patient prognosis (4, 6, 7, 10). It is likely that HSP70 contributes in several ways to the biology of tumors, including as a regulator of apoptosis, and as a mediator of lysosome function and pathways of autophagy. For example, HSP70 promotes the survival of cancer cells by enhancing the integrity of the lysosome membrane (11, 12). The actions and regulation of HSP70-family proteins involve critical and dynamic associations with several co-chaperones, together acting in multi-protein complexes. It is well-recognized, for example, that HSP70 and HSC70 act as key co-chaperones for the HSP90 machinery, at least in part by aiding in client protein recruitment. HSP90 is a 90-kDa molecular chaperone that is abundantly expressed in normal cells, and its expression and activity are upregulated in many tumors (13–15). Its actions regulate the stability, activity and conformational maturation of a very large number and wide variety of client proteins, including certain steroid hormone receptors, kinases, and intracellular signaling molecules. Like HSP70, HSP90 plays an important role in helping to maintain a transformed phenotype in cancer cells. Indeed, HSP70/HSP90 client proteins include many oncogenic proteins that are mutated or over-expressed in cancers, including p53, AKT, HER2/ ERBB2 and EGFR. The HSP70 and HSP90 chaperones also work together to target certain client proteins for degradation by the ubiquitin-proteasome system.
The dependence of cancer cells on the multiple activities of HSP70 and HSP90 have made these molecular chaperones attractive targets for cancer therapy. Over the past decade, several HSP90 inhibitors have been identified (10, 14–16). The use of these compounds has improved the understanding of HSP90 biology, and a few of these are now under clinical evaluation for treating certain forms of cancer. One of the molecular signatures of many HSP90 inhibitors is an upregulation of the anti-apoptotic and cytoprotective HSP70 protein, a consequence that is believed to reduce the overall anti-tumor efficacy of these compounds. This observation has added to a growing interest in HSP70 as a potential anti-cancer target. The identification of small-molecule modulators of HSP70 would be expected not only to promote a better understanding of the diverse actions of this molecular chaperone in normal and transformed cells, but also could provide new avenues for the development of effective anti-cancer strategies. Targeting of the HSP70-family of proteins has proved challenging, however, and few selective small molecule inhibitors, and no drug-like molecules, are yet available to help in assessing the potential of HSP70 as a therapeutic target in cancer cells (10, 17–23). One recently identified inhibitor is 2-phenylethynesulfonamide, here referred to as "PES" (24). PES (also called phenylacetylenylsulfonamide or pifithrin-mu) was originally identified in a screen for molecules that would impair the mitochondrial localization of p53 (25). Our previous work on PES revealed that it acts as a direct inhibitor of the stress-inducible HSP70. It is preferentially cytotoxic to a broad range of solid tumor cell types, regardless of p53 status or elevated expression of the anti-apoptotic BCL-xL protein and, at least in solid tumors, does not depend on caspase activation (23, 24). Rather, PES-mediated cell death in solid tumor cell lines involves an impairment of the autophagy-lysosome system of macromolecule degradation and is characterized by an accumulation of misfolded and aggregated proteins (24). We also determined that PES is able to prolong survival in a mouse model of Myc-induced lymphomagenesis, without obvious toxicity, indicating that it has potential to be developed for therapeutic use (24). Accordingly, we have further explored the cellular consequences of PES-mediated HSP70 inhibition in tumor cells. Here we provide evidence that, in addition to inhibiting the autophagy-lysosome system, PES treatment leads to reduced cellular proteasome activity, thereby impairing the two major protein degradation systems in mammalian cells. The PES-mediated inhibition of HSP70-family proteins indirectly alters the activities of its co-chaperone HSP90. As a consequence, PES exhibits a unique mechanism of anti-cancer action: PES both inhibits autophagic flux and it promotes the accumulation of proteins, including HSP90 clients, in a detergent-insoluble form, consistent with aggregation and inactivation.
MATERIALS and METHODS
Cell culture
Tumor cell lines used were SKBR3 and MDA-MB-468human breast carcinomas, H1299 human non-small cell lung cancer, SKOV3 human ovarian cancer, and FaDu human head and neck cancer. We also employed human IMR90 and WI38 immortalized human fibroblast cells, and MCF10A immortalized human mammary epithelial cells. These cell lines were originally obtained from the American Type Culture Center and grown in DMEM with 10% fetal bovine serum and penicillin/streptomycin, except MCF10A, which were grown as recommended by ATCC in maintained in medium supplemented with 5% heat-inactivated horse serum, 10 µg/mL of insulin, 20 ng/mL of EGF, 0.1 µg/mL of cholera toxin, 0.5 µg/mL of hydrocortisone. HOI118, which are LTAg-immortalized human ovarian surface epithelial cells, were kindly provided by Rugang Zhang (Fox Chase Cancer Center). Our internal authentication has been performed by monitoring growth rates and routinely tracking changes in morphology.
Chemicals and Antibodies
17-allylamino-17-demethoxygeldanamycin (17-AAG) and 2-phenylethyenesulfonamide (PES; also called pifithrin-µ) were purchased from EMD Chemicals Inc. (Gibbstown, NJ). Bortezomib (Velcade) was from LC Laboratories (Woburn, MA). Chloroquine was purchased from Sigma-Aldrich Corp. (St. Louis, MO). The following primary antibodies were used in this work: anti-CHIP, anti-HSP90, anti-HSP40, anti-CHIP, anti-HSP70, anti-Integrin β1, anti-EGFR, anti-HER2/ErbB2, anti-AKT, anti-mTOR, anti-IκBα, anti-HOP and anti-LC3 (Cell Signaling Technology, Inc., Danvers, MA); anti-p53 (AB-6) (EMD Chemicals, Inc., Gibbstown, NJ); anti-p23, anti-HSC70 (sc-71270) and anti-p62/SQSTM1 (D-3, sc-28359) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA); anti-GRP78, anti-MCL1, and anti-BCL-xL (BD Biosciences, San Jose, CA); anti-HSC70 (SPA-819, Stressgen; currently known as ADI-SPA-891, Enzo Life Sciences, Plymouth Meeting, PA); and anti-HSC70 (ab51052, Abcam Inc., Cambridge, MA). Both anti-HSC70 (sc-71270 and SPA-819) antibodies detected an interaction between PES and HSC70 in vivo. The peroxidase-conjugated secondary antibodies (i.e., donkey anti-rabbit, donkey anti-mouse, and donkey anti-goat) were from Jackson ImmunoResearch Laboratories, Inc.
Immunoblotting, immunoprecipitation, and proteasome assay
Immunblotting and immunoprecipitation studies were performed as detailed (24). Detergent (1% NP40)-soluble and detergent-insoluble cell fractions were prepared as detailed previously (24). Measurements of in vivo 20S proteasome activity were performed using cell lysates and the 20S Proteasome Substrate (SUC-LLVY-AMC) as recommended by the manufacturer (Cayman Chemical Company, Ann Arbor, MI).
Animal Studies
The nu/nu athymic female mice were obtained from Taconic Farms, Inc. All experiments were approved by, and conformed to, the guidelines of the Institutional Animal Care and Use Committee of the Fox Chase Cancer Center. For these studies, suspensions of FaDu head and neck squamous cell carcinoma (5×106 cells) were implanted subcutaneously into the flanks of 5–6 week old female nude mice. Tumors reaching a diameter of approximately 5–7 mm were injected intratumorally with 40 mg/kg of PES prepared in 0.1 ml of 20% DMSO solution or with 0.1 ml of 20% DMSO solution. At 24 or 48 hr, tumors were harvested; half of each was fixed in formalin, and half was snap frozen for protein lysates. Tumor lysates and formalin-fixed sections were analyzed for markers of target modulation of HSP70 inhibition, including p62 accumulation and aggregation.
RESULTS
PES Interacts with HSP70 and HSC70 Proteins
We identified HSP70 as a PES-interacting protein in pull-down assays using lysates of cells treated with a biotin-tagged version of this compound, termed B-PES (24). Briefly, PES and its associated proteins were cleaved from the biotin tag coupled to the NeutrAvidin resins, resolved by SDS-PAGE, and visualized by Coomassie staining. The results of this gel analysis revealed only a single prominent band of about 70 kDa, indicating that PES specificity in vivo is highly restricted. The interaction between endogenous HSP70 and PES was confirmed by immunoblotting with an antibody specific for this protein (ref. 24 and data shown below). Moreover, we now show that interaction between B-PES and HSP70 can be competed away with excess untagged PES, providing additional evidence for specificity (Fig. 1A).
Figure 1. PES binds to HSP70 and HSC70.
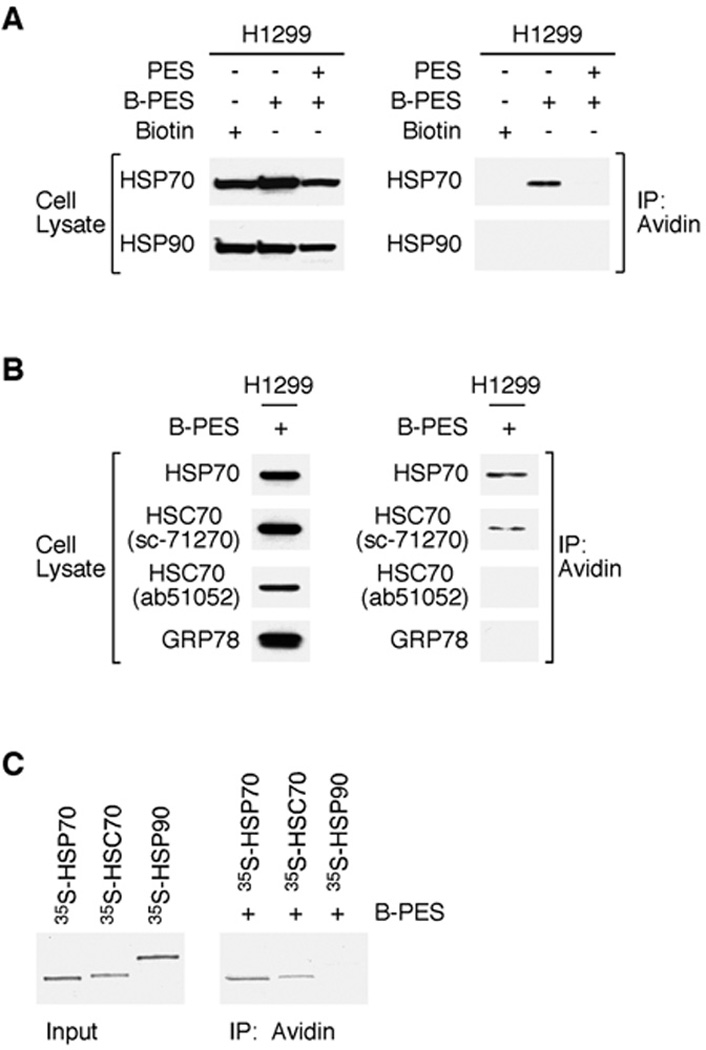
A, Competition supports PES as an HSP70-binding protein. H1299 cells were either pretreated with DMSO or excess (25X) untagged PES for 1 h prior to the addition of 20 µM Biotin-PES (B-PES) for 5 h, and examined for the expression of HSP70 and HSP90 (left). B-PES containing complexes were captured by NeutrAvidin Resins, and immunoblotted using the indicated antibodies. B, Whole cell extracts (WCE) prepared from H1299 cells treated with B-PES, captured using NeutrAvidin agarose resins, and the DTT eluates were immunblotted with the indicated antibodies following SDS-PAGE. C, In vitro evidence for an interaction between B-PES and HSP70 as well as B-PES and HSC70. Full-length human HSP70, HSC70, and human HSP90 proteins were in vitro translated in the presence of 35S-methionine and mixed with B-PES coupled to NeutrAvidin resins. After cleaving PES and its interacting factors from the biotin tag immobilized to the NeutrAvidin resins, the captured PES protein complexes were separated by SDS-PAGE and visualized by autoradiography.
In our initial efforts to identify PES-interacting proteins, the proteomics analysis identified both HSP70 and HSC70 as potential PES targets (ref. 24 and data not shown). However, previous immunoblotting studies using two anti-HSC70 antibodies (abcam ab51052 and R&D System AF4148) did not provide experimental support for HSC70 as a PES-interacting protein (24). In extending our investigations we now realize that this conclusion, which was based on the use of available antibodies, needed to be re-evaluated and that PES appears also to interact with the constitutively expressed HSC70. Specifically, H1299 human lung tumor cells were treated with a biotin-tagged PES and PES-interacting proteins were captured on NeutrAvidin resins; proteins eluted from the resin were assayed by immunoblotting, as previously detailed. When these protein blots were probed with one of the two HSC70 antibodies used previously (Abcam, ab51052), we again found no evidence for an association of HSC70 with PES (Fig. 1B). However, when we employed two new antibodies to HSC70 (sc-71270 and SPA-819), we could consistently detect an interaction between PES and HSC70 in vivo (Fig. 1B and data not shown). The molecular basis for the discrepant findings obtained using the different anti-HSC70 antibodies is not clear; however, it is of note that the HSC70 antibodies that fail to detect a PES interaction map to epitopes at the C-terminus of this protein, a region that is critical for the interaction with PES (24). In contrast to the findings with HSP70 and HSC70, the evidence does not support an interaction of PES with HSP90 or GRP78 (see Fig. 1 and ref. 24). We have not yet tested for interactions between PES and other HSP70-family members. In support of an interaction between both HSC70 and HSP70 with PES, we also find that in vitro-translated proteins consistently interact with biotinylated PES (Fig. 1C). The combined data support the conclusion that PES interacts with the stress-inducible HSP70 as well as with the structurally- and functionally-related HSC70 protein.
Response of Tumor Cells vs. Non-transformed Cells to PES
We previously found that PES exhibits generally greater toxicity for human tumor cell lines relative to immortalized, non-transformed WI38 fibroblasts (24). The half maximal concentration of PES to exert its cytotoxic effect (IC50) in approximately fifty solid tumor cell lines of different histologic type was determined to be in the range of 4 –10 µM (24). To illustrate, there is a dose-dependent loss of viability for H1299 human lung carcinoma cells as well as SKBR3 and MDA-MB-468 human breast carcinoma cell lines treated with PES. In contrast, exposure to this small molecule did not result in a comparable loss of viability for the immortalized IMR90 and WI38 human lung fibroblasts except at the highest concentration (20 µM) used in this experiment (Fig. 2A). Accordingly, we asked if this differential cytotoxicity might correlate with the interaction of PES with HSP70/HSC70. In cells treated with B-PES, both HSP70 and HSC70 are found together with this small molecule in the tumor cells (Fig. 2B). Interestingly, we found negligible PES interacting with these molecular chaperones in the non-tumorigenic IMR90 cells under these conditions.
Figure 2. Preferential binding of PES to HSP70 and HSC70 in tumor cell lines.
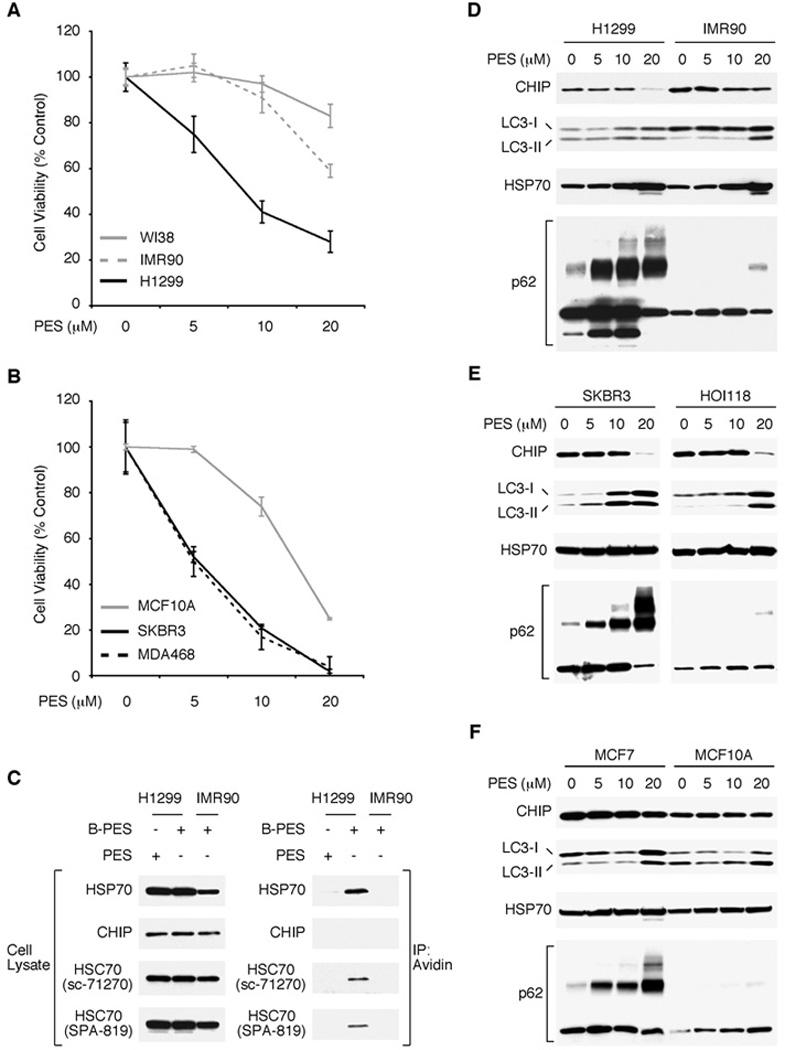
A, B, The cell lines indicated were treated with increasing concentrations of PES for 24 h and cell viability was determined by MTT assays. C, WCE were prepared from the cell lines indicated following 5 hr treatment with 20 µM PES or B-PES and examined for the expression of proteins indicated (left panel) by western blot analysis; note that different exposure times were used to visualize these proteins. B-PES-containing complexes were captured by NeutrAvidin resins, eluted following 50 mM DTT treatment, and immunoblotted using the indicated antibodies. C, D, E, The indicated cell lines (H1299, IMR90, SKBR3, HOI118, MCF7, MCF10A) were treated with the specified amount of PES for 24 h and examined for the proteins indicated.
The HSP70/HSC70 molecular chaperones function in a coordinated manner with proteolytic systems in cells to help maintain protein quality control. PES-mediated inhibition of HSP70 activities impairs the autophagy-lysosome pathway, leading to an accumulation of misfolded and aggregated proteins (24). Given the differential interactions of PES with HSC70/HSP70 in non-transformed and transformed cells, we looked for evidence of altered autophagy in response to PES treatment. Macroautophagy, referred to here as autophagy, is an evolutionarily conserved degradative process in which long-lived proteins, organelles and aggregates are sequestered in double-membrane vesicles, called autophagosomes (26–29). Fusion of the autophagosomes with enzyme-containing lysosomes promotes degradation of the sequestered material. Basal levels of autophagy occur in all cells to promote survival, and this pathway is induced in response to many forms of stress. A sensitive marker of autophagic flux is the adaptor/scaffold protein p62 (also known as sequestosome1 {SQSTM1}, A170, or ZIP), a stress-induced protein implicated in many cellular pathways. Among its activities, p62/SQSTM1 regulates the formation and removal of intracellular aggregates. Because p62/SQSTM1 is degraded by autophagy, an inhibition of that pathway promotes the accumulation and oligomerization of p62/SQSTM1 (30–35). Another marker of autophagy, LC3 (microtubule-associated protein-1 light-chain 3) is converted from an 18 kDa cytoplasmic form (LC3-I) to a smaller (16 kDa) lipidated form (LC3-II), which integrates into newly formed autophagosome membranes along with p62/SQSTM1 (32–35). Accordingly, we used Western blot analysis to determine the effect of PES on p62/SQSTM1 and LC3-II expression. For these studies we used three immortalized human cell lines, namely IMR90 lung fibroblasts, HOI118 ovarian epithelial cells, and MCF10A mammary epithelial cells and three tumor cell lines, namely H1299, SKBR3 and MCF7 breast carcinoma. Untreated H1299, MCF7, and SKBR3 cells exhibit some level of p62 oligomerization, in accord with current evidence indicating that autophagy is induced in established tumors as a survival mechanism in the face of enhanced stress, including chemotherapy. Treatment with increasing concentrations of PES produced a notable dose-dependent increase in p62/SQSTM1 accumulation and oligomerization, as well as some LC3-II accumulation, in all three of these cell lines (Fig. 2C–2E), which is consistent with our previous results that PES impairs autophagic flux in solid tumor cells (24). In contrast, the immortalized IMR90, MCF10A and HOI118 cells displayed a very modest degree of p62 oligomerization and LC3-II accumulation, primarily at the higher PES concentrations employed (Fig. 2C–2E). Because a significant increase in p62/SQSTM1 levels is indicative of an accumulation of aggregated or undegraded proteins (31–35), the combined data reported here indicate that tumor cells are more sensitive than non-tumor cells to this consequence of PES treatment.
To date, there are very few reports comparing the effects of PES on normal, non-transformed and transformed cells, either in culture or in animal models cells. While normal T-lymphocytes and bone marrow hematopoietic progenitor cells have been found to be relatively resistant to PES, chronic lymphocytic leukemia (CLL) cells as well as normal B-cells are more sensitive to PES-mediated cell death (25, 36). During the course of the studies reported here, we noted that the immortalized MCF10A and HOI118 epithelial cells exhibited greater PES-mediated cytotoxicity than IMR90 or WI38 fibroblasts (ref. 24, Fig. 2, and data not shown). It is recognized that immortalized cell lines have genetic and/or epigenetic changes that permit long-term growth in culture, but these changes can complicate interpretation of cytotoxicity studies. For example, the spontaneously immortalized MCF10A cells are null for p15/p16 genes (CDKN2A and CDKN2B on chromosome 9p), a scenario often associated with loss of cell cycle control, and other genetic changes as well (37). The HOI118 cells were immortalized with SV40 T-antigen; HSP70 forms a complex with T-antigen and mediates some of its activities, including nuclear import (38). We have found that PES interferes with the interaction between T-antigen and HSP70 in cells (data not shown), most likely rendering T-antigen immortalized cells more sensitive to PES challenge; however, the specific pathways affected remain to be defined. Accordingly, future studies on PES will need to include a more broad-based assessment of any potential biological effects on normal cells of different histologic type.
PES Alters the Expression Pattern of HSP70/HSP90 Client Proteins
The HSP70 and HSC70 proteins bind to exposed hydrophobic regions of nascent-, translocating-, misfolded- or partially unfolded-proteins. These ATP-dependent molecular chaperones act in multi-protein complexes in conjunction with one or more regulatory co-chaperones, such as HSP40, CHIP and HSP90, to carry out multiple cellular functions. Our data show that, in a dose-dependent fashion, PES exposure results in a reduction in the cellular abundance of at least a subset of these HSP70/HSC70-containing complexes (24). This is illustrated for H1299 cells treated with 10 or 20 µM PES for 24 h. In this experiment, the latter concentration of PES results in lower expression levels of the client protein, integrin β1, as well as the co-chaperone CHIP (Fig. 3A). This correlates with a reduced abundance of detectable complexes containing HSP70, or HSC70, together with client proteins or co-chaperones (Fig. 3A and B). Under these same experimental conditions, PES does not alter the overall abundance of complexes containing HSP70 with its co-chaperones HSC70 or HSP90 (Fig. 3A and B). However, since HSP70/HSC70 act as critical co-chaperones for HSP90, for example in client protein recruitment, PES-mediated alteration in the function of the 70-kDa molecular chaperones could be reflected in an altered behavior or expression of many client proteins. To test this idea, we examined the expression of a few well-established HSP90 client proteins, including the transmembrane tyrosine kinase receptors EGFR and HER2/ERBB2, and the downstream signaling factor AKT. These proteins are overexpressed, or aberrantly expressed, in a variety of human tumors, and have been implicated in cancer etiology and pathology. As a control for these studies, we compared the effects of PES to that of the well-established HSP90 inhibitor, 17-allylamino-17-demethoxygeldanamycin (17-AAG). 17-AAG interacts with the N-terminal region of HSP90 and inhibits its ATPase activity; this leads to client-protein degradation via the ubiquitin-proteasome pathway (14, 15). To examine the expression of some HSP70/HSP90 clients, we used western blot analysis of detergent-soluble (clarified) whole cell extracts. As illustrated by an analysis of SKBR3 breast cancer cells, in the presence of PES, as with 17-AAG, there is a reduction in the overall cellular abundance of EGFR, HER2/ErbB2, and AKT (Fig. 3C). Co-immunoprecipitation assays confirmed that these HSP90 client proteins also are partners of HSP70/HSC70, and that treatment of cells with PES (20 µM) concomitantly reduces the abundance of complexes containing these tumor-promoting substrates and the molecular chaperones (Fig. 3C). Thus, by targeting HSP70/HSC70 functions, PES also impairs critical activities of HSP90.
Figure 3. PES attenuates HSP70, HSC70 and HSP90 interaction with co-chaperones and clients.
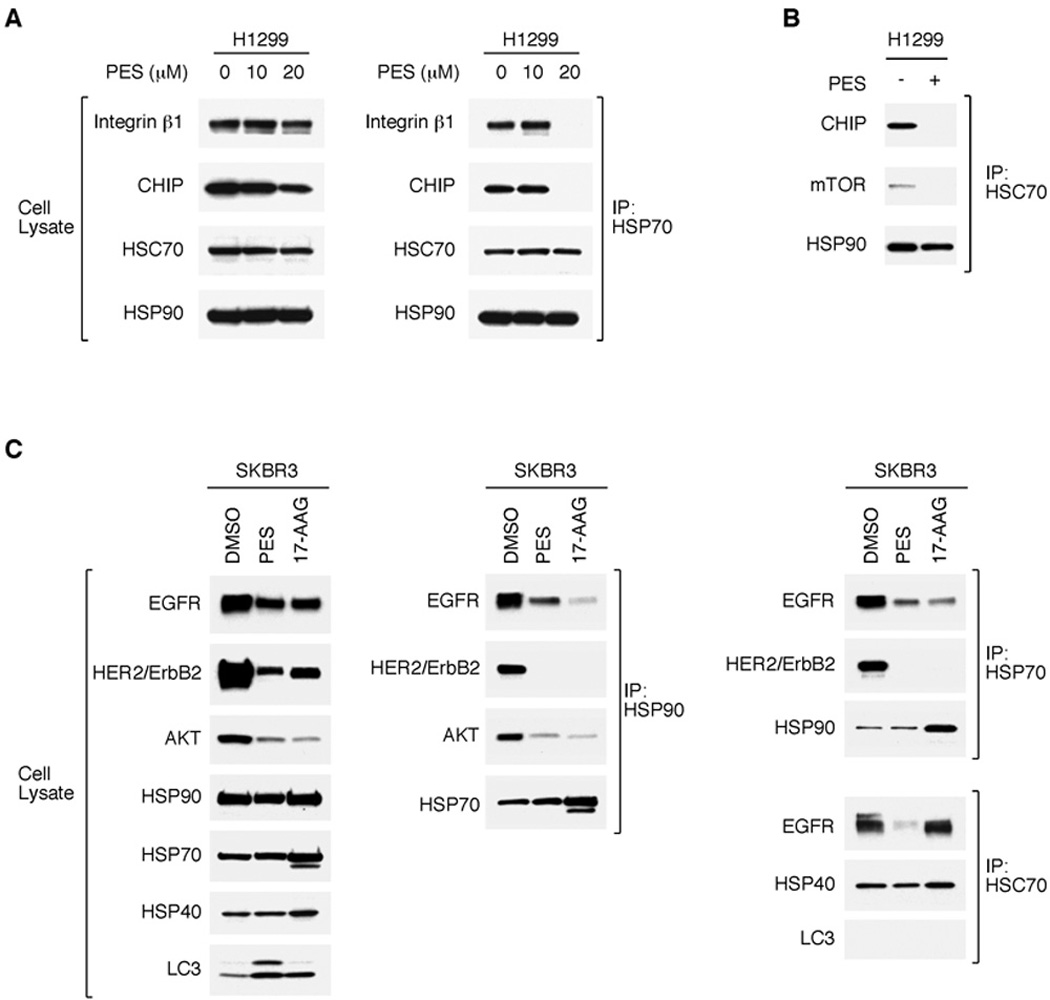
H1299 lung carcinoma cells (A and B) or SKBR3 breast carcinoma cells (C) were treated with DMSO, 20 µM PES, or 1 µM 17-AAG for 24 h. Western blots (WB) assessed the relative protein abundance. Coimmunoprecipitation-western (IP-WB) revealed a reduced degree of interaction between HSP90, HSC70, or HSP70 and the client proteins shown.
PES Interferes with Cellular Proteasome Activity
The autophagy-lysosome system is important for the degradation of long-lived proteins, organelles, and other cytoplasmic cargo, and it aids in the removal of oligomeric and aggregated proteins. The 26S-proteasome system is central to the controlled turnover of short-lived proteins, such as cell cycle regulatory factors, as well as certain misfolded proteins. Perturbing either system can result in the abnormal accumulation of potentially toxic protein aggregates. Recent studies indicate that the autophagy-lysosome and proteasome systems are mechanistically linked. Autophagy is frequently induced in response to proteasome inhibition, and an inhibition of autophagy, or the lysosome, can directly impair the function of the proteasome system, perhaps because the cellular abundance of misfolded or aggregated proteins exceeds the capacity of the system to process these molecules (39–43). Given the key role of HSP70/HSC70 in targeting misfolded proteins and aggregates for elimination through both of these proteolytic pathways, we extended our studies to assess the effect of PES on proteasome function. We employed a widely-used fluorogenic assay to measure the protease activity of the 20S core unit of the 26S proteasome. Equal numbers of H1299 human lung carcinoma cells or A875 human melanoma cells were treated for 24 h with each of the following agents: the HSP70-inhibitor, PES; the proteasome-inhibitor, bortezomib (also called Velcade or PS-341); (−)-epigallocatechin gallate (EGCG), a specific inhibitor of the 20S proteasome); the autophagy inhibitor chloroquine (CQ); the HSP90-inhibitor, 17-AAG. The results of these assays using two different tumor cell lines demonstrate that PES treatment impairs the activity of the 20S proteasome in vivo in a manner comparable to that of the known proteasome-inhibitors Velcade and EGCG (Fig. 4A). In contrast, 17-AAG and CQ have little, if any, effect (Fig. 4A). However, when PES was added directly to control cell lysates it did not inhibit proteasome activity (Fig. 4A). This is consistent with the idea that PES may impair the proteasome pathway indirectly through effects on protein aggregation.
Figure 4. PES inhibits the 20S proteasome system.
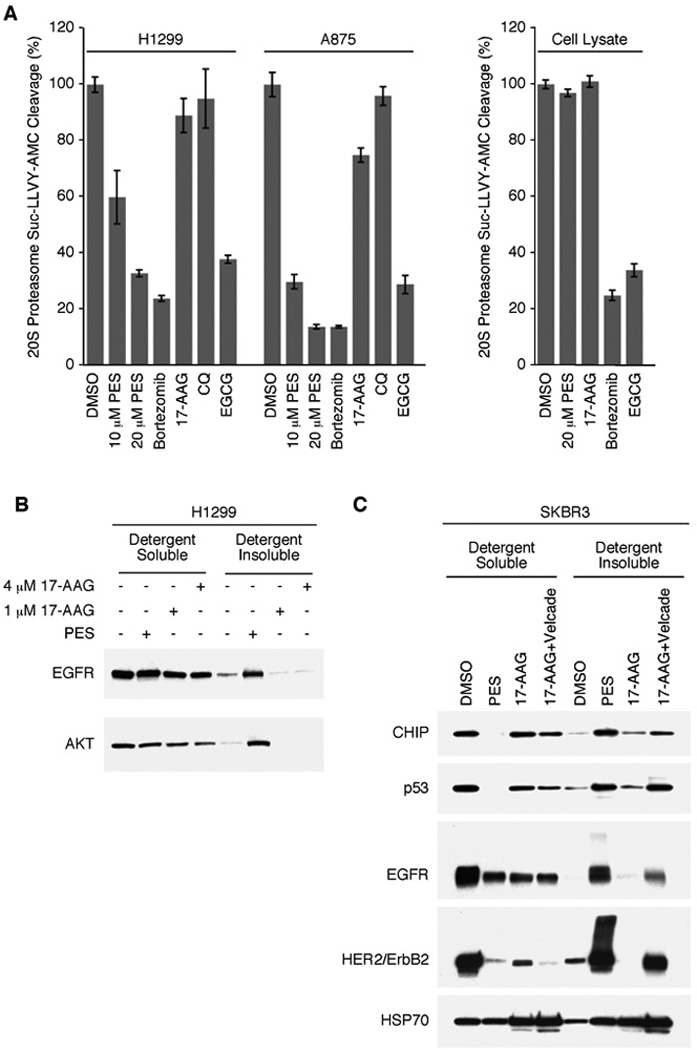
A, H1299 and A875 cells (5 × 103) were plated into 24-well assay plates in 0.5 ml media and allowed to attach overnight. Intact cells (left panel) or cell lysates (right panel) were then treated with DMSO, 20 µM PES, 100 nM bortezomib, 1 µM 17-AAG, 5 µM CQ, or EGCG, as indicated. Cells were then processed for measurement of 20S activity. Relative fluorescent intensity was then measured with a plate reader. B and C, H1299 cells (B) or SKBR3 cells (C) were treated with DMSO, 20 µM PES, 17-AAG, or 17-AAG plus bortezomib (Velcade) as indicated; harvested in 1% NP-40 containing lysis buffer; fractionated into detergent-soluble and detergent-insoluble preparations; and assayed by western blot.
PES Leads to an Accumulation of Proteins in the Detergent-Insoluble Fraction
When the proteasome and/or autophagy-lysosome pathways in cells are impaired, many cellular proteins exhibit increased oligomerization and/or aggregation. These aggregates tend to be poorly soluble, and accumulate in a detergent-insoluble cell fraction (39–43). Therefore, we asked about the fate of HSP70/HSP90 client proteins in PES treated cells. The results of western blot analyses show an increased abundance of EGFR, HER2/ErbB2, and AKT in the detergent-insoluble fraction, even under conditions where there is not an obvious decrease in protein abundance in the detergent-soluble fraction (clarified whole-cell lysates) (Fig. 4B). This may reflect an effect of PES on basal turnover of these proteins. Similar results were obtained when we examined PES-treated SKBR3 cells for the expression of several other proteins, including the HSP70/HSP90 client mutant p53 and the co-chaperone protein CHIP (Fig. 4C). The latter is a ubiquitin ligase that interacts with HSC70/HSP70 as well as HSP90 to promote the ubiquitination and degradation of client proteins, linking the chaperone and proteasome systems (42). In contrast to the effects of PES, 17-AAG causes proteasome-mediated degradation of HSP90 client proteins, as illustrated by client protein depletion (Fig. 4B and 4C). However, preventing the turnover of these substrates by co-treatment of tumor cell lines with an HSP90 inhibitor together with a proteasome-inhibitor leads to their accumulation as detergent-insoluble forms (41, 44). Accordingly, treating cells with PES alone produces an outcome that is similar to that obtained when using 17-AAG in combination with Velcade; namely, there is a reduced abundance of HSP70/HSC70/HSP90 client proteins in the detergent-soluble fraction and an increased abundance of detergent-insoluble forms (Fig. 4C).
The HSP70/HSP90 molecular chaperone machinery has a large number and wide variety of client proteins in tumor cells; thus, disrupting the activity of these pathways would be predicted to affect many cellular proteins, either directly or indirectly. Therefore, we compared the effects of PES and 17-AAG on the expression of several other cellular proteins in SKBR3 breast cancer, or SKOV3 ovarian cancer, cells. The data reveal that many of the proteins examined accumulate in the detergent-insoluble fraction following exposure of the cells to PES (Fig. 5A–5C). Among these are several chaperone proteins, such as HSP40, p23 and HOP, the signaling factor p62/SQSTM1 and the NFkB inhibitor IkBα (Fig. 5A–5C). Thus, one important consequence of PES treatment includes the aggregation of a large number of different proteins, likely associated with chaperones. Accordingly, short-lived proteins such as the apoptosis regulators MCL1 and p53 may accumulate in the detergent insoluble fraction because proteasome activity is compromised. It is reasonable to speculate that the enrichment of a broad range of cellular proteins in the insoluble cell fraction indicates the formation of aggresomes or aggregates, likely in an unsuccessful effort by the molecular chaperones and the protein quality control machinery to rid the cell of misfolded, aggregated, or unwanted macromolecules. Indeed, both p62/SQSTM1 and HSP70 are among a large group of proteins found to be enriched in low-solubility aggregates after proteasome inhibition (45).
Figure 5. Accumulation of HSP90 client proteins in the protein detergent-insoluble fraction following PES.
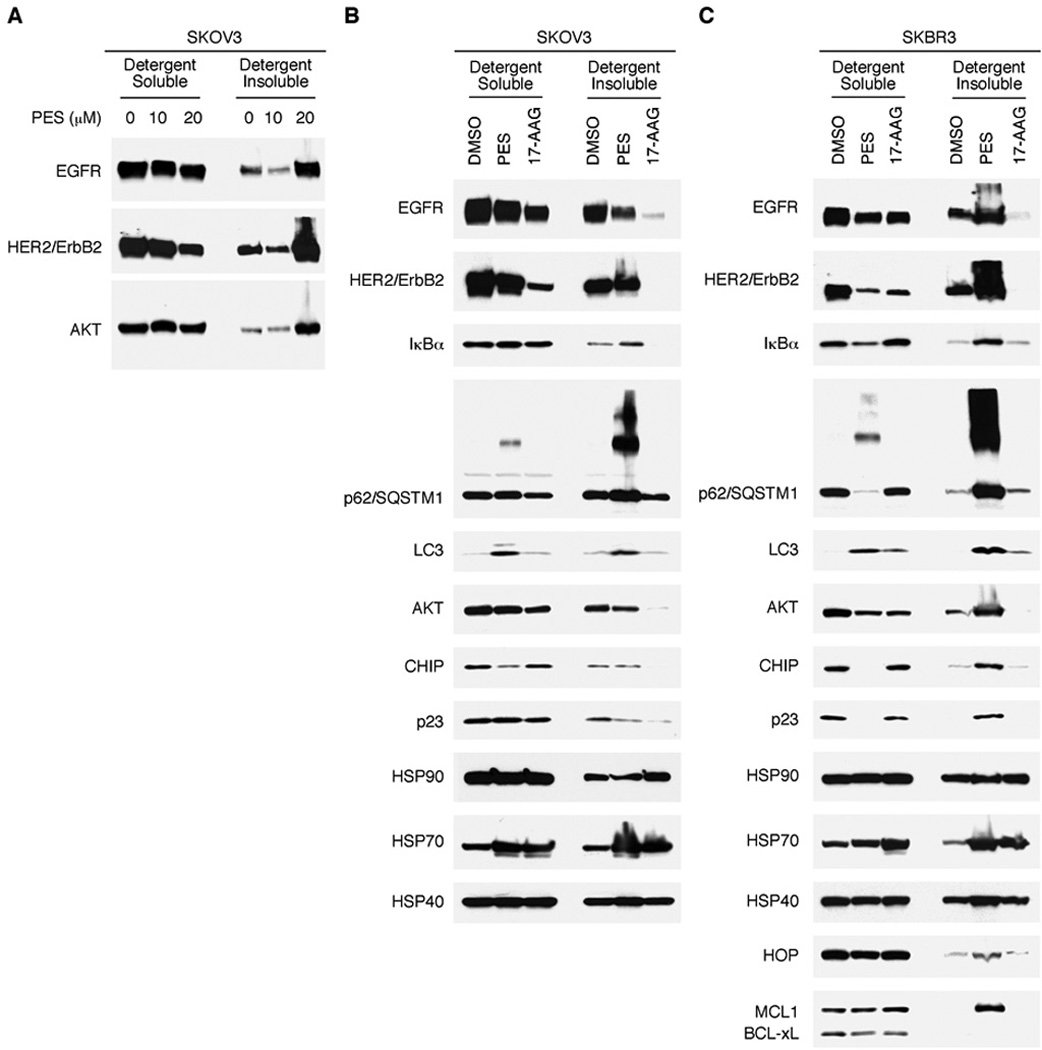
A, SKOV3 cells were treated with DMSO, 10 µM PES, or 20 µM PES for 24 h. Note the marked accumulation of EGFR, HER2/ErbB2, and AKT in the detergent-insoluble fraction following 20 µM PES treatment. B and C, SKOV3 (B) or SKBR3 (C) cells were treated with DMSO, 20 µM PES, or 1 µM 17-AAG, as indicated, for 24 h. Cells were harvested in 1% NP-40 containing lysis buffer, fractionated into detergent-soluble and detergent-insoluble preparations, and assayed by western blot for the proteins indicated.
PES Affects p62/SQSTM1 Expression In Vivo
Using the Eµ-Myc mouse model of B-cell lymphoma, we previously reported that PES treatment effectively suppresses tumor development in vivo (24). To assess whether PES administration leads to impaired autophagy in vivo, we utilized nude mice bearing a human head and neck cancer (FaDu) xenograft. When tumors reached a palpable size (approximately 5–7 mm), they were treated with PES and then harvested 24 or 48 hr later. Tumor lysates were separated into detergent-soluble and detergent-insoluble fractions, and these were examined by western blot analysis for p62/SQSTM1 and LC3 expression. Several lines of evidence presented here and previously (24) indicate that an accumulation and oligomerization of p62/SQSTM1 represents a sensitive molecular signature for PES-mediated HSP70 inhibition and, thus, may serve as a potential biomarker of PES efficacy in vivo. Similar to results obtained with cultured FaDu cells (Fig. 6A), western blot analysis of the tumor xenografts revealed increased p62/SQSMT1 abundance and oligomerization in both detergent-soluble and -insoluble fractions at both 24 and 48 hr following PES administration (Fig. 6B). The western blots also provided evidence of accumulated LC3, EGFR and AKT in the detergent-insoluble fractions in the PES-treated tumors. Thus, a major biological effect of PES treatment in cultured tumor cells is recapitulated in vivo. Together, these results help lay the foundation for ongoing development of PES derivatives and other HSP70 inhibitors for clinical use.
Figure 6. PES enhances p62/SQSTM1 accumulation in xenograft tumors.
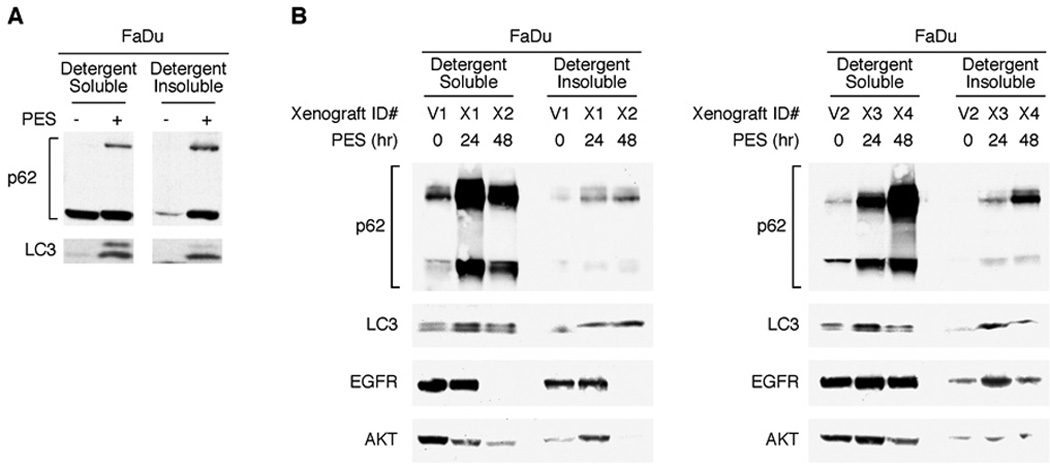
A, Immunoblots showing p62/SQSTM1 oligomerization and LC3-II processing in FaDu cells in culture. B, Nude mice bearing FaDu tumor xenografts were dosed by intratumoral administration with either vehicle for 48 h, or with 40 mg/kg of PES for either 24 or 48 h, as indicated. The dissected tumors were homogenized in 1% NP-40 containing lysis buffer, fractionated into detergent-soluble and detergent-insoluble fractions, and immunoblotted for either p62, LC3, EGFR or AKT. Representative results for two xenografts per treatment condition and time point are presented.
DISCUSSION
Increasing evidence points to the multi-functional HSP70 molecular chaperone as a key factor in the ability of cancer cells to survive the potentially toxic effects of enhanced proteotoxic stress. This stress-induced factor helps to maintain the function and/or stability of a large number of client proteins, including many oncogenes and signaling factors that directly contribute to a tumorigenic phenotype. The close functional interaction among various molecular chaperones and with systems that maintain protein homeostasis helps to explain how disrupting the function of HSP70 simultaneously impacts multiple signaling pathways that sustain cancer cells. Thus, targeting the actions of HSP70 offers a promising therapeutic approach for cancer treatment. PES is a recently identified small molecule that selectively interacts with the closely related HSP70 and HSC70 proteins and impairs their actions.
The autophagy-lysosome system and the proteasome machinery represent the two major routes used by mammalian cells for protein degradation and organelle clearance. One important consequence of PES treatment in solid tumors is a disruption of both of these degradative systems, as shown here and in our previous work (24). As a consequence, potentially toxic misfolded, aggregated, or otherwise altered proteins accumulate in the cell. Among the proteins affected by PES treatment is p62/SQSTM1. The p62/SQSTM1 adapter scaffold protein participates in, and is degraded by, autophagy; however, when autophagy is inhibited this protein accumulates and oligomerizes. Interestingly, overexpression of p62 also can have an inhibitory effect on proteasome function (34), which would be expected to contribute to the cytotoxic effects of PES. A disruption of both the autophagy-lysosome and proteasome systems likely impacts a range of cell signaling pathways that depend on the appropriate expression and activity of both long-lived and short-lived regulatory proteins (39–43). PES promotes an accumulation and oligomerization of p62/SQSTM1 in a time-dependent and dosage-dependent manner (see for example Fig. 2C–2E and ref. 24). Indeed, our investigations suggest that an accumulation of p62/SQSTM1 represents a sensitive molecular signature for PES-mediated alterations in protein quality control pathways.
Like other members of the 70-kDa family of molecular chaperones, HSP70 and HSC70 are ATPases comprised of two major domains, an N-terminal adenine-nucleotide binding domain (NBD or ATPase domain) and a C-terminal substrate/client binding domain (SBD) (5, 7, 8). A conformational change in structure accompanies cycles of substrate binding and release. The HSP70 and HSC70 proteins dynamically interact with a large and diverse array of substrate proteins via the SBD, and with co-chaperones via both the N-terminal and C-terminal regions. Co-chaperone partners, such as HSP90, CHIP and HSP40 are essential in carrying out HSP70/HSC70 activities, such as client protein selection, allosteric cycling and client processing (5, 7, 8). Although the broad outlines of HSP70/HSC70 actions have been determined, key questions remain about the molecular mechanisms involved in these activities. These include how critical conformational changes in HSP70 are modulated and how specific client protein selection, binding, and fate are determined. Given that HSP70/HSC70 participate in many cellular pathways, selective binding of PES to the C-terminal domain of the proteins may alter cellular homeostasis in many ways. PES may promote a dissociation of chaperone complexes and/or prevent the release of client proteins from the complex, thereby preventing the proteins from achieving stable conformation and participating in cellular processes. It is likely that more than one pathway of PES-mediated HSP70/HSC70 inhibition may be operative in the cell, and future studies will be important in clarifying the way in which PES alters chaperone activities.
A critical function of HSP70 and HSC70 involves delivery of clients to the HSP90 chaperone machinery. As assayed by immunoprecipitation-Western blotting, the interactions of HSP70 with HSC70 and with HSP90 are not detectably impaired by PES. Following treatment of cells with different PES concentrations, we noted that the abundance of protein complexes containing these molecular chaperones with certain client proteins is not significantly changed until higher concentrations of PES are employed (Fig. 3). However, some cellular proteins may be more sensitive to HSP70 inhibition than others, and this may also vary among tumor cell types. In either case, a reduction in the overall abundance of chaperone complexes tends to correlate with the accumulation of various cellular proteins in the detergent insoluble cellular fraction, indicative of aggregation (Fig. 4 and 5). These data suggest that when protein turnover is sufficiently impaired, a significant fraction of proteins become denatured or aggregated and subsequently are relocated, together with accompanying molecular chaperones, into the detergent-insoluble fraction perhaps as a means of disposal. If not resolved, the continued accumulation of aggregated macromolecules in the detergent-soluble and detergent-insoluble cellular compartments would result in cell death. Accordingly, PES-mediated inhibition of HSP70/HSC70 results in impaired autophagy-lysosome and proteasome systems, thereby promoting the simultaneous disruption of many tumor-promoting signaling pathways.
In a recent seminal study, Powers and colleagues (18, 46), investigated the effects on HSP90 resulting from the expression knockdown of HSP70, HSC70 or both. Using an siRNA gene silencing approach, they determined that simultaneous silencing of both HSP70 and HSC70 was needed to inhibit proliferation and to induce apoptosis in a consistent manner (18, 46). Although they are highly homologous structurally and functionally, the activities of HSP70 and HSC70 are not altogether equivalent. Some of the observed functional differences between these proteins relate to different interactions with some co-chaperones or substrates (47, 48). Moreover, in certain cancer cell lines, sensitivity to HSP90 inhibitors can be enhanced by the silencing of HSP70, but not of HSC70 (46). The knockdown of expression of HSP70, HSC70, or both does not impair cellular proteasome activity (46). Indeed, studies have shown that the dual knockdown of both HSP70 and HSC70 expression, or the use of an adenosine-derived inhibitor that targets the ATPase domain of these proteins, promotes HSP70/HSC70/HSP90 client proteins for proteasomal degradation (22, 46). This outcome is similar to that achieved by targeting HSP90 with pharmacologic inhibitors like 17-AAG. However, previous work has shown that the simultaneous silencing of HSP70 and HSC70 in non-tumorigenic cell lines does not produce the anti-proliferative or apoptotic effect noted in tumor cells (46). This observation is consistent with the idea that tumor cells seem to be much more dependent on the actions of molecular chaperones in order to cope with constitutively enhanced proteotoxic and physiologic stress associated with tumorigenesis. While certain levels of HSP70/HSC70 inhibitors may be toxic to normal cells, studies on cultured cells and an in vivo Eµ-myc animal tumor model indicate that differential responses to PES between normal and tumor cells can be achieved (24, 25, 36). It should also be noted that, taken together, the data obtained to date indicate that PES-mediated HSP70/HSC70 inhibition may promote a "gain-of-function" phenotype distinct from that achieved by expression silencing of these proteins. Future biophysical studies should provide needed insight regarding the molecular mechanisms by which PES interferes with HSP70/HSC70 actions.
The development of new agents and new approaches to therapy are needed to improve the survival of cancer patients. An inhibition of autophagy and/or of the proteasome is being considered for the treatment of some tumors, including those that are resistant to conventional therapies (49–51). Also, recent studies show that a combination treatment of an HSP90 inhibitor together with a proteasome inhibitor is more effective at killing cultured tumor cells when compared to using either alone (41, 44). The small molecule PES represents a novel selective inhibitor of the HSP70 family proteins that leads to a dysfunctional molecular chaperone machinery and impairs protein homeostasis. These findings support the further development of compounds for targeting HSP70 in human malignancies. The small-molecule PES also represents a valuable new tool to interrogate the critical functions of HSP70 proteins in normal biology and in various pathologies.
Acknowledgments
Grant support
This work was supported by National Institutes of Health (NIH) grants RO1CA118761 (D. George), KO1DK078025 (J. Leu) and RO1CA139319 (M. Murphy). This project also was funded in part under a grant with the Pennsylvania Department of Health (M. Murphy). The Department specifically disclaims responsibility for any analyses, interpretations, or conclusions.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
REFERENCES
- 1.Gregersen N. Protein misfolding disorders: pathogenesis and intervention. J Inherit Metab Dis. 2006;29:456–470. doi: 10.1007/s10545-006-0301-4. [DOI] [PubMed] [Google Scholar]
- 2.Kubota H. Quality control against misfolded proteins in the cytosol: a network for cell survival. J Biochem. 2009;146:609–616. doi: 10.1093/jb/mvp139. [DOI] [PubMed] [Google Scholar]
- 3.Wong E, Cuervo AM. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol. 2010 doi: 10.1101/cshperspect.a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mosser DD, Morimoto RI. Molecular chaperones and the stress of oncogenesis. Oncogene. 2004;23:2907–2918. doi: 10.1038/sj.onc.1207529. [DOI] [PubMed] [Google Scholar]
- 5.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31:164–172. doi: 10.1016/j.tibs.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Rohde M, Daugaard M, Jensen MH, Helin K, Nylandsted J, Jäättelä M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005;19:570–582. doi: 10.1101/gad.305405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daugaard M, Rohde M, Jäättelä M. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 9.Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6:79–87. doi: 10.1038/nrm1552. [DOI] [PubMed] [Google Scholar]
- 10.Powers MV, Workman P. Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett. 2007;581:3758–3769. doi: 10.1016/j.febslet.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 11.Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Høyer-Hansen, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200:425–435. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirkegaard T, Roth AG, Petersen NHT, Mahalka AK, Olsen OD, Moilanen I, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463:549–553. doi: 10.1038/nature08710. [DOI] [PubMed] [Google Scholar]
- 13.Esser C, Alberti S, Höhfeld J. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim Biophys Acta. 2004;1695:171–188. doi: 10.1016/j.bbamcr.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 14.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 15.Neckers L. Heat shock protein 90: the cancer chaperone. J Biosci. 2007;32:517–530. doi: 10.1007/s12038-007-0051-y. [DOI] [PubMed] [Google Scholar]
- 16.Pearl LH, Prodromou C, Workman P. The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem J. 2008;410:439–453. doi: 10.1042/BJ20071640. [DOI] [PubMed] [Google Scholar]
- 17.Brodsky JL, Chiosis G. Hsp70 molecular chaperones: emerging roles in human disease and identification of small molecule modulators. Curr Top Med Chem. 2006;6:1215–1225. doi: 10.2174/156802606777811997. [DOI] [PubMed] [Google Scholar]
- 18.Powers MV, Clarke PA, Workman P. Death by chaperone: HSP90, HSP70 or both? Cell Cycle. 2009;8:518–526. doi: 10.4161/cc.8.4.7583. [DOI] [PubMed] [Google Scholar]
- 19.Evans CG, Chang L, Gestwicki JE. Heat shock protein 70 (hsp70) as an emerging drug target. J Med Chem. 2010;53:4585–4602. doi: 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jego G, Hazoume A, Seigneuric R, Garrido C. Targeting heat shock proteins in cancer. Cancer Lett. 2010 doi: 10.1016/j.canlet.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 21.Massey AJ. ATPases as drug targets: insights from heat shock proteins 70 and 90. J Med Chem. 2010;53:7280–7286. doi: 10.1021/jm100342z. [DOI] [PubMed] [Google Scholar]
- 22.Massey AJ, Williamson DS, Browne H, Murray JB, Dokurno P, Shaw T, et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother Pharmacol. 2010;66:535–545. doi: 10.1007/s00280-009-1194-3. [DOI] [PubMed] [Google Scholar]
- 23.Rerole A-L, Gobbo J, De Thonel A, Schmitt E, Pais de Barros JP, Hammann A, Lanneau D, et al. Peptides and aptamers targeting HSP70: a novel approach to anticancer chemotherapy. Cancer Res. 2011;71:484–495. doi: 10.1158/0008-5472.CAN-10-1443. [DOI] [PubMed] [Google Scholar]
- 24.Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I, et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat Chem Biol. 2006;2:474–476. doi: 10.1038/nchembio809. [DOI] [PubMed] [Google Scholar]
- 26.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20:748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 29.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 32.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 33.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, et al. Structural basis for sorting mechanism of p62/SQSTM1 in selective autophagy. J Biol Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 36.Steele AJ, Prentice AG, Hoffbrand AV, Yogashangary BC, Hart SM, Lowdell MW, et al. 2-Phenylacetylenesulfonamide (PAS) induces p53-independent apoptotic killing of B-chronic lymphocytic leukemia (CLL) cells. Blood. 2009;114:1217–1225. doi: 10.1182/blood-2008-11-190587. [DOI] [PubMed] [Google Scholar]
- 37.Cowell JK, LaDuca J, Rossi MR, Burkhardt T, Nowak NJ, Matsui S. Molecular characterization of the t(3;9) associated with immortalization in the MCF10A cell line. Cancer Genet Cytogenet. 2005;163:23–29. doi: 10.1016/j.cancergencyto.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y, DeFranco DB. Differential roles of heat shock protein 70 in the in vitro nuclear import of glucocorticoid receptor and simian virus 40 large tumor antigen. Mol Cell Biol. 1994;14:5088–5098. doi: 10.1128/mcb.14.8.5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. 2005;17:351–365. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 40.Ding WX, Ni HM, Gao W, Chen X, Kang JH, Stolz DB, et al. Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Mol Cancer Ther. 2009;8:2036–2045. doi: 10.1158/1535-7163.MCT-08-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mimnaugh EG, Xu W, Vos M, Yuan X, Neckers L. Endoplasmic reticulum vacuolization and valosin-containing protein relocalization result from simultaneous Hsp90 inhibition by geldanamycin and proteasome inhibition by Velcade. Mol Cancer Res. 2006;4:6676–6681. doi: 10.1158/1541-7786.MCR-06-0019. [DOI] [PubMed] [Google Scholar]
- 42.McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qiao L, Zhang J. Inhibition of lysosome functions reduces proteasomal activity. Neuroscience Letters. 2009;456:15–19. doi: 10.1016/j.neulet.2009.03.085. [DOI] [PubMed] [Google Scholar]
- 44.Mimnaugh EG, Xu W, Vos M, Yuan X, Isaacs JS, Bisht KS, et al. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol Cancer Ther. 2004;3:551–566. [PubMed] [Google Scholar]
- 45.Wilde IB, Brack M, Winget JM, Mayor T. Proteomic characterization of aggregating proteins after the inhibition of the ubiquitin proteasome system. J Proteome Res. 2011;10:1062–1072. doi: 10.1021/pr1008543. [DOI] [PubMed] [Google Scholar]
- 46.Powers MV, Clarke PA, Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell. 2008;14:250–262. doi: 10.1016/j.ccr.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 47.Goldfarb SB, Kashlan OB, Watkins JN, Suaud L, Yan W, Kleyman TR, Rubenstein RC. Differential effects of Hsc70 and Hsp70 on the intracellular trafficking and functional expression of epithelial sodium channels. Proc Natl Acad Sci USA. 2006;103:5817–5822. doi: 10.1073/pnas.0507903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tutar Y, Song Y, Masison DC. Primate chaperones Hsc70 (constitutive) and Hsp70 (induced) differ functionally in supporting growth and prion propagation in Saccharomyces cerevisiae. Genetics. 2006;172:851–861. doi: 10.1534/genetics.105.048926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams J. The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004;5:417–421. doi: 10.1016/s1535-6108(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 50.Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest. 2007;117:326–336. doi: 10.1172/JCI28833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu K, Dunner K, Jr, McConkey J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene. 2010;29:451–462. doi: 10.1038/onc.2009.343. [DOI] [PMC free article] [PubMed] [Google Scholar]